
Abstract
The Monticola series comprises two anthropophilic and widely distributed species in Brazil: Pintomyia (Pifanomyia) monticola (Costa Lima, 1932) and Pintomyia (Pifanomyia) misionensis (Castro, 1959). They mainly occur in the Atlantic Rainforest, and it is known that Pi. monticola comprises at least two well-structured genetic lineages regarding a fragment of the cytochrome c oxidase subunit I (COI) gene. Here, we aim to elucidate the taxonomic status of this group using integrative taxonomy tools. Collections were performed in nine localities of four Brazilian states, and COI fragments were sequenced and merged with publicly available data. Several single-locus species delimitation algorithms, genetic distance metrics, phylogenetic trees, and haplotype networks were used to uncover cryptic diversity and population structure within Pi. monticola and Pi. misionensis. The resulting genetic clusters were then tested for morphological differences through linear and geometric morphometry of several characters. We analyzed 152 COI sequences, comprising 48 haplotypes. The maximum intraspecific p distances were 8.21% (mean 4.17%) and 9.12% (mean 4.4%) for Pi. monticola and Pi. misionensis, respectively, while interspecific ones ranged from 10.94 to 14.09% (mean 12.33%). Phylogenetic gene trees showed well-supported clades for both species, with clear structuring patterns within them. Species-delimitation algorithms split our dataset into at least three putative species for each taxon. Moreover, population structure analysis showed a strong correlation between Atlantic Forest areas of endemism as sources of molecular variation in Pi. monticola. Morphometric analyses were significant for wing shape variation and some linear measurements (mainly of the head) when comparing specimens of different genetic clusters for both taxa. These results indicate strong genetic structuring of Monticola series species, confirmed by morphometry, indicating two possible cryptic species complexes.
Similar content being viewed by others

Introduction
Sand flies (Diptera, Psychodidae, Phlebotominae) are the main vectors of leishmaniasis agents1. This subfamily comprises about 1060 species distributed worldwide, with an estimated number of 555 occurring in the Neotropical region2. Entomological monitoring at the species level and understanding the diversity of sand flies is a high priority to public health, since this subfamily presents great plasticity of ecological interactions and not all species or lineages have vectorial capacity.
For the Americas, 23 sand fly genera were proposed3,4. The genus Pintomyia Costa Lima, 1932 comprises 80 extant nominal species allocated in two subgenera and several species series, in addition of 13 fossil species2,5. This group is considered one of the most important ones due to its involvement in the leishmaniasis cycle. For instance, Pintomyia (Pintomyia) fischeri (Pinto, 1926) is implicated as one of the vectors of Leishmania infantum in the metropolitan region of Greater São Paulo, Brazil6, and others of the Pintomyia (Pifanomyia) subgenus are proven or suspected vectors of Leishmania spp. in Latin America7. Moreover, the Monticola series stand out, containing only two species – Pintomyia (Pifanomyia) monticola (Costa Lima, 1932) and Pintomyia (Pifanomyia) misionensis (Castro, 1959) – but which are widely distributed. Both species has been recorded for Brazilian northeast/southeast/south/midwest regions, Paraguay, and Argentina, but Pi. monticola has dubious records in Peru and the northern region of Brazil, whereas Pi. misionensis also can be found in the Brazilian northern state of Tocantins4,8.
These two sand fly species are not proven vectors of Leishmania protozoans, although their participation has been considered. Natural infection investigations found Pi. monticola with Leishmania braziliensis and Leishmania infantum DNA in the Southeast region of Brazil9,10, and its participation in the transmission of Leishmania enriettii has been questioned11,12. In addition, this sand fly is highly anthropophilic, with diurnal activity, and can be found in regions of preserved forest or close to urban centers, as well as different climatic regimes8,13,14,15. Its sibling species – Pintomyia misionensis – is also anthropophilic16, and was found with Leishmania spp. DNA17.
The species-level identification of these two taxa is considerably reliable using morphological data. Concerning the female reproductive system, those of Pi. monticola exhibits a vesicular spermathecae, while Pi. misionensis display elongated and transversally striated spermathecae, bearing an apical inverted tapered collar. Males of both species can be identified, respectively, by the presence and absence of the preapical spiniform seta on the gonostyle and a cluster of setae on the gonocoxite4. However, molecular taxonomy approaches show that Pi. monticola may be a complex of species that are indistinguishable by morphology18.
The molecular markers have facilitated the detection of cryptic diversity in cases of low morphological disparity among incipient species. One of the most popular markers in the study of Phlebotominae subfamily include a fragment of the mtDNA cytochrome c oxidase subunit I (COI) gene19. Although it was designed for species identification20, its sequences also have been used in the detection of structured populations and different genetic lineages within species21,22,23,24. Moreover, the relationship between genetic lineages/populations, with morphological differences using morphometry, has been evaluated in Neotropical sand flies25,26, providing reliable evidences regarding the diversification of these insects through integrative taxonomy27,28.
Due to the possible relevance of Pi. monticola and Pi. misionensis to public health, and its widespread distribution in South America, it is important that population-level studies be performed. Here, we aim to evaluate an integrative taxonomy approach in detecting cryptic diversity within sand fly species of the Monticola series of different populations in Brazil.
Materials and methods
Collection sites and sample processing
Entomological collections were performed between 2023 and 2024 in nine localities across four different Brazilian states (São Paulo – SP, Paraná – PR, Minas Gerais – MG, and Mato Grosso do Sul – MS) (Tables 1 and S1). Most of the collection sites were selected in forested environments of the Atlantic Rainforest biome, except for collections carried out in the municipality of Aquidauana, Piraputanga locality, in a transitional environment between the Cerrado and the Brazilian Pantanal, in the state of Mato Grosso do Sul. We used different collection methods, including Centers for Disease Control and Prevention (CDC)-type light traps, active search, and Shannon traps. Most collection efforts were done throughout the night (CDC light traps), and in the early hours of dusk through night (Shannon and active search). Some individuals of the “Parque Estadual da Serra do Mar (PESM)” were collected during daytime activity (9 am), or in the middle of the afternoon (4 pm), at times when the traps were being installed.
Insects were kept in 70% alcohol at − 20 °C, and after screening, sand flies were dissected. Specimens having three or more legs preserved after the initial trial, had them dissected and stored in sterile microtubes, whereas those with fewer or no legs had their thorax dissected and stored, and then the DNA extraction was performed. The remaining parts of sand flies, not stored for molecular procedures, were processed for morphological identification at the species level. For clarification, sand flies were kept for 12 h in KOH (20%), followed by two washes of distilled water for 10 min each, and an alcoholic series for 10 min each, using 70%, 90% and 100% alcohol. Then, sand flies were left in Eugenol oil for at least 24 h until the specimens were slide-mounted in Canada balsam. Sand flies were morphologically identified using the dichotomous keys of Galati (2018), and then deposited in the ‘Collection of Phlebotomine of René Rachou Institute’ (Fiocruz/COLFLEB) and the private entomological collection of EABG at School of Public Health, University of São Paulo (FSP USP).
DNA extraction, PCR, and sequencing
Total DNA was extracted from the legs or thorax of specimens. For this, we performed a salt-based extraction protocol using the Digsol Buffer (ethylenediaminetetraacetic acid [EDTA] 20 mM, Tris–HCl 50 mM, NaCl 117 mM; sodium dodecyl sulfate [SDS] 1%) as described in Rodrigues et al.29. Sand fly DNA was kept in a freezer at − 20 °C until amplification.
We amplified the DNA barcoding fragment of the cytochrome c oxidase subunit I (COI) gene20. For this, we performed the polymerase chain reactions (PCR) using the GoTaq™ Green Master Mix (PROMEGA), and the pair of primers LCO1490 (5′-GGT CAA CAA ATC ATA AAG ATA TTG G-3′) and HCO2198 (5′-TAA ACT TCA GGG TGA CCA AAA AAT CA-3′)30, following the manufactures’ instructions. The PCR conditions were as follows: 95 °C for 2 min; 35 cycles of 95 °C for 1 min, 54 °C for 1 min, and 72 °C for 1 min 30 s; 72 °C for 10 min. We then performed electrophoresis with 1% agarose gel stained with GelRed (Biotium Inc) to check the amplification. Positive reactions were sent to ACTGene Análises Moleculares (Brazil) for quantification, purification, and sequencing of the PCR products in both directions.
