
Abstract
Lines of evidence have indicated that type 2 diabetes mellitus (T2DM) is an independent risk factor for osteoarthritis (OA) progression. However, the study focused on the relationship between T2DM and OA at the transcriptional level remains empty. We downloaded OA- and T2DM-related bulk RNA-sequencing and single-cell RNA sequencing data from the Gene Expression Omnibus (GEO) dataset. Differential expression analysis and weighted gene co-expression network analysis (WGCNA) were performed to screen out hub genes between OA and T2DM, and functional enrichment was done. Single-cell sequencing analysis was further used to screen key genes on OA and T2DM datasets. Rat chondrocytes and human articular cartilage were used to validate biomarkers among OA and T2DM. Sixty-eight hub genes were obtained, which were mainly enriched in the inflammatory response. We found that the hub gene TNFAIP6 is not only closely related to OA and T2DM but also a marker of prehypertrophic chondrocytes, which are closely related to the progression of OA. TNFAIP6 was found to be significantly elevated in CD14 + monocytes in T2DM patients, and this group of cells can promote inflammation. Validation on rat chondrocytes and human cartilage showed that TNFAIP6 was highly expressed in OA and further increased in the presence of T2DM or high glucose. Our study identified several characteristic modules and hub genes in the pathogenesis of T2DM-induced OA, which may facilitate further investigation of its molecular mechanisms. Up-regulated TNFAIP6 may contribute to OA in patients with T2DM by the recruitment of pro-inflammatory CD14 + monocytes in the OA synovium, which provides a potential target for the diagnosis and treatment of T2DM-associated OA.
Similar content being viewed by others


Introduction
As the most common chronic degenerative joint disease, OA attacks approximately 250 million people worldwide1. It is reported that nearly 30% of individuals greater than 45 years old have radiographic evidence of knee OA, about half of whom have symptoms1. Diabetes mellitus is a serious long-term disease that has a significant impact on the lives and well-being of people and societies worldwide. It is estimated by the International Diabetes Federation that 463 million people worldwide are suffering from DM in 2019, and this number will rise to 578 million by 2030 and 700 million by 20452.
Some risk factors, such as aging2, mechanical forces3, and metabolic factors4 are known to be critical for joint damage. Epidemiological research has shown that there may be an association between OA and type 2 diabetes mellitus (T2DM)5,6,7,8,9,10,11,12, in which T2DM could be an independent risk factor for OA. Several lines of studies suggest that hyperglycemia or T2DM could cause cartilage damage13,14,15,16,17,18,19,20,21,22, abnormal subchondral bone remodeling23,24,25,26,27,28, and synovitis29,30,31,32,33,34,35,36, finally leading to OA. In addition, hyperinsulinemia and insulin resistance are also been reported to contribute to cartilage degradation37. However, the molecular mechanisms underlying this association are not well understood, especially in studies based on transcriptome sequencing profiles. To fill the gap, we designed this research based on the Gene Expression Omnibus (GEO) dataset and bioinformatics analysis.
Weighted gene co-expression network analysis (WGCNA) is an excellent method to analyze the relationship between clinical phenotypes and gene modules which include highly correlated genes38. WGCNA has been proven to be a good tool for screening biomarkers in tumors39,40, hematological diseases41, and immune diseases42. Single-cell RNA sequencing (scRNA-seq) is an innovative technology used for whole-transcriptome amplification and high-throughput sequencing of mRNA at the single-cell level. It could reveal the gene structure and expression status of individual cells and reflect the heterogeneity between cells43. In this study, we used the WGCNA method to screen the hub genes between OA and T2DM, performed functional enrichment analysis, and applied scRNA-seq analysis to find marker genes in OA cartilage and T2DM peripheral blood mononuclear cells (PBMCs). Finally, we identified the marker gene TNFAIP6, which was significantly up-regulated in both OA and T2DM datasets. This is probably the first transcriptome study of this research topic.
Materials and methods
Data acquisition
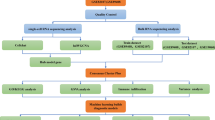
The gene expression profiles of patients with T2DM and OA were downloaded from the GEO database of NCBI (https://www.ncbi.nlm.nih.gov/geo/), and four data sets meeting the experimental requirements were downloaded. During the mRNA expression data selection process, we developed several inclusion criteria as follows: (1) The species studied was Homo sapiens; (2) The sample types of relevant data that will be subjected to the same analysis are consistent; (3) The sequencing platforms that will carry out the relevant data for the same analysis are consistent. The GSE95849 dataset44 is a bulk RNA-sequencing (RNA-seq) data including 6 normal and 6 T2DM PBMC samples. The GSE114007 dataset45 contained 18 normal and 20 OA cartilage samples, of which 10 normal and 10 OA samples were from the same sequencing platform. To avoid the batch effect, these 20 samples were chosen for analysis. The GSE169454 includes 3 normal and 4 OA cartilage scRNA-seq data46. The GSE165816 includes 54 scRNA-seq data (10 PBMC and 44 skin) from 10 non-diabetic subjects and 17 diabetic patients47, of which a total of 10 PBMC samples (3 normal and 7 T2DM) were used for subsequent analysis. An overall flowchart of this study is shown in Fig. 1.

Schematic representation of the present study.
Identifying differentially expressed genes (DEGs)
The “limma” R package (v3.52.4) was used for identifying DEGs from the microarray and high-throughput dataset48. Data from GSE95849 and GSE114007 were normalized using the “quantile” method in the “limma” R package, where DEGs were selected with adjusted P value < 0.05 and |log2fold change (FC)| >1. The heatmap and volcano plot of DEGs were plotted with R package “pheatmap” (v1.0.12) and “ggplot2” (v3.4.1), respectively.
WGCNA
After ranking the genes according to the median absolute deviation (MAD) from largest to smallest, we selected the top 10,000 genes for WGCNA by the “WGCNA” R package (v.1.72-1)49. The adjacency matrix was converted into a topological overlap matrix (TOM). A TOM heatmap was constructed using the “Tomplot” function in the “WGCNA” R package. Subsequently, we screened out the power parameter ranging from 1 to 20 using the “pickSoftThreshold” function in the “WGCNA” package, then genes were divided into different modules. Finally, T2DM-related and OA-related modules were obtained, and the resulting gene network was visualized via a heatmap.
Screening of hub genes and functional analysis
The R package “VennDiagram” (v1.7.3) was used for screening hub genes from T2DM- and OA-related modules and DEGs50. The correlation analysis of these genes was performed using “ggcorrplot” R package (v0.1.4) through the “Pearson correlation coefficient” method on the OA matrix. Gene set enrichment analysis (GSEA) was done using “clusterProfiler” package (v4.6.2) based on the Kyoto Encyclopedia of Genes and Genomes (KEGG)51,52,53 database on both T2DM and OA datasets. Gene set permutations were performed 1000 times for each analysis54. Protein–protein interaction (PPI) network of hub genes was constructed via the STRING online database (https://string-db.org/) for protein interaction analysis, and PPI was visualized using Cytoscape (v3.7.1, https://cytoscape.org) software55. The Cytohubba plugin in the Cytoscape was applied for sorting the hub genes, and the higher the degree value, the darker the gene background color and the larger the circle. The functional enrichment analysis of the Gene Ontology (GO) biological process (GO: BP) and the KEGG analysis were performed using the Metascape56 online tool (https://metascape.org/gp/index.html).
