
Abstract
Magnesium (Mg) plays a pivotal role as an essential component of plant chlorophyll and functions as a critical coenzyme. However, research exploring the regulatory mechanisms of magnesium ions on the synthesis of secondary metabolites is still in its early stages. Sophora tonkinensis is a widely utilized medicinal plant in China, recognized for its diverse secondary metabolites with active properties. This study investigates variations in these ingredients in tissue-cultured seedlings under varying magnesium concentrations. Simultaneously, an omics data analysis was conducted on tissue-cultured seedlings subjected to treatments with magnesium and low magnesium. These comprehensive omics analyses aimed to elucidate the mechanisms through which magnesium influences active components, growth, and development. Magnesium exerts a pervasive influence on various metabolic pathways, forming an intricate network. Research findings indicate that magnesium impacts diverse metabolic processes, including the absorption of potassium and calcium, as well as photosynthetic activity. Consequently, these influences lead to discernible changes in the levels of pharmacologically active compounds and the growth and developmental status.This study is the first to employ a multi-omics data analysis in S. tonkinensis. This methodology allows us to uncover the overarching impact of metabolic networks on the levels of various active ingredients and specific phenotypes.
Similar content being viewed by others

Introduction
Magnesium is an essential element for plant growth and development. Numerous studies have demonstrated its pivotal roles in various physiological processes, including photosynthesis, protein synthesis, chlorophyll biosynthesis, phloem loading, and reactive oxygen species (ROS) generation. Additionally, magnesium serves as a critical coenzyme, playing an irreplaceable role in overall plant metabolism1. Although magnesium plays a crucial role in plant physiology, research on magnesium nutrition in plant species commenced relatively late compared with that on other essential elements2. In recent years, it has garnered increasing attention, leading to many findings regarding the relationship between magnesium and secondary metabolism. A magnesium deficiency can increase cardenolide synthesis in callus cultures of various Digitalis plant species3. Magnesium also influences the distribution of secondary metabolites in tomato (Solanum lycopersicum) plants4. Moreover, magnesium influences tea (Camellia sinensis) metabolism and can impact the contents of secondary metabolites5,6,7. In recent years, various studies have explored the impact of magnesium on the active ingredients of medicinal plants. For example, these studies have investigated the influence of magnesium fertilizer on the total flavonoid content in Phyllanthus niruri8, the effects of different magnesium concentrations on the composition of Morus alba9, and the consequences of excessive magnesium on the components of Perilla frutescens10. Given that metabolism operates as an interconnected network in plants, alterations in specific metabolic nodes may influence downstream metabolite content through network transmission. Consequently, attributing changes in product levels solely to variations in a single related metabolite enzyme-enccoding gene can be challenging; hence, certain traditional genetic methods have limitations when studying secondary metabolism in plants11.
Sophora. tonkinensis is a plant indigenous to the region at the junction of China and the Indochina Peninsula and is typically found in tropical northern areas and nearby subtropical karst landscapes. This plant has a rich history of medicinal use, and recent research has revealed its abundant active ingredients, with a particular focus on compounds such as matrine12, oxymatrine13, maackiain14, and trifolirhizin15. Among them, matrine and oxymatrine are prototypical quinolizidine alkaloids. These two alkaloids are widely distributed within the roots of Sophora plants and have been the subject of extensive research. They exhibit a wide array of pharmacological activities, including anti-inflammatory, antitumor, antiallergic, antiviral, and cardiovascular protective effects16. Maackiain, classified as a pterocarpan flavonoid, is predominantly found in leguminous plants. Research has demonstrated its multifaceted pharmacological properties, including antitumor, antibacterial, and antiallergic activities17. Trifolirhizin, another pterocarpan flavonoid, has been the subject of pertinent research, revealing its antitumor and anti-inflammatory properties18. The successful cultivation of tissue culture-generated seedlings of S. tonkinensis has provided an opportunity to examine the factors influencing the presence of these pharmacological components in a controlled environment. Given that magnesium is a crucial nutrient element, its potential influence on the accumulation of these active components in S.tonkinensis tissue culture-generated seedlings is of interest. Consequently, this project introduced varying concentrations of magnesium to tissue culture-generated seedlings to assess its impact on their growth, development, and the contents of these active components.
Over the past two to three years, several studies have reported the utilization of multi-omics methods to analyze the molecular mechanisms associated with the active components of medicinal plants19,20,21,22. Hence, multi-omics analysis can be an effective approach for assessing the influence of magnesium on the active components of S.tonkinensis. To elucidate the molecular mechanisms underlying the impact of magnesium on the active ingredient content and related phenotypes in S. tonkinensis, this project employed a comprehensive multi-omics approach. The project selected plants subjected to specific magnesium concentrations and gathered transcriptome, proteome, and metabolomic data from these samples, whereas a control group received no additional magnesium supplementation.
Materials and methods
Materials
The tissue culture seedlings of Sophora tonkinensis used in this study were sourced from the in vitro conservation collection at the Medicinal Botanic Garden. These specimens were derived from wild populations collected in the Jingxi Mountainous Area of Guangxi Zhuang Autonomous Region in 2016. The voucher specimen of S. tonkinensis is housed within the Conservation Center of the Botanic Garden, with the specimen number JX00763. Species identification was carried out by the corresponding author, Wei Kunhua (Tables 1, 2).
Mg treatment and experimental setup
Tissue culture seedlings of S. tonkinensis were selected from the in vitro library. Stem segments, approximately 2–3 cm in length, were obtained from these seedlings and propagated in a new medium (MS + 5.0 mg L− 1 6-BA + 0.3 mg L− 1 IAA + 0.3 mg L− 1 KT). Once the explants produced numerous clustered buds, regenerated buds were transferred to a robust seedling culture medium (MS + 1.0 mg L− 1 IAA) for further cultivation. After the tissue culture seedlings had lignified sufficiently, those with a high lignification degree were chosen. The top stem segments, including leaves and measuring 2–3 cm in length, were selected and inoculated into media with varying magnesium ion concentrations for rooting. Each bottle contained 8 explants, and 30 bottles per treatment were used. Six treatments were designated as T0, T0.5, T1, T2, T3, and T4. Building on existing methods for regulating magnesium levels in tissue culture experiments23,24, this study employs varying concentrations of magnesium sulfate to modulate magnesium levels. The magnesium concentration for each treatment is listed in Table 3, and the experiment extended over 60 days. The medium for root culture was supplemented with sucrose (final concentration 30 g L− 1) in addition to the culture medium for promoting the strong seedling. The medium contained 3.4 g/L of agar with a pH = 5.8. After inoculation, the cultures were maintained at a temperature of 25 ℃, with a light intensity of 1500–2000 lx and 12-hours’ light per day. After 60 days of rooting culture, the materials were collected for phenotype determination, biochemical analyses, and omics data collection.