Sequence analysis and phylogenetic inference
We visualized electropherograms quality, removed primer sequences, and assembled contigs using SeqTrace v0.9. COI-barcode sequences were submitted to the Barcode of Life Data System – BOLD31 and NCBI GenBank32, being assigned the Accession numbers MIMO001-24 to MIMO079-24 and PP865228 to PP865306 for BOLD and GenBank, respectively (Table S1).
We added to our dataset publicly available COI sequences of Pi. monticola and Pi. misionensis. All sequences are from the Brazilian state of Espírito Santo, and were processed by Pinto et al.18. Access numbers and full information on specimens can be found in Table S1.
After merging all sequences, we performed the multiple alignment using the Muscle algorithm implemented in the Mega v7 program33. Sequences larger than the original fragment of the DNA barcoding proposal (658 bp) were trimmed, and missing data within this fragment were assigned accordingly. We estimated the pairwise genetic distances of both within (intraspecific) and between (interspecific) species using uncorrected simple distances (p distance) and the ‘pairwise deletion’ option to treat sites with gaps in the BOLD environment.
Phylogenetic gene trees were reconstructed to check the clustering pattern of COI sequences and perform species delimitation analysis. Initially, the best-fit substitution model was checked in the IQ-TREE web server34,35, which selected the TPM2 + F + G4 model. Then, a maximum likelihood analysis was performed in the same web server with 10,000 ultrafast bootstrap pseudoreplicates. We also performed a Bayesian inference using the BEAST v2.7 program36. For this, we selected the best-fit model using the bModelTest37 plugin, with the data partitioned according to the three codon positions. The ‘Coalescent Constant Population’ model and ‘Strict Clock’ were set as priors with the three codon positions linked, but different combinations were checked (see Rodrigues et al.38). We performed two runs in parallel, each with 15,000,000 generations (sampling every 1000). Trace logs were visualized in the Tracer v1.7.1 program to check the convergence of runs and Effective Sample Size values of statistic parameters, which were all above 300. We merged the tree files of both runs using the LogCombinner v2.5.2 program with 10% burn-in each, and then a maximum credibility tree (MCC) was generated with retained trees in TreeAnnotator v2.6.6. Lastly, FigTree v.1.4.4 program (http://tree.bio.ed.ac.uk/software/figtree) was used to visualize, edit, and export the trees.
Single-locus species delimitation
COI-sequences of Pi. monticola and Pi. misionensis were identified at Molecular Operational Taxonomic Unit (MOTU) level. For this, we performed a set of discovery approaches to species delimitation. This method assigns samples to groups without a priori information39. Thus, we seek to discover putative species or different genetic lineages in our COI dataset without assigning populations to specimens. We considered the delimitations generated by the algorithms: Automatic Barcode Gap Discovery (ABGD)40, Assemble Species by Automatic Partitioning (ASAP)41, Refined Single Linkage (RESL)42, Generalized Mixed Yule-Coalescent (GMYC)43, Poisson Tree Processes (PTP)44,45, and TCS haplotype network46.
The ABGD algorithm is a computationally efficient distance-based method and seeks to find the ___location of the barcode gap that separates intra- from interspecific distances at a given pairwise distance matrix. As ABGD requires the specification of input parameter values, we performed our analysis with the following values, according to Ratnasingham and Hebert42: Pmin = 0.005, Pmax = 0.1, X (relative gap width) = 1.0, and simple distance to construct the matrix. We considered the partition with prior maximal distance (P) = 0.013, which is between 1.0% and 2.5%, following Pinto et al.18. All analyses were run in the web server https://bioinfo.mnhn.fr/abi/public/abgd/, lastly accessed on June 20, 2024.
Similarly, ASAP is also a distance-based method that employs threshold values to distinguish between interspecific divergence and intraspecific variation. In this case, the partitions are ranked by a score system. ASAP analysis was run with default parameters and simple distances, and the best partition considered was the one with the lowest ASAP-score (i.e., 2.0), with distance threshold of 0.042. ASAP analysis was run in the web server https://bioinfo.mnhn.fr/abi/public/asap/, lastly accessed on June 20, 2024.
The RESL method employs single linkage clustering as a tool for the preliminary assignment of records into MOTUs. Sequences are initially clustered by employing a fixed 2.2% threshold of uncorrected p-distance, and refined into the final partitions by Markov clustering. This algorithm is implemented directly in the BOLD systems and is used to generate the Barcode Index Numbers (BIN) in this database. We performed RESL analysis by constructing a new dataset with our COI sequences and those processed by Pinto et al.18 directly in BOLD (https://www.boldsystems.org/, accessed on June 20, 2024), and then the ‘cluster sequences’ tool was performed.
Lastly, GMYC and PTP are coalescent-based methods that seek to identify transition points between species-level processes (speciation) and population-level processes (allele coalescence) events in phylogenetic trees. These two methods differ in the input data; GMYC is intended to be performed using ultrametric gene trees, whereas PTP does not require that the input tree be time-calibrated. Thus, we employed the GMYC analysis using the MCC tree generated by BEAST as input data in the web server https://species.h-its.org/gmyc/, lastly accessed on June 20, 2024, with the ‘single threshold’ method. PTP analysis was performed using the maximum likelihood phylogenetic tree by IQ-TREE as input in the web server https://mptp.h-its.org/, lastly accessed on June 20, 2024, using both multi and single rate methods (which provided the same result). Both trees were rooted at the mid-point and no outgroups were included for species delimitation.
Population structure
We calculated the genetic indices such as number of variable sites, nucleotide diversity, haplotype diversity, and neutrality tests for all populations of Pi. monticola and Pi. misionensis in the DnaSP v6.12 program47.
To check the population structure signal within our COI dataset, we performed a hierarchical and non-hierarchical Analysis of Molecular Variance (AMOVA)48 for each species in the Arlequin v3.5 program49. Global Fst and other F-statistics values had their significance tested with 10,000 permutations.
For hierarchical analysis, we defined groups according to biomes, areas of endemism and the main ecoregions of the Atlantic Forest50. Then, Pi. monticola was grouped into the following geographical regions: (i) North of the Doce River [NDR], (ii) South of the Doce River [SDR], (iii) Serra do Mar Coastal Forest [SMC], (iv) Araucaria moist Forest [ARA], (v) Alto Paraná Atlantic Forest [APA], and (vi) Bahia Interior Forest [BIF]. Pintomyia misionensis was grouped as follows: (i) Serra do Mar Coastal Forest [SMC], (ii) South of the Doce River [SDR], and (iii) Piraputanga (transitions of Cerrado and Pantanal) [PIR]. All regions, except for Piraputanga, are in the Atlantic Forest biome, or its transition zone.
The phylogeographic structure was also visualized by inferring a Median-Joining haplotype network based on our COI dataset for each species in the PopART software (Population Analysis with Reticulate Trees)51.
Geometric morphometry
For wing geometric morphometry (GMM) analysis, we selected specimens that were also processed for molecular analysis and had their COI-barcodes generated. Therefore, we only analyzed individuals in which the thorax (and consequently the wings) was not used for DNA extraction. Here, only female specimens were evaluated to avoid allometry bias, and lack of male samples. The groups to be tested were assembled based on the main molecular lineages discovered by an intermediate scenario of species delimitation analysis (i.e., PTP. See below). The final dataset comprises 64 individuals of Pi. monticola (25 for genetic lineage I, and 39 for lineage II), and 27 of Pi. misionensis (22 for lineage I, and five for lineage II).
