
Abstract
People living with human immunodeficiency virus (HIV) (PLWH) might have an increased risk of developing coronavirus disease 2019 (COVID-19); however, the impact of their gut microbiota and metabolites on the progress of COVID-19 is unknown. Herein, we analyzed the temporal changes in the gut microbiota composition and metabolites of PLWH at baseline and during severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. We analyzed the gut microbiota and metabolites by integrating 16 S rRNA gene sequencing and liquid chromatography-mass spectrometry untargeted metabolomics of 36 PLWH at baseline and during SARS-CoV-2 infection periods. Significant changes in the composition of the gut microbiota and an increased ratio of Firmicutes/Bacteroidetes during SARS-CoV-2 infection, relative to the baseline, were observed. At the genus level, the abundances of Lactobacillus and Lactiplantibacillus decreased significantly. Furthermore, seven differentially enriched human metabolic pathways (including protein digestion and absorption, central carbon metabolism in cancer, aminoacyl-tRNA biosynthesis, mineral absorption, ABC transporters, arginine and proline metabolism, and phenylalanine metabolism) were identified by comparing the fecal metabolites at baseline and during SARS-CoV-2 infection. Spearman correlation analysis revealed close relationships between the two differentially abundant microbiota members and the five differentially abundant fecal metabolites that might affect specific human metabolic pathways.This study is the first to characterize the gut microbiota and metabolites in PLWH at baseline and during SARS-CoV-2 infection. The key microbiota and metabolites in the infection process were identified, providing new ideas for treatment.
Similar content being viewed by others

Background
The novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, which causes coronavirus disease 2019 (COVID-19), led to a pandemic and a devastating public health crisis. As of April 2024, the cumulative number of COVID-19 infections has reached over 700 million, with a death toll surpassing 7.01 million globally1. The pathophysiological impact of COVID-19 might persist long after acute infection, leading to prolonged symptoms2,3. Even with the effectiveness of antiretroviral therapy (ART), people living with human immunodeficiency virus (HIV) (PLWH) who have predispositions to underlying diseases still experience chronic inflammation4. Consequently, understanding the distinct conditions of PLWH at baseline and during SARS-CoV-2 infection is important.
The gut microbiota plays an important role in host’s immune responses and homeostasis mechanisms, such as nutrition, metabolism, and immune regulation, and is associated with the progression of various pathological conditions5,6,7. Studies reported alteration of the gut microbiota composition in patients with COVID-1989,. In PLWH, infection-induced changes in the gut microbiota (dysbiosis) have been reported10. These changes were characterized by an increase in opportunistic bacteria and a decrease in beneficial commensal bacteria. The levels of certain specific bacteria correlated with disease severity11.
Gut metabolites, synthesized by both the host and the gut microbiota, play a critical role in maintaining the stability and physiological functionality of the gut microenvironment. Metabolites associated with the gut microbiota, such as short-chain fatty acids (SCFAs), bile acids, and tryptophan metabolites, are key to regulating the host’s metabolic balance and immune responses12,13. The technique of metabolomics captures the phenotypic outcomes of these biological processes and determines the correlations between changes in metabolite profiles and physiological or pathological states. However, no study has reported the correlation between the gut microbiota and metabolites during COVID-19 in PLWH.
Herein, we performed 16 S rRNA gene sequencing and liquid chromatography-mass spectrometry (LC-MS) untargeted metabolomics of the gut microbiota to examine changes in the gut microbiota and metabolites during treatment of PLWH infected with SARS-CoV-2, and searched for metabolites that were closely related to the gut microbiota of PLWH through association analysis. During the 3-week COVID-19 surge window following the initial fecal sampling, all recruited participants, who were PLWH, were simultaneously infected with SARS-CoV-2. This provided a unique and homogeneous cohort with comprehensive, pre-COVID-19 baseline gut microbiome data, as well as COVID-19 data collected during the infection period. Leveraging this cohort, we further investigated the associations between the gut microbiome and metabolites at baseline and during COVID-19. These results highlight the role of the gut microbiota in regulating overall metabolism and provide new insights into the pathological mechanisms of COVID-19.
Methods
Subject recruitment and sample collection
This study includes a cohort of PLWH from our ongoing research, but focuses specifically on those who subsequently contracted SARS-CoV-2. Stool samples were routinely collected in September 2022 from people living with HIV (PLWH), who were under the care of the HIV clinic at the First Affiliated Hospital of Zhejiang University in Zhejiang, China. All individuals had started ART during the chronic phase of HIV infection. Subjects were informed individually about the purpose of the study, including its risks and benefits. Subjects who decided to participate this study signed the informed consent form. All subjects who received antibiotics, probiotics, or both within 4 weeks before enrollment were excluded.