Single-cell RNA sequencing analysis
The R package “Seurat” (v4.3.0)57 was used for OA and T2DM scRNA-seq data processing, cells were filtered by mitochondrial gene percentage < 5% in the OA dataset and < 15% in the T2DM dataset depending on the type of tissue58. Cells with less than 200 gene features or greater than 8000 gene counts were removed to exclude low-quality cells, empty droplets, and doublets. After the principal component analysis, the “Harmony” R package (v0.1.1) was used for data integration and batch effect removal59. The Seurat functions of “FindNeighbors” and “FindClusters” were used to assign cells to the optimal clusters, and cells were displayed using a Uniform Manifold Approximation and Projection (UMAP) method. DEGs were determined using “FindAllMarkers”, genes with log2FC > 0.5 & adjusted P value < 0.05 were considered as significant markers. The top 10 markers of each cluster were presented using the “DoHeatmap” function of Seurat. Cells were annotated according to the previous studies47,60,61,62 and the CellMarker online database (http://bio-bigdata.hrbmu.edu.cn/CellMarker/). A gene set variation analysis (GSVA) was conducted using the “GSVA” R package (v1.48.3) based on the OA dataset63. High dimensional weighted gene co-expression network analysis (hdWGCNA) was performed using the “hdWGCNA” package (v0.3.03) to identify key genes associated with OA in the cartilage samples following the standard pipeline64. Gene expression correlation matrix, weighted gene co-expression networks, and module detection were conducted. Module-trait relationship analysis identified modules significantly associated with the OA group, and hub genes within significant modules were identified based on their intra-module connectivity. The R package “Monocle” (v2.24.0) was used to conduct a pseudotime analysis on the OA matrix65. GSEA was done using “clusterProfiler” package (v4.6.2) based on the Gene Ontology (GO) database on the gene set of CD14 + monocyte.
Materials and reagents
Recombinant human IL-1β was purchased from PeproTech (New Jersey, USA). The cell culture reagents were purchased from Gibco (Grand Island, NY). The primary antibodies against TNFAIP6 (13321-1-AP), MMP13 (18165-1-AP), and β-actin (20536-1-AP) were purchased from Proteintech (Wuhan, China). The primary antibody against IL6 (TD6087) was purchased from Abmart (Shanghai, China). TRIzol™ reagent was purchased from Invitrogen (California, USA).
Cellular culturing
Rat primary chondrocytes were isolated from the articular cartilage of 4-week-old Wistar rats from the Experimental Animal Centre of Wuhan University. To collect fresh articular cartilage, mature rats were euthanized by the inhalation of carbon dioxide. Briefly, cartilage was exposed to type II collagenase (2 mg/mL, Invitrogen, USA) at 37℃ for 6 h after shearing. Then, chondrocytes were harvested after centrifugation at 500 g for 5 min and seeded with DMEM medium (5.5mM glucose) and 10% FBS with 100 mg/ml streptomycin and 100 U/ml penicillin. Il-1β, which is commonly used to mimic in vitro pathological conditions, was employed as a positive control to induce the osteoarthritic phenotype of chondrocytes. Rat chondrocytes were stimulated for 72 h with IL-1β (5 ng/mL) under normal-glucose (5.5 mM) or high-glucose (25 mM) conditions as previously described20. To investigate whether the effect of high glucose was related to its osmotic effect, cells were incubated with mannitol (19.5 mM) (Biofroxx, Einhausen, Germany) instead of glucose (25 mM) for 72 h with or without IL-1β (5 ng/mL) (n = 6). Cell lysates were collected for mRNA extractions. All animal procedures were conducted according to the Guidelines for Care and Use of Laboratory Animals of the National Institutes of Health and approved by the Experimental Animal Welfare Ethics Committee, Zhongnan Hospital of Wuhan University (ZN2022191). For in vitro experiments, similar results were obtained in 6 independent experiments, and there were 3 repeats within an experiment. This study is reported following ARRIVE guidelines.
Human specimens and serum IL6 level
Human OA knee cartilage samples were obtained from 24 patients (12 non-diabetic and 12 T2DM, grade 4 by Kellgren & Lawrence classification for knee OA) undergoing total knee arthroplasty because of OA at Zhongnan Hospital of Wuhan University shortly after surgery from September 2022 to July 2023. The basic patient information is presented in Table S1. We harvested the human outer lateral (intact) and inner medial (damaged) tibial articular cartilages as previously described66, and samples were divided into four groups: intact cartilage of OA patients, intact cartilage of patients with combined T2DM and OA (DMOA), damaged cartilage of OA patients, damaged cartilage of DMOA patients. Samples were fixed in 4% paraformaldehyde for immunohistochemistry (IHC) or lysis for RNA extraction. We conducted a retrospective study to obtain serum IL6 levels in T2DM and non-diabetic OA patients in our department from January 2021 to June 2023. Patients who had serum IL6 testing before surgery, without other inflammatory diseases, and finally underwent total knee arthroplasty were included in the study. A total of 40 patients without diabetes and 22 T2DM patients were brought into the study (Table S1). The protocol used in this study complies with the ethical guidelines of the Helsinki Declaration in 1975 and was approved by the Medical Ethics Committee, Zhongnan Hospital of Wuhan University (2022151 K). The authors ensured that all participants were given the necessary information and signed informed consent forms before proceeding with the study.
RNA extraction and quantitative RT-PCR (qRT-PCR)
Total RNA was isolated from the cartilage samples and chondrocytes with TRIzol reagent following the manufacturer’s protocol. The concentration and purity of the isolated RNA were detected by NanoDrop 2000 micronuclei acid analyzer (ThermoFisher Scientific™, USA), and the A260/A280 ratio of all samples between 1.8 and 2.0 was considered as qualified samples. The cDNA was synthesized from 1 µg total RNA using HiScript® II Q RT SuperMix (Vazyme, Nanjing, China). Finally, qRT-PCR was performed by ChamQ Universal SYBR qPCR Master Mix (Vazyme, Nanjing, China) following the manufacturer’s protocol. The relative mRNA expression was calculated using the 2−ΔΔCt method with β-actin for normalization. The primer sequences are shown in Table S2.
Western blot (WB)
Total intracellular proteins were extracted according to the manufacturer’s instructions. The samples were incubated with respective primary antibodies against TNFAIP6 (1:1000 dilution), IL6 (1:1000 dilution), MMP13 (1:1000 dilution), and β-actin (1:2000 dilution) at 4 °C overnight. After being incubated with HRP-conjugated secondary antibody at room temperature for 1 h, the protein bands were measured using an ECL kit on an Imaging System (BIO-ID VL, Conn France).
Immunohistochemistry
The sections were placed in a humidified box at 4℃ overnight and covered with primary antibody against TNFAIP6 (1:100). After incubation, the sections were incubated with HRP-conjugated secondary antibody (Servicebio, Wuhan, China) for 1 h at 37℃, then counterstained with hematoxylin solution (Servicebio, Wuhan, China) for 3 min.
Statistical analysis
Data were analyzed and plotted with R (v4.2.1, https://www.R-project.org) software and GraphPad Prism 9. The P value was corrected following the false discovery rate (FDR) described by Benjamini & Hochberg. For in vitro and human experiments, data with different treatments were analyzed using one-way analysis of variance (ANOVA) with the post hoc test for multiple comparisons. Unpaired Student’s t-test was applied for comparisons between two groups if the homogeneity of variance was consistent. P value < 0.05 was considered statistically significant for all tests.
Results
Differential expression analysis and construction of weighted gene co-expression network in GSE95849
The GSE95849 dataset is a T2DM-related matrix containing 6 normal samples and 6 T2DM samples. A total of 1535 DEGs were screened using the above criteria, including 1051 up-regulated genes and 484 down-regulated genes (Table S3), and the heatmap visually represents the differences of all DEGs (Fig. S1A). The volcano plot shows DEGs between T2DM patients and healthy controls, DEGs with |log2FC| > 1 are labeled red or blue (Fig. 2A).
