
Abstract
Physical exercise is beneficial to keep physical and mental health. The molecular mechanisms underlying exercise are still worth exploring. The healthy adult mice after six weeks of moderate-intensity exercise (experimental group) and sedentary mice (control group) were used to perform transcriptomic, proteomic, lactylation modification, and metabolomics analysis. In addition, gene sets related to hypoxia, glycolysis, and fatty acid metabolism were used to aid in the screening of hub genes. The mMCP-counter was employed to evaluate infiltration of immune cells in murine liver tissues. Transcriptomics analysis revealed 82 intersection genes related to hypoxia, glycolysis, and fatty acid metabolism. Proteomics and lactylation modification analysis identified 577 proteins and 141 differentially lactylation modification proteins. By overlapping 82 intersection genes with 577 differentially expressed proteins and 141 differentially lactylation modification proteins, three hub genes (Aldoa, Acsl1, and Hadhb) were obtained. The immune infiltration analysis revealed a decreased score for monocytes/macrophages and an increased score for endothelial cells in the experimental group. Then, 459 metabolites in positive mode and 181 metabolites in negative mode were identified. The “Metabolic pathways” (mmu01100) was a common pathway between intersection genes-enriched pathways and metabolites-enriched pathways. These findings highlight the pivotal roles of hub genes in the glycolysis and fatty acid metabolism under the context of chronic exercise.
Similar content being viewed by others


Introduction
Physical activity is one of the important ways to maintain health and can regulate the function of major organs such as heart, brain and liver1. The benefits of scientific exercise are the result of the synergy, interaction, and communication of multiple physiological systems. The positive effects of physical exercise on spatial learning and memory had been reported both in animal and human studies2. Increasing physical activity through exercise can improve fatty liver disease3. Gao et al. reported that exercise promoted lipophagy by activating AMPK/ULK1 pathway to improve nonalcoholic fatty liver disease (NAFLD)4. Hu et al. indicated that exercise alleviated the mitochondrial dysfunction to improve NAFLD by activating Sirt1-mediated Drp1 acetylation5. Regular physical exercise during pregnancy can help preventing pregnancy related disorders, such as gestational diabetes, hypertensive disorders, anxiety and prenatal depression6. Physical exercise had potential roles in prevention, treatment, and prognoses improvement of cancer patients7. The molecular mechanisms underlying exercise are still worth exploring.
Lactate, initially considered as metabolic waste products in glycolysis, not only serves as an energy source for metabolism and a major gluconeogenic precursor, but also has been shown to regulate intracellular signal transduction as a signaling molecule8. Lactate is involved in a variety of physiological and pathological processes, such as energy regulation, regulation of fatty acid metabolism, ischemic injury, inflammatory responses, and tumor growth and metastasis9. Lactylation, as a novel histone modification that can epigenetically regulate gene transcription, was formally proposed for the first time in 201910. Intracellular hypoxia induces mitochondrial protein lactate to limit oxidative phosphorylation to reduce reactive oxygen species damage11. Elevated lactylation level in colorectal cancer tissues was associated with poor prognosis12.
The integration of multi-omics data can enhance the understanding the molecular behavior underlying exercise. Multi-omics data encompasses a diverse array of biological measurements, including genomic, transcriptomics, proteomic, epigenomic, and metabolomics. For instance, transcriptomics utilizes RNA sequencing to identify differential splicing and quantification of the relative levels of gene expression13. Proteomics, on the other hand, employs mass spectrometry to estimate the abundance and alterations of proteins, while metabolomics offers a snapshot of the metabolic state of cells14. Whole genome methylation was decreased in skeletal muscle biopsies obtained from healthy sedentary individuals following one acute bout of exhaustive aerobic exercise15. Metabolomic analysis revealed increased levels of polyunsaturated free fatty acids and lysophospholipids, accompanied by reductions in ceramides, sphingolipids and phospholipids, and alterations in gut microbiome metabolites after enhanced physical activity16. An integrated analysis of metabolomic and transcriptomic data in zebrafish subjected to an exercise intervention revealed alterations in metabolic pathways, metabolites, and expression of core genes, which may contribute to exercise-induced improvements in bone mass17. To date, research has predominantly focused on one or two omics domains. Therefore, multi-omics map of the effects of exercise is needed to understand the molecular basis of exercise training-induced adaptations.
