
Abstract
Malachite green (MG) is used as a dye for materials such as wood, cotton, and nylon, and is used in aquaculture to prevent fungal and protozoan diseases. However, it is highly toxic, with carcinogenic, mutagenic, and teratogenic properties, resulting in bans worldwide. Despite this, MG is still frequently used in many countries due to its efficacy and economy. MG is persistent in the environment and so requires degradative intervention. In this work we isolated Bacillus pacificus ROC1 strain from a salt flat in Pakistan that had the ability to aerobically detoxify MG, as determined by bacterio- and phyto-toxicity assays. We demonstrate immobilized B. pacificus ROC1 can effectively detoxify MG, which highlights a potential method for its biodegradation. Genomic sequencing identified three candidate azo-reductases within B. pacificus ROC1 that could be responsible for the MG-degrading activity. These were cloned, expressed and purified from Escherichia coli, with one (AzrC), catalyzing the reduction of MG to leuco-MG in vitro. AzrC was crystallised and MG was captured within the active site in a Michaelis complex, providing structural insight into the reduction mechanism. Altogether, this work identifies a bacterium capable of aerobically degrading a major industrial pollutant and characterizes the molecular basis for this activity.
Similar content being viewed by others

Introduction
Malachite green (MG), a triphenylmethane dye, is a water-soluble dye that is often used to dye materials such as wood, leather, nylon, and cotton1. MG is also used in aquaculture as it prevents diseases such as saprolegniasis caused by the mould Saprolegnia, and white spot disease caused by the protozoan Ichthyophthirius multifiliis2,3,4. Despite the efficacy of MG, it has been banned in many nations, including China, the EU, and the US, due to negative health effects1,5,6. MG can accumulate in organisms, along with the lipophilic reduced leuco form (LMG), and has been documented to be carcinogenic, teratogenic, and mutagenic1,7,8,9,10,11,12,13,14,15. Low levels of MG (0.1 mg/L) have been shown to be toxic to mammalian cells, lower serum calcium levels, and reduce fertility16,17,18. MG, being a chromophore in visible wavelengths, also has environmental impacts; it decreases the transmission of light through waterbodies, resulting in decreased photosynthetic activity, and thus oxygen19. Yet, MG is still used in some developing nations and is used illegally in others, due to its efficacy and cost efficiency; in the dyeing process 15% of dye is lost directly to wastewater1,9,20,21,22. Indeed, despite bans on MG, eels in German and Belgian waterways were found to contain MG and LMG, and in 2022 and 2023 shrimp imports to Europe were found to contain MG and LMG23,24,25,26.
The complex aromatic structure of MG makes it recalcitrant to degradation, resulting in accumulation in the ecosystem, but consistent with their intended use as a dye needing to be resistant to light, water, oils, and chemicals27. Chemical degradation methods, such as ozonation, the use of photo-Fenton reagents, microwave-assisted catalysis, sono-photolysis, and photoelectrocatalysis have been developed for the degradation of MG28,29,30,31,32,33,34. However, chemical methods are held back by their cost, production of secondary pollutants, or low efficiency, which can result from salinity, pH, or temperature of wastewater.
A promising alternative to chemical degradation methods is biodegradation, using whole bacterial or fungal cells for complete detoxification of MG. This ability has been observed in multiple organisms, including Saccharomyces cerevisiae, Streptomyces exfoliatus, and Bacillus vietnamensis35,36,37. Individual enzymes have also been identified as being able to degrade MG, such as the metalloenzymes laccase and manganese peroxidase, and the NADH dependent triphenyl methane reductase (TMR)38,39,40,41. TMR has been described to reduce MG at the central C10 forming LMG, whereas laccase and manganese peroxidase have been identified as capable of oxidising MG to many smaller molecules. A de novo metalloenzyme has also been developed based off myoglobin for the degradation of MG42.
In this work, we isolated and characterised a bacterial strain from dye-contaminated salt flats in Pakistan, Bacillus pacificus ROC1 (ROC1). We identified ROC1 as being able to completely, and aerobically, decolourise MG. Additional experiments monitoring bacterio- and phyto-toxcity identified that ROC1 was not only able to decolourise, but also detoxify MG, suggesting a substantial breakdown of MG. We performed genomic sequencing of ROC1, alongside cell lysate assays, to identify an azoreductase capable of MG decolourisation. AzrC was recombinantly expressed and was observed to decolourise MG via reduction to LMG, as determined by LCMS analysis. The formation of LMG was supported by the solved AzrC-MG co-crystal structure, which captured MG and AzrC in a Michaelis complex.
Results
Malachite green degradation in bacterial culture and immobilized cells
To identify the ability of ROC1 to utilise MG as a sole carbon or nitrogen source, ROC1 was screened on different minimal media, containing either a carbon source, a nitrogen source, or neither, all with 50 ppm MG. ROC1 was unable to grow on any minimal media, indicating that ROC1 was unable to use MG as a sole carbon or nitrogen source. However, ROC1 grew well with LB media containing 250 ppm MG, and visibly decolourised the dye, indicating that ROC1 likely co-metabolises MG.
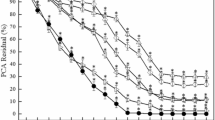
To quantify the ability of ROC1 to decolourise MG, ROC1 was incubated with various concentrations of MG (Fig. 1A). The duration of treatment affected the overall decolourisation of MG and it took more time to decolourise fully when higher concentrations were used. For the 50 ppm MG culture, 100% decolorization was observed after 72 h of treatment, while 100% decolorization for 100 to 500 ppm was observed after 96 h of treatment and for 750 ppm decolourisation required 144 h. However, in the presence of 1,000 ppm only 41% decolourisation was recorded after 144 h or greater, a significant decrease compared to all other concentrations, suggesting MG could have a toxic effect on the cells at such high concentrations.
To investigate the potential inhibitory effect of high MG concentrations on cell growth, the growth of ROC1 was recorded along with dye decolourisation every 24 h. In the presence of all concentrations of MG, the OD600 increased. However, it was found that faster growth was observed at lower concentrations up until 500 ppm (OD600 1), and at higher concentrations of 750 and 1000 ppm the growth was significantly inhibited (Fig. 1B). Similarly, when measuring colony forming units, wet biomass, and dry biomass (Supporting Information 1), the data indicated that all concentrations of MG allowed cell growth, but that growth was slower at higher MG concentrations, suggesting that MG does have an inhibitory effect against ROC1 growth, which becomes apparent at high concentrations.