A total of 10,000 genes were selected for WGCNA following the standard pipeline after the normalization of the raw data in GSE95849. The sample clustering analysis was performed to eliminate the outlier using the dynamic shear tree method, and the cut height of 150 was used for the shear criterion. A total of 12 T2DM and control transcriptome data were converted into colors and plotted in a dendrogram and a trait heatmap (Fig. 2B). We then constructed a scale-free topology model and analyzed the mean connectivity of the adjacency matrix to choose the appropriate soft threshold. As shown in Fig. 2C, when the soft threshold is 13, R67 reaches 0.8, and the mean connectivity is close to 0, which means that the network is good. To identify the key modules related to T2DM, the trait-related genes were clustered by colors to draw a cluster dendrogram (Fig. 2D). A total of 20 co-expression modules were identified as shown in Fig. 2E (details were shown in Table S4), of which the turquoise module has the highest correlation with T2DM (a coefficient of 0.9, a P value of 7e-05). Visualization of the eigengene network also presents the close relationship between the turquoise module and the clinical trait T2DM (Fig. S1B). Therefore, we defined the turquoise module as the hub module, and a total of 1329 genes were filtered according to the standard pipeline (|GS| ≥ 0.2, |MM| ≥ 0.8) as shown in Fig. 2F. Four hundred genes were selected from the network for visualization of the TOM using a heatmap (Fig. 2G).

Differential expression analysis and weighted gene co-expression network analysis (WGCNA) on the type 2 diabetes mellitus (T2DM) bulk RNA-sequencing data. (A) The volcano plot of differentially expressed genes (DEGs) in the T2DM matrix. DEGs with |log2FoldChange| > 1 are labeled red or blue. (B) The sample dendrogram and trait heatmap on the T2DM matrix. (C) Determination of the soft threshold. (D) Cluster dendrogram utilized to detect coexpression clusters with corresponding color assignments. Gene clusters in different colors stand for different coexpression modules, and genes that are not co-expressed in any modules are assigned to the gray module. (E) Correlation matrix of module eigengene values obtained from WGCNA. Twenty modules were identified, and each module eigengene was tested for correlation with a trait. Within each cell, upper values are correlation coefficients between module eigengene and the traits; lower values are the corresponding P value. (F) Scatter plot of gene significance (GS) and module membership (MM) in the turquoise module. Key genes were screened out in the upper-right area where |GS| >0.2 and |MM| >0.8. (G) The heatmap depicts the topological overlap matrix (TOM) among all modules included in the analysis. The light color represents a low overlap, and the progressively darker red color represents an increasing overlap.
Identification of DEGs and hub module in GSE114007
The GSE114007 dataset is an OA-related matrix containing 10 normal human cartilage samples and 10 OA human cartilage samples. After the differential expression analysis according to the above criteria, 1141 DEGs were identified, including 641 up-regulated genes and 500 down-regulated genes (Table S5), which are represented in the volcano plot (Fig. 3A). The differences in DEGs were depicted in the heatmap (Fig. S1C). The same method above was performed to construct a WGCNA network. A total of 10,000 genes were included for analysis. To eliminate outliers, we set the cut height to 120, and all 20 samples were included (Fig. 3B). The scale-free topology model and mean connectivity were stable when the soft threshold was 10 (Fig. 3C). The network was constructed using a “one-step” method, and the result of clustering is shown in Fig. 3D. Finally, a total of 20 modules were identified (Fig. 3E, Fig. S1D), and the turquoise module showed the highest correlation with trait OA (a coefficient of 0.9, and a P-value of 6e-08) (details were shown in Table S6). Using the same method as above, we screened 885 OA-related genes for subsequent analysis (Fig. 3F). Selected genes of TOM were depicted in Fig. 3G via a heatmap.

Identification of differentially expressed genes (DEGs) and construction of weighted gene co-expression network analysis (WGCNA) on osteoarthritis (OA) bulk RNA-sequencing data. (A) The volcano plot of DEGs in the OA matrix. (B) Clustering dendrogram of samples based on their Euclidean distance. (C) Determination of the soft-threshold power. (D) Cluster dendrogram of genes, with dissimilarity based on topological overlap, together with assigned module colors. (E) Module–trait associations. Each row corresponds to a module, and each column corresponds to a trait. Each cell contains the corresponding correlation and P value. (F) Scatter plot analysis of the turquoise module. (G) The heatmap depicts the topological overlap matrix (TOM) among all modules included in the analysis.
Acquisition and analysis of trait-related DEGs and hub genes
To identify the hub genes and underlying molecular mechanism between T2DM and OA, we screened out the intersection of trait-related genes and DEGs. After filtering out the duplicate probes, we found 875 T2DM-related DEGs and 124 OA-related DEGs. As shown in the Venn plot, there are a total of 68 genes in at least 3 gene lists (underlined in Fig. 4A), of which 3 genes are shown in all 4 gene lists. These 68 genes were defined as the hub genes, which were expressed differently and associated with T2DM and OA (Table S7). The correlation between these hub genes is strong, as shown in the correlation heatmap based on the Pearson correlation coefficient in the OA dataset (Fig. 4B). The differential expression of the hub genes in T2DM and OA datasets was shown by heatmaps (Fig. S2A and B).
GSEA and Metascape functional enrichment analyses
We applied GSEA analyses in bulk RNA-seq data of T2DM and OA, respectively, to find the potential molecular mechanisms and biological processes of T2DM and OA, and to understand the underlying links between these two diseases. In the T2DM matrix, the GSEA analysis showed 49 activated pathways and 6 suppressed pathways (Fig. 4C). The up-regulated genes were significantly related to cytokine-cytokine receptor interaction, endocytosis, IL-17 signaling pathway, MAPK signaling pathway, neutrophil extracellular trap formation, NF-kappa B signaling pathway, NOD-like receptor signaling pathway, phagosome, rheumatoid arthritis, TNF signaling pathway, toll-like receptor signaling pathway, etc. In the OA dataset (Fig. 4D), a total of 14 up-regulated terms were identified. The up-regulated genes were mainly enriched in the B cell receptor signaling pathway, cell cycle, natural killer cell mediated cytotoxicity, neutrophil extracellular trap formation, osteoclast differentiation, phagosome, rheumatoid arthritis, etc. According to the above results, we can see that the transcriptome data of both T2DM and OA are enriched in rheumatoid arthritis and immune-related pathways, indicating that DM and OA might be potentially related in these types of pathways.
To further explore the function of hub genes between T2DM and OA, PPI network analysis and functional enrichment analysis were constructed. Our results showed that there were some biological connections between these 68 hub genes (Fig. 4E). The GO and KEGG functional annotation of hub genes was performed by Metascape, and the results are shown in Table S8. Interestingly, enriched pathways are mainly inflammation-, bone differentiation-, and immune-related, which are closely associated with OA. The most statistically significant clusters were presented in Fig. 4F according to the -log10(P). In Fig. 4G, gene enrichment clusters and nodes were colored by cluster ID. The inflammatory response seems to be more enriched based on statistical significance and node number (Fig. 4F and G).

Gene set enrichment analysis (GSEA) of type 2 diabetes mellitus (T2DM) and osteoarthritis (OA) bulk RNA-sequencing data and correlation and functional enrichment of hub genes. (A) The Venn plot showed the identification of intersection genes. (B) Pearson correlation analysis of hub genes in OA matrix. (C, D) GSEA analysis of (C) T2DM and (D) OA matrix. (E) Visualization of PPI network of hub genes. (F) Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment of hub genes using Metascape. (G) Gene enrichment clusters and nodes colored by cluster ID.
ScRNA-seq analysis of GSE169454
The GSE169454 data set is an OA-related single-cell transcriptome data containing 3 normal human cartilage samples and 4 OA human cartilage samples. Our goal was to verify the expression changes of hub genes in OA and identify the corresponding cell types in this data set. Firstly, we performed quality control and filtering of scRNA-seq data based on the number of feature genes detected in each cell, the total number of molecules detected inside the cell, and the percentage of mitochondrial (MT) genome readings. According to the visualization results of quality control, cells with feature counts greater than 8000 or less than 200 and cells with MT counts greater than 5% were filtered out for further analysis (Fig. S3A). The correlation between the number of counts and the feature genes of samples was 0.93, demonstrating a good sequencing depth (Fig. S3B). Secondly, after the normalization of data, we calculated the subset of features (n = 2000) that exhibited high cell-to-cell variation in the data set, and the top 10 genes were labeled (Fig. S3C). Thirdly, a total of 66,209 cells were clustered into 9 subsets using the “UMAP” method (Fig. S3D). Then, according to the above criteria, 821 markers were found for the identification of cell subsets, in which one of the hub genes TNFAIP6 presented as a marker gene of subset 2 (Fig. S3E). In Fig. S3F, subsets were presented by samples, indicating that the batch effect was nicely corrected.