Phenotype determination
Collect materials and measure various parameters of tissue-cultured seedlings, including plant height, stem diameter, leaf count, rooting rate, root length, and root dry weight.
-
Plant Height: Measure the maximum height of the main stem using a ruler.
-
Stem Diameter: Determine the diameter of the lower stem with a vernier caliper.
-
Leaf Count: Count the number of proliferating leaves.
-
Rooting Rate: Calculate the rooting rate as the number of rooting explants divided by the number of inoculated explants, multiplied by 100%.
-
Root Length: Measure root length with a ruler.
-
Root Dry Weight: After collecting S. tonkinensis roots, clean them with water, and then oven-dry until a constant weight is reached. Weigh the roots.
Determination of active ingredient content
Determination of matrine and oxymatrine
The methods for measuring the matrine and oxymatrine content followed the Pharmacopoeia of the People’s Republic of China version 2020 .
Extraction Method: 0.1 g of the sample was mixed with 2 mL of the extraction solution (methylene chloride: methanol: ammonia solution in a ratio of 40:10:1). The mixture was kept at room temperature for 30 min, followed by 30 min of sonication. Afterward, it was centrifuged at 4000 rpm for 10 min at room temperature. After centrifugation, 1 mL of the resulting supernatant was dried under nitrogen at 40 °C, followed by reconstitution with 1 mL of methanol. The solution was subsequently filtered through a 0.22 μm syringe filter before analysis.HPLC conditions: HPLC equipment - Shimadzu LC-2030 Plus(Shimadzu, Kyoto, Japan). The Chromatographic column - Agilent Polaris 5 NH2 (5 μm, 250 × 4.6 mm). The mobile phase consisted of acetonitrile-isopropanol-3% phosphoric acid solution (in a ratio of 80:5:15). The injection volume was 5 µL with a flow rate was 0.5 mL/min, and the column temperature was maintained at 25 °C. Detection was performed at a wavelength of 210 nm.
Determination of maackiain and trifolirhizin
Extraction Method: Approximately 0.1 g of the sample was weighed and mixed with 1 mL of methanol, followed by thorough grinding. The mixture was then subjected to 50 min of ultrasonication. The volume was adjusted to its original size using methanol. After centrifugation at 4000 rpm for 10 min, the supernatant was collected and filtered through a 0.22 μm membrane.
HPLC Conditions: HPLC equipment - Shimadzu LC-2030 Plus(Shimadzu, Kyoto, Japan). (Shimadzu, Kyoto, Japan). The Chromatographic column - Agilent Plus C18 column (5 μm, 250 × 4.6 mm). The mobile phase consisted of acetonitrile and water with a gradient program as follows: The gradient program consisted of the following stages: starting with 25% acetonitrile for 5 min, it was followed by a gradual increase to 50% acetonitrile for 50 min, then a further rise to 95% acetonitrile for 25 min, maintaining 95% acetonitrile for 6 min, returning to the initial 25% acetonitrile for 1 min, and finally equilibrating at this concentration for 5 min. Detection occurred at a wavelength of 205 nm, using a 10 µL injection volume, a flow rate of 1 mL/min, and a column temperature of 30 °C. Detection was carried out at a wavelength of 205 nm. The injection volume was 10 µL, the flow rate was 1 mL/min, and the column temperature was set at 30 °C.
Collecting omics data
Given that only the T2 treatment was able to elevate the content of maackiaian, it was chosen alongside the T0 treatment for the collection of omics data.Each treatment has three replicates. Moreover, the samples were also used for transcriptomic sequencing and proteomic data collection.
Transcriptomic data collection and analysis
Materials were frozen using liquid nitrogen and then transported to the MajorBio (Shanghai, China) for transcriptomesequencing. Transcriptome sequencing was performed using the Illumina Novaseq 6000 platform (San Diego, USA).
The raw paired end reads were processed using fastp25 with default parameters. Then clean reads were separately aligned to the reference genome of S. tonkinensis with orientation mode using HISAT225 software. The transcriptome reference genome data for S.tonkinensis (unpublished) was supplied by the Botanical Garden. The mapped reads for each sample were assembled by StringTie25 in a reference-based approach.
To identify DEGs (differential expression genes), the expression level of each transcript was calculated by the transcripts per million reads (TPM) using RSEM25. Essentially, differential expression analysis was performed using the DESeq226. DEGs with |log2FC|≧1 and FDR ≤ 0.05(DESeq2 ) were considered to be significantly different expressed genes. Furthermore, functional enrichment analysis, incorporating GO and KEGG, was conducted to pinpoint the DEGs significantly enriched in GO terms and metabolic pathways, with a Bonferroni-corrected P-value threshold of ≤ 0.05 compared to the entire transcriptome dataset.GO and KEGG pathway analyses were carried out using Goatools and KOBAS27, respectively.
Proteomic data collection and analysis
Materials were frozen in liquid nitrogen and then transported to MajorBio (Shanghai, China) for proteomic data collection. Trypsin-digested peptides were analyzed by an EASY nLC-1200 system (Thermo, USA) coupled with a timsTOF Pro2 (Bruker, Germany) mass spectrometer. Briefly, the C18-reversed phase column (75 μm x 25 cm, Ionopticks, USA) as equilibrated with solvent A (A:2% ACN with 0.1% formic acid) and solvent B (B: 80% ACN with 0.1% formic acid).The peptides were eluted using the following gradient: 0–45 min, 3-28% B; 45–50 min, 28−44%B; 50–55 min, 44−90% B; 55–60 min,90%−90% B. The tryptic peptides were separated at a flow rate of 250 nL/min. Peptides were separated by an ultrahigh performance liquid phase system subjected to a capillary ion source and then analyzed by timsTOF Pro2 (Bruker, Germany) ; the electrospray voltage was 1.5 kV. The peptide parent ions and their secondary fragments were detected and analyzed using high-resolution TOF. The secondary MS scanning range was 100–1700 m/z. Data acquisition on the timsTOF Pro2 was collected using the parallel accumulation serial fragmentation (PASEF) acquisition mode. After the first MS stage, the second MS stage (charge number of the parent ions was 0–5) was recorded using the 10 PASEF mode. A dynamic exclusion time of 24 s was used for the MS/MS scan.
MS/MS spectra were searched using MaxQuant version 2.0.3.1 software against a self-built database using genomic data constructed by MajorBio. The highest score for a given peptide mass (best match to that predicted in the database) was used to identify parent proteins. The parameters for protein searching were set as follows: tryptic digestion with up to two missed cleavages, carbamidomethylation of cysteines as fixed modification, and oxidation of methionines and protein N-terminal acetylation as variable modifications. False discovery rate (FDR) of peptide identification was set as FDR ≤ 0.01. A minimum of one unique peptide identification was used to support protein identification.