To investigate approaches to prevent MG inhibition of ROC1 growth, LB media was adjusted with 0.5% (w/v) and 1% (w/v) glucose alongside 1,000 ppm MG. 1,000 ppm MG was chosen because this concentration was not fully decolourised and resulted in reduced ROC1 growth, permitting identification of improvement. No significant improvement in decolourisation was observed with the addition of glucose (Supporting Information 2). Alternatively, the continuous addition of 1% (v/v) fresh LB was conducted every 24 h, resulting in 100% decolourisation of MG after 120 h (Supporting Information 2). The addition of fresh LB also affected ROC1 growth positively, with an insignificant difference in OD600 after 120 h between samples with 0 and 1,000 ppm MG, and a significant difference between 1,000 ppm samples with and without media addition (Fig. 1C). These results suggest that the addition of fresh media increased the cell biomass, which had a positive impact on the decolourisation of MG at high concentrations.

ROC1 decolourisation of MG. (A) Decolourisation of 50 − 1,000 ppm MG measured at 620 nm over 144 h in LB by free ROC1 ± SD (N = 2). (B) Optical density measured at 600 nm of ROC1 cultures grown over 144 h in LB in the presence of 0–1,000 ppm MG ± SD (N = 2). (C) Optical density of free ROC1 in LB measured at 600 nm in the addition of 1% (v/v) media with 0 ppm (white) and 1000 ppm (green) MG, and with 1000 ppm without media addition (black) ± SD (N = 2). (D) Decolourisation of 50 − 1,000 ppm MG measured at 620 nm over 144 h in LB by immobilised ROC1 ± SD (N = 2). Data presented with significance in Supporting Information 3.
Immobilised cells (IMC) are an attractive substitute for free cell biodegradation due to their reusability and stability43. IMC of ROC1, prepared using an agar entrapment method previously described44,45, were tested to determine their ability to decolourise MG. After 144 h of treatment, 100% decolourisation was observed for concentrations of 50–400 ppm (Fig. 1D). However, at 500-1,000 ppm MG approximately 20% decolourisation was recorded after 144 h of treatment. This reduced decolourisation, as compared to the free cells, could be a result of reduced biomass.
Toxicity of ROC1 products
Given that ROC1 was capable of MG decolorization, ROC1 could prove useful if it were able to degrade MG into non-toxic molecules. To test for bacteriotoxicity of MG degradation products, both Gram-positive and Gram-negative strains, Staphylococcus aureus and Escherichia coli, respectively, were used. Both strains exhibited growth inhibition when exposed to MG, with a positive relationship between clearance zone and MG concentration, demonstrating the toxicity of MG to bacteria (Fig. 2A, B). Alternatively, MG treated with ROC1 did not inhibit growth of either bacteria.
To test for phytotoxicity, the percentage of seed germination of the commercially important Solanum lycopersicum variety Riogrande (tomato) was assessed, along with the root and shoot length of germinated seeds. A substantial increase in percentage of seed germination was measured when seeds were incubated with water containing MG and treated with ROC1 vs. untreated water, with a negative relationship between percentage of seed germination and MG concentration (Supporting Information 4). At 50 ppm MG there was a 60% germination rate, and 13% at 1,000 ppm. In ROC1 treated water, the germination rate was above 80% for all MG concentrations, indicating that the phytotoxic effect of MG is ameliorated by ROC1-mediated MG degradation. A similar negative relationship was observed for root and shoot length (Fig. 2C, D): at 1,000 ppm MG the root and shoot length were negligible, whereas in ROC1 treated samples the root and shoot lengths were significantly longer, suggesting that any metabolites formed after ROC1-mediated degradation were not phytotoxic. Altogether, these results indicate that ROC1 can completely detoxify MG and that any metabolites produced after treatment by ROC1 were non-toxic to the bacteria and plants tested.

ROC1 treated MG toxicity. (A) Clear zones of E. coli when grown with kanamycin (white), untreated MG (green), and treated MG (red) ± SD. Asterisks indicate significance (N = 3). (B) Clear zones of S. aureus when grown with kanamycin (white), untreated MG (green), and treated MG (red) ± SD. Asterisks indicate significance (N = 3). (C) Shoot length of Solanum lycopersicum when incubated with water (white), untreated MG (green), and treated MG (red) ± SD. Asterisks indicate significance (N = 7). (D) Root length of Solanum lycopersicum when incubated with water (white), untreated MG (green), and treated MG (red) ± SD. Asterisks indicate significance (N = 7).
Identifying the enzymatic basis for MG degradation
The cell free extract (CFE) of ROC1 was used to identify the enzyme involved in MG degradation, with specific testing for laccase and reductase activity since these enzyme families have previously been observed to catalyze MG degradation38,40,41. The reductase assay revealed a gradual decrease in MG absorbance at 620 nm over time with CFE (Fig. 3). When additional NADH was provided, there was a significant increase in the rate of decolourisation. Alternatively, no laccase activity was detected in the ROC1 CFE. This suggested that the enzyme responsible for MG decolourisation was an NADH-dependent reductase.

Reductase activity in the CFE. MG absorbance (620 nm) measured over two hours in buffer (black), buffer with CFE (red), and buffer with CFE and NADH (green) ± SD (N = 3). Data presented with significance in Supporting Information 5.
To identify MG degrading enzymes, the ROC1 genome was sequenced with 98.82% completeness, and revealed a genome length of 5,189,869 bp (Table 1), with a GC content of 35.4%. The ROC1 genome has 5,384 predicted protein encoding genes, 102 predicted tRNA genes, and 32 predicted rRNA genes. The circular genome generated by Proksee (Supporting Information 6) displays the genome characteristics, including gene distribution and GC content46. RAST annotation of the ROC1 genome categorised 5,384 genes into 27 subsystems (Fig. 4A). A total of 23% of its genes are present in the largest subsystem, “amino acids and derivatives” with 326 members. “Metabolism of aromatic compounds” contained 12 predicted genes.

ROC1 genome. (A) Functional annotation of ROC1 genomic genes using RAST. (B) Phylogenetic tree of the relationship between ROC1 (red) and other Bacillus species. The branch numbers are GBDP pseudo-bootstrap values from 100 replicates.