Informed consent was obtained from the participants. All procedures were performed following the principles of the Declaration of Helsinki. The Institutional Review Committee of the First Affiliated Hospital of Zhejiang University has approved this study (ethical number 2020-IIT54). All methods adhered to the relevant guidelines set by the institutional ethics committee.
COVID-19 severity classification
In this study, the severity of COVID-19 among participants was categorized based on clinical symptoms and imaging findings, as outlined in previous research14. The classifications are as follows: Mild: Patients exhibited common symptoms of COVID-19 (such as fever, cough, sore throat, anosmia) without experiencing dyspnea, and chest computed tomography (CT) scans showed no signs of pneumonia. Moderate: Patients presented with fever and respiratory symptoms, and chest CT scans revealed signs of pneumonia. Oxygen saturation levels were maintained at or above 94%. Severe: Patients showed evidence of pneumonia on chest CT scans accompanied by oxygen saturation levels below 94%. All participants enrolled in this study were classified as having mild COVID-19.
Sample collection
Fecal samples were collected following previously described methods15. Briefly, stool samples were obtained using the natural defecation method, ensuring minimal contamination. Urine was drained prior to defecation to prevent sample adulteration. Approximately 2 g of feces were transferred into sterile fecal collection tubes using a sampling spoon. To prevent bacterial overgrowth, the collected samples were immediately placed on ice and transported to the laboratory within 2 h, where they were stored at −80 ℃ for subsequent analyses.
Samples of plasma were collected into 10 mL vacutainers. The blood samples were mixed well and subjected to centrifugation at 3000 × g for 10 min to collect the plasma (supernatant), which was placed at −80 °C.
Cytokine analysis using meso scale discovery kits
Cytokine levels were determined using a Meso Scale Discovery (MSD, Rockville, MD, USA) electrochemiluminescence V-PLEX assay, including interferon (IFN)-γ, tumor necrosis factor alpha (TNF-α), interleukin (IL)−13, IL-12p70, IL-10, IL-8, IL-6, IL-4, IL-2, and IL-1β (pg/mL). Two-fold diluted plasma samples were determined in duplicate. The cytokine concentrations were determined using attached electrochemiluminescent labels on plates in the MSD apparatus (MESO QuickPlex SQ 120). Variations between plates were determined by including high and low level controls, giving an interassay coefficient of variation of <1%. MSD Discovery Workbench analysis software was used to analyze the results.
Extraction of DNA and 16 S rRNA high-throughput sequencing
Total microbiome DNA was extracted from fecal samples using the Omega Mag-Bind Stool DNA Kit (Omega Bio-Tek, Norcross, GA, USA), following the manufacturer’s instructions, and stored at −20 ℃ prior to further analysis. We amplified the V3-V4 hypervariable regions of the 16 S rRNA gene from the extracted DNA. The PCR amplifications employed the forward primer 5′-ACTCCTACGGGAGGCAGCA-3′ and the reverse primer 5′-GGACTACHVGGGTWTCTAAT-3′. Thermal cycling was performed with an initial denaturation at 98 ℃ for 5 min, followed by 25 cycles of denaturation at 98 ℃ for 30 s, annealing at 53 ℃ for 30 s, and extension at 72 ℃ for 45 s. The process concluded with a final extension at 72 ℃ for 5 min. The PCR amplicons were purified using Vazyme VAHTS™ DNA Clean Beads (Vazyme, Nanjing, China) and quantified with the Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen, Carlsbad, CA, USA). After quantification, the purified amplicons were pooled in equal concentrations, and paired-end 2 × 250 bp sequencing was conducted on the Illumina NovaSeq 6000 platform.
For the bioinformatic analysis, we utilized the dada2 pipeline within the QIIME2 software suite (version 2019.4)16. This method allowed for the optimization of sequence quality before further analysis. Taxonomic classification of the sequences was performed using the Greengenes database (http://greengenes.secondgenome.com/) accessed through the QIIME2 feature-classifier plugin, employing a pretrained Naive Bayes classifier (https://github.com/QIIME2/q2-feature-classifier)17,18.
Liquid chromatography-mass spectrometry metabolomic data collection
Metabolites were isolated from fecal samples utilizing the following extraction process19. One gram of the sample was accurately weighed into a 2 mL centrifuge tube, followed by the addition of 600 µL methanol (pre-cooled to −20 ℃) containing 2-Amino-3-(2-chlorophenyl)-propionic acid (4 ppm) as an internal standard. The mixture was vortexed for 30 s. Subsequently, 100 mg of glass beads were added, and the tube was processed using a tissue grinder at 60 Hz for 90 s. The homogenized mixture was then subjected to ultrasonic treatment at room temperature for 10 min. After sonication, the sample was centrifuged at 12,000 rpm for 10 min at 4 ℃. The resulting supernatant was filtered through a 0.22 μm membrane and transferred into a detection vial for LC-MS analysis.