Cells were mainly annotated as effective chondrocyte (EC), prehypertrophic chondrocyte (preHTC), hypertrophic chondrocyte (HTC), proliferative chondrocyte (ProC), regulatory chondrocyte (RegC), fibrocartilage chondrocyte (FC), homeostatic chondrocyte (HomC), and erythrocyte according to the previous research and CellMarker online database (Fig. 5A). After the intersection of hub genes and significant markers (Table S9), 5 genes were found (Fig. 5B) and only TNFAIP6 and UBE2T were significantly up-regulated in both sets of bulk RNA-seq data, and the rest of them were inconsistent in T2DM and OA (Fig. S2A and B). Preliminary validation was carried out on rat chondrocytes using qRT-PCR (Fig. S4A). The result of TNFAIP6 was consistent with the previous data (Fig. S4B), and UBE2T was neither enriched in any pathway above (Table S8) nor differentially expressed in DMOA cell models. Therefore, TNFAIP6 would be used for subsequent studies.
As presented in Fig. 5C, the marker gene TNFAIP6 was significantly up-regulated in OA samples. As a marker gene of preHTCs, TNFAIP6 was mainly expressed in preHTC (Fig. 5D and E) and the ratio of preHTC was significantly increased in OA samples (Fig. 5F and G, Fig. S4).
The function of TNFAIP6 in OA was further studied. A GSVA study was applied to find the enriched pathways of maker genes in each chondrocyte subtype. As shown in Fig. 5H and Table S10, the genes of preHTC mainly enriched in IL6 JAK STAT3 signaling, inflammatory response, apoptosis, TNFA signaling via NFKB, and reactive oxygen species pathway, which were closely related to the progression of OA. Genes were also enriched in pancreatic beta cells, indicating that preHTCs might be associated with the pathological process of DMOA.
We used hdWGCNA to identify the key molecular features of preHTC. During the construction of the co-expression network, we observed that when the scale-free topology fitness index reached 0.90, the soft threshold power β was 9 (Fig. 6A) to build an unweighted cell network, achieving the best connectivity. We identified seven gene modules (Fig. 6B). Subsequently, we assessed the module scores for the seven clusters and found that the M7 was highly activated in preHTC (Fig. 6C, Fig. S6A). We calculated modular connectivity to determine the connectivity of each gene based on the characterized genes (Fig. 6D, Fig. S6B), interestingly, TNFAIP6 presented as one of the characterized genes of preHTC.
Pseudotime analysis is often used to model the differentiation process of cell subtypes. In this study, we extracted data from ProC, preHTC, and HTC to find the change of TNFAIP6 in the differentiation. The result was consistent with the previous study, preHTCs represent a state between ProCs and HTCs (Fig. 6E). Pseudotemporal expression dynamics of TNFAIP6 marked the progression from ProC to HTCs (Fig. 6F). Taken together, these data reveal that TNFAIP6 plays a role in the potential differentiation between these cell types.

Analysis of osteoarthritis (OA) single-cell RNA sequencing data. (A) Eight clusters were annotated and labeled with different colors. (B) The Venn plot showed the intersection of hub genes and significant markers in osteoarthritis (OA) single-cell RNA sequencing data. (C) Violin plots showing the expression levels of TNFAIP6 grouped by samples. (D) Violin plot and (E) dim plot showing the expression of TNFAIP6 in each cell cluster. (F) Stacked Column Chart showing the ratio of each cell cluster grouped by samples. (G) Scatter plot showing the ratio of prehypertrophic chondrocytes between osteoarthritis and control samples (P = 0.011). (H) Heatmap showing the result of gene set variation analysis (GSVA) in each cell cluster.

High dimensional weighted gene co-expression network analysis and pseudotime analysis in osteoarthritis (OA) single-cell RNA sequencing data and analysis of single-cell RNA sequencing data on type 2 diabetes mellitus peripheral blood mononuclear cells (PBMCs). (A) Optimal soft threshold selection. (B) Construction of co-expression network using the optimal soft threshold of 9, with genes divided into 7 modules and resulting in a dendrogram. (C) UMAP plot of module 7 with ME staining. (D) KMes for module 7 characterization gene. (E) Monocle pseudotime trajectory showing the progression of Proliferative chondrocytes, prehypertrophic chondrocytes, and hypertrophic chondrocytes. (F) Pseudotemporal expression dynamics showing the expression level of TNFAIP6. (G) UMAP plot of 10 subpopulations in PBMC samples. (H) Stacked Column Chart showing the ratio of each cell cluster grouped by diseases. (I) Dot plot showing the expression of TNFAIP6 in each cell cluster.
ScRNA-seq analysis of GSE165816
GSE165816 contains 54 scRNA-seq data from 10 non-diabetic subjects and 17 diabetic patients, of which a total of 3 non-diabetic and 7 diabetic PBMC samples were used for subsequent analysis. After the cell filtration and data normalization the same as above (Fig. S7A and B), 2000 high-variation features were figured out (Fig. S7C). Cells were clustered into 13 subtypes using the “UMAP” method (Fig. S7D), and annotated according to the previous study47 as 10 subpopulations including CD14 + monocyte (CD14Mono), CD16 + monocyte (CD16Mono), T cells (T), B cells (B), natural killer T cell (NKT), natural killer cell (NK), dendritic cell (DC), plasma cell (Plasma), platelet, proliferative cell (Pro) (Fig. S7E and Fig. 6G). Interestingly, as the highest proportion of subpopulations in PBMC, the ratio of CD14 + monocyte was increased in T2DM PBMC samples (Fig. 6H) and TNFAIP6 was exclusively expressed in CD14 + monocytes (Fig. S7F). Moreover, CD14 + monocyte from diabetic samples expressed more TNFAIP6 than those from non-diabetic samples (Fig. 6I). In addition, the results of GSEA showed that up-regulated genes in CD14 + monocytes were significantly enriched in chemokine production, interleukin − 1 beta production, interleukin − 1 production, interleukin − 8 production, positive regulation of inflammatory response, etc. (Fig. S7G) These results suggest that there might be a synergistic effect between T2DM and OA, resulting in a higher expression of TNFAIP6 in both articular cartilage and PBMC of DMOA individuals than in OA patients alone.
Validation of the expression of TNFAIP6 on rat chondrocytes
To determine the expression of TNFAIP6 in vitro, a DMOA rat chondrocyte model was constructed based on a previous study20. The chondrocytes were treated or not with IL-1β (5ng/ml) for 72 h under normal glucose (5.5mM) or high glucose (25mM) environment. The expression level of TNFAIP6 was detected by RT-qPCR and WB. As expected, IL-1β stimulated the expression of TNFAIP6 (Fig. 7A and B), as well as the expression of inflammatory factor IL6 and matrix-degrading components MMP13 (Fig. 7B and C). Moreover, a high glucose environment enhanced the response of chondrocytes to IL-1β stimulation on the expression of TNFAIP6, IL6, and MMP13. To exclude the effects of osmotic stress caused by high glucose, we replaced glucose excess (19.5mM) with mannitol at the same molar concentration, as a control for the hyperosmotic effects of high glucose level. The rat chondrocytes stimulated with or without IL-1β in the presence of mannitol released similar amounts of TNFAIP6 to that under normal glucose conditions (Fig. 7D). Hence, the impact of hyperosmotic stress under a high glucose environment could be eliminated. To analyze the relationship between TNFAIP6 and markers of OA cartilage, a correlation analysis was carried out on the mRNA level of TNFAIP6, IL6, and MMP13. The Pearson correlation coefficient method was used to calculate the correlation of ΔCt of each gene in rat chondrocytes under normal or high glucose environments. A total of 24 samples were analyzed, and the mRNA levels of TNFAIP6 were strongly positively correlated with both IL6 (R = 0.82, p = 7.6e-07) and MMP13 (R = 0.71, P = 0.00011) (Fig. 7E), which indicated that TNFAIP6 might also be one of the markers of OA or DMOA cartilage.

