
Abstract
IgA nephropathy (IgAN) is one of the most common immune-related primary glomerular diseases. The pathological mechanism of this disease is complex, and the specific pathogenesis is still unclear. To obtain a comprehensive understanding of its molecular mechanism and to provide new perspectives regarding the detection and treatment of the disease, this study investigated the role of immune cells in IgAN, as well as the role of autophagy-related biomarkers in IgAN development. The original datasets GSE93798, GSE35487, GSE58539, GSE116626 and GSE115857 were downloaded from Gene Expression Omnibus (GEO) and were further integrated and analyzed. The differentially expressed genes (DEGs) between IgAN and healthy control (HC) group were identified by the “limma” R package. The gene ontology (GO) function, Kyoto Encyclopedia of Genes and Genome (KEGG) pathway, GeneSet Enrichment Analysis (GSEA) and DisGeNet enrichment were adopted to analyze the genes from the intersection of DEGs. The hub genes were screened by the square least absolute shrinkage and selection operator (LASSO) and cross validation. Immune cell infiltration was analyzed using CIBERSORT. The correlation between hub genes and infiltrating immune cells was calculated by R software. For the purpose of exploring the value of hub genes for diagnosing IgAN, a receiver operating characteristic (ROC) curve was constructed. Finally, Real-time quantitative polymerase chain reaction (qRT-PCR) was used to verify the relative mRNA level of the AT-DEGs. 12 DEGs were screened out. Enrichment analysis revealed that autophagy-related DEGs (AT-DEGs) were mainly related to intrinsic apoptotic signaling pathway, cellular response to external stimulus, transcription repressor complex and other cellular functions, KEGG pathways enriched by AT-DEGs mainly included biological metabolic pathways related to autophagy, while DisGeNET analysis showed that these AT-DEGs were mainly related to immunological diseases. The optimal six hub genes were obtained by lasso analysis as potential biomarkers for IgAN. ROC curve analysis showed that 4 of the 6 HUB genes had great diagnostic value. Immune infiltration results showed B cells memory, macrophages M2, NK cells activated, T cells CD4+ memory resting, and monocytes are the predominant immune cells with the development of IgAN. The qRT-PCR results showed that, compared to the NC group, SIRT1 mRNA expression in PBMCs from IgAN patients was significantly reduced, while BAG3, CDKN1A, and FOS mRNA levels were markedly elevated. SIRT1, BAG3, COKN1A and FOS can be considered as effective biomarkers related to autophagy for the diagnosis of IgAN. These findings suggest some potential new serum biomarkers for IgAN diagnosis.
Similar content being viewed by others


Introduction
IgA nephropathy (IgAN) is the most common immune-related primary glomerular disease and a leading cause of end-stage renal disease (ESRD), imposing a significant social and economic burden worldwide. Notably, 20% to 40% of IgAN patients progress to ESRD within 10 to 20 years after diagnosis1,2. Persistent proteinuria, hypertension, decline in renal function and renal pathological damage are the main risk factors for the progression of IgAN3. Glucocorticoids and certain immunosuppressants are effective in the clinical management of IgAN, but their side effects should not be overlooked. The 2021 Kidney Disease: Improving Global Outcomes (KDIGO) guidelines recommend strict control of blood pressure and the comprehensive use of angiotensin-converting enzyme inhibitors or angiotensin receptor antagonists as the main supportive treatment model for IgAN4,5.
At present, renal biopsy remains the gold standard for diagnosing IgAN. However, as an invasive procedure, renal biopsy carries various contraindications and risks of complications, making it less acceptable for certain population, especially the elderly and children6,7. Therefore, identifying biomarkers with high sensitivity and specificity for early diagnosis, monitoring and treatment of IgAN is of critical importance.
Autophagy, a ubiquitous physiological and innate immune process in eukaryotic cells, autophagy is indispensable for cell growth, apoptosis and maintenance of homeostasis8. Autophagy has been proven to play a renoprotective role in various chronic kidney diseases (CKDs), including IgAN, diabetic nephropathy (DN), and membranous nephropathy9,10. The accumulation of autophagosomes and tumoral lysosomes in podocytes was observed in renal biopsy tissues from IgAN patients. Immune cell infiltration also play a crucial role in the pathogenesis and progression of IgAN11. In a mouse model of IgAN, SIRT1/SIRT3-mediated autophagy inhibits the activation of NLRP3 inflammasome in macrophages stimulated through IgA immune complexes, suggesting that autophagy activation in immune cells may negatively regulate the pathogenesis of IgAN12. The autophagy process regulates inflammation by interacting with innate immune signaling pathways, clearing endogenous inflammasome agonists, and influencing immune mediator secretion13. Furthermore, autophagy contributes to the presentation of antigens and the maintenance of T cell homeostasis14. Thus, autophagy has multiple immune functions that regulate infection, inflammation, and immunity.
Dysregulation of autophagy plays a pivotal role in the pathogenesis of CKD. Despite significant progress in elucidating the molecular mechanisms underlying IgAN, several critical gaps remain. First, the role of autophagy-related genes (ATGs) in the progression of IgAN has not been systematically investigated, limiting our understanding of their potential as diagnostic or therapeutic targets. Second, current studies on immune cell infiltration in IgAN primarily rely on single datasets, which may not fully capture the disease's heterogeneity. Finally, the diagnostic potential of ATGs in IgAN has not been comprehensively assessed using integrated bioinformatics approaches.