The images were taken using the Zen version 4.7 program, with an Axiocam 105 color (Carl Zeiss MicroImaging GmbH, Jena, Germany) coupled to an optical microscope with a 2.5× objective lens. Then, a total of 13 wing landmarks were selected and set in TpsDig v2.3. Landmarks are located at the margin of the wing (R1; R2; R3; R4; R5; M1; M2; M3; and M4), and in the intersection of these veins (base of R5; R2/R3; R2/R3/R4; and M1/M2), as previously described in Riva et al.52 and Mikery et al.53. We evaluate the shape variation, the generalized procrustes analysis, procrustes analysis of variance (ANOVA), the principal component (PCA), the canonical variate (CVA), and discriminant function (DF) analyses using the MorphoJ v1.07 program54. Also, we exported the log-transformed centroid sizes values from MorphoJ and the mean difference per species was compared by an analysis of variance (ANOVA), followed by pairwise comparisons between lineages within species with tukey’s test in the GraphPad Prism v9.5 program.
Linear morphometry
For linear morphometry (LM), we also used specimens that were processed for molecular analysis and have COI-barcodes available in this study. This time, all genetic lineages could be tested, since all sand flies had at least the head and abdomen available for linear measurements. The total number of specimens analyzed for each species and genetic lineages are available in Tables S3 and S4 for Pi. monticola and Pi. misionensis, respectively. The following morphological characters were assessed when available: head length and width, clypeus, eye length and width, interocular distance, length of flagellomere F1, F13, and F14, labrum-ephipharynx (LE), wing width, wing indices R5, R2, R2 + 3, R2 + 3 + 4 and delta, mesonotum length, genital fork: chamber and stem length, and cerci length. The measurements, given in micrometers, were obtained as described in the GMM section, using objectives of 2.5x, 10x or 40x depending on the length of the structure.
We tested whether the normal distribution is applicable to each character considering the three genetic lineages of each species as independent groups. We performed the Shapiro-Wilk test in the Past v4.17 and GraphPad Prism v9.5 programs. The characters that did not follow a normal distribution in at least one of the groups were considered nonparametric in our subsequent analyses. For characters in which the measurement of structures was performed only in two of the three genetic groups due to the unavailability of the insect segments or the difficulty in visualizing them, we used the unpaired t-test and Mann-Whitney for parametric and non-parametric characters, respectively. For those in which observations were performed in the three groups for each species, we performed an Analysis of Variance (ANOVA) with Tukey’s post-test, and Kruskal-Wallis with Dunn’s post-test for parametric and non-parametric characters, respectively. These analyses were performed in the Past v4.17 and GraphPad Prism v9.5 programs. Boxplots were generated in GraphPad Prism v9.5.
Results
Molecular taxonomy
We generated COI-barcode fragments for 68 and 11 specimens of Pi. monticola and Pi. misionensis, respectively, which ranged from 635 base pairs (bp) to 658 bp (full-length barcode fragment). To set up the final molecular dataset, we included publicly available COI sequences extracted from NCBI GenBank, totaling 152 specimens of both taxa in the final dataset (Table 1 and S1). Following alignment, the visual inspection revealed the absence of pseudogenes and/or nuclear copies of mitochondrial origin. This was achieved by carefully visualizing the alignment, searching for potential indels, and in-frame stop codons.
Pairwise genetic distances ranged from 0 to 8.21% (mean 4.17%) for Pi. monticola, and 0–9.12% (mean 4.4%) for Pi. misionensis specimens. Interspecific comparisons ranged from 10.94 to 14.09% (mean 12.33%), indicating that these two classes of distances do not overlap in our dataset.
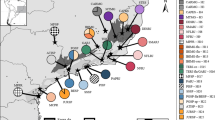
The phylogenetic gene trees generated by both the Bayesian Inference and Maximum Likelihood methods exhibited well-supported clades (posterior probabilities > 0.95; bootstrap > 70) for both species, with clear structuring patterns within them, indicating the presence of multiple lineages for both species (Figs. 1 and 2 and S1). In the case of Pi. monticola, two main clades were identified, with no clear distinction between the geographical localities of the specimens. However, the second clade demonstrated a cluster of related specimens from Intervales Park (south coast of São Paulo state) and Piraquara municipality (coast region of Paraná state) (Fig. 1). Conversely, Pi. misionensis exhibited three highly divergent and well-supported clades, as evidenced by the Brazilian states of Espírito Santo, Mato Grosso do Sul, and São Paulo (Fig. 2).

Phylogeographic analysis of Pintomyia monticola populations based on COI sequences in Brazil. Left: Bayesian maximum credibility (MCC) tree with terminal labels being GenBank accession numbers. The color pattern of tip shapes and labels corresponds to the sampling localities. Values near nodes are posterior probabilities > 0.95. Bars adjacent to terminals represent MOTU delimitation made by corresponding algorithms and are colored according to the intermediate scenario given by PTP. Top right: Median Joining Haplotype network. Bottom right: Map of sample localities. The different locations were grouped and highlighted according to the main geographical regions mentioned in the methods section: NDR North of the Doce River, SDR South of the Doce River, SMC Serra do Mar Coastal Forest, ARA Araucaria moist Forest, APA Alto Paraná Atlantic Forest, BIF Bahia Interior Forest.

Phylogeographic analysis of Pintomyia misionensis populations based on COI sequences in Brazil. Left: Bayesian maximum credibility (MCC) tree with terminal labels being GenBank accession numbers. The color pattern of tip shapes and labels corresponds to the sampling localities. Values near nodes are posterior probabilities > 0.95. Bars adjacent to terminals represent MOTU delimitation made by corresponding algorithms and are colored according to the intermediate scenario given by PTP. Top right: Median Joining Haplotype network. Bottom right: Map of sample localities. The different locations were grouped and highlighted according the main geographical regions mentioned in the methods section: SDR South of the Doce River, SMC Serra do Mar Coastal Forest, PIR Piraputanga.
Single-locus species delimitation partially agrees with the main clades of phylogenetic trees, since some algorithms oversplitted our dataset; ABGD and ASAP were the most conservative ones, and sorted COI sequences into five MOTUs, three for Pi. misionensis and two for Pi. monticola, according to the main clades of the phylogenetic tree (Figs. 1 and 2). Additionally, PTP identified three MOTUs for Pi. monticola, splitting the cluster of specimens from Intervales/Piraquara into a different MOTU, being the second most conservative partition. The species delimitation scenarios of RESL, GMYC, and TCS split our dataset of two nominal species into 12, 41, and seven MOTUs, respectively (Fig. 1), without agreeing with other lines of evidence, and are therefore considered unsuitable for our dataset. Thus, for further morphometric analysis, we considered an intermediate species delimitation scenario generated by the PTP (see Discussion section), which indicated the partition of Pi. monticola and Pi. misionensis into three MOTUs each (Figs. 1 and 2).
Population genetics
We identified 48 haplotypes among the 152 analyzed COI sequences, 38 for Pi. monticola and 10 for Pi. misionensis (Table 1). The haplotype diversity was high for both species: 0.934 and 0.849, respectively, but it varies greatly depending on the sampling ___location, ranging from 0.25 to 1.0 (Table S2). Some haplotypes were shared among localities, in the same sense that different localities have multiple haplotypes (Table 1; Figs. 1 and 2). Neutrality tests were not significant for any of the populations analyzed, indicating the absence of selection evidence.
The hierarchical AMOVA analysis of Pi. monticola populations from different ecoregions of the Atlantic Forest revealed that the highest percentage of variation was observed among geographical groups (34.81%) and among populations within groups (46.2%) (Table 2). Besides that, when groups are not assigned, the highest source of variation is among populations (60.49%), with a global Fst of 0.604 (p < 0.0001). Regarding Pi. misionensis, AMOVA analysis shows even higher rates of population structure, with a global Fst of 0.921 (p < 0.0001) (Table 2).
All the pairwise Fst comparisons between different geographical regions were significant. For Pi. monticola, the highest value was observed between populations of North of the Doce River (NDR) and Alto Paraná Atlantic Forest (APA) (Fst = 0.98), while the lowest was between the South of Doce River (SDR) and Serra do Mar Coastal Forest (SMC) (Fst = 0.28). For Pi. misionensis, the Fst for population comparisons was always greater than 0.9 (Table 3).