The analysis was conducted using a Vanquish Ultrahigh Performance Liquid Chromatography (UHPLC) System (Thermo Fisher Scientific, Waltham, MA, USA). We employed an ACQUITY UPLC HSS T3 column (150 × 2.1 mm, 1.8 μm, Waters, Milford, MA, USA) for chromatography, maintaining the column temperature at 40 ℃. The flow rate was consistently set at 0.25 mL/min. The mobile phase composition varied with the ionization mode: for the positive ion mode, it included 0.1% formic acid in acetonitrile (v/v) and 0.1% formic acid in water (v/v); for the negative ion mode, acetonitrile and 5 mM ammonium formate were used20.
Metabolite detection was conducted using a Q Exactive HF-X mass spectrometer (Thermo Fisher Scientific, USA) equipped with an ESI ion source. Data acquisition was performed in Full MS-ddMS2 mode, enabling simultaneous MS1 and MS/MS detection. Key parameters included a sheath gas pressure of 30 arb, auxiliary gas flow of 10 arb, and spray voltages of 3.50 kV and − 2.50 kV for ESI (+) and ESI (-) modes, respectively. The capillary temperature was set at 325 °C. The MS1 scan range was m/z 100–1000 with a resolving power of 60,000 FWHM, while MS/MS analysis achieved a resolving power of 15,000 FWHM. Each scan cycle included eight data-dependent scans, with normalized collision energy set to 30 eV and dynamic exclusion time set to automatic21.
LC–MS metabolomic data analyses
The raw data were first converted to mzXML format using MSConvert from the ProteoWizard software package (v3.0.8789)22 and processed with XCMS for feature detection23retention time correction, and alignment. Metabolite identification was based on accurate mass measurements (< 30 ppm) and MS/MS data, matched against several databases, including HMDB24MassBank25LipidMaps26mzCloud27and KEGG28. To minimize systematic bias, robust LOESS signal correction (QC-RLSC)29 was applied for data normalization. Only ion peaks with relative standard deviations (RSDs) below 30% in quality control (QC) samples were retained to ensure reliable metabolite identification.
Multivariate data analyses and modeling were performed using the Ropls software30. Data were mean-centered and scaled, with models developed using principal component analysis (PCA), orthogonal partial least-squares discriminant analysis (OPLS-DA), and partial least-squares discriminant analysis (PLS-DA). Variables contributing to classification were identified based on P values, variable importance projection (VIP) scores from OPLS-DA, and fold change (FC) values. Metabolites with P values < 0.05 and VIP scores > 1 were considered statistically significant.
To determine the origins of top 20 differentially abundant metabolites, we utilized MetOrigin31an integrative web-based platform that classifies metabolites based on source information from seven comprehensive databases: KEGG, HMDB, BIGG, ChEBI, FoodDB, DrugBank, and T3DB. This approach enabled us to categorize metabolites as originating from the host, microbiota, both (co-metabolism), food, drugs, or environmental sources.
Statistical and bioinformatic analyses
For central tendency measurements, geometric mean (GM) values were utilized. Significant differentially abundant microbiota were identified using linear discriminant analysis (LDA) effect size (LEfSe), selecting microbiota with an LDA value greater than 2. The predictive functional profiling of microbial communities was performed using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States 2 (PICRUSt2)32referencing the Kyoto Encyclopedia of Genes and Genomes (KEGG) database to analyze KEGG Orthology (KO) and predict microbial metabolic functions. Correlations between differentially abundant bacteria and the top 20 differentially abundant fecal metabolites based on their relative abundances were examined using Spearman correlation analysis. False-discovery rate (FDR) values were estimated using the Benjamini-Hochberg method to control for multiple testing. All statistical computations and data analysis were conducted in R (version 4.2.1; R Foundation for Statistical Computing, Vienna, Austria). For all paired sample comparisons, we employed the Wilcoxon signed-rank test to determine significant differences between baseline and SARS-CoV-2 infection conditions within the same PLWH subjects. All statistical tests were two-tailed. A P value of less than 0.05 was considered statistically significant.
Results
Clinical characteristics of the study participants
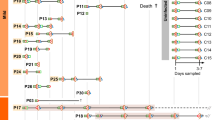
From February 1 to February 28, 2023, these individuals experienced mild COVID-19 symptoms. Their infections were confirmed via SARS-CoV-2 quantitative real-time reverse transcription PCR, and additional stool samples were collected during this time (Fig. 1). All patients enrolled in the study were infected with the omicron variant. The demographic and clinical characteristics of the patients included in the study are presented in Table 1. The cohort comprised 36 adults (median age of 40.25 years; range = 34.00–45.75 years) who provided stool samples and indicated their willingness to participate in the study. In terms of severity classification, all patients had mild disease.