Validation of TNFAIP6 expression level in rat chondrocytes and human articular cartilage, and IL6 levels in osteoarthritis (OA) and combined type 2 diabetes mellitus and osteoarthritis (DMOA) patients. (A) Induction of TNFAIP6 with normal glucose (5.5 mM) and high glucose (25 mM) in rat chondrocytes treated with or without IL-1β (5 ng/mL) at 72 h. Quantitative RT-PCR (qRT-PCR) analysis of mRNA levels of TNFAIP6, IL6, and MMP13 relative to that of β-actin (n = 6). (B) Western blot was used to determine protein expression of TNFAIP6, IL6, and MMP13 (n = 3). (C) QRT-PCR was used to determine the mRNA expression of IL6 and MMP13 (n = 6). (D) Impact of osmotic stress on the expression of TNFAIP6 by rat chondrocytes with or without IL-1β stimulation. Cells were incubated with normal glucose (5.5 mM) or mannitol (19.5 mM) for 72 h. the mRNA level of TNFAIP6 was detected by qRT-PCR (n = 6). (E) Correlation of ΔCt (compared to β-actin) of TNFAIP6 with IL6 (R = 0.82, P = 7.6e-07) and MMP13 (R = 0.71, P = 0.00011) (n = 24). (F) The expression of TNFAIP6 in the articular cartilage shown in immunohistochemical staining (n = 6). (G) The mRNA expression of TNFAIP6 in human articular cartilage was detected using quantitative RT-PCR (n = 12). (H) Concentration of IL6 in serum in OA and DMOA patients. *P < 0.05, **P < 0.01, ***P < 0.001.
Validation of the expression of TNFAIP6 on human articular cartilage samples
To determine the expression of TNFAIP6 in patients with T2DM and OA, qRT-PCR and immunohistochemical staining of articular cartilage were performed in patients who underwent total knee arthroplasty. As presented in Fig. 7F, the immunostaining of TNFAIP6 was mostly detected in chondrocytes and extracellular matrix in the area of damaged cartilage, but rarely expressed in the intact zone. As expected, the TNFAIP6 protein is released more in the lesioned cartilage of T2DM people than normal.
The results of qRT-PCR also showed a high mRNA level of TNFAIP6 in damaged cartilage, and the expression was significantly higher in DMOA individuals (Fig. 7G). In the above study, we found that the expression of IL6 was strongly correlated with TNFAIP6 in chondrocytes, thus, we conducted a retrospective analysis. A total of 62 patients who underwent total knee arthroplasty for OA were included (40 non-diabetic and 22 T2DM, details were shown in Table S1), and their preoperative serum IL6 levels were routinely measured. To our surprise, IL6 levels were significantly elevated in diabetic patients (Fig. 7H), which predicted the expression level of TNFAIP6. Therefore, up-regulated TNFAIP6 might be indicative of OA in T2DM patients.
Discussion
OA is associated with a local and systemic low-grade inflammation state13. Insulin resistance and hyperglycemia, two major features of T2DM, could both induce and aggravate the inflammation of OA. Ribeiro37 et al. found that insulin could reduce the expression of LC3II and increase the phosphorylation of Akt and rpS6 through the Akt/mTOR signaling pathway in immortalized human chondrocytes and cultures of primary human chondrocytes, thereby causing autophagy dysfunction of chondrocytes and aggravating cartilage matrix degradation and inflammation. OA cartilages from T2DM patients showed increased responsiveness to IL-1β-induced inflammation, and high glucose enhanced IL-1β-induced expression of IL6 and COX2 in cultured chondrocytes20. In our previous study, we demonstrated that hyperglycemia caused the accumulation of advanced glycosylation end products in fibroblast-like synoviocytes via the HIF-1α-GLUT1 pathway, which increases the release of inflammatory factors from fibroblast-like synoviocytes, and subsequently induces cartilage degradation and OA progression29. In this study, we further tried to uncover the genomic relationship between T2DM and OA and GSEA analysis showed that up-regulated genes in both T2DM and OA transcriptome data were enriched in inflammatory and immune-related pathways. The enrichment analysis of hub genes by Metascape showed that the top enriched pathway was the inflammatory response, which was consistent with the previous studies. Hub genes including ADM, ALOX5AP, CCR1, FCGR2B, HCK, TNFAIP6, TLR8, TLR9, and PROK2 were enriched in this pathway.
Due to the complexity of chondrocyte differentiation in osteoarthritis and the lack of specific biomarkers to characterize chondrocyte differentiation in osteoarthritis, the differentiation process and cell subtype of chondrocytes in osteoarthritis remain largely unknown. Previously, an scRNA-seq map of knee cartilage was reported from a cohort of 10 patients with OA and outlined several known and new cell populations60. However, only 1464 cells were included in this study, and the samples lacked non-OA cartilage. In the current study, we acquired GSE16945447, which contains 3 normal cartilage samples and 4 OA samples, with a total of 66,209 cells included. After data filtration and normalization, 821 significant markers of subtypes were figured out. By intersecting with the hub genes of DMOA above, we obtained the maker gene TNFAIP6, which may be the key gene in the pathogenesis of DMOA. TNFAIP6 is a secreted protein, which has been used as a novel biomarker of OA cartilage66,68, synovium68, and synovial fluid69. In this study, TNFAIP6 was found to be enhanced in OA cartilage and acted as a marker gene for preHTC, which might also play a role in the differentiation of ProC to preHTC. PreHTC can modulate the onset of hypertrophic differentiation, which regulates the mineralization of the surrounding matrix in cartilage, leading to the progression of OA70. In the current study, we found that the ratio of preHTC in OA cartilage was significantly increased than normal cartilage, and the results of GSVA analysis showed enrichment in the inflammatory response, which was consistent with the result of Metascape analysis. It also supported the role of preHTC in OA progression.
Many studies have demonstrated that diabetes can increase the levels of human pro-inflammatory factors, such as TNF-a, IL-1β, and IL-6. It is well known that immune cells play an important role in inflammatory response. Studies have found that TNFAIP6 can be expressed by immune cells, including neutrophils, mast cells, dendritic cells, and macrophages71,72. In a study of scRNA-seq, TNFAIP6 was found to be released more in CD14 + monocytes of patients with T2DM than in non-diabetic individuals61, this subgroup was defined as pro-inflammatory CD14 + monocytes which were uniquely characterized by the upregulation of inflammatory genes, including inflammatory cytokines (TNF, IL1B, NFKBIA, DUSP2, NLRP3, and TNFAIP6), chemokines (CCL3, CCL3L1, CCL4L2, CXCL2, and CXCL8), and transcriptional factors (FOS, FOSB, and JUNB). Recruitment of monocytes in the OA synovium could lead to synovitis and cartilage damage73. In our study, we found an increased proportion of CD14 + monocytes in T2DM PBMC, and overexpression of TNFAIP6 in diabetic CD14 + monocytes using the scRNA-seq data of PBMC in T2DM and non-diabetic patients, indicating that TNFAIP6 may be involved in the proliferation of CD14 + monocytes in T2DM. In addition, the up-regulated genes in CD14 + monocytes were enriched in pathways related to inflammatory response and production of inflammatory cytokines and chemokines. However, the connections between CD14 + monocytes and chondrocytes need further study.
