
Abstract
Mungbean (Vigna radiata L.) is one of the most important yet genomically under-researched leguminous food crop. Its productivity is low due to the complex nature of yield realization, which is regulated by various yield-contributing traits. Thus, understanding the genetic basis of these traits is essential for developing an ideal genotype with high yield. In this study, mapping of quantitative trait loci (QTLs) was conducted for six yield-contributing traits using a recombinant inbred line (RIL) population (166 number), developed by crossing two contrasting genotypes (Pusa Baisakhi × PMR-1). The genotyping-by-sequencing (GBS) of RILs was used to construct the genetic map using 1347 single nucleotide polymorphism (SNPs). The QTL mapping using multiple interval mapping (MIM) and composite interval mapping (CIM) has identified 17 yield contributing QTLs of which, 4 for number of leaves/plant (NL), 3 for plant height (PH), 2 for SPAD value, 3 for 100 seed weight (SW), 2 for number of pods/plant (NP), and 3 for total grain yield (GY). The Logarithm of Odds (LOD) scores for these QTLs ranged from ~ 3–9, while phenotypic variance explained (PVE) ranged from ~ 9–24%. Several candidate genes with mRNA expression and protein-altering mutations were identified as having a direct role in key processes like growth (LOC106756212, LOC106776425, LOC106777991), flowering (LOC106777903, LOC106768860), metabolism (LOC106757749, LOC106758189), etc. The candidate genes are validated through digital gene expression analysis. In addition, Insertion-Deletion (InDel) markers were also developed for the identified QTLs which hold broad applications for the improvement of yield-related traits in mungbean.
Similar content being viewed by others

Introduction
Mungbean (Vigna radiata (L.) R. Wilczek) or green gram holds immense promise for global food security and sustainable agriculture1. Mungbean (2n = 22) is a self-pollinated crop having a short life cycle (60–70 days to maturity), and a small genome size (579 Mb)2. Mungbean grains contain nearly 20–30% protein and 60–70% carbohydrates along with numerous vitamins, and minerals3. When consumed with cereals, they provide an ideal balance of amino acids required by the human body, especially in Southeast Asian countries3. Mungbean is highly preferred for its protein because it is easily digestible and has no antinutritional components. It has detoxifying, hypolipidemic, hypoglycaemic, anti-cancerous, antihypertensive, and immunomodulation properties3,4. Despite its significant economic importance, research on mungbean is mostly underfunded Internationally, especially in the field of genomics5.
Globally, mungbean is cultivated in ~ 7.3 m ha and is predominantly grown in South, East, and Southeast Asia with an average productivity of 721 kg/ha. India and Myanmar occupy the top positions with a combined contribution of 60% followed by China (16%), and Indonesia (5%) to world production6. India alone accounts for more than 70% of world acreage (~ 5.6 m ha) and produces a meager ~ 3.68 m tones7. The low mungbean productivity in India is mainly due to the use of poor-quality seeds, non-availability of suitable ideotypes for different cropping systems, low harvest index, poor crop management practices, and the susceptibility of crops to various biotic and abiotic stresses8,9.
Grain yield is the primary focus of any breeding program, but its complex nature with high environmental interaction can only be dissected with effective phenotyping and genotyping. Yield of mungbean crop is determined by several parameters like number of pods per plant, number of seeds per pod, plant height, number of leaves, chlorophyll content, maturity duration, grain weight, resistance and tolerance to various biotic and abiotic stresses10. Comprehending the genetics and genomics of yield-related agronomic traits is essential for incorporating these traits into elite crop varieties.
Mapping traits using bi-parental population is a popular approach due to its power and ability to identify the linked QTLs (Quantitative Trait Loci) when allelic frequencies are low compared to Genome-Wide Association Studies (GWAS). The success of QTL mapping depends on factors such as population size, marker type, and marker density11,12. In mungbean, limited reports are available that associate markers with complex yield-contributing traits13,14,15. In addition, these studies have mainly used low-throughput and poorly dispersed markers like SSRs, which do not provide accurate information about the number and ___location of QTLs. The use of RILs is advantageous due to high recombination and were used to map various traits in mungbean10,16.