A phylogenetic tree (Fig. 4B) was constructed using the Type Strain Genome Server (TYGS) for pairwise comparison between genomes using Genome Blast Distance Phylogeny (GBDP) and genetic distance calculator47,48. Both average nucleotide identity (ANI), and average amino acid identity (AAI), were used for phylogenetic analysis. ROC1 had the highest similarity with B. pacificus anQ_h4, with an ANI value of 100%. The lowest similarity was observed with B. badius NBPM_293, with an ANI value of 65.79%. Regarding AAI, ROC1 had the highest similarity with B. pacificus anQ_h4, B. pacificus NCCP_15909, and Bacillus cereus ATTC_10987, with AAI values of 100%. The lowest level of protein similarity was observed with B. badius NBPM_293, with an AAI value of 61.54%.
To identify candidate genes involved in MG degradation, the 12 genes categorised as as being involved in “metabolism of aromatic compounds” by RAST were examined. Of these initial 12, three were identified as putative MG degrading genes, via analysis using reference genes sourced from the Integrated Microbial Genomes (IMG) system, which were compared to the ROC1 genome49. The products of these three genes were annotated as two FMN-dependent NADH-azoreductases (MDF0734648.1 and MDF0737692.1), AzrC and AzrA respectively, and one NADPH-dependent FMN reductase (MDF0734684.1) YhdA; His-tagged sequences, with Tobacco Etch Virus (TEV) cleave sites, are presented in Supporting Information 7. These three enzymes are found throughout the Bacillus genus and consist of 211, 208, and 178 residues, respectively. ROC1 AzrC and AzrA has sequence identities of 98% and 99% with AzrC (PDB-ID 3W79) and AzrA (PDB-ID 3W77) from Bacillus sp. B29, respectively, which have previously been shown to be capable of reducing numerous azo-dyes, including Methyl-Red, Orange I, and Acid Red 8850,51,52. AzrC similarity is displayed in Supporting Information 8 and 9 A. ROC1 AzrC also had 57% sequence identity with Indigo reductase (PDB-ID 6JXN) from Bacillus smithii which could reduce indigo carmine to leuco-indigo carmine, along with reducing multiple azo-dyes53. Whereas ROC1 YhdA had just 59% sequence identity with YhdA (PDB-ID 3GFR) from Bacillus subtilis, which has been demonstrated to reduce quinones and azo-dyes54,55.
Purification of the azo-reductases was achieved by Ni(II)-affinity chromatography for initial assays (Supporting Information 10 A). Elution fractions were orange in colour, indicating the presence of a riboflavin based cofactor, and the SDS-PAGE contained a strong bands at ~ 20–25 kDa for each protein, consistent with the calculated masses of 21.6, 25.2, and 25.0 kDa, for YhdA, AzrC, and AzrA, respectively.
To determine which enzyme was responsible for the degradation of MG, all three enzymes were separately incubated with MG. Only AzrC was capable to decolourising MG and was capable of decolourising MG using both NADH and NADPH (Supporting Information 11). AzrC was then further purified using size exclusion chromatography (Supporting Information 10B). Analysis of the 3D gel in the ImageLab software indicated that AzrC was > 99% pure, with few impurities, demonstrating that AzrC was easily purified to a high extent. Kinetic analysis, in which the NADH and NADPH concentration was varied, was performed to determine a Kd for the cofactors (Supporting Information 12). Both NADH and NADPH were used to determine Michaelis-Menten kinetics for AzrC with MG (Fig. 5A and B). The kcat was determined to be 0.942 s− 1 and 0.611 s− 1 for NADH and NADPH, respectively. The KM was 935.2 µM (95% CI 794.3–1111) and 475.5 µM (95% CI 381.2-598.8) for NADH and NADPH, respectively.

AzrC reactivity. (A) Michaelis-Menten plot of AzrC activity with NADH as a reducing agent. (B) Michaelis-Menten plot of AzrC activity with NADPH as a reducing agent. (C) LC chromatogram of the AzrC reaction with no reducing agent. (D) Mass spectrum of MG at 11.55 min of the AzrC reaction with no reducing agent. (E) Mass spectrum at 13.29 min of the AzrC reaction with no reducing agent. (F) LC chromatogram of the AzrC reaction with reducing agent. (G) Mass spectrum of LMG at 14.89 min of the AzrC reaction with reducing agent. H – Mass spectrum of desmethyl-LMG at 13.28 min of the AzrC reaction with reducing agent.
LCMS analysis was undertaken to determine reaction products of AzrC. The reaction with no NADH displayed a strong peak at 11.5 min on the LC, which was determined to be MG with mass 329.2012 (Fig. 5C and D). Alternatively, the reaction with NADH displayed a peak, not at 11.5 min, but at 14.89 min, which was determined to be LMG with a protonated mass of 331.2164 (Fig. 5F and G). A small peak at 13.28 min was also evident in the LC trace; this was identified to have a mass of 317.2012, consistent with desmethyl-LMG (Fig. 5F and H). Desmethyl-LMG was not identified in the cofactor-free control reaction (Fig. 5E). We are unsure of the mechanism of desmethyl-LMG formation, but due to a retention time difference of over one minute compared to LMG, it appears that desmethyl-LMG was formed in the reaction.
Structural characterization of AzrC-catalyzed MG reduction
Purified AzrC was co-crystallized with MG, and the structure of the complex was solved to 2.35 Å and was deposited to the protein data bank (PDB) under PDB-ID 9C0W (Table 2). Despite a strong CC1/2, data were truncated to 2.35 Å, due to the presence of an ice ring. AzrC formed a dimer, with the FMN binding sites being located at the dimer interface. Structural similarity of ROC1 AzrC and Bacillus sp. B29 AzrC is displayed in Supporting Information 9B. MG was bound at only one active site, due to the other being occupied by the N-terminus of a symmetry mate (Supporting Information 13). MG was modelled into a Polder electron density difference map (Fig. 6A), showing that it was bound within the AzrC active site, with the cationic imine 3.7 Å from N5 of FMN (Fig. 6B). Despite MG being bound within the predominantly hydrophobic and aromatic active site, there were few direct interactions as determined by Flatland Ligand Environment View (FLEV) (Coot) and PoseView (ProteinPlus), with FLEV indicating a π-cation interaction between F172 and the cationic imine of MG, and PoseView indicating π-π interactions between MG and Y151 and Y156 (Fig. 6C and D), suggesting that AzrC likely has promiscuous substrate specificity. The π-cation interaction from F172 likely stands to aid in positioning the cationic imine proximal to FMN, suggesting reduction would occur at the cationic imine, with protonation at the central C10 forming LMG (Fig. 6E).