TNFAIP6 is an important inflammation-related gene, which plays an anti-inflammatory and extracellular matrix remodeling role in immune regulation and tissue repair74. The TNFAIP6 protein has been shown previously to be upregulated in T2DM75 and when produced by mesenchymal stem cells reduces insulitis in models of type 1 diabetes mellitus76. Moreover, the pancreatic beta cells are one of the few sites where TNFAIP6 is constitutively expressed, where this expression decreases during pre-diabetes77. As reported, TNFAIP6 is a double-edged sword in OA. Tellier78 et al. used medial meniscal transection to induce a rat OA model and injected TNFAIP6-loaded microparticles into rats with OA, the cartilage damage was significantly reduced. However, high expression of TNFAIP6 in OA cartilage and the absence of IαI may damage the formation of the cartilage matrix during OA progression66. In the current study, we found that OA chondrocytes under a high glucose environment expressed more TNFAIP6 than normal glucose, which was strongly correlated with inflammation marker IL6 and matrix degradation marker MMP13. The results of qRT-PCR and IHC showed that the damaged cartilage of DMOA patients expressed more TNFAIP6 protein than the normal OA patients. Moreover, DMOA patients had more IL6 in their serum than non-diabetic patients, which indicated a higher inflammatory state. Also, there are some limitations to our study. Firstly, there is still a lack of high-throughput sequencing data on DMOA patients, so we performed a combined analysis with data from two independent diseases. Such work is undergoing in our related study. Secondly, further mechanistic studies, including the association between CD14 + monocytes and chondrocytes are needed to validate our findings.
Conclusions
In conclusion, we identified the marker gene TNFAIP6 via bioinformatic analysis of bulk RNA-seq and scRNA-seq data from T2DM and OA patients. We speculated that up-regulated TNFAIP6 may promote the production of cytokines and chemokines in T2DM patients by the recruitment of pro-inflammatory CD14 + monocytes in the OA synovium, which accelerates synovitis and cartilage damage, leading to OA progression. Our study provided a potential target for the diagnosis and treatment of T2DM-associated OA.
Data availability
The datasets used and analyzed in this study are available from the GEO dataset (https://www.ncbi.nlm.nih.gov/geo/). Data supporting the findings of this study can be obtained from the corresponding author upon reasonable request.
Abbreviations
- T2DM:
-
Type 2 diabetes mellitus
- OA:
-
Osteoarthritis
- GEO:
-
Gene Expression Omnibus
- WGCNA:
-
weighted gene co-expression network analysis
- scRNA-seq:
-
Single-cell RNA sequencing
- PBMC:
-
Peripheral blood mononuclear cell
- RNA-seq:
-
RNA-sequencing
- DEG:
-
Differentially expressed gene
- FC:
-
Fold change
- MAD:
-
Median absolute deviation
- TOM:
-
Topological overlap matrix
- GSEA:
-
Gene set enrichment analysis
- PPI:
-
Protein–protein interaction
- GO:
-
Gene Ontology
- BP:
-
Biological process
- UMAP:
-
Uniform Manifold Approximation and Projection
- GSVA:
-
Gene set variation analysis
- hdWGCNA:
-
High dimensional weighted gene co-expression network analysis
- DMOA:
-
Patients with combined T2DM and OA
- IHC:
-
Immunohistochemistry
- WB:
-
Western blot
- FDR:
-
False discovery rate
- MT:
-
Mitochondrial
- EC:
-
Effective chondrocyte
- preHTC:
-
Prehypertrophic chondrocyte
- HTC:
-
Hypertrophic chondrocyte
- ProC:
-
Proliferative chondrocyte
- RegC:
-
Regulatory chondrocyte
- FC:
-
Fibrocartilage chondrocyte
- HomC:
-
Homeostatic chondrocyte
- CD14Mono:
-
CD14 + monocyte
- CD16Mono:
-
CD16 + monocyte
- T:
-
T cell
- B:
-
B cell
- Plasma:
-
Plasma cell
- NKT:
-
Natural killer T cell
- NK:
-
Natural killer cell
- DC:
-
Dendritic cell
- Pro:
-
Proliferative cell
References
David, J., Hunter, S. B. Z. & Osteoarthritis Lancet 393, 1745–1759, doi:https://doi.org/10.1016/S0140-6736(19)30417-9 (2019).
Lawrence, R. C. et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 58, 26–35. https://doi.org/10.1002/art.23176 (2008).
Visser, A. W. et al. The relative contribution of mechanical stress and systemic processes in different types of osteoarthritis: the NEO study. Ann. Rheum. Dis. 74, 1842–1847. https://doi.org/10.1136/annrheumdis-2013-205012 (2015).
Xie, C., Chen, Q. & Adipokines New Therapeutic Target for Osteoarthritis? Curr. Rheumatol. Rep. 21, 71. https://doi.org/10.1007/s11926-019-0868-z (2019).
Rehling, T., Bjorkman, A. D., Andersen, M. B., Ekholm, O. & Molsted, S. Diabetes Is Associated with Musculoskeletal Pain, Osteoarthritis, Osteoporosis, and Rheumatoid Arthritis. Journal of diabetes research 6324348, doi: (2019). https://doi.org/10.1155/2019/6324348 (2019).
Arellano, P., Vertti, R. D., Muniz, A., Martinez, L. S. M. & Gonzalez Galarza, J. Arguello Astorga, R. Cartilage Oligomeric Matrix Protein Levels in type 2 diabetes Associated with primary knee osteoarthritis patients. Genetic Test. Mol. Biomarkers. 23, 16–22. https://doi.org/10.1089/gtmb.2018.0184 (2019).
Dubey, N. K. et al. Correlation between diabetes Mellitus and knee osteoarthritis: a Dry-To-Wet Lab Approach. Int. J. Mol. Sci. 19, 3021. https://doi.org/10.3390/ijms19103021 (2018).
King, K. B. & Rosenthal, A. K. The adverse effects of diabetes on osteoarthritis: update on clinical evidence and molecular mechanisms. Osteoarthr. Cartil. 23, 841–850. https://doi.org/10.1016/j.joca.2015.03.031 (2015).
Eymard, F. et al. Diabetes is a risk factor for knee osteoarthritis progression. Osteoarthr. Cartil. 23, 851–859. https://doi.org/10.1016/j.joca.2015.01.013 (2015).
Courties, A., Gualillo, O., Berenbaum, F. & Sellam, J. Metabolic stress-induced joint inflammation and osteoarthritis. Osteoarthr. Cartil. 23, 1955–1965. https://doi.org/10.1016/j.joca.2015.05.016 (2015).
Zheng, J. et al. Associations of Diabetes Status and Hyperglycemia with symptomatic knee osteoarthritis. Arthritis care Res. Epub Ahead Print. https://doi.org/10.1002/acr.24872 (2022).
Schett, G. et al. Diabetes is an independent predictor for severe osteoarthritis: results from a longitudinal cohort study. Diabetes care. 36, 403–409. https://doi.org/10.2337/dc12-0924 (2013).
Courties, A. & Sellam, J. Osteoarthritis and type 2 diabetes mellitus: what are the links? Diabetes Res. Clin. Pract. 122, 198–206. https://doi.org/10.1016/j.diabres.2016.10.021 (2016).
Mobasheri, A., Neama, G., Bell, S., Richardson, S. & Carter, S. D. Human articular chondrocytes express three facilitative glucose transporter isoforms: GLUT1, GLUT3 and GLUT9. Cell. Biol. Int. 26, 297–300. https://doi.org/10.1006/cbir.2001.0850 (2002).
Rosa, S. C. et al. Impaired glucose transporter-1 degradation and increased glucose transport and oxidative stress in response to high glucose in chondrocytes from osteoarthritic versus normal human cartilage. Arthritis Res. Therapy. 11, R80. https://doi.org/10.1186/ar2713 (2009).