Bioinformatic analysis of proteomic data was performed with the Majorbio Cloud platform (https://cloud.majorbio.com)28. P-values and Fold change (FC) for the proteins between the two groups were calculated using R package “t-test”. The thresholds of fold change (> 1.2 or < 0.83) and P-value < 0.05 were used to identify differentially expressed proteins (DEPs). Functional annotation of all identified proteins was performed using GO (http://geneontology.org/) and KEGG pathway (http://www.genome.jp/kegg/).Protein-protein interaction analysis was performed using the String v11.5.
Metabolomic data collection and analysis
The metabolomic data was obtained from two sets of treatments, T0 and T2, with each treatment providing 6 samples for data collection, that is, one treatment had 6 repeated data collection experiments. The materials have been submitted to the company (MajorBio, Shanghai, China) for processing and data analysis.
50 mg solid sample was added to a 2 mL centrifuge tube and a 6 mm diameter grinding bead was added. 400 µL of extraction solution (methanol: water = 4:1 (v: v)) containing 0.02 mg/mL of internal standard (L-2-chlorophenylalanine) was used for metabolite extraction. Samples were ground by the Wonbio-96c ( Shanghai wanbo biotechnology co., LTD) frozen tissue grinder for 6 min (-10 °C, 50 Hz), followed by low-temperature ultrasonic extraction for 30 min (5 °C, 40 kHz). The samples were left at -20 °C for 30 min, centrifuged for 15 min (4 °C, 13000 g), and the supernatant was transferred to the injection vial for LC-MS/MS analysis.
As a part of the system conditioning and quality control process, a pooled quality control sample (QC) was prepared by mixing equal volumes of all samples. The QC samples were disposed and tested in the same manner as the analytic samples. It helped to represent the whole sample set, which would be injected at regular intervals (every 5–15 samples) in order to monitor the stability of the analysis.
The LC-MS/MS analysis of sample was conducted on a Thermo UHPLC-Q Exactive HF-X system equipped with an ACQUITY HSS T3 column (100 mm × 2.1 mm i.d., 1.8 μm; Waters, USA) at Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China). The mobile phases consisted of 0.1% formic acid in water: acetonitrile (95:5, v/v) (solvent A) and 0.1% formic acid in acetonitrile: isopropanol: water (47.5:47.5, v/v) (solvent B). Positive ion mode separation gradient: 0–3 min, mobile phase B was increased from 0 to 20%; 3–4.5 min, mobile phase B was increased from 20 to 35%; 4.5–5 min, mobile phase B was increased from 35 to 100%; 5–6.3 min, mobile phase B was maintained at 100%; 6.3–6.4 min, mobile phase B was decreased from 100 to 0%; 6.4–8 min, mobile phase B was maintained at 0%. Separation gradient in negative ion mode: 0–1.5 min, mobile phase B rises from 0 to 5%; 1.5–2 min, mobile phase B rises from 5 to 10%; 2–4.5 min, mobile phase B rises from 10 to 30%; 4.5–5 min, mobile phase B rises from 30 to 100%; 5–6.3 min, mobile phase B linearly maintains 100%; 6.3–6.4 min, the mobile phase B decreased from 100 to 0%; 6.4–8 min, the mobile phase B was linearly maintained at 0%. The flow rate was 0.40 mL/min and the column temperature was 40℃.
The mass spectrometric data were collected using a Thermo UHPLC-Q Exactive HF-X Mass Spectrometer equipped with an electrospray ionization (ESI) source operating in positive mode and negative mode. The optimal conditions were set as followed: source temperature at 425℃ ; sheath gas flow rate at 50 arb; Aux gas flow rate at 13 arb; ion-spray voltage floating (ISVF) at -3500 V in negative mode and 3500 V in positive mode, respectively; Normalized collision energy, 20-40-60 V rolling for MS/MS. Full MS resolution was 60,000, and MS/MS resolution was 7500. Data acquisition was performed with the Data Dependent Acquisition (DDA) mode. The detection was carried out over a mass range of 70–1050 m/z.
The pretreatment of LC/MS raw data was performed by Progenesis QI (Waters Corporation, Milford, USA) software. The metabolites were identified by searching database, and the main databases were the HMDB (http://www.hmdb.ca/), Metlin ( https://metlin.scripps.edu/) and Majorbio Database .
The data matrix obtained by searching database was uploaded to the Majorbio cloud platform (https://cloud.majorbio.com)28 for data analysis. The R package “ropls”(Version 1.6.2)29 was used to perform principal component analysis (PCA) and orthogonal least partial squares discriminant analysis (OPLS-DA), and 7-cycle interactive validation evaluating the stability of the model. The metabolites with VIP > 1, p < 0.05 were determined as significantly different metabolites based on the Variable importance in the projeciton (VIP) obtained by the OPLS-DA model and the p-value generated by student’s t test.
Differential metabolites among two groups were mapped into their biochemical pathways through metabolic enrichment and pathway analysis based on KEGG database (http://www.genome.jp/kegg/). Python packages “scipy.stats” (https://docs.scipy.org/doc/scipy/ )30 was used to perform enrichment analysis to obtain the most relevant biological pathways for experimental treatments.
Multi-omics data analysis and statistical analysis
Data analysis within each omics discipline was primarily conducted on the Majorbio cloud platform (https://cloud.majorbio.com)28. Comprehensive multi-omics data analysis was performed in the R environment version 4.3.1. Data cleaning and visualization were carried out using the tidyverse package31 and pathview package32,33,34,35, while the construction and analysis of regulatory networks utilized the igraph package36. GSEA analysis was conducted offline employing the GSEA software37,38, and the results were enhanced using the R package GseaVis39. Color palettes for visualization were implemented using the ggsci package40. The data of phenotype and active component content were analyzed by one-way ANOVA and LSD (α = 0.05) for multiple comparison tests.
Results
Phenotypic characteristics of tissue culture seedlings cultivated under different magnesium concentrations
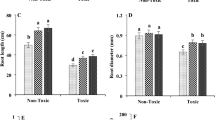
Phenotypic measurements included root dry weight, root length, rooting rate, and root diameter. Furthermore, the leaf count and plant height were also determined to measure the overall growth and development of the tissue-cultured seedlings. The addition of magnesium promoted root development while maintaining the root diameter. The impact on the rooting rate was not significantly pronounced, except in the T0.5 treatment (Fig. 1). In terms of overall developmental aspects, higher concentrations of magnesium were associated with increases in leaf count and plant height.

Phenotypic assessment results of tissue-cultured seedlings under various magnesium ion concentrations treatment (values followed by the same letter are not significantly different at p < 0.05.)