AzrC-MG co-crystal structure. Co-crystal structure of MG-bound AzrC (PDB 9C0W). (A) Polder map at 5σ (green) of MG (grey). (B) MG (grey) and surrounding residues of AzrC (cyan). (C) Flatland ligand environment view (FLEV) of MG in 9C0W. (D) Two-dimensional PoseView of MG in 9C0W. (E) Proposed mechanism of MG reduction to LMG by AzrC.
Discussion
In this work, we utilised a bioprospecting approach, and isolated Bacillus pacificus ROC1 from a MG contaminated site in the Kohat salt range in Pakistan. ROC1 was capable of aerobically degrading MG, and was able to detoxify MG, with the resulting treated water non-toxic to E. coli, S. aureus, and S. lycopersicum variety Riogrande. Despite the ability of ROC1 to degrade MG, it was unable to completely mineralise MG, suggesting that ROC1 works in consortia. The degradation and detoxification of MG suggest that ROC1 could have uses in MG biodegradation processes, due to the aerobic activity and halophilic nature of ROC1.
Bacillus species have been documented to be capable of aerobically degrading dyes: B. subtilis was documented to degrade orange II; B. pseudomycoides was documented to degrade methylene green, acid blue, and basic violet; and B. cereus was documented to degrade reactive orange 16 and reactive black 544,56,57. Whereas, for MG, most identified species have been of other genera, such as Pseudomonas veronii, Streptomyces exfoliatus, Citrobacter sedlakii, and Saccharomyces cerevisiae27,36,37,58. There has been one description of a Bacillus genus member, Bacillus vietnamensis sp. MSB17, which was capable of MG degradation and detoxification. In the work investigating B. vietnamensis, there was an increase in tyrosinase, laccase, and manganese peroxidase activity during MG degradation, leading to the authors concluding the involvement of these enzymes in the process. However, there was no thorough investigation into these enzymes and there was no analysis of reductase activity35.
In our work we investigated three enzymes from ROC1 for their ability to decolourise MG: AzrA, AzrC, and YhdA. Just one of these enzymes, AzrC, was identified as having activity with MG. AzrC from ROC1 was 98% identical to AzrC from Bacillus sp. B29 and 57% identical to Indigo reductase from Bacillus smithii. AzrC from Bacillus sp. B29 was documented to reduce methyl red, 1-(2-pyridylazo)-2-naphthol, acid red 88, and orange I, whereas indigo reductase from B. smithii was documented to reduce indigo carmine, methyl red, and acid red 8850,53. However, neither has been shown to degrade MG. These dyes have similar properties to MG, and are all aromatic molecules, with hydrophobic regions, excluding indigo carmine which was used as a hydrophilic substitute for indigo. Also, the proposed mechanism for AzrC from B. sp. B29 reduction of orange I and acid red 88 involved reduction at an electrophilic carbonyl carbon ~ 3.5 Å from N5 of FMN52. The solved enzyme: substrate Michaelis complex of ROC1 AzrC-MG (Fig. 6; PDB-ID 9C0W) revealed that the distance of FMN N5 to the cationic imine was similar to the carbonyl carbon of orange I and acid red 88 in AzrC from B. sp. B29. This proximity suggested that reduction likely occurs at the imine of MG, resulting in the formation of LMG, for which a mechanism has been proposed. LCMS analysis supported this hypothesis as LMG was the main reaction product; we also noted the formation of desmethyl-LMG, but this formation was unexplained, potentially forming spontaneously. Previous work investigating the breakdown products of whole cell degradation identified LMG and desmethyl-LMG as breakdown products, suggesting that reductases likely play a role in the in vivo degradation of MG36,37,58,59,60,61.
In conclusion, we isolated and characterised the halophilic Bacillus pacificus ROC1 which was capable of aerobically degrading and detoxifying MG. We further identified the enzyme AzrC as capable of reducing MG to LMG, a process potentially involved during in vivo degradation. A potential mechanism for the reduction of MG by AzrC was suggested, supported by mass spectrometry analysis of the reaction products and the structure of the Michaelis complex, as determined by X-ray crystallography.
Materials and methods
Inoculum preparation
Bacillus pacificus ROC1 used in this work was isolated from the Kohat salt range, Kohat, Khyber Pakhtunkhwa, Pakistan. The whole ROC1 genome was submitted under the accession number SAMN33627856 to the NCBI.
Media screen
To determine the ability of ROC1 to use MG as a sole carbon or nitrogen source, ROC1 was streaked onto three different minimal media. These media contained no carbon or nitrogen source (MM), contained glucose as a carbon source but no nitrogen source (MMG), or contained ammonium nitrate as a nitrogen source but no carbon source (MMN), and were composed as described previously, with the addition of 50 ppm MG62. Isolated bacteria was used to inoculate each medIa and the growth was monitored as described previously62.
Biodegradation experiments
To monitor the degradation of MG, both free and immobilized cells were employed. Dye decolourisation experiments were performed in flasks containing different concentrations (0, 50, 100, 250, 400, 500, 750 and 1000 ppm) of MG. In all decolourisation experiments, percentage decolourisation was calculated using the formula:
Malachite green degradation by free cells
E. coli grown in LB media was supplemented with varying concentrations of MG, which was incubated at 37 °C for 6 days. Every 24 h, 3 mL of each sample was centrifuged at 8,000 rcf for 10 min, and the supernatant absorbance at 620 nm was measured, followed by calculation of percentage decolourisation using the previous equation, the pellet was used for mass determination. Free cell growth was monitored by OD600.
The CFU of free cells was measured every 24 h. Five-fold serial dilutions of each sample were prepared and 20 mL of the 5th dilution was spread onto LB agar, followed by incubation at 37 °C overnight.
Free cell wet and dry biomass were also checked every 24 h by weighing the cell pellet to determine the wet biomass, which was then dried at 90 °C overnight using a heat block to determine the dry biomass.
Effect of glucose and LB
For the 1,000 ppm MG samples, 0.5% (w/v) and 1% (w/v) glucose was added to media as an additional nutrient supply every 24 h. Similarly, 1% (v/v) LB was added to the culture every 24 h.
Immobilisation of bacterial isolates
An agar entrapment method described previously was used44,45. Seed cultures (15 mL) of ROC1 were pelleted and washed with water three times. The cell slurry was prepared by resuspending the cell pellet in 1 mL of 0.9% (w/v) NaCl. Next, a 100 mL solution of 0.9% (w/v) NaCl and 0.1 g/mL agar was sterilised in an autoclave, and was cooled to 45 °C, at which point the cell slurry (1 mL) was mixed with the molten agar (15 mL). This mixture solidified in Petri dishes, and was cut into cubes of 10 mm3. The cubes were washed with 100 mM phosphate, pH 7.5 for 1 h at 4 °C.