To address these gaps, this study integrated multiple GEO datasets and employed advanced machine learning algorithms, including LASSO regression and CIBERSORT analysis, to identify autophagy-related biomarkers and delineate immune cell infiltration patterns in IgAN. By combining DEG analysis, functional enrichment, and immune cell profiling, we comprehensively elucidated the molecular mechanisms underlying IgAN pathogenesis and identified novel biomarkers for disease diagnosis, validated through qRT-PCR using peripheral blood samples from both patients and healthy controls. This approach not only enhances the diagnostic accuracy of renal biopsy but also monitors disease progression, evaluates therapeutic efficacy, and helps prevent complications, thereby optimizing IgAN management and improving patient outcomes and quality of life.
Materials and methods
GEO data collection and download
We screened IgAN gene expression profile from an open-access database, GEO database (http://www.ncbi.nlm.nih.gov/geo). Five eligible datasets, GSE93798, GSE35487, GSE58539, GSE116626 and GSE115857, were chosen according to the following criteria: (1) expression profiling array sequencing; (2) renal biopsy tissue sections or peripheral blood mononuclear cells; (3) data contain 15 or more samples. Details are provided in Table 1.
Data processing and merging
The original and serial matrix files for the test dataset (GSE93798, GSE35487) and training dataset (GSE58539, GSE116626, GSE115857) were downloaded respectively. Using "RMA" in the R package (version 4.4.2), the probe expression matrix was extracted and normalized from raw data, and then converted to a gene expression matrix through a platform annotation file. Multiple probes corresponding to the same gene were averaged. The Perl script was used to obtain the transformed gene expression matrix, and after merging the matrices of the two datasets of genes, the R package "SVA" was applied to eliminate the heterogeneity caused by different experimental batches and platforms. Finally, the resulting normalized gene expression matrix was analyzed with the "Limma" package of R.
Screening and analysis of autophagy-related differential genes
In order to identify differentially expressed genes (DEGs) between IgAN and healthy individuals, the "limma" package of R was employed. DEGs were screened according to Adj.P.Val < 0.05 and |log fold change (logFC)|> 1.3. Autophagy genes (ATGs) related to human diseases were obtained from the human autophagy database (HADb) (http://www.autophagy.lu/), and then the intersection between ATGs and DEGs was identified using the VennDiagram package in R software to obtain the DEGs related to autophagy (AT-DEGs). To visualize relevant AT-DEGs, heatmaps and volcano plots were generated via the heatmap and ggplot2 packages, respectively.
Functional enrichment analysis
Gene ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Set Enrichment Analysis (GSEA) were performed on AT-DEGs using R software, and the results were visualized. The screening condition was Adj.P.Val < 0.05. Enrichment analysis of DisGeNet was performed via the Metascape database.
Establishment and evaluation of risk score diagnostic models based on AT-DEGs
Least absolute shrinkage and selection operator (Lasso) regression analysis was performed on AT-DEGs by the "glmnet" package to constrain parameters, prevent overfitting, and minimize bias. Then multivariate logistic analysis was performed on IgAN characteristic genes generated by lasso regression. P < 0.05 was considered statistically significant, and the risk score was calculated by multiplying the expression level of each AT-DEG (α) by the corresponding coefficient (β). Then a nomogram was established to evaluate and analyze the results to predict the incidence of relevant AT-DEGs in IgAN. Finally, the accuracy of the prediction model was evaluated by the "pROC" package which calculated the area under the ROC curve (AUC).
Construction and validation of the nomogram
The "RMS" package was used to construct a nomogram to summarize the significant parameters obtained from logistic regression analysis, and multiple predictors were integrated to evaluate the probability of IgAN. In order to evaluate the predictive ability of the AT-DEGs-based risk score model, the Harrell concordance indexes (C-index) was used to calculate the probability that the predicted result was consistent with the actual observed result, and the calibration curve was illustrated through "Hmisc" and "RMS" package to visualize the results of the analysis.
Evaluation of Infiltration by Immune Cells
An analysis of the relationship between IgAN and immune cell infiltration was performed using the CIBERSORT bioinformatics algorithm. Each sample file was filtered using R software and the data of CIBERSORT’s P < 0.05 were filtered and retained for subsequent analysis ("BiocManager" package). Afterwards, relevant histograms were illustrated to visualize the composition of infiltrated immune cells, and violin plots were produced to visualize the differences in immune cell infiltration between IgAN and HC groups ("reshape2 "and" ggpubr" packages). Finally, Spearman's correlation analysis was performed by R software to explore the correlation between the identified hub AT-DEGs and immune cell levels ("ggplot2" package).
Patients and healthy controls
Peripheral blood was collected from 9 IgAN patients (5 females and 4 males, mean age 42.5 ± 7.1 years) treated at the Kidney Disease Department of Xiyuan Hospital of China Academy of Chinese Medicine from March 2024 to August 2024. Seven healthy volunteers (3 females and 4 males, mean age 38 ± 3.7 years) were included as the control group. This study was approved by the Ethics Committee of Xiyuan Hospital of China Academy of Chinese Medicine (2022XLA088-1), and all participants agreed to participate in the study.
Real-time quantitative polymerase chain reaction
Five milliliters of fasting venous blood were collected from two groups of patients in the morning. Total RNA was extracted from the samples using TRIzol reagent. Subsequently, the total RNA samples were reverse transcribed into complementary DNA (cDNA) using the II First Strand cDNA Synthesis Kit (Geneseed, China). Quantitative reverse transcription polymerase chain reaction (qRT-PCR) was performed on the Thermo Fisher ABI 7500 real-time fluorescence quantitative PCR instrument. The primer sequences for the target genes are detailed in Table 2. The relative mRNA expression levels of each target gene in the samples were calculated using the 2-ΔΔCt method. These studies involving human were approved by the Ethics Committee of Xiyuan Hospital, China Academy of Chinese Medical Sciences (2022XLA088-1). These studies were conducted in accordance with local legislative and institutional requirements. Participants provided written informed consent to participate in this study.