Eaton, C. B. et al. Sex differences in the association of skin advanced glycation endproducts with knee osteoarthritis progression. Arthritis Res. Therapy. 19 https://doi.org/10.1186/s13075-017-1226-z (2017).
Chen, Y. J., Sheu, M. L., Tsai, K. S., Yang, R. S. & Liu, S. H. Advanced glycation end products induce peroxisome proliferator-activated receptor gamma down-regulation-related inflammatory signals in human chondrocytes via toll-like receptor-4 and receptor for advanced glycation end products. PloS One. 8, e66611. https://doi.org/10.1371/journal.pone.0066611 (2013).
Wang, H. et al. Effect of type II diabetes-induced osteoarthritis on articular cartilage aging in rats: a study in vivo and in vitro. Exp. Gerontol. 150, 111354. https://doi.org/10.1016/j.exger.2021.111354 (2021).
Silawal, S. et al. IL-10 could play a role in the interrelation between diabetes Mellitus and Osteoarthritis. Int. J. Mol. Sci. 20 https://doi.org/10.3390/ijms20030768 (2019).
Laiguillon, M. C. et al. Characterization of diabetic osteoarthritic cartilage and role of high glucose environment on chondrocyte activation: toward pathophysiological delineation of diabetes mellitus-related osteoarthritis. Osteoarthr. Cartil. 23, 1513–1522. https://doi.org/10.1016/j.joca.2015.04.026 (2015).
Vaamonde-Garcia, C. et al. The nuclear factor-erythroid 2-related factor/heme oxygenase-1 axis is critical for the inflammatory features of type 2 diabetes-associated osteoarthritis. J. Biol. Chem. 292, 14505–14515. https://doi.org/10.1074/jbc.M117.802157 (2017).
Pineiro-Ramil, M. et al. Reduced Levels of H2S in Diabetes-associated osteoarthritis are linked to hyperglycaemia, Nrf-2/HO-1 signalling downregulation and chondrocyte dysfunction. Antioxidants 11, 628, doi: (2022). https://doi.org/10.3390/antiox11040628
Chen, Y. et al. Abnormal subchondral bone remodeling and its association with articular cartilage degradation in knees of type 2 diabetes patients. Bone Res. 5, 17034. https://doi.org/10.1038/boneres.2017.34 (2017).
Davies-Tuck, M. L. et al. Increased fasting serum glucose concentration is associated with adverse knee structural changes in adults with no knee symptoms and diabetes. Maturitas 72, 373–378. https://doi.org/10.1016/j.maturitas.2012.05.013 (2012).
Wen, C. Y. et al. Bone loss at subchondral plate in knee osteoarthritis patients with hypertension and type 2 diabetes mellitus. Osteoarthr. Cartil. 21, 1716–1723. https://doi.org/10.1016/j.joca.2013.06.027 (2013).
Franke, S. et al. Advanced glycation end products affect growth and function of osteoblasts. Clin. exp. Rheumatol. 29, 650–660 (2011).
Wang, H. J. et al. Diabetes mellitus accelerates the progression of osteoarthritis in streptozotocin-induced diabetic mice by deteriorating bone microarchitecture, bone mineral composition, and bone strength of subchondral bone. Annals Translational Med. 9, 768. https://doi.org/10.21037/atm-20-6797 (2021).
Gui, T. et al. Elevated expression of ICAM-1 in synovium is associated with early inflammatory response for cartilage degeneration in type 2 diabetes mellitus. J. Cell. Biochem. 120, 13177–13186. https://doi.org/10.1002/jcb.28592 (2019).
Li, Q. et al. Hyperglycemia-induced accumulation of advanced glycosylation end products in fibroblast-like synoviocytes promotes knee osteoarthritis. Exp. Mol. Med. 53, 1735–1747. https://doi.org/10.1038/s12276-021-00697-6 (2021).
Tsai, C. H. et al. High glucose induces vascular endothelial growth factor production in human synovial fibroblasts through reactive oxygen species generation. Biochim. Biophys. Acta. 1830, 2649–2658. https://doi.org/10.1016/j.bbagen.2012.12.017 (2013).
Ribeiro, M. et al. Diabetes-accelerated experimental osteoarthritis is prevented by autophagy activation. Osteoarthr. Cartil. 24, 2116–2125. https://doi.org/10.1016/j.joca.2016.06.019 (2016).
Luo, S. et al. Expression and significance of MMPs in synovial fluid, serum and PBMC culture supernatant stimulated by LPS in Osteoarthritis patients with or without diabetes. Experimental and clinical endocrinology & diabetes: official journal. German Soc. Endocrinol. [and] German Diabetes Association. 127, 195–202. https://doi.org/10.1055/s-0043-122223 (2019).
Eitner, A. et al. Pain sensation in human osteoarthritic knee joints is strongly enhanced by diabetes mellitus. Pain 158, 1743–1753. https://doi.org/10.1097/j.pain.0000000000000972 (2017).
Siu, K. K. et al. Increased interleukin 1beta levels in the subacromial fluid in diabetic patients with rotator cuff lesions compared with nondiabetic patients. J. Shoulder Elbow Surg. 22, 1547–1551. https://doi.org/10.1016/j.jse.2013.01.011 (2013).
Eitner, A., Culvenor, A. G., Wirth, W., Schaible, H. G. & Eckstein, F. Impact of diabetes mellitus on knee osteoarthritis pain and physical and mental status: data from the osteoarthritis initiative. Arthritis Care Res. 73, 540–548. https://doi.org/10.1002/acr.24173 (2021).
Hamada, D. et al. Suppressive effects of insulin on Tumor Necrosis factor-dependent early osteoarthritic changes Associated with obesity and type 2 diabetes Mellitus. Arthritis Rheumatol. 68, 1392–1402. https://doi.org/10.1002/art.39561 (2016).
Ribeiro, M., Lopez de Figueroa, P., Blanco, F. J., Mendes, A. F. & Carames, B. Insulin decreases autophagy and leads to cartilage degradation. Osteoarthr. Cartil. 24, 731–739. https://doi.org/10.1016/j.joca.2015.10.017 (2016).
Stuart, J. M., Segal, E., Koller, D. & Kim, S. K. A gene-coexpression network for global discovery of conserved genetic modules. Science 302, 249–255. https://doi.org/10.1126/science.1087447 (2003).
Cheng, S. et al. Identification of IL10RA by Weighted correlation network analysis and in vitro validation of its association with prognosis of metastatic melanoma. Front. Cell. Dev. Biol. 8, 630790. https://doi.org/10.3389/fcell.2020.630790 (2020).
Ding, M., Li, F., Wang, B., Chi, G. & Liu, H. A comprehensive analysis of WGCNA and serum metabolomics manifests the lung cancer-associated disordered glucose metabolism. J. Cell. Biochem. 120, 10855–10863. https://doi.org/10.1002/jcb.28377 (2019).
Xie, Q. et al. An immune-related gene prognostic index for acute myeloid leukemia associated with regulatory T cells infiltration. Hematology 27, 1088–1100. https://doi.org/10.1080/16078454.2022.2122281 (2022).
Yin, L., Chen, Y., Fu, T., Liu, L. & Xia, Q. Identification of candidate blood biomarkers for the diagnosis of septicaemic melioidosis based on WGCNA. Artif. Cells Nanomed. Biotechnol. 50, 252–259. https://doi.org/10.1080/21691401.2022.2126490 (2022).
Paolillo, C., Londin, E. & Fortina, P. Single-cell Genomics. Clin. Chem. 65, 972–985. https://doi.org/10.1373/clinchem.2017.283895 (2019).
Luo, L. et al. Gene expression profiling identifies downregulation of the Neurotrophin-MAPK signaling pathway in Female Diabetic Peripheral Neuropathy patients. J. Diabetes Res. 2017 (8103904). https://doi.org/10.1155/2017/8103904 (2017).