Geometric morphometry
The procrustes ANOVA for the centroid size, and shape, were both significative, with a p value of 0.008 and < 0.0001, respectively. Additionally, for centroid sizes, the ANOVA multiple comparisons of populations pairs showed significant p value (0.0283) for Pi. misionensis I/Pi. misionensis II, but not for Pi. monticola I/Pi. monticola II (p = 0.5717).
The analysis of principal components (PCA) showed major differences in landmark 13 (reduction of R5) for both principal components 1 and 2 (PC1 and PC2), while PC1 also showed great displacement of landmarks 2 and 12 towards the base of the wing affecting the R2 + 3 + 4 (γ).
The CVA showed that our dataset accounted for 93% of the total variance on the first two canonical variates. Pintomyia monticola and Pi. misionensis appears to be completely segregated, as the same for the two genetic lineages of Pi. misionensis (Fig. 3). Additionally, Pi. monticola seems to be clustered into the different groups based on genetic data, but some of the specimens overlap (Fig. 3). In the same sense, Procrustes and Mahalanobis distances among groups were greater among species, and the lowest value was observed for the comparison of Pi. monticola I and P. monticola II (Table 4). After discriminant function analysis, the pairwise cross-validated species reclassification test generally shows high levels of accuracy, with the lowest value for the Pi. monticola I/P. monticola II (Table 5).

Canonical Variate (CV) Analysis of Procrustes coordinates of Pintomyia monticola and Pintomyia misionensis. Left: Phylomorphospace of wing shapes with 95% confidence ellipses for CV1 and CV2; Right: Warp grids depicting wing shapes for CV1 and CV2.
Linear morphometry
Regarding Pi. monticola genetic lineages, there was a significant difference in the measurements of the following characters: head length, clypeus, interocular distance, flagellomere F1, flagellomere F13, labrum-ephipharynx, wing veins R2 + 3, R2 + 3 + 4, and mesonotum (Table S3, Fig. 4). Although there is a visual tendency for differentiation between the groups, the measurement values overlap in all comparisons.

Linear morphometry measurements and comparisons between different genetic lineages of Pintomyia monticola. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Similarly, the genetic lineages of Pi. misionensis were statistically different regarding the following character’s measurements: head length and width, clypeus, eye length and width, interocular distance, flagellomere F1, labrum-ephipharynx, wing width, wing veins R5, R2, R2 + 3 and R2 + 3 + 4, and mesonotum (Table S4, Fig. 5). In general, Pi. misionensis II (São Paulo) are larger than I (Espírito Santo) and III (Mato Grosso do Sul). Also, the measurement values may be diagnostic because do not overlap between lineages while comparing the: head length between Pi. misionensis II and Pi. misionensis III; head width II ≠ I/III; eye length II ≠ I; eye width II ≠ I; interocular distance II ≠ III; labrum-ephipharynx II ≠ I/III; wing width II ≠ I; R2 + 3 + 4 II ≠ I.

Linear morphometry measurements and comparisons between different genetic lineages of Pintomyia misionensis. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Discriminant analysis of linear morphometry shows discrepant results of genetic lineages differentiation of Pi. monticola and Pi. misionensis. Similar to GMM results, the three lineages of Pi. monticola seem to have a tendency towards differentiation, but with some overlapping specimens (Fig. 6). However, in agreement with the linear measurements, the specimens of the different Pi. misionensis lineages did not overlap between them (Fig. 6). The mean of correctly classified specimens was 47% and 94% for Pi. monticola and Pi. misionensis, respectively (Table 6).

Linear discriminant analysis between different genetic lineages in Monticola series.
Discussion
Our findings indicate that species within the Monticola series may be more diverse than previously assumed. The integration of multiple lines of evidence, such as molecular markers and morphometry, can provide assumptions for the elucidation of taxonomic issues27,55. Traditionally, the sole criterion for identifying and describing sand fly species was the morphological characteristics of the diagnostic features. However, this approach is ineffective in distinguishing closely related species, as evidenced by the case of isomorphic Brumptomyia, Trichophoromyia, and Trichopygomyia females4. Additionally, some well-recognized cryptic species complexes, such as Lutzomyia longipalpis sensu lato, are challenging to formally describe due to the absence of discrete morphological attributes that are reliably diagnostic for species identification56. The cryptic diversity uncovered in this study for Pi. monticola and even more so for Pi. misionensis, indicate that these taxa may be cryptic species, but despite the detection of a large morphological variation, none of them are sufficiently diagnostic for the formal description of new taxa. For instance, in some specimens of Pi. monticola there are many bristles on the abdominal pleura, while in others they are few or absent, but this was unrelated to the other lines of evidence observed in our study. Indeed, the taxonomic impediment leads to species descriptions being made at a slower pace than species become extinct57, so proposals have been made to describe new taxa based on DNA barcoding approaches, which can accelerate the discovery and naming of new taxa in hyperdiverse groups58. However, this ‘minimalist’ taxonomy proposal has been harshly criticized, and one of the many issues relies on the usage of single mtDNA markers59. Here, although multiple COI lineages were tested for morphological differences, only this molecular marker and the absence of reliable diagnostic characters prevent the description of new species. However, we argue that both taxa, especially Pi. misionensis, should be further investigated employing a greater number of molecular markers, populations, and individuals. This could potentially lead to the description of distinct taxa within this nominal species.
The genetic divergence of COI barcodes has yielded significant insights into the investigation of cryptic diversity in sand flies. The measurement of intra- and interspecific genetic distances has been performed since the first proposal of DNA barcoding, usually using the Kimura 2-parameter (K2P) model20. It is thus expected that pairwise distances within species are much lower than between species60, but some well-sampled datasets may overlap these classes of distances61,62. The absence of the ‘barcoding gap’ does not make the use of DNA barcoding unfeasible to identify species and discover cryptic diversity, but its presence empowers its assumptions. Here, the maximum genetic divergence within species (9.12%) and the minimum to the nearest neighbor (10.94%) do not overlap, showing that COI sequences can be used as a reliable tool for species identification of both taxa in Monticola series. Moreover, the unexpectedly high intraspecific divergence in both taxa indicates that multiple lineages could be detected. In general, for sand flies, intraspecific pairwise distances do not reach values greater than 3–5%63,64,65, and even different nominal species may have interspecific genetic divergences of less than 3%, such as within Nyssomyia and Trichophoromyia genera66. Thus, our results indicate that the genetic divergences within species of the Monticola series are similar to other genus-level comparisons than species-level as expected. These findings may be seen in other highly widespread taxa where there is an indication that they belong to cryptic species complexes, such as Pintomyia evansi (maximum K2P = 8.92%), Lutzomyia longipalpis (max K2P = 15.4%), and Bichromomyia flaviscutellata (max K2P = 11.8%)66,67,68. The aforementioned results are directly related to the sampling of specimens from different geographic locations, with great divergence in allopatric lineages. Here, multiple genetic lineages were detected in the same sampled locality (i.e., sympatric cryptic diversity), indicating that factors other than geographic ones have influenced the structuring of Pi. monticola.