Result
Identification of DEGs
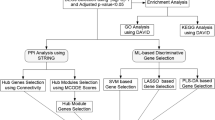
The technology roadmap for this study is shown in Fig. 1.

The technology roadmap of this study.
Combined with GSE93798 and GSE35497 as training dataset, differential expression profile analysis was performed on biological samples of 45 IgAN patients and 28 normal people. 694 DEGs were screened out (Fig. 2A), among which there were 296 up-regulated genes and 398 down-regulated genes. A comparison of these DEGs with 222 ATGs collected from HADb revealed the presence of 12 AT-DEGs with intersections. As shown in Fig. 2C, the expression levels of 12 AT-DEGs were significantly different between IgAN patients and normal individuals, which were visualized by volcano plots (Fig. 2B) and heat maps (Fig. 2C).

Visualization of the results of identification of AT-DEGs. (A) Intersection Venn diagram of DEGs and ATGs. The intersection part is 12 AT-DEGs. (B) Volcano plot of AT-DEGs. Red indicates up-regulated genes, green indicates down-regulated genes, and black indicates genes with no significant difference. (C) Heatmap of AT-DEGs. The red colored grid represents the gene presenting high expression in the sample, and the blue grid represents the gene presenting low expression in the sample.
GSEA enrichment analysis of DEGs
GSEA enrichment analysis (http://gsea-msigdb.org) revealed 51 potential biological pathways. As indicated in Fig. 3, asthma, allograft rejection, autoimmune thyroid disease, type I diabetes mellitus, viral myocarditis were the most active pathways in IgAN group. Arginine and proline metabolism, butanoate metabolism, drug metabolism cytochrome p450, peroxisome, and PPAR signaling pathway were the most active pathways in the HC group.

GSEA enrichment analysis of DEGs. (A) Active pathways in the HC group. (B) Active pathways in the IgAN group.
Enrichment function analysis of AT-DEGs
R software was used to perform an enrichment analysis of AT-DEGs. GO functional enrichment analysis of AT-DEGs consisted of three categories: cellular component (CC), biological process (BP) and molecular function (MF). As shown in Fig. 4A,B, GO functions enriched by AT-DEGs were primarily associated with intrinsic apoptotic signaling pathway, cellular response to external stimulus, transcription repressor complex, core promoter sequence-specific DNA binding and molecular function activator activity. As shown in Fig. 4C, KEGG pathways enriched by AT-DEGs were primarily enriched in biological metabolic pathways related to autophagy, such as lipid and atherosclerosis, apoptosis, cellular senescence and cell cycle and immune and inflammation-related diseases, such as colorectal cancer, chronic myeloid leukemia, Influenza A19. The results of genetic functional analysis of AT-DEGs by Metascape are shown in Fig. 4D. DisGeNET analysis illustrated that AT-DEGs were also associated with infection, HIV coinfection, leukemia, T cell and other diseases related to immune function.

Enrichment analysis of AT-DEGs. These results elucidated the biological processes, signaling pathways, and associated diseases involved in AT-DEGs associated with IgAN. (A) Circleplot of GO functional enrichment of AT-DEGs. (B) Barplot of GO functional enrichment of AT-DEGs. (C) Bubble of KEGG functional enrichment of AT-DEGs19,20,21. (D) Barplot of DisGenNET functional enrichment of AT-DEGs.
Establishment of AT-DEGs related diagnostic model
Lasso regression analysis was used to minimize the bias of the diagnostic model, making the parameters as simple as possible while ensuring the best fit error, reducing the probability of overfitting, and selecting the most suitable tuning parameter as λ (Fig. 5A). Among the 12 candidate genes in the training dataset, a total of six hub DE-ATG genes related to diagnosis were obtained: FOS, CDKN1A, SIRT1, BAG3, EIF2AK3, and SERPINA1. The six hub genes were then used to construct a prediction model (Fig. 5B). The following formula was used for model construction: risk score = FOS *-1.746 + CDKN1A *-0.136 + SIRT1*-6.031 + BAG3 *-2.256 + EIF2AK3 *-3.944 + SERPINA1*-1.509. Afterwards, a nomogram of the diagnostic model was established for further visualization of the results and clinical use. As shown in Fig. 5C, based on the expression levels of the six core AT-DEGs in humans, the physician or patient can locate them on the corresponding scale of the nomogram and calculate the corresponding score. The cumulative expression score for each gene is the total points. Therefore, the risk score projected downward from total points can be calculated to assess the risk probability of IgAN of an individual.

Lasso regression analysis was utilized to screen the appropriate AT-DEGs for constructing the diagnostic prediction model. (A) Lasso regression gene coefficient plot. (B) Lasso regression cross-validation plot. The first dashed line is the optimal λ, corresponding to six hub AT-DEGs. (C) On the basis of core AT-DEGs, a nomogram was developed for predicting the diagnosis of IgAN.
Evaluation and validation of AT-DEGs related diagnostic model
The AUC curve was used to verify the accuracy of the AT-DEGs related diagnostic model. The area under the AUC curve of the riskscore of the training dataset was 0.994, indicating that the model had a high accuracy rate (Fig. 6A). Then the ROC curve of the validation dataset was made, and the AUC area of riskscore was 0.856, which confirmed that the diagnostic model of AT-DEGs had a high degree of prediction accuracy (Fig. 6B). The C-index was utilized to test the nomogram. The results are illustrated in Fig. 6C,D, and the C-indices of the training dataset and the validation dataset are 0.994 and 0.856, respectively, indicating that the predictive ability of the model deserves affirmation. Based on the area under the AUC curve of these six hub AT-DEGs in the training and validation datasets, it is suggested COKN1A and FOS genes have the highest diagnostic accuracy, followed by SIRT1 and BAG3.