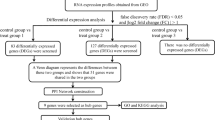
In this present study, transcriptomics, proteomic, lactylation modification and metabolomics analyses were performed on liver tissues in mice after six weeks of moderate-intensity treadmill exercise. Gene sets related to hypoxia, glycolysis, and fatty acid metabolism were used to aid in the screening of hub genes. This multi-omics integration strategy aimed to reveal the potential impact of chronic exercise on the mice liver tissue metabolism, helping to increase the understanding of the molecular mechanisms underlying the physiological changes behind exercise.
Materials and methods
Experimental mice and chronic exercise protocol
Adult C57BL/6J male mice (20–22 g, 8 weeks), purchased from Charles River Laboratory, were used in this study. After 1-week adaptation, mice were randomly divided into two groups: the chronic exercise (Experimental group) and the sedentary control (Control group). All animal procedures were conducted during rodents’ nocturnal phase (6:00 pm to 12:00 pm). The animal experiments were complied with the institutional ethical guidelines, ARRIVE guidelines, and approved by the Ethics Committee on Animal Care and Use of the Capital Institute of Pediatrics (DWLL2021015). The moderate-intensity treadmill exercise was performed as described previously18,19. Mice from each group were anesthetized with isoflurane and sacrificed by cervical dislocation within 24 h after the last exercise. The liver tissues were collected following the same procedures as in the previous study18. Principal component analysis (PCA) was performed with “princomp” function in the R package “stats” (version 4.3.1). Transcriptomics, proteomics, and lactylation modification analysis was performed on liver tissue samples from 6 mice (n = 3, both groups). For metabolomics analysis, liver tissue samples from 12 mice (n = 6, both groups) were used.
Transcriptomics analysis
Total RNA extraction and RNA sequencing procedures were same as in the previous study18. RNA-seq count data were converted to transcripts per million (TPM), and TPM data were log2 (data + 1) processed to normalize the data. Differentially expressed genes (DEGs) were obtained with p < 0.05 & (fold change (FC) > 1.2 or FC < 0.83) using the limma (v 3.46.0), visualized by volcano maps and bar chart with ggplot2 package (version 3.5.1). In total, 200 hypoxia-related genes (HRGs) in HALLMARK_HYPOXIA, 200 glycolysis-related genes (GRGs) in HALLMARK_GLYCOLYSIS, and 158 fatty acid metabolism-related genes (FRGs) in HALLMARK_FATTY_ACID_METABOLISM were obtained from MSigDB (https://www.gsea-msigdb.org/gsea/msigdb/). We identified the intersection of DEGs with 200 HRGs, 200 GRGs, and 158 FRGs, respectively. Subsequently, we took the union of these intersected genes to obtain the intersection genes for further downstream analysis. David (version 2024q1, https://david.ncifcrf.gov/)20 was used to perform Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis for the intersection genes with p < 0.05. The protein-protein interaction (PPI) network of intersection genes was constructed with STRING (version 12.0, https://cn.string-db.org/)21.
Proteomics and lactylation modification analyses
Liquid chromatography-tandem mass spectrometry was performed at PTM Bio lab (Hangzhou, China) as previously described18. The abundance of lactylated protein have been normalized by the protein levels. Differentially expressed proteins (DEPs), differentially lactylation modification proteins (DLPs), and differentially lactylated sites (DLSs) were determined with p < 0.05 & (FC > 1.2 or FC < 0.83) based on a Student’s t-test. The results were visualized by volcano plots (ggplot2, version 3.5.1) and heat maps (pheatmap, version 1.0.12).
Identification of hub genes and transcription factors (TFs) regulatory network
The hub genes were obtained by overlapping the intersection genes with DEPs and DLPs. The gene expression, protein, and lactylation modification levels of hub genes were displayed in box plots with ggpubr package (version 0.6.0). The TFs regulating the hub genes were predicted using the Cistrome DB (http://genemania.org) with RP score > 0.7. Subsequently, the TFs regulatory network was visualized using Cytoscape (version 3.8.2)22.
Immune microenvironment infiltration analysis
The murine Microenvironment Cell Populations counter (mMCP-counter) was employed to evaluate infiltration of immune cells in murine liver tissues. It is a transcriptome-based tool for accurately quantifying tissue-infiltrated immune and stromal cell populations in mouse samples23. mMCP-counter can be accessed as an R package (https://github.com/cit-bioinfo/mMCP-counter). It takes a gene expression profiles matrix as input and returns the abundance of RNA originating from 16 defined cell populations present in the heterogeneous sample.