Determination of the active component contents in tissue-cultured seedlings under different magnesium concentrations
The active ingredient content of Sophora.tonkinensis, a medicinal plant, is a crucial criterion for evaluating its medicinal efficacy. Analyzing the contents of its four active components under different magnesium treatments allows us to infer the influence of magnesium on the medicinal efficacy of this plant. The results (Fig. 2) indicate that 1.5 mmol/L magnesium inhibited the synthesis of matrine and oxymatrine, both of which are quinolizidine alkaloids. In contrast, the content of maackiain actually increased under the 1.5 mmol/L magnesium treatment. The content of trifolirhizin decreased under specific magnesium concentration treatments, but there was no significant difference in content compared to the untreated group for the other magnesium concentration treatments. Maackiain and trifolirhizin are isoflavone derivatives, indicating that the magnesium-induced regulation of flavonoid-related metabolic pathways differs from that of alkaloid metabolic pathways.

The content of active components in tissue-cultured seedlings under different treatments (values followed by the same letter are not significantly different at p < 0.05).
Transcriptomic insights into differential gene expression under magnesium treatment
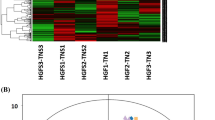
Transcriptome data were acquired from tissue-cultured seedlings subjected to two different treatments: 1.5 mmol/L magnesium (T2) and 0 mmol/L magnesium (T0). Through transcriptomic data analysis, a total of 904 differentially expressed genes were identified, including 641 genes whose expression was upregulated and 303 genes whose expression was downregulated compared with their expression levels in the control group (T0)(Fig. 3A, B). The functional annotation of these differentially expressed genes, as assessed through GO and KEGG annotation analyses, revealed that some of these genes were associated with transcriptional activity and metabolic functions(Fig. 3C, D). Functional enrichment analysis of these differentially expressed genes revealed that numerous genes related to photosynthesis were affected(Fig. 3E, F), possibly reflecting the significant role of magnesium in photosynthetic processes. Furthermore, the KEGG functional enrichment analysis results suggested that, in addition to photosynthesis, certain genes involved in primary and secondary metabolism were also influenced by the treatment(Fig. 3F) .GSEA was performed to assess all genes within the transcriptome, with the gene set chosen from those involved in the KEGG pathway. The outcomes of the analysis for each pathway are illustrated in Fig. 4. The results of the GSEA indicate that under T2 treatment, the expression levels of gene related to all enriched gene sets s were increased. Most of these pathways are metabolic pathways, such as energy metabolism, lipid metabolism, and carbohydrate metabolism. Additionally, a small proportion of these pathways are related to hormone synthesis (brassinosteroid), hormone signaling, and plant-pathogen interactions.

Differential gene expression revealed by transcriptomic analysis. A Differential gene expression volcano plots for Mg0(T0) and Mg2(T2) treatment comparisons. B Hierarchical clustering heatmap of gene sets for 6 samples under Mg0(T0) and Mg2(T2) treatments. C Gene set go classification statistical bar chart. D Gene set KEGG classification statistical bar chart. E GO enrichment analysis bubble chart. F KEGG enrichment analysis bubble chart.

GSEA analysis of the transcriptomic data. A Energy metabolism pathways (Map00195: Photosynthesis; Map00196: Photosynthesis - antenna proteins; Map00710: Carbon fixation in photosynthetic organisms; Map00860: Porphyrin and chlorophyll metabolism). B Metabolism of cofactors and vitamins pathways (Map00740: Riboflavin metabolism; Map00750: Vitamin B6 metabolism; Map00130: Ubiquinone and other terpenoid-quinone biosynthesis). C Glycan biosynthesis and metabolism pathways (Map00531: Glycosaminoglycan degradation; Map00511: Other glycan degradation). D Carbohydrate metabolism pathways (Map00040: Pentose and glucuronate interconversions; Map00051: Fructose and mannose metabolism; Map00030: Pentose phosphate pathway; Map00520: Amino sugar and nucleotide sugar metabolism; Map00010: Glycolysis / Gluconeogenesis). E Lipid metabolism pathways (Map00073: Cutin, suberine and wax biosynthesis; Map00592: alpha-Linolenic acid metabolism; Map00564: Glycerophospholipid metabolism; Map561: Glycerolipid metabolism; Map00591: Linoleic acid metabolism). F Other pathways(Map04626: Plant-pathogen interaction; Map00905: Brassinosteroid biosynthesis; Map00430: Taurine and hypotaurine metabolism; Map04075: Plant hormone signal transduction).
Differential protein profiling revealed by proteomic analysis
Through proteinomic data collection, a total of 377 proteins with differential abundance were identified. Among these proteins, 234 were upregulated, whereas 143 were downregulated compared with those in the control group (T0) (Fig. 5). The functional annotation of these differentially abundant proteins, based on GO and KEGG annotation analyses, revealed a substantial number of proteins associated with transcription and metabolic functions, including catalytic activity(Fig. 4). To further elucidate the roles of individual proteins, GSEA revealed that under T2 treatment, half of the significantly enriched gene pathways presented increased protein content, whereas the other half presened decreased protein content. The table shows that pathways associated with increased protein content are involved mainly in protein and macromolecule degradation as well as transmembrane transport of substances. Conversely, pathways with decreased protein content are related primarily to ribosomes (Fig. 6; Table 1). Additionally, functional enrichment analysis of the differentially abundant proteins indicated that magnesium may also play regulatory roles in various metabolic processes.

Differential protein revealed by proteomic analysis. A Differential protein volcano plots for T0 (Mg0) and T2 (Mg2) treatment comparisons. B Protein set GO classification statistical bar chart. C Protein set KEGG classification statistical bar chart. D GO enrichment analysis bubble chart for protein. E KEGG enrichment analysis bubble chart for protein.

GSEA analysis of the proteomic data.
Metabolomic data analysis of plants under magnesium treatment
Metabolomic analysis focuses primarily on differentially abundant metabolites. Based on the metabolomic data, a total of 543 differentially abundant metabolites were identified between the magnesium ion-treated group and the control group, including 121 metabolites with increased synthesis and 422 metabolites with decreased synthesis (Fig. 7A, B). Functional analysis of these differentially abundant metabolites and their associated KEGG compounds revealed alterations in the synthesis of 8 amino acids, indicating a shift in protein synthesis during primary metabolism. Based on the annotated results of differential compounds in the metabolomic analysis, it was primarily the concentrations of a multitude of secondary metabolite-related compounds that underwent significant changes. Moreover, there were substantial variations in the concentrations of compounds associated with amino acid metabolism (Fig. 7C). Considering that precursor compounds in various secondary metabolic pathways originate from the metabolism of amino acids, the T2 treatment has induced considerable alterations in the secondary metabolic status of Sophora tonkinensis. Pathway analysis based on the KEGG results revealed that magnesium treatment had the most pronounced effect on plant metabolism, with many metabolites involved in the synthesis of secondary metabolites. KEGG pathway enrichment analysis and HMDB compound classification revealed that flavonoid biosynthesis was most profoundly influenced by magnesium (Fig. 8).