Cell viability was determined by dissolving the agar blocks in 100 mM phosphate, pH 7.5, and the dissolved solution was spread onto agar plates. Cells numbers were counted after overnight incubation at 37 °C.
Malachite green degradation by immobilised cells
Three agar cubes containing ROC1 were added to LB media and varying MG concentrations were incubated with the cells for 6 days. Analysis was performed as for free cells. Agar cubes containing no cells were used as a negative control.
Bacteriotoxic effects
Bacteriotoxic effects of untreated and treated MG samples were assess by the agar diffusion methods with some modifications as described previously63,64. E. coli and S. aureus were used, and were cultures in LB at 37 °C at 220 rpm. Aliquots (OD600 equal to 0.5 MacFarland’s turbidity standard) were spread onto LB agar. Wells, 6 mm in diameter, were prepared and 50 µL sample from treated and untreated MG were loaded into the wells. Kanamycin (500 mg/mL) was used as a positive control. Plates were incubated at 37 °C for 24 h, and the clear zone around each well was measured.
Phytotoxic effects
The commercially important tomato plant Solanum lycopersicum variety Riogrande was used for phytoxicity investigation of treated and untreated MG. Seeds (15) were sterilised with 1.2% (v/v) sodium hypochlorite, then were washed three times with water. Seeds were placed into Petri dishes containing filter paper moistened with treated or untreated MG solutions, followed by incubation at 22–24 °C under light. Total seed germination was counted on days 3 and 5, the root and shoot length was measured, and percentage germination was calculated using the formula:
Cell free extract enzyme assay
Reductase activity was determined by providing MG as a substrate and the CFE as an enzyme source. Reactions contained 20 mM Tris pH 7.4, 100 mM NADH, and CFE; MG decolourisation was monitored spectrophotometrically at 620 nm.
Laccase activity was determined by the ABTS method using 2,2’-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)65. Reactions contained 0.5 mM ABTS, 0.1 M sodium acetate pH 4.5, and CFE. ABTS oxidation was monitored at 620 nm.
Whole genome sequencing and analysis
DNA preparation and sequencing
ROC1 genomic DNA was extracted using the PureLink™ genomic DNA mini kit. DNA was quantified by measuring absorbance at 260 nm using a NanoDrop®, followed by 1% (w/v) agarose gel electrophoresis. The ROC1 genome was sequenced by Illumina sequencing technology with 23X genome coverage.
Assembly and annotation
The genome was assembled using Geneious de novo assembler and gaps wre removed by rearrangement of contigs using Mauve multiple genome alignment software, the default progressive Mauve was used with the reference strain Bacillus pacificus NCCP 15,909 (Accession number CP041979.1)66. The sequenced genome was submitted to the NCBI under the accession number JARGCX000000000.1.
Phylogenetic analysis
The list of related species for ROC1 was obtained from LPSN67. All accession numbers and genome sequences required were sourced from the NCBI. The list of all organisms used in phylogenetic analysis are displayed in Supporting Information 14. For phylogenetic tree construction, TYGS was used47. Trees were constructed based on the whole genome of the selected organisms. The genome sequence data and related species accession numbers were uploaded to TYGS. Average nucleotide identity and AAI were calculated by the EDGAR platform, using all species included in the tree68.
Prediction of dye degrading genes
Two approaches were used to predict genes involved in MG degradation. First, RASTtk was used for annotation of ROC1, which annotated and divided the genome into sub-systems, from which the required genes were selected69. Second, the local BLASTp feature of BioEdit 7.2 was also used for identification of MG degrading genes. Protein sequences of enzymes involved in degradation from reference strains were sourced from IMG49,70. These IMG sequences were used as a BLAST query against the ROC1 genome, using BLOSUM62 and an E-value 10. To construct a phylogenetic tree for AzrC sequences were sourced using UniProt BLAST71. Sequences were clustered with a sequence identity of 99% using the easy-cluster function of MMseqs72. A phylogenetic tree was constructed using IQ-TREE273.
Protein production and purification
MDF0734648.1 (AzrC), MDF0737692.1, and MDF0734684.1 were cloned into pET29-b(+) with a N-terminal hexahistidine tag and TEV cleavage site, under the control of a T7 promoter. The plasmids were transformed into E. coli BL21 (DE3) and transformants were cultured in LB containing 50 µg/mL kanamycin at 30 °C at 180 rpm to OD600 0.7–0.8, at which point gene expression was induced with 500 µM IPTG and 500 µM riboflavin, followed by incubation at 30 °C at 180 rpm overnight. Cells were harvested by centrifugation at 4,730 rcf for 30 min at 4 °C and were resuspended in 20 mM Tris, 200 mM NaCl, pH 8.0, followed by lysis via sonication and lysate clarification by centrifugation at 17,400 rcf for 60 min at 4 °C.
Lysate was filtered through a 0.45 μm syringe filter and loaded onto a HisTrap™ HP 5 mL column, followed by washing with 20 mM Tris, 200 mM NaCl, pH 8.0, 20 mM imidazole, and elution with 20 mM Tris, 200 mM NaCl, pH 8.0, 500 mM imidazole with a 0-100% B gradient over 20 CV. Fractions containing enzyme were concentrated and filtered through a 0.2 μm syringe filter, then loaded onto a HiLoad 26/600 Superdex® 200 pg SEC column. Protein purification was analysed by SDS-PAGE and Image Lab version 6.1 (Bio-Rad).
Azoreductase assay
All enzymes were buffer exchanged into 20 mM sodium phosphate, 200 mM NaCl, pH 5.0. Assays were performed using 50 µM enzyme, 2 mM NAD(P)H, 2 mM FMN, and 1 mM MG in different combinations, and absorbance at 620 nm was monitored, due to a MG absorbance maximum, with no absorbance by NAD(P)H (Supporting Information 15).
NADH and NADPH kinetics
The Kd of NADH and NADPH was determined using 100 µM AzrC, 500 µM MG, and cofactor concentrations of 0, 125, 250, 500, 1000, and 2000 µM, and absorbance at 620 nm was monitored. Data were processed using the hyperbola model in GraphPad Prism.