High-density markers like single nucleotide polymorphisms (SNPs) are the most abundant form of DNA variation which can be identified by genotyping-by-sequencing (GBS)17. The use of high-density linkage maps can enhance the precision of effect estimates for detected QTLs, provide accurate QTL positions, and facilitate the identification of candidate genes underlying complex traits. Recently GWAS has been gaining importance, but in comparison to QTL mapping, it may lack precision due to multiple testing, prone to false positives, and can only identify common variants that influence a trait, despite the advantage of mapping with high resolution18.
Traditional breeding methods have played a central role in advancing mungbean breeding; however, their efficiency can be enhanced by integrating molecular approaches. The identification of QTLs associated with various traits, along with their candidate genes can unravel the genomic basis of specific traits. The development of user-friendly markers within the QTLs regions holds great promise for expediting mungbean breeding efforts. This study aims to identify the QTLs governing various yield-contributing traits and to develop the InDel markers closely linked to the QTLs for their use in marker-assisted selection (MAS).
Results
Variation, heritability, and correlation analysis
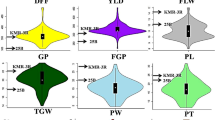
The phenotypic data for the parental genotypes and RILs displayed a remarkable range of diversity across all traits (Table S1; Table S2). The number of leaves per plant ranged from 8.3 to 37.3 (mean = 20.3); plant height varied from 49 to 100.6 cm (mean = 67.59 cm); 100 seed weight spanned from 2036 to 4632 mg (mean = 3178.05 mg); number of pods per plant ranged between 2.8 and 80.3 (mean = 30.29) and grain yield varied from 3.8 to 43.6 g (mean = 15.77 g). The skewness (range: -0.9 to 1.9) and kurtosis (range: -0.6 to 1.2) indicated a nearly symmetrical distribution around the mean (Fig. 1).

Distribution of yield-related traits in the RIL population. (a) Number of leaves; (b) Plant height in cm; (c) SPAD value measured after 25 days of sowing; (d) 100 seed weight in mg; (e) Number of pods; (f) Total yield of three plants in g. Where blue line indicates mean.
Both genotypes and seasons showed a highly significant effect on the traits as revealed by ANOVA (Table S3). It means, the genetic composition of RILs and environmental conditions significantly influenced the trait expression. Additionally, significant interaction was noted between genotype and season for NL, PH, and SPAD values. Although, replications were significant for NL and GY, yet their contribution to the total variation was minimal. The broad-sense heritability estimates ranged from 64% (chlorophyll content) to 98% (seed weight). A significant correlation was recorded between total yield and NL (0.45**), NP (0.86**), and SPAD value (25 DAS 0.26** & 45 DAS 0.21*). Significant correlation was also recorded between SPAD values and NL (0.17*), NP (0.24**), and SW (0.16*) (Figure S1).
Linkage map
The linkage map was generated using 1374 filtered SNPs with Kosambi function. SNP markers were found evenly distributed across 11 chromosomes, with an average density of one SNP per 250 kb. Chromosome 7 exhibited the highest number of markers (278), while chromosome 4 had the lowest (69). Chromosome 1 exhibited lowest marker density (2.9 SNPs/Mb) and chromosome 3 displayed the highest density (8.7 SNPs/Mb) (Table 1, Figure S2-S4).
QTL mapping for grain yield-related traits
QTL mapping was conducted for six yield-related traits namely, plant height (PH in cm), number of leaves per plant (NL), number of pods per plant (NP), chlorophyll content (as SPAD value at 25 and 45 days after sowing), seed weight per 100 seeds (SW in mg), and total grain yield (GY in g) using Multiple Interval Mapping (MIM) and Composite Interval Mapping (CIM) approaches. BLUPs were used for the partitioning of phenotypic variance into its genetic and environmental components. Based on the LOD permutations, a threshold of 3.0 was set for identifying a QTL. The MIM detected 5 QTLs across all the traits, while CIM could identify 18 QTLs, including 5 QTLs which were detected by MIM (Table 2; Figs. 2 and 3; Figure S5-S16). These QTLs were located across 9 chromosomes (except on chr 1 and 8; Fig. 4) having LOD scores ranging from 3.05 to 9, and PVE ranged from 9 to 24%. Chromosome 7 had the highest number of QTLs (6 No), followed by chromosome 11 (3 No), chromosomes 2 and 4 (2 each), and chromosomes 3, 5, 6, 9, and 10 (1 each) (Table 2). The additive effect (‘a’, Table 2) represents the contribution of a QTL allele to the trait variation. A higher absolute value indicates a stronger genetic influence, while a positive effect indicates that the allele from Pusa Baisakhi enhances the trait value, and a negative direction suggests the allele from PMR-1, contributes to the increase. Yield-related QTLs were inherited from both parents but the QTLs associated with higher seed weight, pod number, and yield predominantly carried favourable alleles from Pusa Baisakhi, while those linked to taller plant height, increased chlorophyll content, and greater number of leaves were inherited from PMR-1, aligning with their respective agronomic characteristics.