Correlation analysis
In addition, the correlation analysis between gene expression, protein, and lactylation modification levels of hub genes and immune cells was performed. Pearson correlation analysis was performed using psych package (version 2.4.6) in R to calculate the correlation coefficient and its corresponding p-value. The visual presentation of their correlations was shown by a heat map using the ggplot2 package (version 3.5.1).
Metabolomics analysis
Similarly, the metabolomics analysis was performed as previously described18. The normalization of metabolites was performed using MetaboAnalyst 5.0 (https://www.metaboanalyst.ca/MetaboAnalyst/docs/RTutorial.xhtml) to make metabolites concentration distribution is close to normal distribution. Differential metabolites (DEMs) were determined by variable importance value (VIP) > 1 with p < 0.05 based on a Student’s t-test. KEGG enrichment analysis was performed using Metabolite Biological Role (MBROLE) 2.0 (http://csbg.cnb.csic.es/mbrole2/index.php) with p < 0.05. In addition, the correlation analysis between hub genes and DEMs was performed. The psych package (version 2.4.6) in R was used to calculate the Pearson correlation coefficient and its corresponding p-value.
Statistical analysis
The PCA using the “princomp” function in the R package “stats” (version 4.3.1) was performed to evaluate the accuracy of grouping. The “limma” (3.46.0) package was used to obtain the DEGs. David (https://david.ncifcrf.gov/) was used to perform functional enrichment analysis. DEPs, DLPs, DLSs, and DEMs were determined with p < 0.05 & (FC > 1.2 or FC < 0.83) based on a Student’s t-test. mMCP-counter was used to perform immune infiltration analysis. Pearson correlation analysis was performed using psych package (version 2.4.6) in R to calculate the correlation coefficient and its corresponding p-value. Unless otherwise stated, a p-value < 0.05 was deemed statistically significant. The software packages used for each omics analysis with details were summarized in Table 1.
Results
Identification of intersection genes
The PCA plot showed that there was a significant separation between the experimental and control groups (Figure S1A). Compared to the control group, 2034 DEGs, including 1456 up-regulated mRNAs and 578 down-regulated mRNAs, were detected in the experimental group (Fig. 1A, Table S1). Among them, 40 genes overlapped with HRGs, 23 genes overlapped with GRGs, and 36 genes overlapped with FRGs, resulting in a total of 82 intersection genes (Fig. 1B). GO analysis indicated that these intersection genes were enriched in lipid metabolic process, fatty acid metabolic process, and negative regulation of apoptotic process (Fig. 2A, Table S2). KEGG enrichment analysis revealed several significantly enriched pathways, such as metabolic pathways, fatty acid metabolism, PPAR signaling pathway, and HIF-1 signaling pathway (Fig. 2B, Table S2). PPI network was displayed in Fig. 3, including 72 nodes and 274 edges. The number of proteins interacting with Fasn was the highest (up to 21 proteins).

Identification of intersection genes. (A) Volcano and Venn plots of the DEGs. (B) Venn diagram displaying the DEGs and HRGs/GRGs/FRGs. DEG, differentially expressed genes; HRG, hypoxia-related gene; GRG, glycolysis-related gene; FRG, fatty acid metabolism-related gene.

Functional enrichment of intersection genes. (A) GO analysis for intersection genes. (B) KEGG enrichment analysis for intersection genes. GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes.

PPI network of intersection genes. Node size and color depth are proportional to degree.
Proteomics and lactylation modification analyses
The PCA plot showed that there was a significant separation between the experimental and control groups (Figure S1B). Compared to the control group, 577 DEPs, including 253 up-regulated proteins and 324 down-regulated proteins, were detected in the experimental group (Fig. 4A-B, Table S3). Lactylation modification analysis identified 264 DLSs of 141 DLPs, including 7 up-regulated lactylated sites and 257 down-regulated lactylated sites (Fig. 4C-D, Table S4).

Proteomics and lactylation modification analyses. (A) Volcano plot of the DEPs. (B) Heatmap of the DEPs. (C) Volcano plot of the DLSs. (D) Heatmap of the DLSs. DEP, differentially expressed protein; DLS, differentially lactylated site.