Michaelis-Menten kinetics
Michaelis-Menten kinetics were determined using 50 µM AzrC, 2 mM NADPH, and MG concentrations of 20, 40, 80, 160, 320, 640, and 1280 µM, and absorbance at 620 nm was monitored. Data were processed using the Michaelis-Menten model – nonlinear regression in GraphPad Prism.
Reaction product determination
Reactions containing 50 µM AzrC, 3 mM NADH, and 1 mM MG were incubated at room temperature for two hours. The reaction was quenched with addition of 2X volume acetonitrile with 0.1% (v/v) formic acid. Quenched reactions were then filtered via centrifugation at 13,000 rcf for 15 min using an Amicon® Ultra Centrifugal Filter, 10 kDa MWCO. This was then diluted 1:100 in MQ with 0.1% (v/v) formic acid.
Reaction products were determined using LCMS using a gradient of 5–95% (v/v) acetonitrile with 0.1% (v/v) formic acid over 20 min with an ACQUITY™ Premier BEH C18 1.7 μm VanGuard™ FIT 2.1 × 50 mm Column attached to an UltiMate 3000 HPLC and a Q Exactive Plus mass spectrometer. 1 µL sample was run at 200 µL/min through the LC column, and was analysed by positive mode ESI with spray voltage 3.5 kV. Compounds were fragmented with 30 eV normalised collision energy.
Protein crystallization
AzrC was purified as in Sect. 4.11 with the addition of excess FMN to solution immediately prior to loading onto the SEC column.
AzrC (37 mg/mL) was incubated with 500 µM MG and 500 µM NAD+ before crystallisation. Crystals were grown at 19 °C using 0.1 M bis-tris pH 5.5, 0.1 M sodium acetate, 20% (w/v) PEG 10 K as mother liquor (ML) by hanging drop diffusion. AzrC was crystallised at a 1:1 protein: ML ratio. All crystals were manually looped and flash-cooled in liquid nitrogen.
X-ray crystallography data collection and processing
Diffraction data were collected on beamline MX2 at the Australian Synchrotron74. Data were collected over 360° with 0.1° rotation at a distance of 200 mm and 80% attenuation of the beam.
Data were indexed and integrated using XDSGUI, followed by resolution estimation and data truncation using AIMLESS as implemented in CCP475,76,77. The structure was solved by molecular replacement using Phaser MR as implemented in CCP4, using PDB entry 3W7A as the search model52,76,78,79. MG was identified in difference density, and a Polder map was used to confirm MG coordinates80. The model was iteratively refined and optimised using phenix.refine and Coot version 0.9.8.781,82. The structure was validated using MolProbity. Phenix version 1.21.1 produced the refinement statistics (Table 2)83,84. The final structure was visualised in PyMOL version 3.0.385,86. The MG environment and interactions were visualised using FLEV, as integrated into Coot, and PoseView87,88,89.The structure factor CIF file was generated using pdb_extract and was validated using the wwPDB Validation System90.
Statistical testing
All significance values were determined using a two-way ANOVA followed by a Tukey’s multiple comparisons test for when multiple datasets were being compared, or a Sidak’s multiple comparisons test for when two datasets were being compared.
Data availability
The whole ROC1 genome was submitted under the accession number SAMN33627856 to the NCBI.
References
Srivastava, S., Sinha, R. & Roy, D. Toxicological effects of malachite green. Aquat. Toxicol. 66 (3), 319–329 (2004).
West, P. V. Saprolegnia parasitica, an oomycete pathogen with a fishy appetite: New challenges for an old problem. Mycologist 20, 99–104 (2006).
Scholz, T. Parasites in cultured and feral fish. Vet. Parasitol. 84 (3–4), 317–335 (1999).
Bruno, D. W., West, P. V. & Beakes, G. W. Saprolegnia and Other Oomycetes, in Fish Diseases and Disorders (CABI, 2011).
EC. Commission Decision of 22 December 2003 amending Decision 2002/657/EC as regards the setting of minimum required performance limits (MRPLs) for certain residues in food of animal origin (Text with EEA relevance) (notified under document number C(2003) 4961). Off. J. L 006, 38–39 (2004).
Liu, H., Kerr, W. A. & Hobbs, J. E. A review of Chinese food safety strategies implemented after several food safety incidents involving export of Chinese aquatic products. Br. Food J. 114, 372–386 (2012).
Culp, S. J. et al. Carcinogenicity of malachite green chloride and leucomalachite green in B6C3F1 mice and F344 rats. Food Chem. Toxicol. 44 (8), 1204–1212 (2006).
WHO. Evaluation of Certain Veterninary Drug Residues in Food (Food and Agriculture Organization of the United Nations, 2008).
WHO. Evaluation of Certain Veterninary Drug Residues in Food (Food and Agriculture Organization of the United Nations, 2009).
Allen, J. L. & Hunn, J. B. Fate and distribution studies of some drugs used in aquaculture. Vet. Hum. Toxicol. 28, 21–24 (1986).
Fessard, V. et al. Mutagenicity of malachite green and leucomalachite green in in vitro tests. J. Appl. Toxicol. 19 (6), 421–430 (1999).
Vilhena, A. O. D. et al. The synthetic dye malachite green found in food induces cytotoxicity and genotoxicity in four different mammalian cell lines from distinct tissues. Toxicol. Res. 12, 693–701 (2023).
Stammati, A. et al. Effects of malachite green (MG) and its major metabolite, leucomalachite green (LMG), in two human cell lines. Toxicol. Vitro. 19 (7), 853–858 (2005).
Andersen, W. C., Turnipseed, S. B. & Roybal, J. E. Quantitative and confirmatory analyses of malachite green and leucomalachite green residues in fish and shrimp. J. Agric. Food Chem. 54 (13), 4517–4523 (2006).
Gavrilenko, N. A. et al. Visual determination of malachite green in sea fish samples. Food Chem. 274, 242–245 (2019).
Clemmensen, S. et al. Toxicological studies on malachite green: A triphenylmethane dye. Arch. Toxicol. 56 (1), 43–45 (1984).
Srivastava, S. J. et al. Acute toxicity of Malachite Green and its effects on certain blood parameters of a catfish, heteropneustes-fossilis. Aquat. Toxicol. 31 (3), 241–247 (1995).
Chaturvedi, V. et al. Biodetoxification of high amounts of malachite green by a multifunctional strain of Pseudomonas mendocina and its ability to metabolize dye adsorbed chicken feathers. J. Environ. Chem. Eng. 1, 1205–1213 (2013).
