
Abstract
This study aimed to identify the potential pathogenic genes associated with the comorbidity of rheumatoid arthritis (RA) and renal fibrosis (RF). Transcriptomic data related to RA and RF were retrieved from the GEO database. Differential expression gene analysis (DEGs) and weighted gene co-expression network analysis (WGCNA) were carried out to identify the RA-RF-DEGs. Subsequently, functional enrichment analysis was performed to clarify the biological functions of these genes. Machine learning algorithms were used to screen for the hub RA-RF differential expression genes, and then a Logistic Regression (LR) model was constructed. The accuracy of the model was evaluated using the ROC curve. At the same time, single-sample gene set enrichment analysis (ssGSEA) was applied to conduct immune infiltration analysis on the RF dataset. Gene set enrichment analysis (GSEA) was further performed on the hub genes to explore their underlying mechanisms in RF. Finally, a miRNA-TF-mRNA regulatory network centered around the hub genes was constructed.The results showed that 10 RA-RF-DEGs were identified through a comprehensive screening process. Enrichment analysis indicated that these differential expression genes were mainly involved in inflammatory responses and immune regulation. Subsequently, two hub genes, namely BIRC3 and PSMB9, were identified. A LR model was developed, and its predictive accuracy was verified using the ROC curve derived from an external independent dataset. Immune infiltration analysis revealed a significant correlation between the two hub genes and immune dysregulation in RF. Gene set enrichment analysis (GSEA) clarified the potential biological pathways through which BIRC3 and PSMB9 might function in RF. The constructed miRNA-TF-mRNA regulatory network provided a comprehensive overview of the post-transcriptional and transcriptional regulatory mechanisms. In conclusion, this study identified two candidate risk genes for RA-RF, providing new insights for the early diagnosis and treatment of RA complicated with RF.
Similar content being viewed by others


Introduction
Rheumatoid arthritis (RA) and renal fibrosis (RF) are two complex and debilitating conditions that have garnered significant attention in the medical and scientific communities. Despite their seemingly distinct anatomical locations and initial manifestations, an increasing body of evidence suggests a potential connection between RA and RF1.
RA is an autoimmune disease primarily characterized by erosive arthritis and can afflict individuals at any age. One of the common extra-articular manifestations in RA patients is renal disorders2. Until a few years ago, glomerulonephritis remained the most prevalent renal disease in RA3. The prevalence rate of renal diseases among RA patients ranges from 5 − 50%4. Over time, the cumulative incidence of renal function decline in the RA population is significantly higher than that in the non-RA population5. Taking chronic kidney disease (CKD) as an example, compared with the non-RA population, RA patients have an increased risk of developing CKD6; moreover, the comorbidity of CKD is a risk factor for RA patients failing to achieve clinical remission, which can augment the risks of hospitalization due to infection and mortality7.
In RA, recent studies have leveraged transcriptomic and proteomic data to identify novel diagnostic biomarkers. For example, machine learning analysis of autophagy-related genes in peripheral blood identified EEF2, HSP90AB1, and TNFSF10 as promising diagnostic markers for RA, demonstrating excellent diagnostic accuracy8. Similarly, in RF, integrative bioinformatic analyses of kidney transcriptomes from unilateral ureteral obstruction (UUO) models revealed hub genes such as Bmp1 and CD74, which may serve as biomarkers for chronic kidney disease progression9. These studies highlight the power of ML-driven approaches in dissecting disease heterogeneity and prioritizing candidate biomarkers from complex datasets.
Previous studies have primarily focused on identifying biomarkers for RA or RF in isolation. Existing diagnostic paradigms rely primarily on clinical symptoms, serological markers, and histological assessments, which may fail to detect comorbid conditions at their earliest stages.
In the present study, we utilize bioinformatics and machine learning techniques to investigate potential common pathogenic genes underlying the comorbidity of RA and RF, thereby addressing this critical research gap. By analyzing transcriptomic datasets from patients with RA and RF, we aim to identify common pathogenic genes driving their pathogenesis and elucidate the intricate molecular networks interconnecting these two diseases. Our hypothesis posits that a subset of genes are differentially regulated in both RA and RF, serving as pivotal regulators within the shared pathogenic pathways. The identification of shared biomarkers could enable preemptive interventions, thereby improving outcomes for patients at risk of dual organ damage. The discovery of these genes will not only deepen our fundamental understanding of the relationship between these diseases but also provide valuable insights for potential biomarkers and therapeutic targets.
Methods
Dataset screening and processing
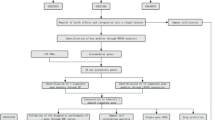
Figure 1 presents the analysis process undertaken in this study.Gene expression profiling data were retrieved from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo). In this study, five disparate datasets were employed. For rheumatoid arthritis (RA), two independent gene expression profiles were used for gene screening, namely GSE12021 and GSE55457. In GSE12021, there were 21 synovial membrane samples, with 9 originating from normal controls and 12 from RA patients10. Correspondingly, GSE55457 consisted of 23 synovial membrane samples, where 10 were from normal controls and 13 were from RA patients11. GSE89408 was used as the external validation set for RA, among which 12 were from normal controls and 152 were from RA patients12. Regarding renal fibrosis, two datasets were also incorporated. GSE76882 encompassed 99 control samples and 175 samples of renal fibrosis13 while GSE22459 comprised 25 control samples and 40 renal fibrosis samples14. Subsequently, the gene expression matrix was extracted, and the probes were annotated in accordance with the annotation file of the platform.Detailed information of the dataset samples can be found in the supplementary material table S1.