The species delimitation algorithms differ in detecting different MOTUs within the same species, but all agree that, Pi. monticola and Pi. misionensis comprises multiple lineages. We performed a set of single-locus approaches that perform differently in the interpretation of molecular data, but none of them uses a priori information for species delimitation (i.e., discovery methods). Using multiple methods and checking congruence between them is recommended, but more conservative delimitations are preferable when checked against other lines of evidence39,69. Here, GMYC clearly oversplitted our dataset into 41 unrealistic MOTUs. This method is widely disseminated and used in several taxa to discover cryptic diversity using ultrametric gene trees43, but these are generally more prone to inference errors than the maximum likelihood ones, and GMYC seems to overestimate the real number of MOTUs with discontinued sampling efforts of populations/species70,71,72,73. Not to the same extent, but RESL and TCS also overestimated the real number of MOTUs in our dataset. These two methods have been used to identify cryptic diversity in sand flies, and they seem to perform better when analyzing less divergent taxa, tending to overspilt highly divergent COI datasets (e.g., Goulpeau et al.74). For instance, RESL is the only method able to correctly delineate close-related Nyssomyia spp.66, and TCS reliably assign multiple lineages of Psychodopygus davisi24, but were not useful in delimiting MOTUs in the Monticola series. Thus, two distance-based (ABGD and ASAP), and one tree-based (PTP) species delimitation methods were best suited to our dataset. The distance-based algorithms split Pi. monticola and Pi. misionensis into two and three MOTUs, respectively, whereas PTP agrees with this partition and split one of these Pi. monticola lineages, totaling three MOTUs for each species. In this case, we consider the PTP partitions as more reliable because (i) empirical observations and simulated datasets indicate that ABGD may overlump well-structured populations/lineages or even morphospecies69,73,75,76, including sand fly COI datasets66,68; and (ii) the additional MOTU of Pi. monticola identified by PTP agrees with the geographical localities of samples, which are from ‘Piraquara’ in Paraná state (South Brazil), and ‘Intervales State Park’ in the South region of São Paulo state. These two localities are the only ones having influence of Araucaria moist Forest, Paraná/Araucária area of endemism50, and were clustered together in the phylogenetic gene tree and species discovery analysis.
Moreover, the different biogeographical regions of samples have influenced the detection of high rates of genetic divergence. The AMOVA results show strong genetic structure between populations, with all comparisons being significant. This is especially true in the case of Pi. misionensis, where 88% of the variation source was observed among groups and Fst values between geographical regions were greater than 0.9. For sand fly COI datasets, it has been shown that high percentage of variation between populations can be detected in well-recognized species complexes, such as Psathyromyia shannoni sensu lato23 and Nyssomyia umbratilis77,78, which may be associated with geographic and climatic barriers in South America, isolation by distance and the major Amazonian rivers. Regarding our findings for Pi. misionensis, the discontinued sampling effort may have also influenced the high genetic structure. In contrast, the sampling of Pi. monticola was better explored, counting on several populations from different biogeographic regions of the Atlantic Forest biome. In this case, hierarchical AMOVA showed that 35% of variation can be found among groups and 46% among populations within groups, with a global Fst of 0.6, indicating that the genetic structure of Pi. monticola is moderate compared to Pi. misionensis. This probably happened because some populations have individuals that have been allocated in different genetic lineages detected by PTP partitions, as in the case of ‘Piraquara – PR’, ‘PESM Santa Virgínia – SP’, and ‘South of the Doce River’, so that the within-population variation was computed. Although the multiple lineages of Pi. monticola are in sympatry, population structure values remain high, indicating that the heterogeneity of this biome has a significant influence on the diversity of this species.
The Atlantic Forest is one of the most biodiverse and threatened biomes worldwide, and has been historically devastated by human actions, since more than half of Brazil’s population lives in regions of this biome, and concentrates the main metropolitan centers, such as São Paulo79. It is a very complex biome with a vast diversity of geographical features, including different climatic regimes, and a large altitude and latitude range, favoring high diversity and different endemism centers50. The historical factors that drove its lineages and species diversity are still in debate. However, the Doce River, located in the center of Espírito Santo state seems to be a relevant transition site between different bioclimatic domains in the Atlantic Forest, which is related to strong biological turnover80. Although the rivers in this biome are not as large as the Amazonian ones, they can influence the genetic structure of mtDNA lineages81,82,83. The pairwise Fst of Pi. monticola populations across different sides of the Doce River (within Espírito Santo state) is high (0.71), supporting this assumption. There is a tendency for these populations to belong to distinct genetic lineages discovered by PTP, but individuals from the South of the Rio Doce occur in multiple lineages, sometimes related to the North population, indicating some degree of gene flow or retention of ancestral features, which need further confirmation using genomic datasets and demographic analysis. Furthermore, the diversification south of Doce River, including the full extension of this biome, has been shaped by other processes. Here, the populations of ‘Cássia dos Coqueiros – SP’ and ‘Serra do Cipó – MG’ are in the interior plateau of the biome, in two different transitions areas with the Brazilian Savanah (Cerrado biome), and is expected that these populations may have some degree of genetic structure concerning coastal populations due to variations in phytophysiognomies, as has been shown for Opiliones84. Finally, coastal populations of ‘Serra do Mar’ and ‘Araucaria’ areas of endemism showed moderate but significative genetic structure (Fst = 0.3) between them, and almost all samples from ‘Araucaria’ clustered into the same MOTU which was here analyzed as a different molecular lineage (Pi. monticola III). The population of ‘Piraquara – PR’ and ‘Intervales State Park – SP’ are below the ‘São Paulo subtropical gap’ (SPSG), which has a major effect on the genetic structure of Morpho butterflies85, and may have also influenced the genetic structure of Pi. monticola. In general, the biodiversity of the Atlantic Forest has been shaped during periods of glaciation in the Pleistocene – which appears to affect most of the southern region of the biome – when there were intense retractions and expansions of forest fragments viable for these insects, and habitat shifts, which resulted in structured populations and absence of gene flow for many generations50. To confirm these prerogatives and propose more reliable scenarios of the biogeography of Pi. monticola, datasets that include non-mitochondrial genes, populations and time-calibrated analysis are necessary.
As the different genetic lineages were not entirely related to biogeographical regions, we explicitly tested them for morphological differences using only DNA-barcoded sand flies, to check whether COI lineages are related to morphological variation. Qualitative characters such as spermathecae shape, presence/absence of Newstead’s sensilla, setae in abdominal pleura, and ascoidal formula were checked across lineages of both species. No concrete diagnostic characteristics that justify the description of new species could be identified, despite the lineages are statistically different when analyzing linear and geometric morphometry, showing a tendency towards incipient morphological differentiation. Regarding the wing GMM, much of the variation was detected in the shape rather than centroid sizes, which are in accordance with mosquitoes’ observations that indicate the wing shape as an indicator of population structure, while wing size tends to be more sensitive to environmental changes86. Pintomyia misionensis represents two distinct GMM clusters, the first comprising specimens from Espírito Santo (South of the Doce River) and the second with sand flies from São Paulo (PESM – Santa Virgínia). Both clusters are in the coastal Atlantic Forest, but in different areas of endemism, which may have impacted this shape variation. In contrast, Pi. monticola GMM clusters overlap and have the smallest distance values of Mahalanobis and Procrustes distances. This also had an impact on the reclassification test, which correctly classified at least 52% of Pi. monticola specimens in the correct lineage. Correct classifications after discriminant analysis may be lower than 50% while comparing close-related species87. The geometric morphometry of sand fly wings struggles to identify different nominal species even for distinct nominal species, as in the case of the close-related pairs Brumptomyia avellari/Brumptomyia brumpti and Lutzomyia longipalpis/Lutzomyia cruzi87,88. However, GMM appears to be an effective method for classifying geographically distinct populations within taxa24,53,89. This may be related to distinct genetic lineages25 and/or epidemiological relevance89. Therefore, the two genetic lineages of Pi. monticola that presented some degree of morphological differentiation should be further investigated.
The linear morphometry analysis could be evaluated in all genetic lineages identified in this study, as all individuals had at least the head and abdomen mounted on slides for microscopy. Several characters show significant differences between the groups, especially in the head, demonstrating that it can be a valuable body segment for other geometric morphometry analyses within Monticola series (e.g., Godoy et al.90). For instance, the measurement values of labrum-ephipharynx and head width of Pi. misionensis II (São Paulo Serra do Mar) did not even overlap in comparison to the other lineages, such is the discrepancy. Similar results were observed for Migonemyia migonei while comparing geographically distinct populations, but this time the length of flagellomere F1 was different among populations26. Moreover, reclassification accuracy for Pi. misionensis groups were much higher than Pi. monticola, which is in accordance with GMM results. The two methods, linear and geometric, typically yield concordant results concerning the inference of morphological tendencies, thereby enhancing the robustness and reliability of the findings53. Nevertheless, both interpretations should be carried out with caution, given that morphometry can be significantly affected by environmental pressures86,91,92, which were not evaluated in this study. Many factors such as altitude, temperature, population sizes and food sources may have influenced the high morphological disparity of Pi. misionensis, but certainly does not explain the morphological variation of Pi. monticola, since the groups tested for morphological differences in this species had a sympatric distribution, with specimens collected under the same environmental conditions. Besides that, the discrepant clustering pattern of Pi. monticola and Pi. misionensis may also be a consequence of the discontinuous sampling of the later species, leading to the absence of intermediate forms in the analysis.