Composite interval mapping of Yield-related traits in Mungbean. (a) Number of leaves; (b) Plant height; (c) SPAD value; (d) Seed weight; (e) Number of pods; (f) Total grain yield.

Effect Plot of important QTLs identified for Yield-related traits in mungbean. Where AA represents PMR-1 and BB represents Pusa Basakhi genotypes.

Distribution of QTLs for yield-related traits on chromosomes of mungbean identified using composite interval mapping (CIM).
Gene ontology and candidate gene analysis
Genes located within the QTL intervals were subjected to gene ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. To identify the candidate genes, polymorphic variants detected by WGRS of the parental genotypes were annotated. WGRS produced nearly 220 million raw reads (151 bp), with an average depth of 34× coverage. Very high variants were recorded between the parents (1.2 million; density: 1 variant per 300 bp), and of these, 3% were Insertions/Deletions (InDels). Nearly 66,000 variants fell within the functional class, of which 53% were missense, 1% were nonsense, and 46% were silent mutations. Genes exhibiting non-synonymous mutations in the exons were identified which include premature start codon gain variants, conservative in-frame deletions and insertions, disruptive in-frame deletions and insertions, missense mutations, frameshifts, exon loss, premature stop codons, start loss, and start gain.
In the QTL region, 298 genes showed non-synonymous variations in the exons, and of these, 6 candidate genes each for PH and GY, 4 each for NL, SW, and NP, and 1 for SPAD value were identified (Table 3). Some of the important genes were identified as candidates like pentatricopeptide repeat-containing protein (NL), Inositol-tetrakisphosphate 1-kinase 1-like (PH), PYR-1 (PH), ferredoxin-like (SW) (Figure S17), Amino acid transporter AVT3B-like (SPAD), etc. (Figure S18).
Validation and development of indel markers
Arabidopsis is one of the most extensively studied model organisms in plant genetics, with a fully sequenced genome and a wealth of functional genomics data. Hence, well-characterized orthologs of Arabidopsis were identified and their pattern of expression was analyzed. For traits like plant height, two genes were identified through digital gene expression among the six identified candidate genes (Fig. 5). This digital gene expression can provide hypotheses for further experimental validation. For further use of these identified QTLs, InDel primers with more than 15 bp of insertions or deletions were developed (Table 4).

Digital gene expression of plant height candidate genes (a) Inositol-tetrakisphosphate 1-kinase 1-like (LOC106777318) and (b) Abscisic acid receptor PYR1 (LOC106776425).
Discussion
Despite its significant economic importance, mungbean is ignored at both national and international levels, in the field of genomics. Mungbean was one of the first legumes in which mapping for bruchid resistance using RFLP markers was reported in 199219. However, advances in mungbean genomics lag behind other legume crops like chickpea and soyabean20. Available literature shows mapping in mungbean for only a few traits like seed size, seed dormancy, bruchid, bean bug, MYMV, and powdery mildew resistance20. Detailed QTL identification using RIL population for yield contributing traits is still missing in mungbean.
Typically, the mean performance of RILs is expected to fall within the range defined by the parental means for quantitative traits. However, the mean values of RILs were recorded lower than the lower parent, despite the presence of transgressive segregants displaying higher performance than both parents. This could be because of highly contrasting nature of the parental lines having co-adapted gene complexes due to the selection21. In the process of RIL formation, it seems that the co-adapted gene complexes got disrupted upon crossing and recombination22. After a few generations of selfing, heterozygosity is lost, which might have disrupted the coadapted gene complexes in certain RILs, as also reported for Avena barbeta23.