The flowchart of this study.
Identification of differentially expressed genes (DEGs)
Within the R software environment, the “limma” package was employed to perform an analysis aimed at identifying differentially expressed genes (DEGs)15. For the comparison between RA patients and healthy controls, the datasets GSE55457 and GSE12021 were utilized. Additionally, for the comparison between RF patients and healthy controls, the dataset GSE76882 was used. The significance of the DEGs was determined based on the criteria of a |log2FC)| >1 and an adjusted p-value < 0.05.
Weighted gene co‑expression network analysis (WGCNA) for identifying trait‑related genes
In the present study, we initially employed the R package “WGCNA” to establish a weighted co-expression network on the datasets16 which served as the fundamental infrastructure for subsequent genetic analyses. Subsequently, our attention was directed towards the genes exhibiting the top 30% of expression variance across all profiles. By doing so, we were able to identify specific gene sets and further explore their associations with diseases. In particular, we determined the co-expression modules related to RA and RF. Subsequently, the weighted adjacency matrix was transformed into a topological overlap matrix (TOM), enabling a different perspective on the relationships among genes and providing a more suitable data format for subsequent clustering analyses. Based on the dissimilarity measure (dissTOM = 1 - TOM) derived from the topological overlap, the genes were hierarchically clustered. This clustering approach grouped the genes according to their similarities, facilitating the discovery of potential functional modules and interactions among them. Subsequently, the hierarchical clustering tree method was utilized to select modules containing more than 100 genes. This step further refined the clustering results, focusing on larger modules that likely possess more significant biological implications for in-depth investigation. Finally, from these selected modules, genes that exhibited strong correlations with clinical features were extracted by computing the gene significance (GS) and module membership (MM).
Protein–protein interaction analysis
In this study, the STRING database (version 12.0) was utilized to establish a protein - protein interaction (PPI) network for the RA - RF related DEGs that had been previously identified17. To enhance the reliability and biological relevance of the network, a confidence score threshold of greater than 0.4 was applied during the construction process.
Functional enrichment analysis
In the research, the R package “clusterProfiler” was utilized to conduct KEGG pathway and Gene Ontology (GO) enrichment analyses on the RA-RF related DEGs18. To ensure the reliability and significance of the results, a P - value threshold of less than 0.05 was adopted, following the application of the Benjamini - Hochberg correction for multiple testing. This correction method was crucial in minimizing the false discovery rate and identifying only those results that were truly biologically relevant. Subsequently, for a more intuitive and comprehensive understanding of the significant findings, the R packages “ggplot2” and “Goplot” were employed to visually represent the data.
Identification of disease-related feature genes
For the further identification of hub RA-RF DEGs, two machine - learning methodologies, namely the least absolute shrinkage and selection operator (LASSO) and the random forest algorithm, were adopted. The LASSO algorithm was applied with the aim of minimizing regression coefficients, thereby eliminating redundant and uncorrelated genes from the analytical process19. This step was crucial in mitigating the risk of overfitting, ensuring that the selected genes were more likely to be truly associated with the RA - RF phenotype and enhancing the overall robustness and accuracy of the gene selection model.
The random forest algorithm, on the other hand, is a highly potent tool for gene selection in microarray analyses20. It is renowned for its robustness, which enables it to effectively handle noisy and high - dimensional datasets. Additionally, it provides accurate measures of variable importance, facilitating the identification of genes that play significant roles in the context of RA - RF. This characteristic is particularly valuable in complex genomic studies where the presence of confounding factors and high data dimensionality pose challenges to traditional analytical methods.
The implementation of the LASSO and random forest algorithms was carried out using the “glmnet” and “randomForest” packages in R, respectively. The hub RA - RF DEGs for both RA and RF were ultimately defined as those genes that were consistently identified by all utilized algorithms.
Construction and evaluation of binary logistic regression (LR) model
The binary LR model was used to find the matching coefficients of hub RA-RF-DEGs and compute the risk score for each sample. The nomogram was constructed using the “rms” program.To comprehensively assess the discriminatory power of the scores derived from the nomogram, a series of statistical approaches were implemented. Decision Curve Analysis (DCA) was conducted to evaluate the net benefit of the model across different threshold probabilities, providing insights into its practicality in clinical decision-making. Calibration curve analysis was carried out to examine the agreement between the predicted and observed values, thereby gauging the calibration accuracy of the model. Moreover, the Area Under the Curve (AUC) values were calculated from the Receiver Operating Characteristic (ROC) curves to quantitatively measure the discriminatory ability of the model, with a higher AUC value indicating a better performance.
Assessment of immune cell infiltration in RF
The single-sample gene set enrichment analysis (ssGSEA) was employed to evaluate the infiltration of immune cells in both RF and control samples within the GSE76882 dataset.