Evaluation of the accuracy of the AT-DEGs diagnostic prediction model. (A) ROC curve of IgAN diagnostic model based on the training dataset. (B) ROC curve of IgAN diagnostic model based on validation dataset. (C) Calibration curve of the nomogram of the training dataset. (D) Calibration curve of validation dataset nomogram prediction.
The six hub AT-DEGs were selected to draw heatmaps in the training dataset and validation dataset to determine their expression levels. As shown in Fig. 7, the expression of SIRT1, BAG3, COKN1A and FOS was upregulated in IgAN group compared with normal control group.

Heatmap of the expression of hub AT-DEGs in the training and validation datasets. (A) The training dataset. (B) The validation dataset. The red grid represents the upregulation of genes and the blue grid represents downregulated genes.
Analysis of immune infiltration in biological samples from IgAN and healthy individuals
CIBERSORT analysis was used to evaluate and analyze the infiltration of immune cells in IgAN and HC biological samples. Results are presented as histograms with different colors representing the percentage of different types of immune cells in each sample. As shown in Fig. 8A,B cells memory, macrophages m2, NK cells activated, T cells CD4+ memory resting, and monocytes are the predominant immune cells.

Immune infiltration analysis of IgAN biomarkers. (A) Histogram of the distribution ratio of 22 immune cells in IgAN and healthy individuals. (B) Boxplot of the distribution of 22 immune cells in IgAN and healthy individuals. (C) Differences in the distribution of hub genes in different immune cells.
As shown in Fig. 8B, the correlation between IgAN and 22 immune cells was analyzed. Compared with HC group, the expression of plasma cells, macrophages M1, neutrophils in IgAN group was the most different, which was significantly up-regulated with P < 0.001. Figure 8C reveals the correlation of AT-DEGs with 20 differential immune cells. SIRT1 expression was negatively correlated with mast cells resting (P < 0.05); FOS expression was positively correlated with neutrophils and T cells CD4 memory activated (P < 0.01 and P < 0.05), the expression of T cells CD8+ and Tregs was negatively correlated (P < 0.01 and P < 0.05); CDKN1A showed positive correlation in T cells gamma delta (P < 0.05); BAG3 was positively expressed in neutrophils (P < 0.01).
Validation of the expression of AT-DEGs by qRT-PCR
We performed qRT-PCR analyses on the PBMC samples from the enrolled IgAN patients and healthy controls to assess the four previously identified AT-DEGs. The results demonstrated that, in comparison to healthy controls, SIRT1 expression was significantly reduced in the PBMCs of IgAN patients, with a statistically significant difference (P < 0.01). Conversely, BAG3, CDKN1A, and FOS expressions were markedly elevated in these patients, also showing statistical significance (P < 0.01), as illustrated in Fig. 9.

qRT-PCR was used to determine the relative mRNA level of five AT-DEGs. NC invisible control group, IgAN IgA nephropathy patient group; ∗∗∗*P < 0.01.
Discussion
IgAN is an immune-related primary glomerular disease mainly characterized by IgA deposition in the mesangial region of the kidney. The specific etiology and pathological mechanism of this disease have not been elucidated yet. Apart from kidney biopsy, there is a lack of effective diagnostic measures. Continuous studies have shown that autophagy plays a pivotal role in the pathogenesis of IgAN, and its dysfunction may contribute to renal cell injury, immune complex deposition, exacerbated oxidative stress, and inflammatory responses22,23. Studies have suggested that treatment with rapamycin and dihydroartemisinin can inhibit the mTOR/S6k1 pathway, thus enhancing autophagy and reducing the proliferation of glomerular mesangial cells24, while triptolide can enhance autophagy through CARD9/p38 MAPK pathway to inhibit the proliferation of IgAN mesangial cells and alleviate renal pathological injury25.
In conclusion, autophagy is essential to maintain renal cell function and homeostasis, and its activation may represent a potential treatment for renal diseases26. However, there are few studies on the relationship between IgAN and autophagy, and even fewer reports on diagnostic markers of autophagy and IgAN. In recent years, many scholars have found that autophagy is related to human immune function and the occurrence and development of a variety of immune diseases8,27. Recent studies have highlighted the role of autophagy in immune regulation, including the homeostasis, survival, and differentiation of T cells and B cells under stress conditions such as nutrient deficiency, hypoxia, and endoplasmic reticulum stress28,29,30. This study systematically investigated the roles of AT-DEGs and immune cell infiltration in IgAN by integrating bioinformatics analysis and experimental validation, providing novel insights into disease diagnosis and mechanistic research.
We intersected 694 DEGs obtained from the GEO database with 222 ATGs published by HADb and then identified 12 AT-DEGs. Heatmap and volcano map of DEGs between the HC group and IgAN group were generated to visualize the results. GO, KEGG, and DisGeNET enrichment analyses were performed on these 12 AT-DEGs. GO analysis revealed that these genes were enriched in functions such as apoptotic signaling pathway, cellular response to external stimulus, transcription repressor complex, core promoter sequence-specific DNA binding and molecular function activator activity. KEGG pathways highlighted their involvement in biological metabolism, such as lipid and atherosclerosis, apoptosis, cellular senescence and cell cycle and immune and inflammation-related diseases, such as colorectal cancer, chronic myeloid leukemia, influenza A. DisGeNET analysis showed that AT-DEGs were also associated with infection, HIV co-infection, leukemia, T cells and other immune function-related diseases. LASSO regression identified six hub genes: FOS, CDKN1A, SIRT1, BAG3, EIF2AK3, and SERPINA1. Further validation was performed using a validation dataset. Notably, the AUC values for SIRT1, BAG3, CDKN1A, and FOS in the ROC curve analysis exceeded 0.5, indicating their diagnostic potential. Meanwhile, the expression of SIRT1, BAG3, COKN1A and FOS was increased in IgAN in both validation and training datasets compared to HC.