Nelson, C. R. & Hites, R. A. Aromatic-Amines in and Near the Buffalo River. Vol. 14. 1147–1149 (Environmental Science & Technology, 1980).
Bilandzic, N. et al. Malachite green residues in farmed fish in Croatia. Food Control. 26 (2), 393–396 (2012).
Conti, G. O. et al. Determination of illegal antimicrobials in aquaculture feed and fish: An ELISA study. Food Control. 50, 937–941 (2015).
Carmen, Z. & Daniela, S. Textile Organic Dyes – Characteristics, Polluting Effects and Separation/Elimination Procedures from Industrial Effluents – A Critical Overview. In Organic Pollutants Ten Years After the Stockholm Convention - Environmental and Analytical Update (2012).
EC. Leuco-malachite green in shrimps from Bangladesh. (2022). https://webgate.ec.europa.eu/rasff-window/screen/notification/582605. Accessed 11 July 2024.
EC. Unauthorized substances malachite green and leucomalachite green in balck tiger shrimp from Bangladesh (2023). https://webgate.ec.europa.eu/rasff-window/screen/notification/630147. Accessed 11 July 2024.
Belpaire, C. et al. Toxic textile dyes accumulate in wild European eel. Chemosphere 138, 784–791 (2015).
Schuetze, A., Heberer, T. & Juergensen, S. Occurrence of residues of the veterinary drug malachite green in eels caught downstream from municipal sewage treatment plants. Chemosphere 72 (11), 1664–1670 (2008).
Mnif, I., Fendri, R. & Ghribi, D. Malachite green bioremoval by a newly isolated strain RI11; Enhancement of the treatment by biosurfactant addition. Water Sci. Technol. 72 (8), 1283–1293 (2015).
Bouafia-Chergui, S. et al. A photo-fentontreatment of a mixture of three cationic dyes. Proc. Eng. 33, 181–187 (2012).
Venkatesh, S., Venkatesh, K. & Quaff, A. R. Dye decomposition by combined ozonation and anaerobic treatment: cost effective technology. J. Appl. Res. Technol. 15, 340–345 (2017).
Mishra, S. et al. Microwave-assisted catalytic degradation of brilliant green by spinel zinc ferrite sheets. Acs Omega. 4 (6), 10411–10418 (2019).
Berberidou, C. et al. Sonolytic, photocatalytic and sonophotocatalytic degradation of malachite green in aqueous solutions. Appl. Catal. B-Environ.. 74 (1–2), 63–72 (2007).
Kusvuran, E. et al. Decolorization of malachite green, decolorization kinetics and stoichiometry of ozone-malachite green and removal of antibacterial activity with ozonation processes. J. Hazard. Mater. 186 (1), 133–143 (2011).
Liu, X. Y. et al. Microwave-induced catalytic oxidation of malachite green under magnetic Cu-ferrites: New insight into the degradation mechanism and pathway. J. Mol. Catal. A-Chem.. 395, 243–250 (2014).
Yang, J. et al. Mechanism of photocatalytic degradation of dye MG by TiO2-film electrode with cathodic bias potential. Chin. Sci. Bull. 55, 131–139 (2010).
Kabeer, F. A., John, N. & Abdulla, M. H. Biodegradation of malachite green by a newly isolated Bacillus vietnamensis sp. MSB17 from continental slope of the Eastern Arabian Sea: Enzyme analysis, degradation pathway and toxicity studies. Bioremediat. J. 23, 334–342 (2019).
Abu-Hussien, S. H. et al. Microbial Degradation, spectral analysis and toxicological assessment of Malachite Green dye by Streptomyces exfoliatus. Molecules 27(19) (2022).
Jadhav, J. P. & Govindwar, S. P. Biotransformation of malachite green by Saccharomyces cerevisiae MTCC 463. Yeast 23 (4), 315–323 (2006).
Kim, M. H. et al. Structural insight into bioremediation of triphenylmethane dyes by Citrobacter sp. triphenylmethane reductase. J. Biol. Chem. 283, 31981–31990 (2008).
Yang, X. et al. Degradation and detoxification of the triphenylmethane dye malachite green catalyzed by crude manganese peroxidase from Irpex Lacteus F17. Environ. Sci. Pollut Res. Int. 23 (10), 9585–9597 (2016).
Yang, J. et al. Laccase-catalyzed decolorization of malachite green: performance optimization and degradation mechanism. PLoS One. 10 (5), e0127714 (2015).
Xu, K. Z. et al. Extracellular expression of mutant CotA-laccase SF in Escherichia coli and its degradation of malachite green. Ecotoxicol. Environ. Saf. 193, 110335 (2020).
Xiang, H. F. et al. Efficient biodegradation of malachite green by an artificial enzyme designed in myoglobin. RSC Adv. 11, 16090–16095 (2021).
Kulkarni, M. & Chaudhari, A. Microbial remediation of nitro-aromatic compounds: An overview. J. Environ. Manag. 85 (2), 496–512 (2007).
Fareed, A. et al. Decolorization of azo dyes by a novel aerobic bacterial strain Bacillus cereus strain ROC. PLoS One. 17 (6), e0269559 (2022).
Woodward, J. Methods of immobilization of microbial cells. J. Microbiol. Methods. 8, 91–102 (1988).
Grant, J. R. et al. Proksee: In-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 51 (W1), W484–W492 (2023).
Meier-Kolthoff, J. P. & Goker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 10 (1), 2182 (2019).
Meier-Kolthoff, J. P. et al. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 14 (2013).
Markowitz, V. M. et al. The integrated microbial genomes (IMG) system. Nucleic Acids Res. 34 (Database issue), D344–D348 (2006).
Ooi, T. et al. Comparative enzymatic analysis of azoreductases from Sp B29. Biosci. Biotechnol. Biochem. 73 (5), 1209–1211 (2009).
Ooi, T. et al. An azoreductase, aerobic NADH-dependent flavoprotein discovered from sp.: Functional expression and enzymatic characterization. Appl. Microbiol. Biotechnol. 75 (2), 377–386 (2007).
Yu, J. et al. Structures of AzrA and of AzrC complexed with substrate or inhibitor: insight into substrate specificity and catalytic mechanism. Acta Crystallogr. D Biol. Crystallogr. 70 (Pt 2), 553–564 (2014).
Yoneda, K. et al. Structural and biochemical characterization of an extremely thermostable FMN-dependent NADH-indigo reductase from Bacillus smithii. Int. J. Biol. Macromol. 164, 3259–3267 (2020).