Gene set enrichment analysis
The functional roles of the hub RA-RF-DEGs were examined through Gene Set Enrichment Analysis (GSEA). This analysis was performed using the “clusterProfiler” package, with the “C2.cp.kegg.v7.0.symbols.gmt” gene set serving as the reference.
Construction of TF-miRNA regulatory networks for hub PRG-DEGs
For the prediction of microRNAs (miRNAs) that target the hub RA-RF-DEGs, the miRTarBase database was employed21. Subsequently, the Enrichr database was utilized to identify the transcription factors (TFs) associated with these hub RA-RF-DEGs, with a significance threshold set at p < 0.05. Following the identification of miRNAs and TFs, miRNA-TF-mRNA networks were constructed. These networks were then visualized using Cytoscape software, facilitating a more intuitive understanding and in-depth analysis of the complex regulatory relationships among these genetic elements.
Results
Identification of DEGs in RA and RF
In this study, we initially conducted a differential expression gene analysis (DEGs). Volcano plots and heatmaps of the differentially expressed genes were constructed for visualization purposes. In the two RA datasets, namely GSE55457 (Fig. 2A, B) and GSE12021 (Fig. 2C, D), we obtained 341 and 253 DEGs, respectively. In the RF dataset GSE76882 (Fig. 2E, F), 315 DEGs were identified.

Identification of the DEGs in RA and RF. (A) A volcano map illustrating the DEGs in RA patients compared to healthy controls (GSE55457). (B) heatmap of the top 25 upregulated and downregulated DEGs (GSE55457). (C) A volcano map illustrating the DEGs in RA patients compared to healthy controls (GSE12021). (B) heatmap of the top 25 upregulated and downregulated DEGs (GSE12021). (E) A volcano map illustrating the DEGs in RF patients compared to healthy controls (GSE76882). (B) heatmap of the top 25 upregulated and downregulated DEGs (GSE76882).
WGCNA uncovers trait-related genes
For the identification of hub module genes associated with RA and RF, the WGCNA was employed. In the RA datasets, GSE55457 and GSE12021, the analysis revealed that the optimal soft threshold was determined to be 5 (Fig. 3A, D). Concurrently, as the average connectivity approached zero, an increase in the scale-free topology model fit index was observed, with the signed R2 value = 0.9 threshold. Moreover, by setting minModuleSize to 100, a total of 7 co-expression modules were generated (Fig. 3B, E). Notably, the turquoise module exhibited a significant correlation with RA. Subsequently, through the application of stringent cut-off criteria (|MM| > 0.8 and | GS| > 0.5) (Fig. 3C, F), 126 and 117 genes with high connectivity within the clinically relevant RA-related module were respectively identified as hub module genes for further investigation.

WGCNA. (A, D) Determine the best soft threshold. The soft threshold value of 5 was determined as the optimal choice for constructing a scale-free network based on the position of the redline (R²=0.9).(B, E) Correlations between different modules and clinical traits(RA). The turquoise module has the highest correlation with RA. (C, F) Scatterplot of correlations between gene significance (GS) > 0.5 and module membership (MM) > 0.8 in the blue module. ABC corresponds to the RA dataset GSE55457, while DEF refers to the RA dataset GSE12021.(G) Determine the best soft threshold. The soft threshold value of 15 was determined as the optimal choice for constructing a scale-free network based on the position of the redline (R²=0.9).(H)Correlations between different modules and clinical traits(RF), the blue module has the highest correlation with RF.(I) Scatterplot of correlations between GS > 0.4 and MM > 0.8 in the blue module.
Similarly, in the RF dataset GSE76882, the optimal soft threshold was established at 15 (Fig. 3G), ultimately leading to the generation of 6 co-expression modules. The blue module demonstrated a significant correlation with RF (Fig. 3H). By applying the cut-off criteria (|MM| > 0.8 and |GS| > 0.4) (Fig. 3I), 250 genes with high connectivity within the clinically relevant RF-related module were identified as hub module genes.
Identification of RA-RF-DEGs and functional analysis
The intersection operation was performed on the four gene sets of previously obtained DEGs and WGCNA significant module genes. As a result, a total of 10 RA-RF-DEGs were obtained, namely: PSMB90, BIRC3, GZMA, TAP1, CD8A, CD2, CCL5, CD27, GZMK, and LCK (Fig. 4A). Subsequently, a PPI network was constructed with the parameter of medium confidence set to be greater than 0.4(Fig. 4B). KEGG pathway analysis22 demonstrated that the RA-RF-DEGs were predominantly implicated in pathways such as Primary immunodeficiency, NF-kappa B signaling pathway, and TNF signaling pathway. The top 10 pathways are illustrated in Fig. 4C. GO enrichment analysis further disclosed that these RA-RF-DEGs were remarkably enriched in various aspects, namely biological processes (Fig. 4D), cellular components Fig. 4E), and molecular functions (Fig. 4F). In the figures, the top 10 enriched terms were presented, which mainly encompassed T cell activation, membrane raft, MHC class I protein binding, among others.