Conclusions
In this study, we showed that the molecular and morphological diversity in the Monticola Series is greater than expected. Our integrative taxonomy effort detected at least three genetic lineages for each of the analyzed species. The hidden diversity within Pi. monticola is partially related to geographically distinct populations of Atlantic Forest, but multiple genetic lineages may be found in the same collection site, showing a sympatric cryptic diversity pattern. In contrast, Pi. misionensis diversity is highly impacted by biogeographical regions, although discontinued sampling of this taxon may have led to this divergent pattern. The COI genetic lineages generated in this study were partially or completely confirmed by linear and geometric morphometry, demonstrating that species of the Monticola Series may be a complex of cryptic species. These results are insufficient for the description of new taxa, mainly due to biases related to the use of single-locus mtDNA datasets and the absence of diagnostic reliable morphological traits. Due to the wide distribution of these taxa, future studies analyzing a larger number of populations, individuals, and nuclear molecular markers are needed to confirm these findings and elucidate the natural history of the Monticola series.
Data availability
The data that support the findings of this study are openly available in the GenBank database at https://www.ncbi.nlm.nih.gov/genbank/, under accession numbers PP865228-PP865306.
Change history
09 December 2024
A Correction to this paper has been published: https://doi.org/10.1038/s41598-024-81429-4
References
Cecílio, P., Cordeiro-da-Silva, A. & Oliveira, F. Sand flies: basic information on the vectors of leishmaniasis and their interactions with Leishmania parasites. Commun. Biology. 5, 305 (2022).
Galati, E. A. B. & Rodrigues, B. L. A review of historical Phlebotominae taxonomy (Diptera: Psychodidae). Neotrop. Entomol. 52, 539–559 (2023).
Galati, E. A. B. phylogenetic systematics of the Phlebotominae (Diptera, Psychodidae) with emphasis on American groups. Boletín De Malariología Y Salud Ambiental. 35, 133–142 (1995).
Galati, E. A. B. & Phlebotominae (Diptera, Psychodidae): classification, morphology and terminology of adults and identification of American Taxa. In: (eds Rangel, E. & Shaw, J.) Brazilian Sand Flies. Springer, Cham (2018).
Ibáñez-Bernal, S., Kraemer, M. S., Stebner, F. & Wagner, R. A new fossil species of Phlebotominae sand fly from Miocene amber of Chiapas, Mexico (Diptera: Psychodidae). PalZ. 88, 227–233 (2014).
Galvis-Ovallos, F. et al. Detection of Pintomyia Fischeri (Diptera: Psychodidae) with Leishmania infantum (Trypanosomatida: Trypanosomatidae) promastigotes in a focus of visceral leishmaniasis in Brazil. J. Med. Entomol. 58 (2), 830–836 (2021).
ReadyP.D. Biology of phlebotomine sand flies as vectors of disease agents. Ann. Rev. Entomol. 58, 227–250 (2013).
Andrade, A. J. & Gurgel-Gonçalves, R. New record and update on the geographical distribution of Pintomyia monticola (Costa Lima, 1932) (Diptera: Psychodidae) in South America. Check List. 11 (2), 1566 (2015).
Margonari, C. et al. Phlebotomine sand flies (Diptera: Psychodidae) and Leishmania infection in Gafanhoto park, Divinópolis, Brazil. J. Med. Entomol. 47 (6), 1212–1219 (2010).
Donalisio, M. R. et al. Visceral leishmaniasis in an environmentally protected area in southeastern Brazil: epidemiological and laboratory cross-sectional investigation of phlebotomine fauna, wild hosts and canine cases. PLoS Negl. Trop. Dis. 11 (7), e0005666 (2017).
Luz, E., Giovannoni, M., Borba, A. M. & de Infecção Lutzomyia monticola por Leishmania enriettii. Anais da Faculdade de Medicina da Universidade Federal do Paraná 9, 121–128 (1967).
Lainson, R. On Leishmania Enriettii and other enigmatic Leishmania species of the neotropics. Memórias do Instituto Oswaldo Cruz. 92, 377–387 (1997).
Galati, E. A. B., Marassá, A. M., Gonçalves-Andrade, R. M., Consales, C. A. & Bueno, E. F. Phlebotomines (Diptera, Psychodidae) in the Ribeira Valley Speleological Province-1. Parque Estadual Intervales, state of São Paulo, Brazil. Revista Brasileira De Entomol. 54, 311–321 (2010).
Pinto, I. S. et al. Sand fly vectors (Diptera, Psychodidae) of American visceral leishmaniasis areas in the Atlantic Forest, State of Espírito Santo, southeastern Brazil. J. Vector Ecol. 37, 90–96 (2012).
Serra et al. Phlebotominae Fauna (Diptera: Psychodidae) and molecular detection of Leishmania (Kinetoplastida: Trypanosomatidae) in Urban caves of Belo Horizonte, Minas Gerais, Brazil. J. Med. Entomol. 59 (1), 257–266 (2022).
Infran, J. O. Nycthemeral rhythm of phlebotominae (Diptera: Psychodidae) in a craggy region, transitioning between the Wetland and the Plateau, Brazil. J. Med. Entomol. 54 (1), 114–124 (2017).
Silva, O. S. & Grunewald, J. Contribution to the sand fly fauna (Diptera: Phlebotominae) of Rio Grande do sul, Brazil and Leishmania (Viannia) infections. Memórias do Instituto Oswaldo Cruz. 94, 579–582 (1999).
Pinto, I. S. et al. DNA barcoding of neotropical sand flies (Diptera, Psychodidae, Phlebotominae): species identification and discovery within Brazil. PLoS One 10(10), e0140636 (2015).
Rodrigues, B. L. & Galati, E. A. Molecular taxonomy of phlebotomine sand flies (Diptera, Psychodidae) with emphasis on DNA barcoding: a review. Acta Trop. 238, 106778 (2023).
HebertP.D., Cywinska, A., Ball, S. L. & DeWaardJ.R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. B Biol. Sci. 270(1512), 313–321 (2003).
Guernaoui, S., Hamarsheh, O., Garcia, D., Fontenille, D. & Sereno, D. Population genetics of Phlebotomus papatasi from endemic and nonendemic areas for zoonotic cutaneous leishmaniasis in Morocco, as revealed by cytochrome oxidase gene subunit I sequencing. Microorganisms. 8(7), 1010 (2020).
Wedage, W. M., Harischandra, I. N., Weerasena, O. J. & Silva, B. G. Genetic diversity and phylogeography of Phlebotomus argentipes (Diptera: Psychodidae, Phlebotominae), using COI and ND4 mitochondrial gene sequences. Plos One. 18(12), e0296286 (2023).
Lozano-Sardaneta, Y. N., Viveros-Santos, V., Colunga-Salas, P., Rodríguez-Rojas, J. J. & Becker, I. Is psathyromyia shannoni (Diptera: Psychodidae: Phlebotominae) a species complex? Retrospective study of genetic diversity of COI gene, pathogens and geographic distribution. Acta Trop. 238, 106807 (2023).
Rodrigues, B. L. Molecular and morphometric study of Brazilian populations of Psychodopygus Davisi. Med. Vet. Entomol. 38(1), 83–98 (2024).
Souza Freitas, M. T. Phenotypic and genotypic variations among three allopatric populations of Lutzomyia Umbratilis, main vector of Leishmania guyanensis. Parasites Vectors. 8, 448 (2015).