Binter, A. et al. A single intersubunit salt bridge affects oligomerization and catalytic activity in a bacterial quinone reductase. FEBS J. 276 (18), 5263–5274 (2009).
Deller, S. et al. Characterization of a thermostable NADPH:FMN oxidoreductase from the mesophilic bacterium. Biochemistry 45 (23), 7083–7091 (2006).
Ikram, M. et al. Bacillus subtilis: As an efficient bacterial strain for the reclamation of Water loaded with Textile Azo Dye, Orange II. Int. J. Mol. Sci., 23(18). (2022).
Hossain, M. S. et al. Mixed dye degradation by Bacillus pseudomycoides and Acinetobacter haemolyticus isolated from industrial effluents: A combined affirmation with wetlab and in silico studies. Arab. J. Chem. 15(9). (2022).
Song, J. L. et al. Pathway and kinetics of malachite green biodegradation by Pseuodomonas Veronii. Sci. Rep., 10(1). (2020).
Du, L. N. et al. Efficient degradation of malachite green by Aeromonas sp. strain DH-6. Appl. Environ. Biotechnol. 3, 2–8 (2018).
Parshetti, G. et al. Biodegradation of malachite green by MTCC 1532. Acta Chim. Slov. 53 (4), 492–498 (2006).
Kobayashi, T. et al. Malachite-green-removing properties of a bacterial strain isolated from fish ponds in Thailand. Fish. Sci. 83 (5), 827–835 (2017).
Fareed, A. et al. Biodegradation of N-methylated carbamates by free and immobilized cells of newly isolated strain Enterobacter cloacae strain TA7. Bioremediat. J. 21, 119–127 (2017).
Phulpoto, A. H. et al. Ecotoxicological assessment of oil-based paint using three-dimensional multi-species bio-testing model: Pre- and post-bioremediation analysis. Environ. Sci. Pollut Res. Int. 25 (17), 16567–16577 (2018).
Jamil, M. et al. Isolation of antibacterial compounds from Quercus Dilatata L. through bioassay guided fractionation. Ann. Clin. Microbiol. Antimicrob. 11, 11 (2012).
Roopa, K. B. & Usha, M. S. Partial purification and assay of azoreductase and laccase in the degradation of reactive black 5 dye from Aeromonas punctata. Int. J. Latest Res. Eng. Techol. 3, 16–19 (2017).
Darling, A. C. et al. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14 (7), 1394–1403 (2004).
Parte, A. C. et al. List of prokaryotic names with standing in nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 70 (11), 5607–5612 (2020).
Dieckmann, M. A. et al. EDGAR3.0: Comparative genomics and phylogenomics on a scalable infrastructure. Nucleic Acids Res. 49 (W1), W185–W192 (2021).
Brettin, T. et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 5, 8365 (2015).
Chen, I. A. et al. The IMG/M data management and analysis system v.7: Content updates and new features. Nucleic Acids Res. 51 (D1), D723–D732 (2023).
Zaru, R., Orchard, S. & UniProt, C. UniProt Tools: BLAST, align, peptide search, and ID Mapping. Curr. Protoc. 3 (3), e697 (2023).
Hauser, M., Steinegger, M. & Soding, J. MMseqs software suite for fast and deep clustering and searching of large protein sequence sets. Bioinformatics 32 (9), 1323–1330 (2016).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37 (5), 1530–1534 (2020).
Aragao, D. et al. MX2: A high-flux undulator microfocus beamline serving both the chemical and macromolecular crystallography communities at the Australian Synchrotron. J. Synchrotron Radiat. 25 (Pt 3), 885–891 (2018).
Brehm, W. et al. XDSGUI: A graphical user interface for XDS, SHELX and ARCIMBOLDO. J. Appl. Crystallogr. 56 (Pt 5), 1585–1594 (2023).
Agirre, J. et al. The CCP4 suite: Integrative software for macromolecular crystallography. Acta Crystallogr. D Struct. Biol. 79 (Pt 6), 449–461 (2023).
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69 (Pt 7), 1204–1214 (2013).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40 (Pt 4), 658–674 (2007).
McCoy, A. J. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63 (Pt 1), 32–41 (2007).
Liebschner, D. et al. Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr. D Struct. Biol. 73 (Pt 2), 148–157 (2017).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68 (Pt 4), 352–367 (2012).
Emsley, P. et al. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66 (Pt 4), 486–501 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 75 (Pt 10), 861–877 (2019).
Afonine, P. V. et al. phenix.model_vs_data: A high-level tool for the calculation of crystallographic model and data statistics. J. Appl. Crystallogr. 43 (Pt 4), 669–676 (2010).
Schrodinger, L. The PyMOL Molecular Graphics System, Version 3.0. (2010).
DeLano, W. L. The PyMOL Molecular Graphics System. (2004).
Emsley, P. Tools for ligand validation in Coot. Acta Crystallogr. D Struct. Biol. 73 (Pt 3), 203–210 (2017).
Stierand, K. & Rarey, M. Drawing the PDB: Protein-ligand complexes in two dimensions. ACS Med. Chem. Lett. 1 (9), 540–545 (2010).
Stierand, K., Maass, P. C. & Rarey, M. Molecular complexes at a glance: Automated generation of two-dimensional complex diagrams. Bioinformatics 22 (14), 1710–1716 (2006).
Yang, H. et al. Automated and accurate deposition of structures solved by X-ray diffraction to the Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 60 (Pt 10), 1833–1839 (2004).
Acknowledgements
This research was undertaken in part using the MX2 beamline at the Australian Synchrotron, which is part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector.
Funding
We acknowledge the ARC Centre of Excellence for Innovations in Peptide and Protein Science (CE200100012) and the ARC Centre of Excellence in Synthetic Biology (CE200100029) for funding.
Author information
Authors and Affiliations
Contributions
S.B., V. J., A.F., and A.S. performed microbiology experiments and prepared Fig. 4.C.B. and R.F. performed crystallography experiments.C.B. performed biochemistry experiments and prepared Figs. 1, 2, 3 and 5-6.H.Z., M.A., I.Z, C.J.J., and T.A.N. provided supervision and funding.All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bibi, S., Breeze, C.W., Jadoon, V. et al. Isolation, identification, and characterisation of the malachite green detoxifying bacterial strain Bacillus pacificus ROC1 and the azoreductase AzrC. Sci Rep 15, 3499 (2025). https://doi.org/10.1038/s41598-024-84609-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-84609-4