PPI and fnctional enrichment analysis of RA-RF-DEGs. (A) A Venn diagram of the DEGs and WGCNA module genes identifying 11 RA-RF-DEGs. (B) PPI network of RA-RF-DEGs. (C) Bubble plots showing the top 10 results of KEGG enrichment analysis22 and GO enrichment analysis with (D) BP, (E) CC, and (F) MF.
Identification of hub RA-RF-DEGs through machine learning
Hub RA-RF-DEGs were screened using machine learning (ML) algorithms. LASSO and Random Forest analyses were respectively conducted on the RA dataset and the RF dataset. Through the LASSO analysis, three characteristic genes were obtained from the GSE55457 dataset, as depicted in Fig. 5A, with an Area Under the Curve (AUC) value of 0.985. Additionally, seven characteristic genes were derived from the GSE76882 dataset, shown in Fig. 5C, with an AUC value of 0.861. In the random forest analysis, four characteristic genes were identified for GSE55457, illustrated in Fig. 5B, with an AUC value of 0.908, and ten characteristic genes were determined for GSE76882, as presented in Fig. 5D, with an AUC value of 0.797.

Machine Learning Screening hub RA-RF-DEGs. (A) LASSO coefficient analysis identifies the optimal lambda marked by vertical dashed lines. This determination is based on five cross-validations of adjustment parameters, and the ROC curve is also provided for evaluation(GSE55457). (B) The relationship between the number of random forest trees and the error rate is explored, along with the ranking of RA-RF-DEGs based on their relative importance(importance > 1), and the ROC curve is also provided for evaluation(RA: GSE55457). (C) LASSO coefficient analysis identifies the optimal lambda marked by vertical dashed lines. This determination is based on five cross-validations of adjustment parameters, and the ROC curve is also provided for evaluation(RF: GSE76882). (D) The relationship between the number of random forest trees and the error rate is explored, along with the ranking of RA-RF-DEGs based on their relative importance(importance > 5), and the ROC curve is also provided for evaluation(RF: GSE76882).
Construction and evaluation of the LR model
Firstly, a venn diagram was employed to identify the intersection of genes derived from the ML methods, leading to the discovery of two hub RA-RF-DEGs, namely BIRC3 and PSMB9 (Fig. 6A). Subsequently, further in-depth analyses were carried out to investigate the differential expression patterns of these two hub RA-RF-DEGs. Across the RF training set (GSE76882), it was observed that the expression levels of the two hub RA-RF-DEGs were significantly elevated in comparison to the control group((Fig. 6B).

A nomogram designed for RF. (A) A Venn diagram of the LASSO and RF features identifying two hub RA-RF-DEGs. Violin plots show hub PRG-DEGs expression in control and RF tissues in (B) training set GSE76882.(C) Risk distribution in the training set between RF patients and healthy controls. (D) A nomogram presents the risk distribution within the training set. (E) shows a calibration curve, which evaluates the predictive accuracy of the nomogram for the training set. (F) employs DCA to assess the clinical utility of the nomogram for the training set. Additionally, ROC curves are utilized to evaluate the diagnostic performance of the LR model in the (G) training set. ***P < 0.001;****P < 0.0001.
Thereafter, the LR model was constructed using the hub RA-RF-DEGs, with the final risk score calculated as (1.3095 × BIRC3)+ (0.1785 × PSMB9). In the training set, the risk scores of RF patients were substantially greater than those of the healthy controls(Fig. 6C). A risk nomogram for RF was devised based on the expression levels of these hub RA-RF-DEGs, aiming to formulate a more clinically relevant risk model(Fig. 6D). By constructing decision curves(Fig. 6E) and clinical calibration curves(Fig. 6F), it became evident that our proposed model possessed high predictive capabilities for RF. The LR model achieved an AUC of 0.829 through 10-fold cross-validation (Fig. 6G).
Additionally, the risk scores of RF patients were significantly higher than those of the healthy controls in the validation set GSE22459(Fig. 7A). The calibration and DCA curves exhibited strong diagnostic prediction ability (Fig. 7B and C), with an AUC value of 0.743 (Fig. 7D). External validation using the GSE89048 dataset revealed that the risk score of RA patients was significantly higher (Fig. 7E). The calibration curves, and DCA further exhibited the robust diagnostic prediction ability of the LR model (Fig. 7F and G).The LR model obtained an AUC value of 0.952 for the validation set (Fig. 7H).

Verification of diagnostic efficacy of model in the test set. (A) Risk distribution of RF and healthy control group in the RF test set. (B) Use calibration curves to evaluate the predictive ability of the model in the RF test set. (C) DCA evaluates the clinical benefits of the model for the RF test set. (D) ROC curve evaluates the diagnostic efficacy of the model in the RF test set. (E) Risk distribution of RA and healthy control group in the RA test set. (F) Use calibration curves to evaluate the predictive ability of the model in the RA test set. (G) DCA evaluates the clinical benefits of the model for the RA test set. (H) ROC curve evaluates the diagnostic efficacy of the model in the RA test set. ***P < 0.001;****P < 0.0001.