Differential metabolites selected from metabolomic data under different treatments. A Volcano plot displaying differential metabolites (blue indicating decreased synthesis, red indicating increased synthesis). B OSLA-DS dimensionality reduction depicting differences in sample metabolites between T0 and T2. C Hierarchical clustering analysis and functional annotation of differential metabolites in a heatmap.

Metabolite types and functional annotations. A KEGG compound annotations of differential metabolites. B KEGG functional annotations of differential metabolites. C KEGG functional enrichment analysis of differential metabolites. D Classification statistics of differential metabolites at the class level in HMDB.
Multi-omics data analysis
Based on the omics data mentioned above, KEGG pathway enrichment analysis was conducted for all differentially expressed genes, differentially abundant proteins, and differentially synthesized metabolites, as shown in Fig. 8. The results indicate that a significant number of differential factors are enriched in various metabolic processes, suggesting alterations in the metabolic state of plants under magnesium treatment. Because metabolites detected in the metabolome itself are metabolic products, it is possible that metabolic products may be enriched in specific metabolic processes during enrichment analysis. When avoiding interference from the functional enrichment of metabolites and focusing solely on genes and proteins, the results also showed that the enriched functional pathways sere predominantly associated with metabolic processes. Therefore, these results confirmed that magnesium treatment can alter multiple metabolic pathways in plants, possibly through the role of magnesium as an important magnesium-catalyzed cofactor. Simultaneously, under the GO analysis, there was enrichment of pathways shared by both genes and proteins. This finding not only indicates a connection with metabolic processes but also suggests that many pathways are related to ion absorption and balance. A comprehensive analysis of the enriched functions mentioned above revealed that the impact of magensium on plants is multifaceted, and affects various functions and metabolic pathways in plants (Fig. 9).

Integrated analysis of differential genes, proteins, and metabolites. A KEGG functional pathway annotations for differential genes, proteins, and metabolites. B KEGG functional annotations for differential genes and proteins. C GO functional annotations for differential genes and proteins.
The transcriptome data provided information on a series of transcription factor genes and their regulatory pathways under the action of magnesium. Using these data, a transcriptional regulatory network corresponding to magnesium action was constructed. First, differentially expressed genes were selected from the transcriptome, and from these genes, transcription factors that participate in the regulation of upstream genes were further identified. The igraph package was employed to construct the regulatory networks. Notably, some nodes in these regulatory networks presented not only differential gene expression but alsosignificant differences in protein abundance, as indicated by the nodes colored in red and blue in Fig. 10. Based on the topological structure of the directed network, the Page Rank algorithm was used to evaluate the importance of each node, and node size was used to represent the node importance score (Fig. 10A).The majority of nodes with significantly different protein abundances are also important regulatory nodes. Moreover, when considering only differentially expressed proteins as nodes in the network, the network shown in Fig. 10A was obtained by removing all white nodes and subsequently eliminating the remaining isolated nodes lacking network connections, resulting in a network consisting solely of nodes with significantly differential protein content (Fig. 10B). A simplified network composed of seven nodes was extracted from the original network(Fig. 10B), The color-coding of the network and nodes suggested that the protein evm.model.1.6239, which functions as a transcription factor similar to the Cell Division Cycle 5-like protein (CDC5L), may play a central role in a regulatory network within Sophora tonkinensis. As the concentration of this protein increases, the synthesis of proteins from the six genes it regulates decreases, indicating transcriptional repression of these six gene nodes.The types of these six gene nodes and the central proteins are detailed in Table 2.

Construction and analysis of the transcriptional regulatory network. (White nodes represent genes with differential expression only in the transcriptome, while colored nodes represent proteins with differential synthesis in the proteome, red indicating upregulation and blue indicating downregulation.) A The overall transcriptional regulatory network. B The transcriptional regulatory network consisting solely of differential proteins.
The correlation between differential genes in the transcriptome and metabolites can be explored through shared KEGG pathways between the two. Considering the pharmacologically active components, such as alkaloids and flavonoids, the metabolic pathways related to flavonoids and isoflavonoids were examined. Currently, the synthetic pathways of quinolizidine alkaloids such as metrine and oxymetrine are not well understood. Therefore, other alkaloid pathways and nitrogen metabolism pathways were also investigated to determine if these pathways had any impact. These changes might indirectly affect the synthesis of metrine and oxymetrine, as well as the status of the synthesis of other secondary metabolites. An analysis of biosynthetic pathways for secondary metabolites in plants using the KEGG database revealed alterations in both metabolites and genes. Specifically, seven genes were differentially expressed, with six being upregulated and one being downregulated. Concurrently, three metabolites exhibited changes in concentration, with two exhibiting decreasing concentrations and one increasing concentration. These genes and metabolites are involved in the biosynthesis of mugineic acid, benzoxazinoids, coumarins, scopolin, sideretin, podophyllotoxin, and secoisolariciresinol (see Fig. 11A). In the nitrogen metabolism pathway, the expression levels of three relevant genes decreased, and the levels of two nitrogen-related metabolites, glutamine and glutamate, decreased (see Fig. 11B). In the flavonoid biosynthetic pathway, significant changes in gene expression were observed, with 19 genes upregulated and 5 genes downregulated. Additionally, the concentrations of nine metabolites shifted, including an increase in pinocembrin and decreases in the levels of the remaining compounds (Fig. 11C). In the isoflavonoid biosynthesis pathway, all five genes presented downregulated expression, and the concentrations of the four metabolites decreased (Fig. 11D). In the KEGG pathway for alkaloid biosynthesis, no changes in gene expression levels were detected in the transcriptome. However, vanillylamine and tryptophan concentrations decreased, whereas anthranilate concentrations increased (Fig. 11E). In the flavone and flavonol biosynthesis pathway, gene expression remained unchanged; however, changes in the concentrations of eight metabolites were noted: seven decreased and one increased (Fig. 11F).

Differential gene expression and differential synthesis of metabolites depicted in KEGG pathway maps (red and green indicate transcriptional fold change differences for genes, while blue and yellow indicate fold change differences in metabolites). A Synthesis pathways of various plant secondary metabolites. B Nitrogen metabolism pathway. C Flavonoid biosynthesis pathway. D Isoflavonoid biosynthesis pathway. E Biosynthesis pathways of various alkaloids. F Biosynthesis pathway of flavone and flavonol.