Grain yield was found positively associated with the number of pods, number of leaves, and SPAD values; whereas, it was found not significantly affected by the seed weight and plant height. Pulses typically begin flowering after reaching a specific number of nodes, suggesting that plant height and leaf count indirectly affect pod number and seed yield24,25. Leaves and their chlorophyll content play crucial roles in photosynthesis, light interception, and also as a sign for disease and pest resistance26. More number of leaves per plant with higher chlorophyll content enhances photosynthetic activity, resulting in more assimilates for pod formation and grain filling. Previous studies also reported a positive correlation between single-leaf photosynthetic rates, biomass production, and yield in pulses like cowpea, soybean27,28, and mungbean14,29.
Number of leaves per plant (NL)
The NL in mungbean plays a pivotal role in determining the yield, as it directly influences the pod yield by increasing the number of nodes and photosynthate accumulation26. The number of nodes was mapped on LG3 and LG10 using InDel markers30; while number of primary branches was mapped using DArT markers on chr.7 and 11 in a RIL population with 26% and 27.7% PVE, respectively31. Recently, 3 SNPs were found associated with primary branches on chr. 4, 9, and 11 using GWAS5. This study identifeid 4 QTLs (qNL-7-1, qNL-7-2, qNL-9-1, and qNL-11-1) for leaf number. Both MIM and CIM detected a qNL-7-1 (9.9% PVE) located in 37 Mb region on chr. 7, as also reported previously31. Within the 4 QTL regions, 5 candidate genes were identified exhibiting non-synonymous mutations and having a role in leaf growth, regulation of flowering, and cellular development (Table 3). The LOC106777903 gene, a pentatricopeptide repeat-containing protein, functions by directly interacting with the protein GIGANTEA, which regulates various physiological processes like flowering time, leaf senescence, hypocotyl elongation, chlorophyll accumulation, etc. Moreover, GIGANTEA may negatively influence leaf number by promoting flowering32.
Plant height (PH)
Plant height influences the overall architecture of the mungbean plant. The varieties with appropriate plant height can optimize light interception and resource utilization, leading to higher yield30. Two major QTLs (qPH-11-1 and qPH-7-1) and one minor QTL (qPH-2-1) controlling plant height were identified on chromosome numbers 11, 7, and 2 respectively. The qPH-11-1 accounted for 21.5% PVE, with a negative additive effect (-2.3), suggesting its inheritance from PMR-1. Within the three QTL regions, 32 genes are identified with mutations, and of these, six genes [LOC106778804 (chr.2), LOC106756212 (chr.2), LOC106777391 (chr.11), LOC106777318 (chr.11), LOC106776425 (chr.11), LOC106777991 (chr.11)] were having role in growth, flower development, energy metabolism, and environmental adaptation.
The LOC106756212 protein contains a Calmodulin binding protein-like N-terminal ___domain, having a role in protein localization to ER exit site, cellulose biosynthesis, and salicylic acid metabolic process. The Inositol-tetrakisphosphate 1-kinase 1-like (LOC106777318) gene is involved in the biosynthesis of inositol phosphates (IPs), which act as secondary messengers and plays a crucial role in auxin-related processes33. The digital gene expression analysis showed that this gene was primarily expressed during pollen maturation and seed maturation stages (Fig. 5). Similarly, the PYR1 (LOC106776425) gene regulates plant responses to abscisic acid (ABA) and it is highly expressed in the second internode of stem and also in the imbibed seeds34. ABA at its basal level promotes plant growth, however, at elevated levels, it impairs plant growth35. When PYR1 binds to ABA, it inhibits ABI1 by forming a complex thereby preventing ABI1 from dephosphorylating (and thus inhibiting) the SnRK2 kinases. This activation of the SnRK2 kinases leads to the expression of ABA-responsive genes, promoting stress responses inhibiting shoot growth and increasing root growth34. LOC106777991 is involved in the synthesis of porphyrin and chlorophyll metabolism, which are crucial in determining plant height. Similarly, various reports of QTL identification for stem length on various chromosomes are known like six QTLs on six chromosomes (1, 2, 3, 6, 9, 10)36, three QTLs on chr. 4, 5, 8 (2.9–20% PVE)37, four QTLs on chr. 3, 4, 8, 11 (4.2–16.6% PVE)31, four QTLs on four chromosomes (1, 3, 7, 10)30, and 2 QTLs on two chromosomes (1, 7)5.