The role of hub RA-RF-DEGs in RF immune microenvironment
To explore the disparities in the immune microenvironment between RF patients and healthy controls, we evaluated the correlations among 28 immune cell types, as depicted in Fig. 8A. Our findings demonstrated that RF patients exhibited a higher degree of infiltration of several immune cell subsets, including activated B cells, activated CD4 T cells, activated CD8 T cells, activated dendritic cells, central memory CD4 T cells, effector memory CD8 T cells, and myeloid-derived suppressor cells (MDSC), etc. Subsequently, we further probed into the associations between the hub RA-RF-DEGs and the 28 immune cell types. As illustrated in Fig. 8B, BIRC3 and PSMB9 were significantly correlated with a multitude of immune cells.

The immune characteristics in the RF. (A) Boxplot showing the variations in immune cell distribution in the RF patients compared to controls. (B) Correlation analysis between BIRC3 ,PSMB9 and infiltrating immune cells in RF.*p < 0.05; **p < 0.01; ***p < 0.001.
Single-gene GSEA of the hub RA-RF-DEGs
To explore the underlying mechanisms governing the significance of the expression patterns of RA-RF-DEGs in RF, we implemented a single-gene GSEA. The resultant analysis unveiled six pathways that demonstrated statistically significant positive correlations with the expression of two hub RA-RF-DEGs. More specifically, the expression of BIRC3 was positively associated with pathways including chemokine signaling pathway, cytokine cytokine receptor interaction, hematopoietic ccell lineage, natural killer cell mediated cytotoxicity, and systemic lupus erythematosus (Fig. 9A). Likewise, the expression of PSMB9 exhibited positive correlations with chemokine signaling pathway, hematopoietic cell lineage, systemic lupus erythematosus, and toll like receptor signaling pathway (Fig. 9B). These findings not only provide crucial insights into the molecular underpinnings of RA-RF-DEGs in RF but also offer potential targets for future therapeutic interventions.

GSEA and regulatory networks of hub RA-RF-DEGs. The correlation of (A) BIRC3, (B) PSMB9 with the top six significantly enriched pathways. (C) A diagram representing the miRNA-TF-RNA regulatory network. Pink diamond signifies genes, blue V-shapes represent miRNAs, and green squares denote TFs.
Regulatory networks of hub RA-RF-DEGs
In an endeavor to acquire a more profound understanding of the regulatory mechanisms governing the expression of hub RA-RF-DEGs, we carried out a predictive analysis with a specific focus on miRNAs and TFs associated with these genes. After this analysis, Cytoscape was utilized to construct a visual representation of the intricate regulatory network that emerged. This network was composed of 42 miRNAs, 4 TFs, and 2 genes, affording a comprehensive panorama of the interacting elements in play(Fig. 9C).
Discussion
Studies have indicated that the primary cause of renal insufficiency in patients with rheumatoid arthritis (RA) is the nephrotoxicity associated with RA treatment, such as the use of nonsteroidal anti-inflammatory drugs (NSAIDs) and disease-modifying antirheumatic drugs (DMARDs). Additionally, inadequate control of systemic inflammation may lead to mesangial proliferative glomerulonephritis (GN) or secondary amyloidosis. Given the high prevalence and frequent co-occurrence of RA and renal diseases23 which severely impact patients’ quality of life, early diagnosis of these comorbidities is particularly imperative.
In the present study, we aimed to uncover the potential pathogenic genes underlying the comorbidity of RA and RF-related conditions, with the goal of identifying early diagnostic markers. Utilizing transcriptomic data from the GEO database and employing a combination of bioinformatics analysis and machine learning algorithms, we identified BIRC3 and PSMB9 as hub RA-RF-DEGs. Functional enrichment analysis revealed that these genes are primarily associated with immune responses. Given the role of the immune system in the pathogenesis of both RA and RF, this finding is not entirely unexpected. In rheumatoid arthritis, chronic inflammation driven by aberrant immune responses leads to joint damage, while in RF-related conditions, immune-mediated processes are increasingly recognized as key drivers of fibrosis progression. A Logistic Regression model constructed based on the expression profiles of these genes in RF-related contexts further highlights their significance, demonstrating robust predictive performance for RF risk assessment.
Baculoviral IAP Repeat Containing 3 (BIRC3), which belongs to the conserved IAP protein family, is characterized by the presence of one-to-three N-terminal tandem baculovirus IAP repeats24. Previous investigations have revealed that BIRC3 constitutes a part of a signature gene cluster associated with hypoxia-induced inflammation in glioblastoma multiforme25. Additionally, it has been documented that BIRC3 functions as an inhibitor of non-traditional NF - κB signaling in chronic lymphocytic leukemia26. BIRC3 has been identified as a crucial regulator that suppresses cell death and serves as a significant mediator in inflammatory signaling and immunity27. Numerous studies have underlined the essential role of BIRC3 in a variety of diseases28. For instance, Jiang et al. reported that BIRC3 promoted the growth and dissemination of cancer cells in liver cancer via the NF - κB signaling pathway29. Conversely, tissue samples from deceased patients infected with H7N9 demonstrated a reduction in BIRC3 accompanied by an increase in necroptosis30.