Discussion
Magnesium, a versatile element, profoundly influences various dimensions of plant biology, affecting individual molecules, photosynthesis, metabolic processes, and nutrient absorption41. It also serves as a cofactor for numerous enzymes in plants. Its pivotal role in photosynthesis is emphasized by the indispensable binding of magnesium ions, which are essential for the proper functioning of tetrapyrrole rings in chlorophyll a and b42. Notably, chloroplasts boast the highest magnesium content among plant cell components1,43. At the transcriptome level, differential gene expression analysis revealed alterations in genes directly associated with magnesium. These genes fall into two main categories: those involved in chlorophyll synthesis, such as magnesium protoporphyrin IX methyltransferase and other enzymes. The second category comprises magnesium-dependent enzymes, including serine/threonine protein phosphatases and pyruvate kinase. Serine/threonine protein phosphatases can play crucial roles in plant growth, stress response, and nutrition44,45,46. Because magnesium functions as a linchpin in metabolic processes critical for plant growth and development, magnesium levels have a direct influence, particularly in the aerial parts of plants. The experimental findings revealed a positive correlation between leaf count and magnesium concentration.
In our experiments, the T0 treatment lacked additional magnesium supplementation, and the culture medium, althouth not supplemented, was prepared with distilled water rather than deionized water. Consequently, a trace amount of magnesium from water sources may exist, ensuring the normal growth of the tissue-cultured seedlings in the T0 treatment. Nonetheless, discernible differences in leaf color and size are evident in the images compared with those of the seedlings supplemented with extra magnesium(Supplementary Fig. 1).
Given the direct impact of magnesium on plant photosynthesis and enzyme activity, our study employs a multi-omics approach to elucidate the specific mechanisms through which magnesium influences plants. Multi-omics approaches, which are increasingly prevalent in medicinal plant studies47, leverage advancements in omics technologies as indispensable tools for molecular-level investigations of medicinal plants. These technologies generate extensive data, contributing to publicly accessible databases housing substantial medicinal plant omics datasets48.
Our aim was to analyze the functional pathways through which magnesium influences the pharmacologically active constituents of plants using an integrative multi-omics analysis. Functional pathway data were obtained through enrichment analysis across the transcriptome, proteome, and metabolome.As modules, these pathways collectively shape the overall physiological state of plants through intermodule relationships, thereby impacting plant growth, development, and secondary metabolism. Gene set annotation analysis, which is based on the KEGG pathway of the transcriptome, revealed five principal functional pathways (Fig. 3D) governing the plant state through the influence of magnesium. These pathways include metabolism, genetic and environmental information processing, transportation and catalysis, and environmental adaptation. Considered as interconnected modules, these pathways are influenced by upstream factors such as photosynthesis, alterations in enzyme activities, and intermodule interactions. These processes collectively regulate the growth, developmental status, and active ingredient content of plants affected by magnesium.
Magnesium, which often function as a crucial cofactor for enzymes, directly influences diverse metabolic processes in plants. Its impact on energy metabolism is explained through gene set enrichment analysis of the KEGG pathways in the transcriptome, which affect various metabolic pathways, particularly those associated with photosynthesis. The importance of magnesium in photosynthesis is underscored by alterations in the expression levels of genes directly linked to photosynthesis following treatment. Moreover, it is essential to consider the influence of other ions, notably the absorption of specific mineral elements closely associated with magnesium. Elements such as potassium and calcium can hinder magnesium absorption by plants49,50. Given that magnesium serves as a cofactor for numerous enzymes, and under T2 treatment, metabolomic data indicate changes in the levels of some cofactor enzymes (Fig. 8B), fluctuations in magnesium concentration may affect the activity of various metabolic enzymes within the plant. In summary, magnesium may influence plant conditions through photosynthesis, calcium and potassium absorption, and alterations in enzyme activity (Fig. 12).

Exploring the multifaceted mechanisms through which magnesium influences medicinal compounds and growth and development.
These external shifts, coupled with alterations in metabolism, may modify the genetic information processing of plants, subsequently influencing the expression and translation of multiple genes. Consequently, a transcriptional regulatory network was constructed in this study, and key nodes within the network were identified through binding with proteomics, data using the PageRank algorithm. Owing to the intricate nature of regulatory networks, their topological characteristics often necessitate a joint representation of multiple parameters. The PageRank algorithm, renowned for identifying nodes crucial for network regulation51, has found widespread applications in biological network analysis, including temporal changes in transcriptional regulatory networks and the examination of disease-related protein-protein interaction networks52. In the plant ___domain, this algorithm has proven useful in analyzing critical factors in various physiological processes, particularly in Arabidopsis thaliana53. The identification of these nodes underscores their significant influence on the overall network.
This investigation identified regulated enzymes as critical nodes, with the most prominent protein being xyloglucan endotransglucosylase/hydrolase (XTH). XTH plays a pivotal role in hydrolyzing and reshaping polysaccharide components in the cell wall, which are crucial for plant growth and development. Its functions include regulating of the cell wall to accommodate changes in cell size, responding to stress, and influencing on physiological processes such as fruit ripening54,55,56. Notably, observed changes in root diameter may be directly linked to variations in this gene. Another poteintially node protein is ferroxidase, with its decrease possibly associated with photosynthesis given the critical role of iron in this process57,58,59.Another significant node protein may inhibit protease activity, potentially influencing protein modifications. Under magnesium treatment, these three proteins presented decreased levels, whereas the sole noteworthy node protein showing increased synthesis was Rubisco activase, which is directly linked to carbon fixation in photosynthesis60. These proteins collectively impact plant growth and development through their influence on photosynthesis and cell wall regulation, indirectly shaping the plant’s metabolic state.
Based on the comprehensive experimental results and analyses presented in this study, it can be concluded, as depicted in Fig. 12, that magnesium primarily influences plant functions by affecting photosynthesis, calcium and potassium absorption, and the activites of various enzymes. These influences modulate key processes such as metabolism, environmental information processing, transport, and catabolism. Consequently, these modulated processes impact the plant’s genetic information processing and environmental adaptation, ultimately affecting its growth, development, and content of active compounds.
In the next step, to further investigate the impact of magnesium on the synthesis of active components in plants, more metabolomic dataare needed. Analyzing the entire metabolic network from a systems biology perspective is necessary to understand the effects of various metabolic changes on secondary metabolism. Although metabolic system analysis is more common in microbial research61,62,63, its application in plants has been relatively limited, possibly due to the scarcity of relevant large-scale biological data. Therefore, the accumulation of more data is essential to better elucidate how a single element affects plant secondary metabolism through multiple pathways, especially when many of these effects are often indirect rather than direct. Combining systems biology with multi-omics analyses may provide a more comprehensive research approach.
The primary objective of this study was to utilize tissue culture techniques to propagate Sophora tonkinensis, thereby facilitating the direct extraction of active ingredients from cultured seedlings. Given that wild populations of Sophora tonkinensis are predominantly found in karst mountainous regions, analyzing the elemental composition of the soil and researching the optimal culture medium formulation are highly important for realizing the aforementioned goals. Concurrently, while focusing on magnesium, future studies will investigateon the impact of the calcium and selenium content’s on the activity of medicinal substances.