SPAD (soil-plant analysis development) values
The SPAD value is an indirect measure of chlorophyll content in the leaves. It holds considerable importance in determining plant growth, physiological status, and yield12. A consistent QTL (qSPAD25-7-1) on chr. 7 at 3 Mb position (15.8% PVE) was identified for both 25 and 45 DAS having a role in chlorophyll accumulation, which is inherited from PMR-1. Another major QTL (qSPAD25-5-1) was identified regulating SPAD value on chr. 5 (12.2% PVE). Similarly, a QTL on chr. 7 (5.5 Mb position, 12% PVE) was identified12, while another QTL on chr. 1 was identified through association mapping in mungbean5. In soybean, QTLs for SPAD value were also found located on chr. 738,39. Within these two QTL regions, 22 candidate genes were identified with mutations, and LOC106765948 was found coding for the amino acid transporter AVT3B-like having a role in the transport of amino acids, mediating nitrogen distribution, which is crucial for plant chlorophyll and growth40.
Seed weight (SW)
Seed weight is a critical trait which directly affects the amount of reserves present in the seeds to provide energy for early seedling growth and establishment. Seed weight is also associated with seed quality attributes and bigger seeds are often preferred by consumers and processors41. It is one of the most extensively mapped traits where researchers have identified a number of QTLs on different chromosomes42,43,44.
This study identified 3 QTLs for SW (qSW-10-1, qSW-11-1, qSW-6-1) on chromosomes 10, 11, and 6 with a PVE of 24.4%, 17.5%, and 10.8%, respectively. Similarly, 11 QTLs (PVE: 5.2–15.1%) have been identified for SW using RFLP markers45, while 5 QTLs were identified on chr.1, 2, 10 and 11 (PVE: 12.6%) using SSR markers15. SW is also mapped using an association mapping approach5,11,14. The most significant QTL (qSW-10-1) was identified from the parent Pusa Baisakhi at 20.6 Mb on chr. 10 which had 7 genes with variations between the parents. Among these only one gene, ferredoxin-like was found to have a role in the biological processes related to seed development. It is functional in chloroplasts of photosynthetic tissues, participates in carbon assimilation, plays a role in photosynthetic electron transport of PS1, and affects other FD-dependent genes46. The expression of its ortholog in Arabidopsis thaliana showed that it was highly expressed in the torpedo stage of embryo development. As seed formation progresses, the expression of this gene increases, reaching a peak around seed stage 6, after which its expression declines during the maturation phase. The involvement of ferredoxin in early and mid-stage seed formation seems linked to energy transfer and metabolic processes required for seed growth. It is also expressed in other plant tissues, like leaves and flowers, having a role in electron transport and redox reactions. However, its expression in siliques is low47.
In addition, three more candidate genes identified include, ATP-dependent zinc metalloprotease FTSH 4 (LOC106763742), L-type lectin-___domain containing receptor kinase (LOC106763733), and alpha-L-fucosidase 2-like (LOC106777853 respectively). Of these, LOC106763742 codes for a cytoplasmic-membrane protein that controls photosynthesis, chloroplast, and mitochondrial development. The ATPase and proteolytic activities were reported in the ATP-dependent zinc metalloprotease FTSH 4 protein which influences overall seed weight48. Similarly, L-type lectin-___domain-containing receptor kinases (LecRLKs) are plasma membrane-located proteins having a carbohydrate recognition ___domain (CRD), needed for the seed development49. Also, the LecRK-VIII.2 functions as a regulator of both seed size and number50. Similarly, an Alpha-L-fucosidase 2-like enzyme is likely to have its role in seed weight regulation by modulating the cell wall composition, especially xyloglucans51.