Schematic overview of the study design.
Analysis of the diversity of the gut microbiota of PLWH
The alpha diversity (Wilcoxon signed-rank test) was analyzed based on the Chao1 and Shannon indexes; however, no significant changes were observed (Fig. 2A). Nonmetric multidimensional scaling (NMDS) analysis based on the weighted UniFrac distance algorithm and the analysis of similarity (ANOSIM) method showed that there was a significant change in the beta diversity (P = 0.001) after SARS-CoV-2 infection (Fig. 2B). This indicated that SARS-CoV-2 infection altered the composition of the PLWH’s gut microbiota. At the phylum level, Firmicutes, Actinobacteria, Proteobacteria, and Bacteroidota accounted for the major components of the gut microbiota (Fig. 2C). The Firmicutes/Bacteroidota ratio increased after SARS-COV-2 infection, but not significantly (P = 0.2581, by a Wilcoxon signed-rank test). At the genus level, the most common genus in the two groups was Blautia. Compared with the baseline group, the proportion of Bifidobacterium, Catenibacterium, and Holdemanella in the COVID-19 group was significantly higher, while the abundance of Megamonas was significantly lower (Fig. 2D).
The LEfSe algorithm was applied to further evaluate the differences in abundances between the baseline and SARS-CoV-2 infection groups (Fig. 2E). As shown in the differential species score chart, Catenibacterium, Turicibacter, and Staphylococcus were among the relatively high-abundance species during SARSCoV-2 infection, while Megamonas, Lactiplantibacillus, and Lactobacillus were among the relatively high-abundance species at baseline.

(A) Chao1 indicates species richness between the two groups. The Shannon index was employed to assess species diversity, accounting for both richness and evenness within the microbial communities. (B) Nonmetric multidimensional scaling (NMDS) analysis based on the weighted UniFrac distance algorithm and the ANOSIM method between the Baseline and COVID-19 groups. The horizontal axis (NMDS1) and the vertical axis (NMDS2) are the two sorting axes. (C) Community composition histogram of the Baseline and COVID-19 groups at the phylum and genus levels (D). (E) LDA score diagram and the significantly differentially abundant genera. The color represents groups, and the length of the histogram represents the LDA score. The higher the score, the more significant difference in the differentially abundant microbiota. (LDA threshold of 2). (F) Liquid chromatography-mass spectrometry analysis of fecal metabolites. Volcano plots indicating the discrimination of fecal metabolites between the two groups. The ordinate shows the -log10 (P-value), and the abscissa is the log2 (fold change) value. The screening threshold for significantly differentially abundant metabolites was fold change > 2, VIP value > 1.5, and P-value < 0.05 or fold change < 0.5, VIP value > 1.5, and P < 0.05. Every dot represents a kind of metabolite, with the blue dots representing the significantly downregulated metabolites, and the red dots represent the significantly upregulated metabolites. The gray dots represent the metabolites with no significant difference between the two groups. The bar chart displays the statistics of the differentially abundant metabolites in PLWH during COVID-19.
Liquid chromatography-mass spectrometry analysis of fecal metabolic profiles
A total of 437 metabolites were identified, including 199 differentially abundant metabolites, with 56 upregulated and 143 downregulated between the two groups (Fig. 2F). The differentially abundant metabolites in the LC-MS analysis were then annotated according to the KEGG pathway mapper. Analysis of the top 20 differential metabolites using MetOrigin revealed that one metabolite was classified as host-derived, two as microbiota-derived, and nine as co-metabolized by both host and microbiota (TableS1, Fig.S1). Additionally, metabolites related to food, drugs, and environmental sources were identified and are presented in TableS1. A comparative analysis of the human metabolic profiles showed significant changes in protein digestion and absorption, as well as changes in major metabolic pathways, including carbohydrate and amino acid pathways (Fig. S2). Specifically, in carbohydrate metabolism, central carbon metabolism in cancer was affected. In amino acid metabolism, phenylalanine, tyrosine, tryptophan, and arginine biosynthesis; lysine degradation; and arginine, proline, glycine, serine, threonine, and phenylalanine metabolism were affected. We focused on seven differentially enriched human metabolic pathways. Based on the P values, protein digestion and absorption, central carbon metabolism in cancer, aminoacyl-tRNA biosynthesis, mineral absorption, ABC transporters, arginine and proline metabolism, and phenylalanine metabolism were characterized as significantly relevant pathways affected by SARS-CoV-2 infection (Fig. S2).