Data availability
The analysis data table for omics can be downloaded from ScienceDB (https://doi.org/10.57760/sciencedb.13728). The primary data on the transcriptome, proteome, and metabolome of Sophora tonkinensis utilized in this experimental project have been publicly released and are accessible at (https://doi.org/10.57760/sciencedb.13743) for download of the raw data.
References
Cakmak, I. & Yazici, A. M. Magnesium: a forgotten element in crop production. Better Crops. 94, 23–25 (2010).
Guo, W., Nazim, H., Liang, Z. & Yang, D. Magnesium deficiency in plants: an urgent problem. Crop J. 4, 83–91 (2016).
Sahin, G., Verma, S. K. & Gurel, E. Calcium and magnesium elimination enhances accumulation of cardenolides in callus cultures of endemic Digitalis species of Turkey. Plant Physiol. Biochem. 73, 139–143 (2013).
Kwon, M. C. et al. Comparative metabolomics unravel the effect of magnesium oversupply on tomato fruit quality and associated plant metabolism. Metabolites. 9, 231 (2019).
Zhang, Y. et al. Effects of Magnesium on Transcriptome and Physicochemical Index of Tea leaves. Plants. 12, 1810 (2023).
Jayaganesh, S., Venkatesan, S. & Senthurpandian, V. K. Impact of different sources and doses of magnesium fertilizer on biochemical constituents and quality parameters of black tea. Asian J. Biochem. 6, 273–281 (2011).
Xu, J. et al. Magnesium supplementation alters leaf metabolic pathways for higher flavor quality of oolong tea. Agriculture. 11, 120 (2021).
Hanudin, E., Wismarini, H., Hertiani, T. & Sunarminto, B. H. Effect of shading, nitrogen and magnesium fertilizer on phyllanthin and total flavonoid yield of Phyllanthus niruri in Indonesia soil. J. Med. Plants Res. 6, 4586–4592 (2012).
Jin, X. et al. Magnesium Nutrient Application induces metabolomics and physiological responses in Mulberry (Morus alba) plants. Int. J. Mol. Sci. 24, 9650 (2023).
Mun, H. I. et al. Metabolomic response of Perilla frutescens leaves, an edible-medicinal herb, to acclimatize magnesium oversupply. PLos One. 15, e0236813 (2020).
Stitt, M. & Joost Keurentjes. Metabolic networks: how to identify key components in the regulation of metabolism and growth. Plant Physiol. 152, 428–444 (2010).
Huang, J., Xu, H. & Matrine Bioactivities and structural modifications. Curr. Top. Med. Chem. 16, 3365–3378 (2016).
Tang, L. et al. Pharmacokinetic characterization of oxymatrine and matrine in rats after oral administration of radix Sophorae tonkinensis extract and oxymatrine by sensitive and robust UPLC–MS/MS method. J. Pharm. Biomed. Anal. 83, 179–185. https://doi.org/10.1016/j.jpba.2013.05.003 (2013).
Yoo, H. et al. SKI3301, a purified herbal extract from Sophora tonkinensis, inhibited airway inflammation and bronchospasm in allergic asthma animal models in vivo. J. Ethnopharmacol. 206, 298–305. https://doi.org/10.1016/j.jep.2017.05.012 (2017).
Jiang, X. et al. Trifolirhizin inhibits proliferation, migration and invasion in nasopharyngeal carcinoma cells via PI3K/Akt signaling pathway suppression. Biochem. Biophys. Res. Commun. 667, 111–119. https://doi.org/10.1016/j.bbrc.2023.05.030 (2023).
Liu, Y. et al. Anti-tumor activities of matrine and oxymatrine: literature review. Tumor Biol.. 35, 5111–5119 (2014).
Mizuguchi, H. et al. Maackiain is a novel antiallergic compound that suppresses transcriptional upregulation of the histamine H1 receptor and interleukin-4 genes. Pharmacol. Res. Perspect. 3, e00166 (2015).
Zhou, H., Lutterodt, H., Cheng, Z. & Yu, L. Anti-inflammatory and antiproliferative activities of trifolirhizin, a flavonoid from Sophora flavescens roots. J. Agric. Food Chem. 57, 4580–4585 (2009).
Li, C. et al. Single-cell multi-omics in the medicinal plant Catharanthus roseus. Nat. Chem. Biol. 19, 1031-1041 (2023).
Zhong, C. et al. Multi-omics profiling reveals comprehensive microbe–plant–metabolite regulation patterns for medicinal plant Glycyrrhiza Uralensis Fisch. Plant Biotechnol. J. 20, 1874–1887 (2022).
Wang, Y., Tong, Y., Adejobi, O. I., Wang, Y. & Liu, A. Research advances in Multi-omics on the traditional Chinese Herb Dendrobium officinale. Front. Plant Sci. 12, 808228 (2022).
Zhou, P. et al. Multi-omics analysis of the bioactive constituents biosynthesis of glandular trichome in Perilla frutescens. BMC Plant Biol. 21, 277 (2021).
Hank, H. et al. Effect of magnesium on the growth and alkaloid production of hairy root cultures. Acta Hort. 597, 271–274 (2003).
Podwyszyńska, M. & Goszczyńska, D. M. Effect of inhibitors of ethylene biosynthesis and action, as well as calcium and magnesium on rose shoot rooting, shoot-tip necrosis and leaf senescence in vitro. Acta Physiol. Plant. 20, 91–98 (1998).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 9, 357–359. https://doi.org/10.1038/nmeth.1923 (2012).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 12, 323. https://doi.org/10.1186/1471-2105-12-323 (2011).
Xie, C. et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–322. https://doi.org/10.1093/nar/gkr483 (2011).
Ren, Y. et al. Majorbio cloud: a one-stop, comprehensive bioinformatic platform for multiomics analyses. IMeta. 1, e12 (2022).
Thévenot, E. A., Roux, A., Xu, Y., Ezan, E. & Junot, C. Analysis of the human adult urinary metabolome variations with age, body mass index, and gender by implementing a comprehensive workflow for univariate and OPLS statistical analyses. J. Proteome Res. 14, 3322–3335 (2015).
Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods. 17, 261–272 (2020).
Wickham, H. et al. Welcome to the Tidyverse. J. open. Source Softw. 4, 1686 (2019).
Luo, W. & Brouwer, C. Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics. 29, 1830–1831 (2013).
Kanehisa, M. et al. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592 (2023).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951 (2019).
Csardi, G. & Nepusz, T. The igraph software. Complex Syst. 1695, 1-9 (2006)
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. 102, 15545–15550. https://doi.org/10.1073/pnas.0506580102 (2005).
Mootha, V. K. et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273. https://doi.org/10.1038/ng1180 (2003).