Identification of hub genes
Venn diagram shows the overlap among 577 DEPs, 141 DLPs, and 82 intersection genes (Figure S2A). There were 24 shared genes between DEPs and DLPs, 19 shared genes between DEPs and intersection genes, 10 shared genes between DLPs and intersection genes. Finally, three hub genes (Aldoa, Acsl1, and Hadhb) were obtained by overlapping 82 intersection genes with 577 DEPs and 141 DLPs. Among them, aldolase A, fructose-bisphosphate (Aldoa) is a gene related to glycolysis and hypoxia, and also a gene related to fatty acid metabolism. The gene expression levels of three hub genes were up-regulated in the experimental group (Fig. 5A). The protein expression levels of acyl-CoA synthetase long-chain family member 1 (Acsl1) and hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit beta (Hadhb) were up-regulated in the experimental group, while Aldoa was down-regulated (Fig. 5B). Lactylation modification analysis revealed significantly decreased lactylation levels of Aldoa_K42 and Hadhb_K273 in the experimental group, as well as decreased lactylation level of Acsl1_K676 without significance (Fig. 5C). The TF regulatory network, including 3 hub genes and 55 TFs, was constructed (Figure S2B). Aldoa was regulated by 45 TFs, Acsl1 was regulated by 14 TFs, and Hadhb was regulated by 6 TFs. In particular, peroxisome proliferator activated receptor gamma (Pparg) regulated all three hub genes. Among the 55 TFs, the expression levels of 12 TFs (Brd2, Btaf1, Chd4, Creb1, Esr1, Ets1, Fos, Gata4, Max, Myc, Rxrg, and Stat1) were significantly different between the experimental and control groups (Figure S2C).

Characterization of hub genes. (A) Gene expression levels of hub genes. (B) Protein levels of hub genes. (C) Lactylation modification levels of hub genes. ns indicated not significant, *indicated p < 0.05, **indicated p < 0.01, ***indicated p < 0.001, ****indicated p < 0.0001.
Immune microenvironment infiltration analysis
The immune infiltration analysis revealed a decreased score for monocytes/macrophages and an increased score for endothelial cells in the experimental group (Fig. 6A). The correlation analysis indicated that the gene expressions of three hub genes were negatively correlated with monocytes/macrophages and positively correlated with endothelial cells (Fig. 6B). The protein expressions of Acsl1 and Hadhb were negatively correlated with monocytes/macrophages and positively correlated with endothelial cells, while Aldoa was positively correlated with monocytes/macrophages and negatively correlated with endothelial cells (Fig. 6C). The lactylation levels of Aldoa_K42 and Hadhb_K273 were positively correlated with monocytes/macrophages and negatively correlated with endothelial cells (Fig. 6D).

Analysis of immune microenvironment. (A) The immune infiltration levels of two immune cells. (B) The correlation analysis between gene expression levels of hub genes and two immune cells. (C) The correlation analysis between protein expression levels of hub genes and two immune cells. (D) The correlation analysis between lactylation modification levels of hub genes and two immune cells. * indicated p < 0.05, ** indicated p < 0.01.
Metabolomics analysis
The PCA score plot clearly showed a complete separation between the experimental and control groups in positive (Figure S1C) and negative (Figure S1D) mode. In positive mode, 459 DEMs were identified, among which 255 metabolites were significantly increased and 204 metabolites were significantly decreased, in the experimental group (Fig. 7A-B, Table S5). Of these 459 DEMs, KEGG enrichment analysis was performed for 81 metabolites with KEGG_ID using MBROLE 2.0. These 81 DEMs were significantly enriched in metabolic pathways, purine metabolism, alpha-linolenic acid metabolism, caffeine metabolism, and sphingolipid metabolism (Fig. 7C, Table S6). The “metabolic pathways” was a common pathway between intersection genes-enriched pathways and DEMs-enriched pathways in positive mode. The correlation analysis was performed between 26 metabolites in “metabolic pathways” and the gene and protein expression of hub genes (Fig. 7D-E). The gene expressions of Acsl1 was the most negatively correlated with PTM_3508 [(-)-Jasmonic acid] (r = -0.994) and the gene expressions of Aldoa was the most positively correlated with PTM_43 (Uridine 5’-diphosphate) (r = 0.973) (Fig. 7D). The protein expression of Acsl1 was the most negatively correlated with PTM_3508 [(-)-Jasmonic acid] (r = -0.996) and the protein expressions of Aldoa was the most positively correlated with PTM_3508 [(-)-Jasmonic acid] (r = 0.998) (Fig. 7E).