Correlations between 16 S rRNA gene sequencing and LC-MS untargeted metabolomics
Overall correlation analysis demonstrated that interleukin (IL)−13, IL-4, and tumor necrosis factor alpha (TNF-α) correlated significantly with the gut microbiota and metabolites (Fig. 3A). To investigate the associations among the gut microbiota, fecal metabolites, and human metabolic pathways, Spearman correlation analysis was employed to evaluate the links between the differentially abundant bacteria and the top 20 differentially abundant fecal metabolites according to their relative abundances (Fig. 3B). Among the top 20 differentially abundant fecal metabolites, five could be annotated according to the human KEGG pathway mapper. The correlations among the differentially abundant fecal metabolites, altered bacteria, and related human metabolic pathways were then tabulated (Table 2). Close relationships were observed between two differentially abundant microbiota members and five differentially abundant fecal metabolites that might affect specific human metabolic pathways. Specifically, increases in L-asparagine correlated with a decrease in Lactobacillus levels, impacting pathways such as protein digestion and absorption, central carbon metabolism in cancer, aminoacyl-tRNA biosynthesis, and mineral absorption. Elevated spermidine levels associated with reduced Lactobacillus levels might influence ABC transporters and arginine and proline metabolism. Additionally, decreases in creatine correlated with lower levels of both Lactobacillus and Lactiplantibacillus, primarily affecting arginine and proline metabolism. The reduction in phenylethylamine, observed alongside a decline in Lactobacillus and Lactiplantibacillus, could potentially alter phenylalanine metabolism. Moreover, a decrease in L-phenylalanine linked to reduced Lactobacillus might impact several pathways, including protein digestion and absorption, central carbon metabolism in cancer, ABC transporters, phenylalanine metabolism, aminoacyl-tRNA biosynthesis, and mineral absorption. Lactobacillus was found to be associated with the greatest number of decreased differentially abundant metabolites Specifically, Lactobacillus correlated with creatine, L-Asparagine, L-Phenylalanine, phenylethylamine, and spermidine. In other words, these species could perform pivotal and pleiotropic roles in the microbiota-host interaction (Table 2).

Correlations among gut genera, metabolites, and cytokines. (A) Comparisons of the gut genera and metabolites using Spearman’s correlation coefficient and the correlation between them and cytokines, determined using the Mantel test. The color gradient denotes Spearman’s correlation coefficients. Cytokines were correlated with the gut microbiota and metabolites using the Mantel test. Edge width corresponds to Mantel’s r statistic for the corresponding distance correlations. The edge color denotes the statistical significance of Mantel’s P value. (B) Heat map visualization of the correlation analysis for the top 20 differentially abundant metabolites and the differentially abundant bacteria between the Baseline and COVID-19 groups. Spearman correlation analysis was used to examine correlations between the differentially abundant bacteria and the top 20 differentially abundant fecal metabolites according to their relative abundances. Genera/metabolites enriched post-infection are highlighted in red, while those depleted are shown in blue.
Discussion
This study aimed to assess the relationship between the gut microbiota and metabolites in PLWH following SARS-CoV-2 infection. The major findings of this study are as follows: (i) SARS-CoV-2 infection significantly altered the composition of the gut microbiota; (ii) seven differentially enriched human metabolic pathways are worthy of attention; and (iii) Spearman correlation analysis showed close relationships between two differentially abundant microbiota members and five differentially abundant fecal metabolites, which might affect specific human metabolic pathways.
During SARS-CoV-2 infection, PLWH were confronted with various physical and psychological stressors. Consistent with a previous report33the current study revealed that the gut microbial composition of PLWH was significantly altered after SARS-CoV-2 infection. The LC-MS untargeted metabolomics analysis identified seven differentially enriched metabolic pathways (protein digestion and absorption, central carbon metabolism in cancer, aminoacyl-tRNA biosynthesis, mineral absorption, ABC transporters, arginine and proline metabolism, and phenylalanine metabolism) in the PLWH. To obtain more detailed information on the microbiota-host interaction, Spearman correlation analysis of the differentially abundant microbiota members and differentially abundant metabolites was performed. The results revealed that two differentially abundant microbiota members correlated closely with five differentially abundant fecal metabolites, which might have specific effects on the metabolic pathways of PLWH. Specifically, decreases in various fecal metabolites (including creatine, phenylethylamine, and L-Phenylalanine) correlated with various differentially abundant bacteria (including Lactobacillus and Lactiplantibacillus), which might impact the human metabolic pathways related to arginine and proline metabolism, phenylalanine metabolism, protein digestion and absorption, and aminoacyl-tRNA biosynthesis.