Fisch, K. M. et al. Identification of transcription factors responsible for dysregulated networks in human osteoarthritis cartilage by global gene expression analysis. Osteoarthr. Cartil. 26, 1531–1538. https://doi.org/10.1016/j.joca.2018.07.012 (2018).
Fu, W. et al. 14-3-3 epsilon is an intracellular component of TNFR2 receptor complex and its activation protects against osteoarthritis. Ann. Rheum. Dis. 80, 1615–1627. https://doi.org/10.1136/annrheumdis-2021-220000 (2021).
Theocharidis, G. et al. Single cell transcriptomic landscape of diabetic foot ulcers. Nat. Commun. 13, 181. https://doi.org/10.1038/s41467-021-27801-8 (2022).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559. https://doi.org/10.1186/1471-2105-9-559 (2008).
Chen, H. & Boutros, P. C. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 12, 35. https://doi.org/10.1186/1471-2105-12-35 (2011).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592. https://doi.org/10.1093/nar/gkac963 (2023).
Wu, T. et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innov. (Camb). 2, 100141. https://doi.org/10.1016/j.xinn.2021.100141 (2021).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. https://doi.org/10.1101/gr.1239303 (2003).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523. https://doi.org/10.1038/s41467-019-09234-6 (2019).
Stuart, T. et al. Comprehensive Integration of single-cell data. Cell 177, 1888–1902 e1821, doi: (2019). https://doi.org/10.1016/j.cell.2019.05.031
Ferreira, P. G. et al. The effects of death and post-mortem cold ischemia on human tissue transcriptomes. Nat. Commun. 9, 490. https://doi.org/10.1038/s41467-017-02772-x (2018).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 16, 1289–1296. https://doi.org/10.1038/s41592-019-0619-0 (2019).
Ji, Q. et al. Single-cell RNA-seq analysis reveals the progression of human osteoarthritis. Ann. Rheum. Dis. 78, 100–110. https://doi.org/10.1136/annrheumdis-2017-212863 (2019).
Ma, P. et al. Immune cell landscape of patients with diabetic macular edema by single-cell RNA analysis. Front. Pharmacol. 12, 754933. https://doi.org/10.3389/fphar.2021.754933 (2021).
Kang, X., Zhang, K., Wang, Y., Zhao, Y. & Lu, Y. Single-cell RNA sequencing analysis of human chondrocytes reveals cell-cell communication alterations mediated by interactive signaling pathways in osteoarthritis. Front. Cell. Dev. Biol. 11, 1099287. https://doi.org/10.3389/fcell.2023.1099287 (2023).
Hanzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14 https://doi.org/10.1186/1471-2105-14-7 (2013).
Morabito, S., Reese, F., Rahimzadeh, N., Miyoshi, E. & Swarup, V. hdWGCNA identifies co-expression networks in high-dimensional transcriptomics data. Cell. Rep. Methods. 3, 100498. https://doi.org/10.1016/j.crmeth.2023.100498 (2023).
Trapnell, C. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386. https://doi.org/10.1038/nbt.2859 (2014).
Chou, C. H., Attarian, D. E., Wisniewski, H. G., Band, P. A. & Kraus, V. B. TSG-6 - a double-edged sword for osteoarthritis (OA). Osteoarthr. Cartil. 26, 245–254. https://doi.org/10.1016/j.joca.2017.10.019 (2018).
Saeedi, P. et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res. Clin. Pract. 157, 107843. https://doi.org/10.1016/j.diabres.2019.107843 (2019).
Bayliss, M. T. et al. Up-regulation and differential expression of the hyaluronan-binding protein TSG-6 in cartilage and synovium in rheumatoid arthritis and osteoarthritis. Osteoarthr. Cartil. 9, 42–48. https://doi.org/10.1053/joca.2000.0348 (2001).
Wisniewski, H. G. et al. TSG-6 activity as a novel biomarker of progression in knee osteoarthritis. Osteoarthr. Cartil. 22, 235–241. https://doi.org/10.1016/j.joca.2013.12.004 (2014).
Dy, P. et al. Sox9 directs hypertrophic maturation and blocks osteoblast differentiation of growth plate chondrocytes. Dev. Cell. 22, 597–609. https://doi.org/10.1016/j.devcel.2011.12.024 (2012).
Mahoney, D. J. et al. TSG-6 inhibits osteoclast activity via an autocrine mechanism and is functionally synergistic with osteoprotegerin. Arthritis Rheum. 63, 1034–1043. https://doi.org/10.1002/art.30201 (2011).
Qi, Y. et al. TSG-6 released from intradermally injected mesenchymal stem cells accelerates wound healing and reduces tissue fibrosis in murine full-thickness skin wounds. J. Invest. Dermatol. 134, 526–537. https://doi.org/10.1038/jid.2013.328 (2014).
Raghu, H. et al. CCL2/CCR2, but not CCL5/CCR5, mediates monocyte recruitment, inflammation and cartilage destruction in osteoarthritis. Ann. Rheum. Dis. 76, 914–922. https://doi.org/10.1136/annrheumdis-2016-210426 (2017).
Li, R. et al. An update on the role of tumor necrosis factor alpha stimulating gene-6 in inflammatory diseases. Mol. Immunol. 152, 224–231. https://doi.org/10.1016/j.molimm.2022.11.008 (2022).
Elhadad, M. A. et al. Deciphering the plasma proteome of type 2 diabetes. Diabetes 69, 2766–2778. https://doi.org/10.2337/db20-0296 (2020).
Kota, D. J., Wiggins, L. L., Yoon, N. & Lee, R. H. TSG-6 produced by hMSCs delays the onset of autoimmune diabetes by suppressing Th1 development and enhancing tolerogenicity. Diabetes 62, 2048–2058. https://doi.org/10.2337/db12-0931 (2013).
Day, A. J. & Milner, C. M. TSG-6: a multifunctional protein with anti-inflammatory and tissue-protective properties. Matrix Biol. 78–79, 60–83. https://doi.org/10.1016/j.matbio.2018.01.011 (2019).
Tellier, L. E. et al. Intra-articular TSG-6 delivery from heparin-based microparticles reduces cartilage damage in a rat model of osteoarthritis. Biomaterials Sci. 6, 1159–1167. https://doi.org/10.1039/C8BM00010G (2018).
Acknowledgements
We would like to thank the study participants for voluntarily participating in this study.
Funding
This research was supported by the National Natural Science Foundation of China (No. 81972036, 81603214, 82002265), the Key Research and Development Project of Hubei Province (No. 2020BCA071), Youth Interdisciplinary Special Fund of Zhongnan Hospital of Wuhan University (No. ZNQNJC2022001), and Research fund from medical Sci-Tech innovation platform of Zhongnan Hospital, Wuhan University (No. PTXM2021033).
Author information
Authors and Affiliations
Contributions
Siyi Liu participated in the conception and design of the study. Siyi Liu and Haitao Chen carried out the analysis and interpretation of data and writing of the manuscript. Yinxian Wen, Xu Yang, and Liaobin Chen have been involved in the examination and critical manuscript revisions. All authors have read and approved the final version of the manuscript. Siyi Liu and Yinxian Wen have directly accessed and verified the underlying data reported in the manuscript. Haitao Chen and Liaobin Chen were responsible for the decision to submit the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
All animal procedures were conducted according to the Guidelines for Care and Use of Laboratory Animals of the National Institutes of Health and approved by the Experimental Animal Welfare Ethics Committee, Zhongnan Hospital of Wuhan University (ZN2022191). The studies involving human specimens were reviewed and approved by the Medical Ethics Committee, Zhongnan Hospital of Wuhan University (2022151 K). The authors ensured that all participants were given the necessary information and signed informed consent forms before proceeding with the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, S., Chen, H., Yang, X. et al. Identification and validation of up-regulated TNFAIP6 in osteoarthritis with type 2 diabetes mellitus. Sci Rep 14, 31450 (2024). https://doi.org/10.1038/s41598-024-82985-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-82985-5