Metabolomics analysis in positive mode. (A) Volcano plot of the DEMs in positive mode (B) Heatmap of the DEMs in positive mode. (C) KEGG pathway annotation for DEMs in positive mode. (D) The correlation analysis between gene expression levels of hub genes and DEMs in Metabolic pathways. (E) The correlation analysis between protein expression levels of hub genes and DEMs in Metabolic pathways. DEM, Differential metabolite.
In negative mode, 181 DEMs were identified, among which 84 metabolites were significantly increased and 97 metabolites were significantly decreased, in the experimental group (Fig. 8A-B, Table S7). Of these 181 DEMs, KEGG enrichment analysis was performed for 65 metabolites with KEGG_ID using MBROLE 2.0. These 65 DEMs were significantly enriched in metabolic pathways, purine metabolism, arachidonic acid metabolism, primary bile acid biosynthesis and pantothenate and CoA biosynthesis (Fig. 8C, Table S8). The “metabolic pathways” was a common pathway between intersection genes-enriched pathways and DEMs-enriched pathways in negative mode. The correlation analysis was performed between 33 metabolites in “metabolic pathways” and the gene and protein expression of hub genes (Fig. 8D-E). The gene expressions of Acsl1 was the most negatively correlated with PTM_104 (s-adenosylhomocysteine (r = -0.999) and the gene expressions of Aldoa was the most positively correlated with PTM_631 (4-pyridoxic acid) (r = 0.984) (Fig. 8D). The protein expression of Acsl1 was the most negatively correlated with PTM_104 (s-adenosylhomocysteine (r = -0.994) and the protein expressions of Aldoa was the most positively correlated with PTM_104 (s-adenosylhomocysteine (r = -0.995) (Fig. 8E).

Metabolomics analysis in negative mode. (A) Volcano plot of the DEMs in negative mode. (B) Heatmap of the DEMs in negative mode. (C) KEGG pathway annotation for DEMs in negative mode. (D) The correlation analysis between gene expression levels of hub genes and DEMs in Metabolic pathways. (E) The correlation analysis between protein expression levels of hub genes and DEMs in Metabolic pathways. DEM, Differential metabolite.
Discussion
The importance of physical exercise in the treatment of various diseases and improvement of organ functions has been extensively reported24,25,26. The bioinformatics analysis of omics data helps to deepen the understanding of the molecular mechanism underlying exercise. A proteomics study indicated that exercise training could alter the hepatic protein expression profiles of aged rat, thereby regulating the key liver metabolic pathways27. Analysis of transcriptomic and metabolomics data revealed that early active exercise had significant effects on bone development and metabolism in mice28. In the present study, we identified three hub genes (Hadhb, Aldoa, and Acsl1) whose mRNA and protein expression levels and lactylation levels were significantly altered during chronic exercise based on mutil-omics data (transcriptome, proteome and lactatomics data).
Mitochondrial trifunctional protein (MTP), consisting of two α subunits (HADHA) and two β subunits (HADHB), locates on the inner mitochondrial membrane and plays important roles in fatty acid β-oxidation to participate in and regulate fatty acid metabolism29. ACSL1 (acyl-CoA synthetase long chain family member 1) contributes the majority of total acyl-CoA synthetase (ACS) activity in human skeletal muscle and also plays important roles in fatty acid metabolism30. Acsl1 deficiency in skeletal muscle of mice impairs fatty acid oxidation31,32. Elevated Acsl1 induced by exercise training has potential to increase the utilization of fatty acids through β-oxidation30. It has been reported that one genome-wide significant polymorphism (rs116143768) within the ACSL1 gene was associated with fat loss efficiency in response to aerobic exercise33. Stierwalt et al. suggested that the expression of Acsl1 may undergo regulation by exercise training34. In this study, both the gene and protein expression level of Acsl1 and Hadhb were increased in the liver tissues of experimental mice, suggesting that chronic exercise may elevate the expressions of enzymes involved in fatty acid β-oxidation, thereby enhancing the utilization of fatty acids as an energy source. This underscores the importance of exercise in promoting healthy fatty acid metabolism and potentially enhancing fat loss efficiency.