Two differentially abundant bacteria, Lactobacillus and Lactiplantibacillus, identified in our study might have insidious effects on PLWH during SARS-CoV-2 infection. Notably, Lactobacillus is involved in the production and utilization of SCFAs34. SCFAs influence the immune response through two primary mechanisms. Firstly, unmetabolized SCFAs can travel directly to the lungs via the bloodstream, where they activate G protein-coupled receptors (GPCRs) or inhibit histone deacetylases, thus modulating immune functions35. Secondly, SCFAs can migrate to the bone marrow through the circulation, where they promote the differentiation of macrophage and dendritic progenitor cells (MDPs) into Ly6C– monocytes. These monocytes then travel to the lungs and differentiate into anti-inflammatory alternatively activated macrophages (AAMs). These AAMs reduce the recruitment of neutrophils and stimulate regulatory T cells (Tregs) to produce anti-inflammatory cytokines such as IL-10 and transforming growth factor beta (TGF-β), thereby decreasing lung injury and inflammation36,37. Our findings indicate a reduction in Lactobacillus abundance during SARS-CoV-2 infection in PLWH, which may suggest a potential role in the gut microbiome-host interaction. However, further research is needed to explore its therapeutic implications.
Additionally, Lactiplantibacillus might be another potential probiotic for PLWH. Research involving animal models and human clinical trials have shown that some Lactiplantibacillus plantarum strains, when administered orally, effectively modulate respiratory immunity and boost resistance to bacterial and viral infections. This enhancement in the immune response was observed across various age groups, including children, adults, and the elderly, helping to protect them against respiratory infections, such as pneumococcal pneumonia, the common cold, and influenza-like illnesses38. Notably, L. plantarum, and its exopolysaccharides (EPSs), induces an antagonistic effect against human rotavirus. MA104 cells (a rotavirus-susceptible cell line) treated with the strain or its EPSs showed significantly lower numbers of cytopathic alterations and reduced viral replication when compared with untreated controls39. Our study showed that the significant decrease in Lactiplantibacillus correlated with the abundance of metabolites (e.g., creatine and phenylethylamine) in feces, which might impact human metabolic pathways, including arginine and proline metabolism and phenylalanine metabolism.
The observed decrease in TNF-α and IL-13, coupled with an increase in IL-4, in SARS-CoV-2–infected PLWH has important immunological implications in the context of chronic HIV infection. People living with HIV often exhibit a baseline immune skew characterized by elevated Th2 cytokines (e.g., IL-4) and suppressed Th1 responses40. IL-4 could down-regulate proinflammatory cytokine production – for example, it suppresses TNF-α release from activated monocytes/macrophages, which might partly explain the attenuated TNF-α levels observed in co-infected PLWH41. Meanwhile, as another Th2 cytokine, IL-13 has been implicated in COVID-19 immunopathology, as higher IL-13 levels are associated with worse outcomes (e.g., need for mechanical ventilation)42. Thus, the reduction of IL-13 in our PLWH cohort could potentially limit hyperinflammatory damage during COVID-19. Notably, we found that TNF-α levels correlated with multiple metabolites, underscoring that HIV-associated inflammation is intertwined with metabolic pathways. Furthermore, we expanded the discussion of gut microbiome differences between PLWH and HIV-negative individuals. Even with long-term ART and viral suppression, PLWH maintain an altered gut microbiota with reduced diversity and a distinct composition compared to HIV-negative controls15,43. In particular, beneficial commensals (such as Bifidobacterium, Lactobacillus, and Faecalibacterium) tend to be depleted, whereas potentially pro-inflammatory taxa (like Prevotella and Enterobacterales family bacteria) are enriched in the gut microbiome of PLWH43. This persistent dysbiosis – driven by residual HIV in tissues that sustains chronic immune activation - could lead to microbial translocation and ongoing inflammation43. Such microbiome alterations, and their associated metabolic byproducts, likely contribute to the distinctive cytokine and metabolite profile observed in PLWH during SARS-CoV-2 co-infection.
This study provides initial insights into gut microbiota-host interactions in PLWH following SARS-CoV-2 infection by integrating gut microbiome and metabolomic analyses. While the findings highlight potential changes in specific microbiota and metabolites, further research is necessary to explore their roles and therapeutic implications. It is important to recognize the limitations of this study. First, while our study identified changes in the abundance of Lactobacillus and Lactiplantibacillus, the data do not allow definitive conclusions regarding their roles in SARS-CoV-2 infection. Further large-scale studies and experiments are required to validate these observations. Second, our analysis identified several metabolic pathways altered during infection, yet the directionality (up- or down-regulation) of these pathways was not determined, warranting further mechanistic investigation. Third, our study did not include a long-term follow-up period. Therefore, we could not determine whether the observed alterations in the gut microbiome and metabolites are associated with long-term COVID-19 sequelae (long-COVID) in PLWH. Future studies should incorporate longitudinal designs with extended follow-up to address this important aspect. The primary focus of this study was on the overall impact of SARS-CoV-2 infection on the gut microbiome and metabolome in PLWH. This research provides valuable insights into the physiological disorders that might arise during SARS-CoV-2 infection, as evidenced by changes in the gut microbiome and metabolome. Future prospective studies on SARS-CoV-2 infection should incorporate comprehensive analyses using multiple psychological and physiological scales. Additionally, cellular and animal studies are needed to verify the underlying causal relationships between SARS-CoV-2 infection, physical disorders, and alterations in the gut microbiome and metabolome.