As a type of nuclear protein transcription factors, FOS is essential in regulating cell growth, division, proliferation, differentiation and apoptosis31. C-FOS can induce apoptosis in germinal center B cells32 and is involved in DNA destruction, telomere damage, and neutrophil activity, all of which are closely associated with the pathogenesis of IgAN33. One study has found that FOS protein is associated with the disappearance of foot processes in podocytes34. Gao et al. reported that the Syk/Ras/c-Fos pathway was upregulated in adriamycin-treated rats, both in vitro and in vivo, leading to suppressed mesangial cell viability35. Silent information regulator (SIRT)1, a nuclear protein widely expressed in mammalian organs, regulates energy metabolism and exerts antioxidant, anti-inflammatory, and anti-apoptotic effects, thereby protecting cells and maintaining organ function36,37,38. For example, SIRT1 inhibits IL-1b expression and ROS production, promoting autophagy39,40. Wu et al. found that compound K treatment benefits IgAN by inhibiting NF-B/NLRP3 inflammasomes and enhancing autophagy and SIRT112. Bcl2-related immortalized gene 3 (BAG3), a stress-induced protein with BAG, WW, and PXXP domains, regulates apoptosis, autophagy, and protein homeostasis41. The expression of BAG3 mRNA and protein is significantly down-regulated in renal clear cell carcinoma tissues, which is related to poor clinicopathological factors and prognosis42. Knockdown of BAG3 can inhibit FGF-2-mediated epithelial-mesenchymal transition (EMT), migration and invasion of HK2 cells43. These studies reveal a potential role of BAG3 in tubular epithelial cell EMT and renal fibrosis. Tumor suppressor Cyclin-dependent kinase inhibitor 1A (CDKN1A) is a gene closely related to tumor, which encodes an effective cyclin-dependent kinase inhibitor44. In chromophobe renal cell carcinoma, loss of CDKN1A expression predicts poor prognosis45, while miR-93-5p-mediated CDKN1A inhibition prevents renal interstitial fibrosis in DN46. By downregulating CDKN1A, lncRNA SNHG7 promotes proliferation and inhibits apoptosis in renal cell cancer cells47.
In addition, we investigated the correlation between IgAN and immune genes in the HC group. The results showed that the expression levels of plasma cells, Tregs, NK cells activated, monocytes, macrophages M1, dendritic cells resting were all significantly higher than those in HC group. Studies have indicated that the production of galactose-deficient IgA1 (Gd-IgA1) and subsequent anti-Gd-IgA1 antibodies are important factors leading to IgAN48,49. Plasma cell is considered to be the main producer of antibodies, and an increasing number of studies have suggested that Gd-IgA1 may be derived from cited plasma cells in the mucosa, which is the basis for IgAN initiation and progression50,51,52. Regulatory T cells (Tregs) are a subset of T cells with immunosuppressive function, modulating immune responses to both self-antigens and foreign antigens53. Previous studies have shown that Treg dysfunction contributes to IgAN pathogenesis54, and CD4 + CD25 + Tregs can ameliorate kidney injury in IgAN rats54. Macrophages are also involved in the occurrence and development of IgAN55, and their deposition in the kidney may indicate disease progression and prognosis56. M1 macrophage is able to promote an inflammatory response and eliminate bacteria or tumor cells by secreting pro-inflammatory factors such as TNF and IL-1, IL-6 and reactive ROS. Inflammation, however, can eventually lead to tissue fibrosis if it persists57. Some studies indicated that compared with normal kidney, the expression of M0, M1 and M2 macrophages in kidneys of IgAN paients was significantly increased, indicating that there was obvious macrophage infiltration in the renal tissue of IgAN patients58.
Furthermore, the qRT-PCR results indicated that, in comparison to the NC group, the mRNA expression level of SIRT1 in PBMCs from IgAN patients was significantly reduced, whereas the mRNA expression levels of BAG3, CDKN1A, and FOS were markedly elevated. These findings corroborate the aforementioned statements and suggest potential new serum biomarkers for diagnosing IgAN.
There are inevitably some limitations to this study. First, only mRNA expression levels of the core genes were validated by qRT-PCR, without protein-level validation (e.g., Western blot or immunohistochemistry). Changes in mRNA levels do not fully reflect alterations in protein function, necessitating further experimental validation. Second, the data primarily originated from the GEO database, and differences in sample size, ethnic diversity, and technical platforms across other public datasets may affect the generalizability of the results. Third, larger-scale animal and clinical studies are needed to validate and enhance the clinical utility of our proposed model.
Conclusion
This study, for the first time, systematically analyzed the relationship between AT-DEGs and immune cell infiltration by integrating multiple GEO datasets, exploring the role of immune-related autophagy biomarkers in IgAN, thereby addressing a critical gap in this field. Through LASSO regression and cross-validation, four hub genes (SIRT1, BAG3, CDKN1A, and FOS) were identified as potential biomarkers, offering new possibilities for the non-invasive diagnosis of IgAN. The reliability of the bioinformatics analysis was preliminarily confirmed using ROC curve analysis and qRT-PCR, laying the foundation for further research and generally providing a reference for early diagnosis and targeted drug research of IgAN.
Data availability
The datasets used and analysed during the current study available from the corresponding author on reasonable request. Furthermore, the datasets (GSE93798, GSE35487, GSE58539, GSE116626 and GSE115857) generated and analysed during the current study are available in the GEO dataset repository. https://www.ncbi.nlm.nih.gov/geo.