In addition, up to 21 proteins, including Acsl1 and Hadhb, interacted with Fasn in the PPI network. In this study, both the gene and protein expression levels of Fasn were increased in the liver tissues of experimental mice. Fatty acid synthase (FASN) is a key lipogenic enzyme catalysing the terminal steps in the de novo biogenesis of fatty acids35. In the short-term high-fat diet experiment of female C57BL/6 mice, the expression of Fasn was increased in the setting of exercise36. Hippocampal-specific up-regulation of FASN was induced by voluntary running in 20 week-old male mice, which plays an important role in exercise-mediated cognitive enhancement37. Taken together, these findings may provide new perspectives for understanding the health benefits of exercise.
ALDOA (aldolase A, fructose-bisphosphate) is a highly conserved enzyme that plays a role in the fourth step of glycolysis and participates in a variety of cellular functions and biological processes38,39. The genome resequencing on buffaloes revealed that ALDOA was involved in pathways associated with the response to exercise39. Compared to sedentary men, ALDOA was expressed at lower levels in the left vastus lateralis muscles of sprinters40. In this study, the gene expression level of Aldoa were up-regulated and protein expression level of Aldoa were down-regulated in the liver tissues of experimental mice. Taken together, these findings suggested the multifaceted role of Aldoa in cellular metabolism, particularly its involvement in glycolysis and the response to exercise. Further research into the specific mechanisms underlying the regulation and function of Aldoa may yield significant insights into human health and disease.
In the TF regulatory network, Pparg regulated all three hub genes (Hadhb, Acsl1, and Aldoa). Peroxisome proliferator activated receptor gamma (PPARG/PPARγ) is a member of the nuclear hormone receptor superfamily. Tong et al. reported that PPARγ could be the direct target of IRF6 in high-fat diet (HFD)-induced fatty liver41. Swimming exercise partly suppresses lipid accumulation in HFD-induced non-alcoholic fatty liver disease in mice by down-regulating PPARγ-mediated pathways42. Based on function enrichment analysis for the intersection genes, three hub genes were enriched in Metabolic pathways (mmu01100), which was a common pathway between intersection genes-enriched pathways and DEMs-enriched pathways. The genes, Hadhb and Acsl1, were enriched in Fatty acid degradation (mmu00071) and Fatty acid metabolism (mmu01212). These findings further illustrated that chronic exercise has a certain effect on mice liver tissue metabolism. In addition, the lactylation levels of hub genes significantly different between the control and experimental groups, which need to be further explored.
The immune cell infiltration analysis revealed a decreased infiltration of monocytes/macrophages and an increased infiltration of endothelial cells in the experimental group. The high level of CD68+ monocytes/macrophages was detected in muscles of muscular dystrophy, X-linked mouse model (mdx) mice, and low-intensity training of mdx mice further increased the CD68 level43. Intense aerobic exercise training increased glycolysis and reduced the formation of reactive oxygen species to alter the muscle microvascular endothelial cells properties44. The specific role of immune cells in the response to exercise needs to be further explored to add new insights into the effects of exercise on the immune microenvironment.
Some limitations exist in this study. First, the sample size used for sequencing of each omics was small. Second, there was a lack of multi-omics level validation and in-depth functional exploration of the three hub genes. Third, the association study among different omics was relatively superficial and not deep enough.
In summary, the findings of this study highlight the pivotal roles of three hub genes in the glycolysis and fatty acid metabolism, particularly in the context of chronic exercise. The moderate-intensity exercise may modulate the mice liver tissue metabolism by regulating the expression of Hadhb, Aldoa, and Acsl1. Future research exploring the molecular mechanisms underlying these findings may provide further insights into the role of exercise in promoting metabolic health. In the future, we will consider more in-depth studies on the correlation among different omics.
Data availability
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession numbers can be found below: The transcriptomics data have been deposited to the GSE280324 dataset (GSE280324, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi? acc=GSE280324). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProX partner repository with the dataset identifier PXD057080 (https://proteomecentral.proteomexchange.org/cgi/GetDataset? ID=PXD057080). Metabolomics data have been deposited to the EMBL-EBI MetaboLights database with the identifier MTBLS11477 (https://www.ebi.ac.uk/metabolights/MTBLS11477).