Costa, P. L. et al. Morphological and phylogenetic analyses of Lutzomyia migonei from three Brazilian states. Acta Trop. 187, 144–150 (2018).
Padial, J. M., Miralles, A., De la Riva, I. & Vences, M. The integrative future of taxonomy. Front. Zool. 7, 16 (2010).
Struck, T. H. et al. Finding evolutionary processes hidden in cryptic species. Trends Ecol. Evol. 33(3), 153–163 (2018).
Rodrigues, B. L., Pinto, I. S. & Galati, E. A. Morphological and DNA-based description of Trichophoromyia peixotoi n. sp. (Diptera: Psychodidae), a new sand fly species from the Brazilian Amazon. Parasites Vectors. 16(1), 240 (2023).
Folmer, O., Black, M., Hoeh, W., Lutz, R. & Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3, 294–299 (1994).
Ratnasingham, S. et al. BOLD v4: a centralized Bioinformatics platform for DNA-Based Biodiversity Data. In: DNA Barcoding: Methods and Protocols (403–441) (Springer US, New York, NY, 2024).
Sayers, E. W. et al. GenBank 2023 update. Nucleic Acids Res. 51(D1), D141–D144 (2023).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33(7), 1870–1874 (2016).
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32 (1), 268–274 (2015).
Trifinopoulos, J., Nguyen, L. T., von Haeseler, A. & Minh, B. Q. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44(W1), W232–W235 (2016).
Bouckaert, R. et al. BEAST 2.5: an advanced software platform for bayesian evolutionary analysis. PLoS Comput. Biol. 15(4), e1006650 (2019).
Bouckaert, R. R., Drummond, A. J. & bModelTest Bayesian phylogenetic site model averaging and model comparison. BMC Evol. Biol. 17, 42 (2017).
Rodrigues, B. L., Baton, L. A. & Shimabukuro, P. H. Single-locus DNA barcoding and species delimitation of the sandfly subgenus Evandromyia (Aldamyia). Med. Vet. Entomol. 34 (4), 420–431 (2020).
Carstens, B. C., Pelletier, T. A., Reid, N. M. & Satler, J. D. How to fail at species delimitation. Mol. Ecol. 22 (17), 4369–4383 (2013).
Puillandre, N., Lambert, A., Brouillet, S., Achaz, G. J. & ABGD Automatic barcode gap Discovery for primary species delimitation. Mol. Ecol. 21 (8), 1864–1877 (2012).
Puillandre, N., Brouillet, S. & Achaz, G. ASAP: assemble species by automatic partitioning. Mol. Ecol. Resour. 21 (2), 609–620 (2021).
Ratnasingham, S. & Hebert, P. D. N. A DNA-based registry for all animal species: the Barcode Index Number (BIN) system. PLoS One. 8, e66213 (2013).
Fujisawa, T. & Barraclough, T. G. Delimiting species using single-locus data and the generalized mixed Yule Coalescent approach: a revised method and evaluation on simulated data sets. Syst. Biol. 62 (5), 707–724 (2013).
Zhang, J., Kapli, P., Pavlidis, P. & Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics. 29 (22), 2869–2876 (2013).
Kapli, P. et al. Multi-rate Poisson tree processes for single-locus species delimitation under maximum likelihood and Markov chain Monte Carlo. Bioinformatics. 33 (11), 1630–1638 (2017).
Clement, M., Posada, D. C. & Crandall, K. A. TCS: a computer program to estimate gene genealogies. Mol. Ecol. 9 (10), 1657–1659 (2000).
Rozas, J. et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34 (12), 3299–3302 (2017).
Excoffier, L., Smouse, P. E. & Quattro, J. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 131 (2), 479–491 (1992).
Excoffier, L. & Lischer, H. E. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10 (3), 564–567 (2010).
Peres, E. A. et al. Patterns of species and lineage diversity in the Atlantic Rainforest of Brazil. In V. Rull & A. C. Carnaval (Eds.), Neotropical diversification: Patterns and processes (pp. 415–447). Springer International Publishing (2020).
Leigh, J. W., Bryant, D. & Nakagawa, S. POPART: full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116 (2015).
Riva, J. et al. Wing geometry as a tool for studying the Lutzomyia longipalpis (Diptera: Psychodidae) complex. Memórias do Instituto Oswaldo Cruz. 96, 1089–1094 (2001).
Mikery, O. F., Rebollar-Téllez, E. A., Cruz-López, L. C., Marina, C. F. & Castillo, A. Traditional and geometric morphometry analyses of Lutzomyia cruciata (Diptera: Psychodidae: Phlebotominae) populations of Chiapas, Mexico. J. Med. Entomol. 56(3), 697–707 (2019).
Klingenberg, C. P. MorphoJ: an integrated software package for geometric morphometrics. Mol. Ecol. Resour. 11 (2), 353–357 (2011).
Pante, E., Schoelinck, C. & Puillandre, N. From integrative taxonomy to species description: one step beyond. Syst. Biol. 64 (1), 152–160 (2015).
Souza, N. A., Brazil, R. P. & Araki, A. S. The current status of the Lutzomyia longipalpis (Diptera: Psychodidae: Phlebotominae) species complex. Memórias do Instituto Oswaldo Cruz. 112, 161–174 (2017).
Boehm, M. M. A. & Cronk, Q. C. B. Dark extinction: the problem of unknown historical extinctions. Biol. Lett. 17, 20210007 (2021).
Sharkey, M. J. et al. Minimalist revision and description of 403 new species in 11 subfamilies of Costa Rican braconid parasitoid wasps, including host records for 219 species. ZooKeys. 1013, 1–665 (2021).
Zamani, A. et al. The future of zoological taxonomy is integrative, not minimalist. Syst. Biodivers. 20 (1), 1–14 (2022).
Hebert, P. D. N., Stoeckle, M. Y., Zemlak, T. S. & Francis, C. M. Identification of birds through DNA barcodes. PLoS Biol. 2, 1657–1663 (2004).
Meyer, C. P. & Paulay, G. DNA barcoding: error rates based on comprehensive sampling. PLoS Biol. 3, 1–10 (2005).
Wiemers, M. & Fiedler, K. Does the DNA barcoding gap exist? - a case study in blue butterflies (Lepidoptera: Lycaenidae). Front. Zool. 4, 8 (2007).
Contreras Gutierrez, M. A., Vivero, R. J., Velez, I. D., Porter, C. H. & Uribe DNA barcoding for the identification of sand fly species (Diptera, Psychodidae, Phlebotominae) in Colombia. PloS One. 9(1), e85496 (2014).
Rodrigues, B. L., Carvalho-Costa, L. F., Pinto, I. S. & Rebêlo, J. M. M. DNA barcoding reveals hidden diversity of sand flies (Diptera: Psychodidae) at fine and broad spatial scales in Brazilian endemic regions for leishmaniasis. J. Med. Entomol. 55(4), 893–901 (2018).
Pinto, I. S. et al. DNA barcoding of sand flies (Diptera, Psychodidae, Phlebotominae) from the western Brazilian Amazon. Plos One. 18(2), e0281289 (2023).
Posada-López, L., Rodrigues, B. L., Velez, I. D. & Uribe, S. Improving the COI DNA barcoding library for neotropical phlebotomine sand flies (Diptera: Psychodidae). Parasites Vectors. 16 (1), 198 (2023).
Contreras Gutierrez, M. A. et al. DNA barcoding of Lutzomyia longipalpis species complex (Diptera: Psychodidae), suggests the existence of 8 candidate species. Acta Trop. 221, 105983 (2021).
Rodrigues, B. L. & Galati, E. A. New sand fly (Diptera, Psychodidae) records and COI DNA barcodes in the state of Maranhão, Eastern Amazon, Brazil. Acta Trop. 250, 107095 (2024).
Dellicour, S. & Flot, J. F. The hitchhiker’s guide to single-locus species delimitation. Mol. Ecol. Resour. 18(6), 1234–1246 (2018).