Abbreviations
- GEO:
-
Gene Expression Omnibus
- DEGs:
-
Differentially expressed genes
- HC:
-
Healthy control
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genome
- GSEA:
-
GeneSet Enrichment Analysis
- LASSO:
-
Least absolute shrinkage and selection operator
- ROC:
-
Receiver operating characteristic
- DEG:
-
Differentially expressed gene
- ATG:
-
Autophagy gene
- AT-DEG:
-
Autophagy-related DEG
- qRT-PCR:
-
Real-time quantitative polymerase chain reaction
- IgAN:
-
IgA nephropathy
- ESRD:
-
End-stage renal disease
- CKD:
-
Chronic kidney disease
- KDIGO:
-
Kidney disease: Improving Global Outcomes
- DN:
-
Diabetic nephropathy
- HADb:
-
Human autophagy database
- AUC:
-
Area under the receiver operating characteristic curve
- C-index:
-
Harrell concordance indexes
- CC:
-
Cellular component
- BP:
-
Biological process
- MF:
-
Molecular function
- mTOR:
-
Mechanistic target of rapamycin
- S6k1:
-
Ribosomal protein s6 kinase beta-1
- CARD9:
-
Caspase recruitment ___domain family, member 9
- SIRT1:
-
Silent information regulator 1
- BAG3:
-
Bcl2-related immortalized gene 3
- CDKN1A:
-
Cyclin-dependent kinase inhibitor 1 A
- Syk:
-
Spleen tyrosine kinase
- Ras:
-
Renin-angiotensin system
- IL-:
-
Interleukin-
- Bcl-2:
-
B-cell lymphoma-2
- JNK:
-
Jun N-terminal kinase
- NF-κB:
-
Nuclear factor kappa-B
- NLRP3:
-
NOD-like receptor thermal protein ___domain associated protein 3
- TGF:
-
Transforming growth factor
- EMT:
-
Epithelial-mesenchymal transition
- FGF-2:
-
Basic fibroblast growth factor
- Gd-IgA1:
-
Galactose-deficient IgA1
- Tregs:
-
Regulatory T cells
- NK:
-
Natural killer cell
- TNF:
-
Tumor necrosis factor
- ARDS:
-
Acute respiratory distress syndrome
- MAPK:
-
Mitogen-activated protein kinase
References
Ohyama, Y. et al. Racial heterogeneity of IgA1 hinge-region O-glycoforms in patients with IgA nephropathy. iScience 25, 105223. https://doi.org/10.1016/j.isci.2022.105223 (2022).
Tang, C. et al. Time-varying proteinuria and progression of IgA nephropathy: A cohort study. Am. J. Kidney Dis. 84, 170-178.e1. https://doi.org/10.1053/j.ajkd.2023.12.016 (2024).
Floege, J., Rauen, T. & Tang, S. C. W. Current treatment of IgA nephropathy. Semin. Immunopathol. 43, 717–728. https://doi.org/10.1007/s00281-021-00888-3 (2021).
Rovin, B. H. et al. Executive summary of the KDIGO 2021 guideline for the management of glomerular diseases. Kidney Int. 100, 753–779. https://doi.org/10.1016/j.kint.2021.05.015 (2021).
Pattrapornpisut, P., Avila-Casado, C. & Reich, H. N. IgA nephropathy: Core curriculum 2021. Am. J. Kidney Dis. 78, 429–441. https://doi.org/10.1053/j.ajkd.2021.01.024 (2021).
Moresco, R. N., Speeckaert, M. M. & Delanghe, J. R. Diagnosis and monitoring of IgA nephropathy: The role of biomarkers as an alternative to renal biopsy. Autoimmun. Rev. 14, 847–853. https://doi.org/10.1016/j.autrev.2015.05.009 (2015).
Duan, Z. Y. et al. Urinary miR-185-5p is a biomarker of renal tubulointerstitial fibrosis in IgA nephropathy. Front. Immunol. 15, 1326026. https://doi.org/10.3389/fimmu.2024.1326026 (2024).
Levine, B. & Kroemer, G. Biological functions of autophagy genes: A disease perspective. Cell 176, 11–42. https://doi.org/10.1016/j.cell.2018.09.048 (2019).
Choi, M. E. Autophagy in kidney disease. Annu. Rev. Physiol. 82, 297–322. https://doi.org/10.1146/annurev-physiol-021119-034658 (2020).
Tang, C., Livingston, M. J., Liu, Z. & Dong, Z. Autophagy in kidney homeostasis and disease. Nat. Rev. Nephrol. 16, 489–508. https://doi.org/10.1038/s41581-020-0309-2 (2020).
Du, W. et al. Increased proportion of follicular helper T cells is associated with B cell activation and disease severity in IgA nephropathy. Front. Immunol. 13, 901465. https://doi.org/10.3389/fimmu.2022.901465 (2022).
Wu, C. Y. et al. IgA nephropathy benefits from compound K treatment by inhibiting NF-κB/NLRP3 inflammasome and enhancing autophagy and SIRT1. J. Immunol. 205, 202–212. https://doi.org/10.4049/jimmunol.1900284 (2020).
Zhang, H., Puleston, D. J. & Simon, A. K. Autophagy and Immune Senescence. Trends Mol. Med. 22, 671–686. https://doi.org/10.1016/j.molmed.2016.06.001 (2016).
Deretic, V. Autophagy in inflammation, infection, and immunometabolism. Immunity 54, 437–453. https://doi.org/10.1016/j.immuni.2021.01.018 (2021).
Liu, P. et al. Transcriptomic and proteomic profiling provides insight into mesangial cell function in IgA nephropathy. J. Am. Soc. Nephrol. 28, 2961–2972. https://doi.org/10.1681/asn.2016101103 (2017).