References
Zamanian-Azodi, M., Khatoon Hajisayah, S., Razzaghi, M. & Rezaei-Tavirani, M. Introducing physical exercise as a potential strategy in liver cancer prevention and development. Gastroenterol. Hepatol. Bed Bench. 14, 317–322 (2021).
Cassilhas, R. C., Tufik, S. & de Mello, M. T. Physical exercise, neuroplasticity, spatial learning and memory. Cell. Mol. Life Sci. 73, 975–983 (2016).
van der Windt, D. J., Sud, V., Zhang, H., Tsung, A. & Huang, H. The effects of Physical Exercise on fatty liver disease. Gene Expr. 18, 89–101 (2018).
Gao, Y. et al. Exercise and dietary intervention ameliorate high-fat diet-induced NAFLD and liver aging by inducing lipophagy. Redox Biol. 36, 101635 (2020).
Hu, Z. et al. Exercise activates Sirt1-mediated Drp1 acetylation and inhibits hepatocyte apoptosis to improve nonalcoholic fatty liver disease. Lipids Health Dis. 22, 33 (2023).
Ribeiro, M. M., Andrade, A. & Nunes, I. Physical exercise in pregnancy: benefits, risks and prescription. J. Perinat. Med. 50, 4–17 (2022).
Wang, Q. & Zhou, W. Roles and molecular mechanisms of physical exercise in cancer prevention and treatment. J. Sport Health Sci. 10, 201–210 (2021).
Brooks, G. A. Lactate as a fulcrum of metabolism. Redox Biol. 35, 101454 (2020).
Li, X. et al. Lactate metabolism in human health and disease. Signal. Transduct. Target. Ther. 7, 305 (2022).
Zhang, D. et al. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580 (2019).
Mao, Y. et al. Hypoxia induces mitochondrial protein lactylation to limit oxidative phosphorylation. Cell. Res. 34, 13–30 (2024).
Xie, B. et al. KAT8-catalyzed lactylation promotes eEF1A2-mediated protein synthesis and colorectal carcinogenesis. Proc. Natl. Acad. Sci. U S A. 121, e2314128121 (2024).
Wang, Z., Gerstein, M. & Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63 (2009).
Nicholson, J. K., Lindon, J. C. & Holmes, E. Metabonomics’: understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 29, 1181–1189 (1999).
Barrès, R. et al. Acute exercise remodels promoter methylation in human skeletal muscle. Cell. Metab. 15, 405–411 (2012).
Gaitán, J. M. et al. Effects of Aerobic Exercise training on systemic biomarkers and cognition in late middle-aged adults at risk for Alzheimer’s Disease. Front. Endocrinol. (Lausanne). 12, 660181 (2021).
Hou, J-L. et al. Integration of Metabolomics and Transcriptomics to reveal the metabolic characteristics of Exercise-Improved Bone Mass. Nutrients 15, 1694 (2023).
Lu, Z. et al. Multi-omics analysis explores the effect of chronic exercise on liver metabolic reprogramming in mice. Front. Cell. Dev. Biol. 11, 1199902 (2023).
Qian, P. et al. Chronic exercise remodels the lysine acetylome in the mouse hippocampus. Front. Mol. Neurosci. 15, 1023482 (2022).
Sherman, B. T. et al. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221 (2022).
Szklarczyk, D. et al. The STRING database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–D646 (2023).
Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Petitprez, F. et al. The murine Microenvironment Cell Population counter method to estimate abundance of tissue-infiltrating immune and stromal cell populations in murine samples using gene expression. Genome Med. 12, 86 (2020).
Paillard, T., Rolland, Y. & de Souto Barreto, P. Protective effects of physical exercise in Alzheimer’s disease and Parkinson’s disease: A narrative review. J. Clin. Neurol. 11, 212–219 (2015).
Lombardi, G., Ziemann, E. & Banfi, G. Physical activity and bone health: what is the role of Immune System? A narrative review of the third way. Front. Endocrinol. (Lausanne). 10, 60 (2019).
Ubaida-Mohien, C. et al. Physical activity Associated Proteomics of skeletal muscle: Being physically active in Daily Life May protect skeletal muscle from aging. Front. Physiol. 10, 312 (2019).