Fibroblast-like synoviocytes (FLS) are recognized as pivotal contributors to the pathogenesis of RA31. These cells are capable of producing substantial quantities of inflammatory mediators, encompassing TNF-α and IL-6, which serve to intensify inflammatory processes within the joint environment32. The elevated expression of the BIRC3 gene and its corresponding protein product, cIAP2, within the context of RA, orchestrates a diverse array of cellular functions, including the modulation of apoptosis, inflammatory signaling pathways, immune response dynamics, mitogen-activated protein kinase (MAPK) signaling cascades, and cell proliferation rates33. These regulatory activities collectively foster the survival of FLS and amplify inflammatory responses. Targeted suppression of BIRC3 expression has been demonstrated to curtail the secretion of inflammatory cytokines by RA-FLS under both resting and inflammatory conditions, while also impeding their proliferative capacity.Despite the promising therapeutic potential of BIRC3 inhibitors in the management of RA, careful consideration must be given to their potential adverse effects. Continued investigation into the intricate molecular mechanisms underlying BIRC3 function, particularly its involvement in cell signaling networks, apoptotic regulation, and immune evasion strategies, is imperative for the identification of novel therapeutic targets and the development of innovative treatment modalities34. However, the role of BIRC3 in kidney diseases, particularly in renal fibrosis, remains poorly understood. In kidney disease, a significant upregulation of BIRC3 expression was observed, which was associated with renal ischemia–reperfusion injury35. Recent research by Chen S et al.36 indicated that Endothelial BIRC3 promotes renal fibrosis through modulating Drp1-mediated mitochondrial fission via the MAPK/PI3K/Akt pathway, suggesting that targeting BIRC3 could offer a promising therapeutic strategy to enhance endothelial cell survival and alleviate the progression of CKD.
PSMB9, a subunit of the proteasome, is intimately associated with antigen processing and immune activation37. A research investigation has indicated that a genetic variation at the rs17587 locus within the PSMB9 gene exhibits an association with RA in the Han Chinese population38. Despite the utilization of diverse bioinformatics methodologies to uncover potential biomarkers for RA39,40 limited literature exists regarding the involvement of PSMB9 in the pathological mechanisms underlying RA. However, recent studies have shed light on the possibility that PSMB9 may serve as a critical factor in the development of various immune-mediated disorders, such as systemic lupus erythematosus, and Parkinson’s disease41,42. Research findings have demonstrated that the deletion of Psmb9 in Apoe− / − mice leads to a reduction in atherosclerotic lesion size, plaque vulnerability, and vascular inflammation. Significantly, the endothelial overexpression-induced atherosclerosis and vascular inflammation, which are augmented by PSMB8-AS1, can be attenuated through knocking out Psmb9. Although currently, there is relatively scant research on PSMB9 in the kidney, its established roles in endothelial cells and vascular inflammation may pave the way for further investigations in the future.
Finally, the construction of the miRNA-TF-mRNA regulatory network centered around the hub RA-RF-DEGs added another layer of complexity and understanding. miRNAs and TFs are known to play pivotal roles in post-transcriptional and transcriptional regulation, respectively. By mapping out this network, we were able to identify potential upstream regulators (miRNAs and TFs) that could control the expression of BIRC3 and PSMB9. This knowledge could be exploited to develop innovative therapeutic strategies, such as using miRNA mimics or inhibitors to modulate the expression of the hub genes and ultimately influence the course of RF in RA patients.
However, several limitations of our study should be acknowledged. First, the transcriptomic data obtained from the GEO database, although a valuable resource, may have inherent biases due to differences in sample collection, processing, and experimental conditions across different studies. Second, while our in silico analyses provided strong hypotheses, further experimental validation, including in vitro and in vivo studies, is essential to confirm the functional roles of BIRC3 and PSMB9 and the regulatory mechanisms we proposed. Third, the complexity of the comorbidity between RA and RF likely involves additional factors and genes that were not captured in our current analysis. Future studies should consider integrating multi-omics data and larger patient cohorts to achieve a more comprehensive understanding.
Conclusion
In conclusion, our study has identified BIRC3 and PSMB9 as hub genes in the comorbidity of RA and RF and provided a framework for understanding their roles in the context of immune responses and fibrotic progression. The insights gained from this research offer promising avenues for future investigations and the development of targeted therapies to improve the management of patients with this challenging comorbidity. Continued efforts to bridge the gap between computational predictions and experimental validations will be crucial in translating these findings into clinical practice.
Data availability
The datasets analysed during the current study are available in the GEO database https://www.ncbi.nlm.nih.gov/geo/.
References
Zhang, T. et al. Spectrum and prognosis of renal histopathological lesions in 56 Chinese patients with rheumatoid arthritis with renal involvement. Clin. Exp. Med. 20, 191–197. https://doi.org/10.1007/s10238-019-00602-6 (2020).
Figus, F. A., Piga, M., Azzolin, I., McConnell, R. & Iagnocco, A. Rheumatoid arthritis: extra-articular manifestations and comorbidities. Autoimmun. Rev. 20, 102776. https://doi.org/10.1016/j.autrev.2021.102776 (2021).