Reid, N. M. & Carstens, B. C. Phylogenetic estimation error can decrease the accuracy of species delimitation: a bayesian implementation of the general mixed yule-coalescent model. BMC Evol. Biol. 12, 196 (2012).
Talavera, G., Dincă, V. & Vila, R. Factors affecting species delimitations with the GMYC model: insights from a butterfly survey. Methods Ecol. Evol. 4 (12), 1101–1110 (2013).
Tang, C. Q., Humphreys, A. M., Fontaneto, D. & Barraclough, T. G. Effects of phylogenetic reconstruction method on the robustness of species delimitation using single-locus data. Methods Ecol. Evol. 5 (10), 1086–1094 (2014).
Renner, M. A., Heslewood, M. M., Patzak, S. D., Schäfer-Verwimp, A. & Heinrichs, J. By how much do we underestimate species diversity of liverworts using morphological evidence? An example from Australasian Plagiochila (Plagiochilaceae: Jungermanniopsida). Mol. Phylogenet. Evol. 107, 576–593 (2017).
Goulpeau, A. et al. OTU delimitation with earthworm DNA barcodes: a comparison of methods. Diversity. 14(10), 866 (2022).
Hofmann, E. P. et al. Cryptic diversity, but to what extent? Discordance between single-locus species delimitation methods within mainland anoles (Squamata: Dactyloidae) of northern central America. Front. Genet. 10, 11 (2019).
Kaiser, S. et al. Species boundaries and phylogeographic patterns in new species of Nannoniscus (Janiroidea: Nannoniscidae) from the equatorial Pacific nodule province inferred from mtDNA and morphology. Zool. J. Linn. Soc. 193(3), 1020–1071 (2021).
Scarpassa, V. M. & Alencar, R. B. Lutzomyia Umbratilis, the main vector of Leishmania guyanensis, represents a novel species complex? PloS One 7(5), e37341 (2012).
Scarpassa, V. M., Cunha-Machado, A. S. & Alencar, R. B. Multiple evolutionary lineages for the main vector of Leishmania guyanensis, Lutzomyia Umbratilis (Diptera: Psychodidae), in the Brazilian Amazon. Sci. Rep. 11, 15323 (2021).
Scarano, F. R., Ceotto, P. & Brazilian Atlantic forest: impact, vulnerability, and adaptation to climate change. Biodivers. Conserv. 24(9), 2319–2331 (2015).
Carnaval, A. C. et al. Prediction of phylogeographic endemism in an environmentally complex biome. Proc. R. Soc. B Biol. Sci. 281(1792), 20141461 (2014).
Mello Martins, F. Historical biogeography of the Brazilian Atlantic forest and the Carnaval–Moritz model of pleistocene refugia: what do phylogeographical studies tell us? Biol. J. Linn. Soc. 104(3), 499–509 (2011).
Paz, A. et al. Phylogeography of Atlantic Forest Glassfrogs (Vitreorana): when geography, climate dynamics and rivers matter. Heredity. 122(5), 545–557 (2019).
Myers, E. A., Folly, H., Ragalzi, E., Feio, R. N. & Santana, D. J. Late pliocene population divergence and persistence despite pleistocene climatic fluctuations in the Rio Doce snouted Treefrog (Ololygon carnevallii). J. Zoological Syst. Evolutionary Res. 59(3), 680–690 (2021).
Peres, E. A., DaSilva, M. B., Antunes, M. & Pinto-Da-Rocha, R. A short-range endemic species from south-eastern Atlantic Rain Forest shows deep signature of historical events: phylogeography of harvestmen Acutisoma longipes (Arachnida: Opiliones). Syst. Biodivers. 16(2), 171–187 (2018).
Pablos, J. L. et al. North-South and climate-landscape-associated pattern of population structure for the Atlantic Forest White Morpho butterflies. Mol. Phylogenet. Evol. 161, 107157 (2021).
Lorenz, C. et al. Geometric morphometrics in mosquitoes: what has been measured? Infect. Genet. Evol. 54, 205–215 (2017).
Giordani, B. F., Andrade, A. J., Galati, E. A. & Gurgel-Gonçalves, R. The role of wing geometric morphometrics in the identification of sandflies within the subgenus Lutzomyia. Med. Vet. Entomol. 31 (4), 373–380 (2017).
Santos, M. F. et al. Morphometric analysis of Longipalpis (Diptera: Psychodidae) complex populations in Mato Grosso do sul, Brazil. J. Med. Entomol. 52 (3), 359–367 (2015).
Prudhomme, J. et al. Wing size and shape variation of Phlebotomus papatasi (Diptera: Psychodidae) populations from the south and north slopes of the Atlas Mountains in Morocco. J. Vector Ecol. 37 (1), 137–147 (2012).
Godoy, R. E. et al. Geometric morphometry of the head in sand flies (Diptera: Psychodidae: Phlebotominae), an alternative approach to taxonomy studies. Zootaxa. 4504(4), 566–576 (2018).
Dujardin, J. P. Morphometrics applied to medical entomology. Infect. Genet. Evol. 8, 875–890 (2008).
Prudhomme, J. et al. Ecology and morphological variations in wings of Phlebotomus Ariasi (Diptera: Psychodidae) in the region of Roquedur (Gard, France): a geometric morphometrics approach. Parasites Vectors. 9, 578 (2016).
Acknowledgements
We thank to Jucelei O. M. Infran (UFMS), Matheus E. P. Barbosa (UFMS), and Waldemar C. da Silva Filho (Laboratório Regional de Entomologia e Controle de Vetores de Aquidauana) for fieldwork assistance in Piraputanga—MS; to Clara Benetti de Lemos Cordeiro (UFPR), Mariana Funchal Gusso (UFPR), Lucas Rossito de Carvalho (UFPR), and to the ‘Central de Transportes’ (CENTRAN/UFPR) for fieldwork assistance in Piraquara—PR; to Vanessa C. Helfstein (SUCEN) for fieldwork assistance in Parelheiros—SP; and to Fredy Galvis-Ovallos (FSP USP) for helping to obtain specimens from Jundiaí—SP.
Funding
This study was financially supported by São Paulo Research Foundation (FAPESP), grant #2023/03715-2; Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) for scholarship grants to BLR—financial code 001; Conselho Nacional de Desenvolvimento Científico e Tecnológico—CNPq for research fellowship to JDAF (314260/2023-4) and AGO (172169/2023-2); and ‘Pró-Reitoria de Pesquisa e Pós-graduação’ of UFPR (Project number: 3527).
Author information
Authors and Affiliations
Contributions
B.L.R.: Conceptualization; methodology; validation; formal analysis; investigation; data curation; writing—original draft; writing—review and editing; visualization; project administration. A.G.O.: Validation; investigation; data curation; writing—review and editing. L.E.H.S.: Validation; formal analysis; writing—review and editing. T.V.S.: Validation; formal analysis; writing—review and editing. L.N.C.O.: Validation; formal analysis; writing—review and editing. F.D.R.: Resources; writing—review and editing. A.J.A.: Resources; writing—review and editing. G.B.M.: Investigation; writing—review and editing. I.S.P.: Resources; writing—review and editing. J.D.A.F.: Resources; writing—review and editing. E.A.B.G.: Conceptualization; validation; investigation; data curation; writing—review and editing; supervision; project administration; funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The entomological collections were performed under the authorization for activities with the scientific purpose of the Brazilian Sistema de Autorização e Informação em Biodiversidade (SISBIO) (no. 7500-1, 90339-1, 86644-1, and 25596-4).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article omitted an affiliation for Alessandra Gutierrez de Oliveira. In addition, the original version of this Article contained an error in the Funding section. Full information regarding the corrections made can be found in the correction for this Article.
Electronic supplementary material
Below is the link to the electronic supplementary material.

Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rodrigues, B.L., de Oliveira, A.G., da Silva, L.E.H. et al. Hidden diversity in anthropophilic sand flies of the Monticola Series (Diptera, Psychodidae). Sci Rep 14, 27215 (2024). https://doi.org/10.1038/s41598-024-77249-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-77249-1

{kind=link}