Reich, H. N. et al. A molecular signature of proteinuria in glomerulonephritis. PLoS ONE 5, e13451. https://doi.org/10.1371/journal.pone.0013451 (2010).
Cox, S. N. et al. Altered monocyte expression and expansion of non-classical monocyte subset in IgA nephropathy patients. Nephrol. Dial. Transplant 30, 1122–1232. https://doi.org/10.1093/ndt/gfv017 (2015).
Cox, S. N. et al. Formalin-fixed paraffin-embedded renal biopsy tissues: An underexploited biospecimen resource for gene expression profiling in IgA nephropathy. Sci. Rep. 10, 15164. https://doi.org/10.1038/s41598-020-72026-2 (2020).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592. https://doi.org/10.1093/nar/gkac963 (2023).
Bhatia, D. & Choi, M. E. Autophagy in kidney disease: Advances and therapeutic potential. Prog. Mol. Biol. Transl. Sci. 172, 107–133. https://doi.org/10.1016/bs.pmbts.2020.01.008 (2020).
Ma, S. et al. Effects and mechanisms of Chinese herbal medicine on IgA nephropathy. Phytomedicine 117, 154913. https://doi.org/10.1016/j.phymed.2023.154913 (2023).
Liu, D. et al. Rapamycin enhances repressed autophagy and attenuates aggressive progression in a rat model of IgA nephropathy. Am. J. Nephrol. 45, 293–300. https://doi.org/10.1159/000456039 (2017).
Zhao, L. et al. Triptolide promotes autophagy to inhibit mesangial cell proliferation in IgA nephropathy via the CARD9/p38 MAPK pathway. Cell Prolif. 55, e13278. https://doi.org/10.1111/cpr.13278 (2022).
Kimura, T., Isaka, Y. & Yoshimori, T. Autophagy and kidney inflammation. Autophagy 13, 997–1003. https://doi.org/10.1080/15548627.2017.1309485 (2017).
Wu, D. J. & Adamopoulos, I. E. Autophagy and autoimmunity. Clin. Immunol. 176, 55–62. https://doi.org/10.1016/j.clim.2017.01.007 (2017).
Xiao, J. et al. TM9SF1 expression correlates with autoimmune disease activity and regulates antibody production through mTOR-dependent autophagy. BMC Med. 22, 502. https://doi.org/10.1186/s12916-024-03729-w (2024).
Al-Kuraishy, H. M. et al. Defective autophagy and autophagy activators in myasthenia gravis: A rare entity and unusual scenario. Autophagy 20, 1473–1482. https://doi.org/10.1080/15548627.2024.2315893 (2024).
White, E., Lattime, E. C. & Guo, J. Y. Autophagy regulates stress responses, metabolism, and anticancer immunity. Trends Cancer 7, 778–789. https://doi.org/10.1016/j.trecan.2021.05.003 (2021).
Chang, D., Li, L., Xu, Z. & Chen, X. Targeting FOS attenuates malignant phenotypes of breast cancer: Evidence from in silico and in vitro studies. J. Biochem. Mol. Toxicol. 37, e23358. https://doi.org/10.1002/jbt.23358 (2023).
Li, Q. et al. Dihydroartemisinin imposes positive and negative regulation on Treg and plasma cells via direct interaction and activation of c-Fos. Commun. Biol. 6, 52. https://doi.org/10.1038/s42003-023-04454-5 (2023).
Zhou, X., Wang, N., Zhang, Y. & Yu, P. Expression of CCL2, FOS, and JUN may help to distinguish patients with IgA nephropathy from healthy controls. Front. Physiol. 13, 840890. https://doi.org/10.3389/fphys.2022.840890 (2022).
Park, H. J., Kim, J. W., Cho, B. S. & Chung, J. H. Association of FOS-like antigen 1 promoter polymorphism with podocyte foot process effacement in immunoglobulin A nephropathy patients. J. Clin. Lab. Anal. 28, 391–397. https://doi.org/10.1002/jcla.21699 (2014).
Gao, J. et al. In vitro and in vivo study of the expression of the Syk/Ras/c-Fos pathway in chronic glomerulonephritis. Mol. Med. Rep. 18, 3683–3690. https://doi.org/10.3892/mmr.2018.9355 (2018).
Chao, Y. M. et al. Anomalous AMPK-regulated angiotensin AT(1)R expression and SIRT1-mediated mitochondrial biogenesis at RVLM in hypertension programming of offspring to maternal high fructose exposure. J. Biomed. Sci. 27, 68. https://doi.org/10.1186/s12929-020-00660-z (2020).
Mirshafa, A. et al. Tropisetron protects against brain aging via attenuating oxidative stress, apoptosis and inflammation: The role of SIRT1 signaling. Life Sci. 248, 117452. https://doi.org/10.1016/j.lfs.2020.117452 (2020).
Wang, K. et al. Resveratrol attenuates aortic dissection by increasing endothelial barrier function through the SIRT1 pathway. J. Cardiovasc. Pharmacol. 76, 86–93. https://doi.org/10.1097/fjc.0000000000000837 (2020).
Islam, A., Chang, Y. C., Tsao, N. W., Wang, S. Y. & Chueh, P. J. Calocedrus formosana essential oils induce ROS-mediated autophagy and apoptosis by targeting SIRT1 in colon cancer cells. Antioxidants (Basel) 13, 284. https://doi.org/10.3390/antiox13030284 (2024).
Yan, D. et al. SIRT1/FOXO3-mediated autophagy signaling involved in manganese-induced neuroinflammation in microglia. Ecotoxicol. Environ. Saf. 256, 114872. https://doi.org/10.1016/j.ecoenv.2023.114872 (2023).
Kirk, J. A., Cheung, J. Y. & Feldman, A. M. Therapeutic targeting of BAG3: Considering its complexity in cancer and heart disease. J. Clin. Invest. https://doi.org/10.1172/jci149415 (2021).