Ponticelli, C., Doria, A. & Moroni, G. Renal disorders in rheumatologic diseases: the spectrum is changing (part 2. Arthridides). J. Nephrol. 34, 1081–1090. https://doi.org/10.1007/s40620-020-00776-3 (2021).
Couderc, M. et al. Prevalence of renal impairment in patients with rheumatoid arthritis: results from a Cross-Sectional multicenter study. Arthritis Care Res. (Hoboken) 68, 638–644. https://doi.org/10.1002/acr.22713 (2016).
Hickson, L. J., Crowson, C. S., Gabriel, S. E., McCarthy, J. T. & Matteson, E. L. Development of reduced kidney function in rheumatoid arthritis. Am. J. Kidney Dis. 63, 206–213. https://doi.org/10.1053/j.ajkd.2013.08.010 (2014).
Raksasuk, S. & Ungprasert, P. Patients with rheumatoid arthritis have an increased risk of incident chronic kidney disease: a systematic review and meta-analysis of cohort studies. Int. Urol. Nephrol. 52, 147–154. https://doi.org/10.1007/s11255-019-02346-4 (2020).
Tokoroyama, T., Ando, M., Setoguchi, K., Tsuchiya, K. & Nitta, K. Prevalence, incidence and prognosis of chronic kidney disease classified according to current guidelines: a large retrospective cohort study of rheumatoid arthritis patients. Nephrol. Dial Transpl. 32, 2035–2042. https://doi.org/10.1093/ndt/gfw315 (2017).
Dong, G., Gao, H., Chen, Y. & Yang, H. Machine learning and bioinformatics analysis to identify autophagy-related biomarkers in peripheral blood for rheumatoid arthritis. Front. Genet. 14, 1238407. https://doi.org/10.3389/fgene.2023.1238407 (2023).
Yuan, Y., Xiong, X., Li, L. & Luo, P. Novel targets in renal fibrosis based on bioinformatic analysis. Front. Genet. 13, 1046854. https://doi.org/10.3389/fgene.2022.1046854 (2022).
Huber, R. et al. Identification of intra-group, inter-individual, and gene-specific variances in mRNA expression profiles in the rheumatoid arthritis synovial membrane. Arthritis Res. Ther. 10, R98. https://doi.org/10.1186/ar2485 (2008).
Woetzel, D. et al. Identification of rheumatoid arthritis and osteoarthritis patients by transcriptome-based rule set generation. Arthritis Res. Ther. 16, R84. https://doi.org/10.1186/ar4526 (2014).
Guo, Y. et al. CD40L-Dependent pathway is active at various stages of rheumatoid arthritis disease progression. J. Immunol. 198, 4490–4501. https://doi.org/10.4049/jimmunol.1601988 (2017).
Modena, B. D. et al. Gene expression in biopsies of acute rejection and interstitial fibrosis/tubular atrophy reveals highly shared mechanisms that correlate with worse Long-Term outcomes. Am. J. Transpl. 16, 1982–1998. https://doi.org/10.1111/ajt.13728 (2016).
Park, W. D., Griffin, M. D., Cornell, L. D., Cosio, F. G. & Stegall, M. D. Fibrosis with inflammation at one year predicts transplant functional decline. J. Am. Soc. Nephrol. 21, 1987–1997. https://doi.org/10.1681/ASN.2010010049 (2010).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559. https://doi.org/10.1186/1471-2105-9-559 (2008).
Szklarczyk, D. et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–D646. https://doi.org/10.1093/nar/gkac1000 (2023).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. ClusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Frost, H. R. & Amos, C. I. Gene set selection via LASSO penalized regression (SLPR). Nucleic Acids Res. 45, e114. https://doi.org/10.1093/nar/gkx291 (2017).
Yan, B. et al. Association of aging related genes and immune microenvironment with major depressive disorder. J. Affect. Disord. 369, 706–717. https://doi.org/10.1016/j.jad.2024.10.053 (2025).
Huang, H. Y. et al. MiRTarBase update 2022: an informative resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 50, D222–D230. https://doi.org/10.1093/nar/gkab1079 (2022).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–d677. https://doi.org/10.1093/nar/gkae909 (2025).
Anders, H. J. & Vielhauer, V. Renal co-morbidity in patients with rheumatic diseases. Arthritis Res. Ther. 13, 222. https://doi.org/10.1186/ar3256 (2011).
Wang, Y. et al. Drp1-mediated mitochondrial fission promotes renal fibroblast activation and fibrogenesis. Cell. Death Dis. 11, 563. https://doi.org/10.1038/s41419-019-2218-5 (2020).
Murat, A. et al. Modulation of angiogenic and inflammatory response in glioblastoma by hypoxia. PLoS One. 4, e5947. https://doi.org/10.1371/journal.pone.0005947 (2009).
Diop, F. et al. Biological and clinical implications of BIRC3 mutations in chronic lymphocytic leukemia. Haematologica 105, 448–456. https://doi.org/10.3324/haematol.2019.219550 (2020).