Li, F., Li, T. & Liu, Y. Proteomics-based identification of the Molecular signatures of Liver tissues from aged rats following eight weeks of medium-intensity Exercise. Oxid. Med. Cell. Longev. 3269405 2016 (2016).
Yu, S. et al. Time of exercise differentially impacts bone growth in mice. Nat. Metab. (2024).
Wang, X., Song, H., Liang, J., Jia, Y. & Zhang, Y. Abnormal expression of HADH, an enzyme of fatty acid oxidation, affects tumor development and prognosis (review). Mol. Med. Rep. 26, (2022).
Poppelreuther, M. et al. Acyl-CoA synthetase expression in human skeletal muscle is reduced in obesity and insulin resistance. Physiol. Rep. 11, e15817 (2023).
Li, L. O. et al. Compartmentalized acyl-CoA metabolism in skeletal muscle regulates systemic glucose homeostasis. Diabetes 64, 23–35 (2015).
Zhao, L. et al. Defective fatty acid oxidation in mice with muscle-specific acyl-CoA synthetase 1 deficiency increases amino acid use and impairs muscle function. J. Biol. Chem. 294, 8819–8833 (2019).
Bojarczuk, A. et al. Genome-wide Association Study of Exercise-Induced Fat loss efficiency. Genes (Basel) 13, (2022).
Stierwalt, H. D., Ehrlicher, S. E., Robinson, M. M. & Newsom, S. A. Diet and Exercise Training influence skeletal muscle long-chain acyl-CoA synthetases. Med. Sci. Sports Exerc. 52, 569–576 (2020).
O’Farrell, M. et al. FASN inhibition targets multiple drivers of NASH by reducing steatosis, inflammation and fibrosis in preclinical models. Sci. Rep. 12, 15661 (2022).
Morton, T. L. et al. Exercise increases and browns muscle lipid in High-Fat Diet-Fed mice. Front. Endocrinol. (Lausanne). 7, 80 (2016).
Chorna, N. E. et al. Fatty acid synthase as a factor required for exercise-induced cognitive enhancement and dentate gyrus cellular proliferation. PLoS One. 8, e77845 (2013).
Pirovich, D. B., Da’dara, A. A. & Skelly, P. J. Multifunctional fructose 1,6-Bisphosphate aldolase as a therapeutic target. Front. Mol. Biosci. 8, 719678 (2021).
Sun, T. et al. Selection signatures of Fuzhong Buffalo based on whole-genome sequences. BMC Genom. 21, 674 (2020).
Yoshioka, M., Tanaka, H., Shono, N., Shindo, M. & St-Amand, J. Gene expression profile of sprinter’s muscle. Int. J. Sports Med. 28, 1053–1058 (2007).
Tong, J. et al. Hepatic Interferon Regulatory Factor 6 alleviates liver steatosis and metabolic disorder by transcriptionally suppressing peroxisome proliferator-activated receptor γ in mice. Hepatology 69, 2471–2488 (2019).
Zhang, Y. et al. Swimming exercise ameliorates insulin resistance and nonalcoholic fatty liver by negatively regulating PPARγ transcriptional network in mice fed high fat diet. Mol. Med. 29, 150 (2023).
Hyzewicz, J. et al. Low-intensity training and the C5a complement antagonist NOX-D21 rescue the mdx phenotype through modulation of inflammation. Am. J. Pathol. 187, 1147–1161 (2017).
Norregaard, L. B. et al. Exercise training alters skeletal muscle microvascular endothelial cell properties in recent postmenopausal females. J. Physiol. (2024).
Acknowledgements
This work was supported by Beijing Finance Bureau (CIP2024-0040) and the National Natural Science Foundation of China, Beijing, China (82271731).
Author information
Authors and Affiliations
Contributions
FW, HG, and TZ designed the study and drafted the manuscript. FW, WW, XH, and PQ carried out the mouse study. FW, JC, ZL, JG, YB, HG and TZ contributed to the data acquisition, interpretation, and discussion. All authors contributed to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Statement of ethics
The study was conducted according to the guidelines of the management and utility of experimental animals, and approved by the Ethics Committee of Capital institute of Pediatrics and all animal manipulations were strictly performed following the relevant laws of China.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, F., Wu, W., He, X. et al. Effects of moderate intensity exercise on liver metabolism in mice based on multi-omics analysis. Sci Rep 14, 31072 (2024). https://doi.org/10.1038/s41598-024-82150-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-82150-y