Gong, B. et al. BAG3 as a novel prognostic biomarker in kidney renal clear cell carcinoma correlating with immune infiltrates. Eur. J. Med. Res. 29, 93. https://doi.org/10.1186/s40001-024-01687-w (2024).
Du, F. et al. Implication of Bcl-2-associated athanogene 3 in fibroblast growth factor-2-mediated epithelial-mesenchymal transition in renal epithelial cells. Exp. Biol. Med. (Maywood) 240, 566–575. https://doi.org/10.1177/1535370214558023 (2015).
Alhalabi, O. & Msaouel, P. Truncating CDKN1A mutations: An insight into the biology of urinary tract carcinomas?. Am. J. Cancer Res. 11, 6214–6217 (2021).
Ohashi, R. et al. Loss of CDKN1A mRNA and protein expression are independent predictors of poor outcome in chromophobe renal cell carcinoma patients. Cancers (Basel) https://doi.org/10.3390/cancers12020465 (2020).
Yang, J. et al. Silencing of long noncoding RNA XIST protects against renal interstitial fibrosis in diabetic nephropathy via microRNA-93-5p-mediated inhibition of CDKN1A. Am. J. Physiol. Renal Physiol. 317, F1350-f1358. https://doi.org/10.1152/ajprenal.00254.2019 (2019).
Dong, J. S., Wu, B. & Jiang, B. LncRNA SNHG7 promotes the proliferation and inhibits apoptosis of renal cell cancer cells by downregulating CDKN1A. Eur. Rev. Med. Pharmacol. Sci. 24, 7556. https://doi.org/10.26355/eurrev_202007_22205 (2020).
Knoppova, B. et al. The origin and activities of IgA1-containing immune complexes in IgA nephropathy. Front. Immunol. 7, 117. https://doi.org/10.3389/fimmu.2016.00117 (2016).
Rodrigues, J. C., Haas, M. & Reich, H. N. IgA nephropathy. Clin. J. Am. Soc. Nephrol. 12, 677–686. https://doi.org/10.2215/cjn.07420716 (2017).
He, J. W., Zhou, X. J., Lv, J. C. & Zhang, H. Perspectives on how mucosal immune responses, infections and gut microbiome shape IgA nephropathy and future therapies. Theranostics 10, 11462–11478. https://doi.org/10.7150/thno.49778 (2020).
Maixnerova, D., El Mehdi, D., Rizk, D. V., Zhang, H. & Tesar, V. New treatment strategies for IgA nephropathy: Targeting plasma cells as the main source of pathogenic antibodies. J. Clin. Med. https://doi.org/10.3390/jcm11102810 (2022).
Tang, Y., He, H., Hu, P. & Xu, X. T lymphocytes in IgA nephropathy. Exp. Ther. Med. 20, 186–194. https://doi.org/10.3892/etm.2020.8673 (2020).
Zhang, A. et al. Regulatory T cells in immune checkpoint blockade antitumor therapy. Mol. Cancer 23, 251. https://doi.org/10.1186/s12943-024-02156-y (2024).
Shen, B. L. et al. Study on the effects of regulatory T cells on renal function of IgAN rat model. Eur. Rev. Med. Pharmacol. Sci. 19, 284–288 (2015).
Xie, D. et al. Intensity of macrophage infiltration in glomeruli predicts response to immunosuppressive therapy in patients with IgA nephropathy. J. Am. Soc. Nephrol. 32, 3187–3196. https://doi.org/10.1681/asn.2021060815 (2021).
Wen, Y., Yan, H. R., Wang, B. & Liu, B. C. Macrophage heterogeneity in kidney injury and fibrosis. Front. Immunol. 12, 681748. https://doi.org/10.3389/fimmu.2021.681748 (2021).
Zhou, D. et al. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal 26, 192–197. https://doi.org/10.1016/j.cellsig.2013.11.004 (2014).
Yang, M., Liu, J. W., Zhang, Y. T. & Wu, G. The role of renal macrophage, AIM, and TGF-β1 expression in renal fibrosis progression in IgAN patients. Front. Immunol. 12, 646650. https://doi.org/10.3389/fimmu.2021.646650 (2021).
Acknowledgements
We would like to thank the authors who generously shared their data and all study participants and the anonymous reviewers for their useful comments on the manuscript.
Funding
This work was supported by Scientific and Technological Innovation Project China Academy of Chinese Medical Science (grant numbers CI2021B001); and the Fundamental Research Funds for the Central Public Welfare Research Institutes (grant numbers YZX-202409); and the Fundamental Research Funds for the Central Public Welfare Research Institutes (grant numbers YZX-202345).
Author information
Authors and Affiliations
Contributions
Conceptualization: Sijia Ma, Shijie Xu, Yu Zhang. Data curation: Sijia Ma, Xiaoxin Tian. Formal analysis: Hangyu Duan, Xiaoxin Tian, Chao Wang. Methodology: Sijia Ma, Jing Liu, Yu Zhang. Validation: Chao Wang, Shijie Xu. Writing – original draft: Sijia Ma, Chao Wang, Jing Liu. Writing – review & editing: Shijie Xu, Yu Zhang.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
These studies involving human were approved by the Ethics Committee of Xiyuan Hospital, China Academy of Chinese Medical Sciences (2022XLA088-1). All methods were carried out in accordance with relevant guidelines and regulations. Participants provided written informed consent to participate in this study.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, S., Wang, C., Liu, J. et al. To establish and validate autophagy related biomarkers for the diagnosis of IgA nephropathy. Sci Rep 15, 13944 (2025). https://doi.org/10.1038/s41598-025-98591-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-98591-y