Number of pods per plant (NP)
A higher number of pods per plant generally results in an increased yield. This study identified 2 QTLs for NP on chr. 2 (qNP-2-1) and 4 (qNP-4-1), from the parent Pusa Baisakhi having substitution effects of 2.9 and 2.8, respectively. Both the QTLs collectively explained 30% PVE. Similarly, previous studies have also reported QTLs for NP on chromosomes 1, 3, and 915, and on chromosomes 2, 4, and 736, collectively explaining 6.7–12.7% PVE. The identified QTLs are having 4 candidate genes (MADS-box transcription factor, UDP-glycosyltransferase 92A1, ADP & ATP carrier protein 1, and E3 ubiquitin-protein ligase RDUF1). The MADS-box transcription factors regulate plant, seed, and pod development in soybean52. Other candidate genes need further confirmation for their role in the determination of NP in mungbean.
Grain yield per plant (GY)
The total yield of a genotype represents a complex quantitative trait influenced by various qualitative traits and a multitude of interconnected genetic, environmental, and management domains. This study identified 3 QTLs (qGY-7-1, qGY-3-1, and qGY-4-1) governing GY (30% collective PVE) on chr. 7, 3 and 1, respectively. Similarly, 11 QTLs are identified on various chromosomes (8.3–63% PVE) and qGY-4-1 was found linked with qNP-4-1, suggesting their pleiotropic effects on both GY and NP31. In the 2.7 Mb QTL region (qGY-7-1, qGY-3-1, and qGY-4-1), 67 genes are identified having mutations and are also part of several pathways like pollination, reproduction, embryo development, post-embryonic development, and fruit ripening. Of these, 6 candidate genes viz., endoglucanase 1 (LOC106757749), squamosa promoter-binding-like protein 13 A (SPL protein) (LOC106768860), ARF guanine-nucleotide exchange factor GNOM (LOC106757622), E3 ubiquitin-protein ligase RDUF1 (LOC106758074), glutamate–cysteine ligase chloroplastic-like (LOC106767912), and bidirectional sugar transporter (LOC106766939) seems having role in GY determination. SPL genes encode plant-specific TF, influencing phase transition from vegetative stage to flowering to fruit development. Under the influence of hormones, SPL genes may increase yield by efficient distribution of energy and nutrients53. Ubiquitin-ligase RDUF1 regulates yield by controlling the degradation of key regulatory proteins involved in stress responses, hormone signaling, and growth pathways54.
Conclusions
The knowledge of QTLs associated with yield and its component traits is crucial for the development of high-yielding mungbean varieties. This study revealed a significant influence on yield-related traits by both environmental and genetic factors. Grain yield was found positively influenced by parameters like number of pods per plant, number of leaves per plant, and chlorophyll content; while seed weight and plant height did not show any noticeable effect. Two QTLs with very high PVE (> 20%) for plant height (qPH-11-1) and seed weight (qSW-10-1) were identified as the ideal candidates for introgression into mungbean breeding programs. Chromosome 7 of mungbean harbored 5 novel QTLs for plant height, grain yield, chlorophyll content, and number of leaves. This makes chromosome 7 as the key chromosome for the development of markers and marker-assisted introgression of various yield-related traits in mungbean. QTLs for grain yield (qGY-7-1 at 54.43 Mb) and number of leaves (qNL-7-2 at 50.67 Mb) are found linked as evident from correlation analysis. The QTLs and the InDel markers identified could serve as a tools for improving the mungbean yield through molecular approaches. This study is the first to employ high-density SNP markers for QTL mapping of yield-related traits in mungbean, leading to the development of QTL-specific markers.
Materials and methods
Mapping population
A RIL population (F9:10; 166 No) was developed by crossing two contrasting genotypes (Pusa Baisakhi × PMR-1). Pusa Baisakhi - A released variety having high yield, high hundred seed weight, lower chlorophyll content with lesser height and number of leaves. PMR1 – An advanced breeding line is having low yield, less hundred seed weight, higher chlorophyll content, and taller plants with a greater number of leaves than Pusa Baisakhi. The RILs were advanced via single seed descent (SSD) method (F2 to F9:10) and evaluated for six yield contributing traits at the Indian Agricultural Research Institute (IARI), New Delhi (28.7°N, 77.1°E, 228 m AMSL). The experiment followed a randomized complete block design (RCBD) with two replications, adhering to the standard agricultural practices. Each genotype was sown in a 5.0 m row, maintaining a spacing of 20 cm between plants and 30 cm between rows.