Zhang, J. & GseaVis An Implement R Package to visualize GSEA results. (2022). https://github.com/junjunlab/GseaVis, https://github.com/junjunlab/GseaVis/wiki
Xiao, N. ggsci: scientific journal and Sci-Fi themed color palettes for’ggplot2’. R Package Version 2 (2018).
Kleczkowski, L. A. & Igamberdiev, A. U. Magnesium signaling in plants. Int. J. Mol. Sci. 22, 1159 (2021).
Wilkinson, S. R. et al. Magnesium in plants: uptake, distribution, function, and utilization by man and animals (1990).
Karley, A. J. & White, P. J. Moving cationic minerals to edible tissues: potassium, magnesium, calcium. Curr. Opin. Plant. Biol. 12, 291–298 (2009).
País, S. M., Téllez-Iñón, M. T. & Capiati, D. A. Serine/threonine protein phosphatases type 2A and their roles in stress signaling. Plant Signal. Behav. 4, 1013–1015 (2009).
Banerjee, A. & Roychoudhury, A. Deciphering the roles of protein phosphatases in the regulation of Salt-Induced signaling responses in plants. in Protein Phosphatases and Stress Management in Plants: Functional Genomic Perspective (ed Girdhar K. Pandey) 149-162 (Springer International Publishing, 2020).
Sathee, L. et al. Role of protein phosphatases in the regulation of nitrogen nutrition in plants. Physiol. Mol. Biol. Plants. 27,2911-2922 (2021).
Yang, L. et al. From single-to multi-omics: future research trends in medicinal plants. Brief. Bioinform. 24, bbac485 (2023).
He, S. et al. MPOD: applications of integrated multi-omics database for medicinal plants. Plant Biotechnol. J. 20, 797 (2022).
Yan, B. & Hou, Y. Effect of soil magnesium on plants: a review. In IOP Conference Series: Earth and Environmental Science. IOP Publishing 170, p. 022168 (2018).
Tränkner, M., Tavakol, E. & Jákli, B. Functioning of potassium and magnesium in photosynthesis, photosynthate translocation and photoprotection. Physiol. Plant. 163, 414–431 (2018).
Wolf, I. R., Simões, R. P. & Valente, G. T. Three topological features of regulatory networks control life-essential and specialized subsystems. Sci. Rep. 11, 24209 (2021).
Ding, H. et al. Prioritizing transcriptional factors in gene regulatory networks with PageRank. Iscience 24, 102017 (2021).
Omranian, N., Mueller-Roeber, B. & Nikoloski, Z. PageRank-based identification of signaling crosstalk from transcriptomics data: the case of Arabidopsis thaliana. Mol. Biosyst. 8, 1121–1127. https://doi.org/10.1039/C2MB05365A (2012).
Tiika, R. J. et al. Transcriptome-wide characterization and functional analysis of Xyloglucan endo-transglycosylase/hydrolase (XTH) gene family of Salicornia europaea L. under salinity and drought stress. BMC Plant Biol. 21, 491. https://doi.org/10.1186/s12870-021-03269-y (2021).
Ishida, K. & Yokoyama, R. Reconsidering the function of the xyloglucan endotransglucosylase/hydrolase family. J. Plant. Res. 135, 145–156 (2022).
Witasari, L. D. et al. Higher expression of the strawberry xyloglucan endotransglucosylase/hydrolase genes FvXTH9 and FvXTH6 accelerates fruit ripening. Plant J. 100, 1237–1253. https://doi.org/10.1111/tpj.14512 (2019).
Rochaix, J. D. Regulation of photosynthetic electron transport. Biochim. Biophys. Acta (BBA) Bioenergetics. 1807, 375–383 (2011).
Kroh, G. E. & Pilon, M. Regulation of iron homeostasis and use in chloroplasts. Int. J. Mol. Sci. 21, 3395 (2020).
Kamalanathan, M. et al. Molecular mechanism of oil induced growth inhibition in diatoms using Thalassiosira pseudonana as the model species. Sci. Rep. 11, 19831. https://doi.org/10.1038/s41598-021-98744-9 (2021).
Waheeda, K., Kitchel, H., Wang, Q. & Chiu, P. L. Molecular mechanism of Rubisco activase: dynamic assembly and Rubisco remodeling. Front. Mol. Biosci. 10, 1125922 (2023).
Yilmaz, L. S. & Walhout, A. J. M. Metabolic network modeling with model organisms. Curr. Opin. Chem. Biol. 36, 32–39. https://doi.org/10.1016/j.cbpa.2016.12.025 (2017).
Mendoza, S. N., Olivier, B. G., Molenaar, D. & Teusink, B. A systematic assessment of current genome-scale metabolic reconstruction tools. Genome Biol. 20, 158. https://doi.org/10.1186/s13059-019-1769-1 (2019).
Fang, X., Lloyd, C. J. & Palsson, B. O. Reconstructing organisms in silico: genome-scale models and their emerging applications. Nat. Rev. Microbiol. 18, 731–743. https://doi.org/10.1038/s41579-020-00440-4 (2020).
Acknowledgements
The authors extend their gratitude to the staff at the ex vivo warehouse of Guangxi Botanical Garden of Medicinal Plants for providing materials and preservation services.
Funding
This research was funded by the National Natural Science Foundation of China (82260747); Central Guiding Local Science and Technology Development Special Project (Guike ZY20198018); China Agriculture Research System (CARS-21) and Key Techniques Research and Promotion of Guangxi Medicinal Materials Varieties (GZKJ2314).
Author information
Authors and Affiliations
Contributions
Kun-Hua Wei and Ling-Yun Chen designed and formulated the comprehensive research protocol. Fan Wei and Ying Liang contributed to the design of the experimental protocol and were responsible for the provision of tissue culture materials. Zhan-Tao Fan and Peng-Cheng Zhou executed specific experimental tasks and collected corresponding data in accordance with the research protocol. Shuang- Shuang Wang was tasked with gathering genomic data from S. tonkinensis and supplying data for the multi-omics analysis. Zhu Qiao and Jing Wang participated in the analysis of experimental data and the manuscript writing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
The plant material collection and research experiments conducted by this institute comply with the laws and regulations of China, as well as all national treaties related to nature reserves and plant protection. We affirm that our work involves no illegal activities. We strictly adhere to principles of scientific integrity, rationality, and sustainability during plant material collection, ensuring no adverse impact on the environment or biodiversity. We prioritize cooperation with local communities and authorities to ensure all collections have legal authorization.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Qiao, Z., Zhou, PC., Fan, ZT. et al. Multi-omics analysis uncovers the transcriptional regulatory mechanism of magnesium Ions in the synthesis of active ingredients in Sophora tonkinensis. Sci Rep 14, 25527 (2024). https://doi.org/10.1038/s41598-024-76575-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-76575-8