Conclusion
Taken together, our findings indicate that the SARS-CoV-2 infection could induce gut dysbiosis and might influence human metabolic pathways in PLWH. However, it is critical to note that this study is a preliminary pilot project with a limited sample size. Thus, these results should be verified in future studies.
Data availability
The raw sequence data reported in this research have been deposited in the National Center for Biotechnology Information database with accession number PRJNA1249258.
References
Worldometer. https://www.worldometers.info/coronavirus/.
Huang, C. et al. 6-month consequences of COVID-19 in patients discharged from hospital: a cohort study. Lancet 397 (10270), 220–232 (2021).
Blanco, J. R. et al. Pulmonary long-term consequences of COVID-19 infections after hospital discharge. Clin. Microbiol. Infect. 27 (6), 892–896 (2021).
Cribbs, S. K., Crothers, K. & Morris, A. Pathogenesis of HIV-Related lung disease: immunity, infection, and inflammation. Physiol. Rev. 100 (2), 603–632 (2020).
Wastyk, H. C. et al. Gut-microbiota-targeted diets modulate human immune status. Cell 184 (16), 4137–4153e4114 (2021).
Nagata, N. et al. Human gut microbiota and its metabolites impact immune responses in COVID-19 and its complications. Gastroenterology 164 (2), 272–288 (2023).
Zheng, D., Liwinski, T. & Elinav, E. Interaction between microbiota and immunity in health and disease. Cell. Res. 30 (6), 492–506 (2020).
Zuo, T. et al. Alterations in gut microbiota of patients with COVID-19 during time of hospitalization. Gastroenterology 159 (3), 944–955e948 (2020).
Ren, Z. et al. Alterations in the human oral and gut microbiomes and lipidomics in COVID-19. Gut 70 (7), 1253–1265 (2021).
Xie, Y. et al. Altered gut microbiota correlate with different immune responses to HAART in HIV-infected individuals. BMC Microbiol. 21 (1), 11 (2021).
Talukdar, D. et al. Association of gut microbial dysbiosis with disease severity, response to therapy and disease outcomes in Indian patients with COVID-19. Gut Pathog. 15 (1), 22 (2023).
Bunnett, N. W. Neuro-humoral signalling by bile acids and the TGR5 receptor in the Gastrointestinal tract. J. Physiol. 592 (14), 2943–2950 (2014).
Jeong, J. J. et al. Multi-strain probiotics alleviate loperamide-induced constipation by adjusting the microbiome, serotonin, and short-chain fatty acids in rats. Front. Microbiol. 14, 1174968 (2023).
Gandhi, R. T., Lynch, J. B. & Del Rio, C. Mild or moderate Covid-19. N Engl. J. Med. 383 (18), 1757–1766 (2020).
Tian, X. et al. Analysis of the gut microbiota and fecal metabolites in people living with HIV. Microbiol. Spectr. 12 (11), e0023824 (2024).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 7 (5), 335–336 (2010).
DeSantis, T. Z. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72 (7), 5069–5072 (2006).
Bokulich, N. A. et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6 (1), 90 (2018).
Turroni, S. et al. Fecal metabolome of the Hadza hunter-gatherers: a host-microbiome integrative view. Sci. Rep. 6, 32826 (2016).
Zelena, E. et al. Development of a robust and repeatable UPLC-MS method for the long-term metabolomic study of human serum. Anal. Chem. 81 (4), 1357–1364 (2009).
Want, E. J. et al. Global metabolic profiling of animal and human tissues via UPLC-MS. Nat. Protoc. 8 (1), 17–32 (2013).
Smith, C. A., Want, E. J., O’Maille, G., Abagyan, R. & Siuzdak, G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 78 (3), 779–787 (2006).
Navarro-Reig, M., Jaumot, J., Garcia-Reiriz, A. & Tauler, R. Evaluation of changes induced in rice metabolome by cd and Cu exposure using LC-MS with XCMS and MCR-ALS data analysis strategies. Anal. Bioanal Chem. 407 (29), 8835–8847 (2015).