Silke, J. & Meier, P. Inhibitor of apoptosis (IAP) proteins-modulators of cell death and inflammation. Cold Spring Harb Perspect. Biol. 5, 452. https://doi.org/10.1101/cshperspect.a008730 (2013).
Asslaber, D. et al. BIRC3 expression predicts CLL progression and defines treatment sensitivity via enhanced NF-kappaB nuclear translocation. Clin. Cancer Res. 25, 1901–1912. https://doi.org/10.1158/1078-0432.CCR-18-1548 (2019).
Jiang, X. J. et al. cIAP2 via NF-kappaB signalling affects cell proliferation and invasion in hepatocellular carcinoma. Life Sci. 266, 118867. https://doi.org/10.1016/j.lfs.2020.118867 (2021).
Qin, C. et al. Close Relationship between cIAP2 and Human ARDS Induced by Severe H7N9 Infection. Biomed Res Int 2019, 2121357. https://doi.org/10.1155/2019/2121357 (2019).
Wu, Z. et al. Fibroblast-like synoviocytes in rheumatoid arthritis: surface markers and phenotypes. Int. Immunopharmacol. 93, 107392. https://doi.org/10.1016/j.intimp.2021.107392 (2021).
Zhao, J. et al. Autophagy, netosis, necroptosis, and pyroptosis mediated programmed cell death as targets for innovative therapy in rheumatoid arthritis. Front. Immunol. 12, 809806. https://doi.org/10.3389/fimmu.2021.809806 (2021).
Ge, L. et al. ATF6α contributes to rheumatoid arthritis by inducing inflammatory cytokine production and apoptosis resistance. Front. Immunol. 13, 965708. https://doi.org/10.3389/fimmu.2022.965708 (2022).
Meng, Q., Wei, K. & Shan, Y. E3 ubiquitin ligase gene BIRC3 modulates TNF-induced cell death pathways and promotes aberrant proliferation in rheumatoid arthritis fibroblast-like synoviocytes. Front. Immunol. 15, 1433898. https://doi.org/10.3389/fimmu.2024.1433898 (2024).
Wang, S. et al. Birc3 and Tip1 are upregulated in renal ischemia reperfusion injury. Gene 876, 147492. https://doi.org/10.1016/j.gene.2023.147492 (2023).
Chen, S., He, Q., Yang, H. & Huang, H. Endothelial Birc3 promotes renal fibrosis through modulating Drp1-mediated mitochondrial fission via MAPK/PI3K/Akt pathway. Biochem. Pharmacol. 229, 116477. https://doi.org/10.1016/j.bcp.2024.116477 (2024).
Kloetzel, P. M. Antigen processing by the proteasome. Nat. Rev. Mol. Cell. Biol. 2, 179–187. https://doi.org/10.1038/35056572 (2001).
Yu, L. et al. Association between polymorphisms of PSMB8, PSMB9 and TAP2 genes with rheumatoid arthritis in ethnic Han Chinese from Yunnan. Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chines. J. Med. Genet. 30, 222–226. https://doi.org/10.3760/cma.j.issn.1003-9406.2013.04.023 (2013).
Li, Z. et al. Identification of PSMB9 and CXCL13 as Immune-related diagnostic markers for rheumatoid arthritis by machine learning. Curr. Pharm. Design. 28, 2842–2854. https://doi.org/10.2174/1381612828666220831085608 (2022).
Wu, Y. K., Zhou, L., Chang, G. & Wang, R. Q. Identification and validation of fibroblast-related biomarkers in rheumatoid arthritis by bulk RNA-seq and single-cell RNA-seq analysis. Clin. Exp. Rheumatol. https://doi.org/10.55563/clinexprheumatol/x6am51 (2025).
Nakamura, K. et al. The role of PSMB9 upregulated by interferon signature in the pathophysiology of cutaneous lesions of dermatomyositis and systemic lupus erythematosus. Br. J. Dermatol. 174, 1030–1041. https://doi.org/10.1111/bjd.14385 (2016).
Sun, C., Jia, G., Wang, X., Wang, Y. & Liu, Y. Immunoproteasome is up-regulated in rotenone-induced parkinson’s disease rat model. Neurosci. Lett. 738, 135360. https://doi.org/10.1016/j.neulet.2020.135360 (2020).
Acknowledgements
We acknowledge GEO database and contributors.
Funding
This work received generous support from the National Natural Science Foundation of China (No.81804208); the Scientific Research Program of Southwest Medical University (No.2023ZYYJ04, No.2021ZKMS052), and Science and Technology Strategic Cooperation Program of Luzhou Municipal People’s Government and Southwest Medical University (No.2021LZXNYD-J15).
Author information
Authors and Affiliations
Contributions
Jiao Qiu, Yalin Xu: Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. Luyuan Tong, Xingchun Yang: Methodology, Visualization. Xiao Wu: Funding, Supervision, Writing – review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Qiu, J., Xu, Y., Tong, L. et al. Identification of potential pathogenic genes associated with the comorbidity of rheumatoid arthritis and renal fibrosis using bioinformatics and machine learning. Sci Rep 15, 21686 (2025). https://doi.org/10.1038/s41598-025-05757-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-05757-9