Wishart, D. S. et al. HMDB 5.0: the human metabolome database for 2022. Nucleic Acids Res. 50 (D1), D622–D631 (2022).
Horai, H. et al. MassBank: a public repository for sharing mass spectral data for life sciences. J. Mass. Spectrom. 45 (7), 703–714 (2010).
Sud, M. et al. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 35 (Database issue), D527–532 (2007).
Abdelrazig, S. et al. Metabolic characterisation of Magnetospirillum gryphiswaldense MSR-1 using LC-MS-based metabolite profiling. RSC Adv. 10 (54), 32548–32560 (2020).
Ogata, H. et al. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 27 (1), 29–34 (1999).
Gagnebin, Y. et al. Metabolomic analysis of urine samples by UHPLC-QTOF-MS: impact of normalization strategies. Anal. Chim. Acta. 955, 27–35 (2017).
Thevenot, E. A., Roux, A., Xu, Y., Ezan, E. & Junot, C. Analysis of the human adult urinary metabolome variations with age, body mass index, and gender by implementing a comprehensive workflow for univariate and OPLS statistical analyses. J. Proteome Res. 14 (8), 3322–3335 (2015).
Yu, G. et al. MetOrigin 2.0: advancing the discovery of microbial metabolites and their origins. Imeta 3 (6), e246 (2024).
Douglas, G. M. et al. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38 (6), 685–688 (2020).
Ishizaka, A. et al. Association of gut microbiota with the pathogenesis of SARS-CoV-2 infection in people living with HIV. BMC Microbiol. 24 (1), 6 (2024).
Rastogi, S. & Singh, A. Gut Microbiome and human health: exploring how the probiotic genus Lactobacillus modulate immune responses. Front. Pharmacol. 13, 1042189 (2022).
Koh, A., De Vadder, F., Kovatcheva-Datchary, P. & Backhed, F. From dietary Fiber to host physiology: Short-Chain fatty acids as key bacterial metabolites. Cell 165 (6), 1332–1345 (2016).
Anand, S. & Mande, S. S. Diet, microbiota and Gut-Lung connection. Front. Microbiol. 9, 2147 (2018).
Du, T., Lei, A., Zhang, N. & Zhu, C. The beneficial role of probiotic Lactobacillus in respiratory diseases. Front. Immunol. 13, 908010 (2022).
Villena, J. & Kitazawa, H. The modulation of mucosal antiviral immunity by immunobiotics: could they offer any benefit in the SARS-CoV-2 pandemic?? Front. Physiol. 11, 699 (2020).
Kim, K. et al. Exopolysaccharide from Lactobacillus plantarum LRCC5310 offers protection against rotavirus-induced diarrhea and regulates inflammatory response. J. Dairy. Sci. 101 (7), 5702–5712 (2018).
Assogba, Y. P. et al. Advanced in immunological monitoring of HIV infection: profile of immune cells and cytokines in people living with HIV-1 in Benin. BMC Immunol. 25 (1), 22 (2024).
Hart, P. H., Cooper, R. L. & Finlay-Jones, J. J. IL-4 suppresses IL-1 beta, TNF-alpha and PGE2 production by human peritoneal macrophages. Immunology 72 (3), 344–349 (1991).
Donlan, A. N. et al. IL-13 is a driver of COVID-19 severity. JCI Insight ; 6(15). (2021).
Gaspar, Z., Nagavci, B., Szabo, B. G. & Lakatos, B. Gut Microbiome alteration in HIV/AIDS and the role of antiretroviral Therapy-A scoping review. Microorganisms 12(11), 2221 (2024).
Acknowledgements
We extend our gratitude to Ying Huang, Dating Han, Haijing Fu, Xiaodi Zhang, and Wanpeng Yin from the First Affiliated Hospital of Zhejiang University for their help with sample collection. We also appreciate the assistance of Suzhou Panomix (http://www.panomix.com/) for their support.
Funding
This work was supported by the National Key Research and Development Program [grant number 2020YFE0204300, 2023YFC2506004], the Shandong Provincial Laboratory Project [grant number SYS202202] and Zhejiang Province Public Health (Disease Prevention and Control) Project (G202503).
Author information
Authors and Affiliations
Contributions
N. W. and X. L. designed the study. Y. X., X. L., and Y. Z. assisted with the experiments. X. T. and X. L. analyzed the data. X. T. drafted and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tian, X., Xie, Y., Zhao, Y. et al. Multiomics analyses of gut microbiota and metabolites in people living with HIV before and during SARS-COV-2 infection. Sci Rep 15, 22144 (2025). https://doi.org/10.1038/s41598-025-06815-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-06815-y