Phenotypic evaluation
The parental genotypes and RILs were phenotyped for three consecutive seasons during 2022 (Sept-Nov) and 2023 (March-May) and 2023 (Sept-Nov) for six traits viz., plant height (PH, cm), number of leaves/plant (NL), number of pods/plant (NP), chlorophyll content as SPAD value at 25 and 45 days after sowing (DAS), 100 seed weight (SW, mg) and total grain yield (GY, g). The data from three plants per replicate (NL, PH, SPAD, and NP) were averaged, while 100 seed weight was measured thrice per replication and averaged. Each tri-foliate leaf was counted as a single leaf, the SPAD value was recorded between 09:00 to 11:00 AM on the matured third leaflet from the top of the plant. The analysis of variance (ANOVA) was measured using Agricole package in R55, the descriptive statistics and genetic parameters were estimated using Variability package56, combined best linear unbiased predictor values (BLUPs) were calculated using MetaR57 and correlation was measured using R base58,59. The studied traits were also tested for normality using the Shapiro-Wilks test.
Sequencing and variant calling
Genotyping by sequencing (GBS) was performed by isolating genomic DNA from the leaves of RILs using DNeasy Plant Mini Kit (Qiagen, Germany) (NCBI BioProject: PRJNA914895, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA914895). The quality and quantity of genomic DNA were assessed using Nanodrop (Thermo Fisher Scientific, Waltham, MA, USA). Libraries were prepared using TruSeq® DNA Nano LP Library Prep Kit (Illumina, San Diego, CA, USA) and were sequenced to produce raw 2 × 150-bp reads (Novaseq 6000, Illumina, San Diego, CA, USA). Trimmomatic software was used to process the raw reads by removing the adapters and low-quality bases60. Following trimming, ≥ 85 bp reads were retained having Q30 (base calling accuracy > 99.9%). Cleaned FASTQ files were then mapped to the reference genome of Vigna radiata (mungbean), assembly GCF_000741045.1 corresponding to cultivar VC1973 (https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000741045.1/)61. Variant calling was performed using the Unified Genotyper in the Genome Analysis Toolkit (GATK) version 3.6 for identification of SNPs from RILs; and whole-genome resequencing (WGRS) was employed to sequence the parents Pusa Baisakhi (NCBI BioProject: PRJNA1191361, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1191361) and PMR-1 (NCBI BioProject: PRJNA1191361, https://www.ncbi.nlm.nih.gov/sra/PRJNA1191375).
High-density map construction and QTL mapping
A total of 38,931 SNPs were identified through GBS across 11 chromosomes of 166 RILs. The SNPs and individuals (taxa) were filtered based on a missing data threshold (> 30%). Then monomorphic, heterozygous (> 5%), and significantly distorted (p > 0.05) SNPs were removed using Tassel62. Between parental lines, 5494 filtered polymorphic SNPs were identified and those significantly deviating from the expected 1:1 ratio were eliminated based on a Chi-square test (p ≤ 0.05). Finally, the linkage map was constructed using 1374 SNPs (146 RIL sequence data) with 8% missing proportion and 0.46 as the minor allele frequency (MAF) (Table S4). The linkage map was generated using Kosambi function having fixed genomic physical positions.
QTL mapping was performed using both MIM and CIM through Rqtl package in R Studio63 and genetic distance between markers was converted into centiMorgan (cM). Combined BLUP values were generated for accurate mapping. Genotypic data were imputed using a hidden Markov model (HMM) (error probability: 0.001; step size: 0.5 cM; simulations: 64). MIM was conducted using the Haley-Knott regression method and penalties were calculated (p = 0.05). For CIM, up to 10 marker covariates were included (window size of 1.0 cM). To find the significant threshold for Logarithm of Odds (LOD) scores, 1000 permutations were conducted and a predefined LOD score threshold of 3.0 was used to identify the QTLs. Additionally, the confidence intervals were calculated using the LOD drop-off method with a decrease of 1.0 LOD unit on either side of the QTLs. PVE (%) was estimated using the following formula: \(\:PVE=1-{10}^{\left(\frac{-2\times\:LOD}{N}\right)}\). Where, n: sample size and LOD: Peak LOD value64. The QTLs were classified as major if they explained > 10% of phenotypic variance, while those with PVE < 10% were considered minor12.
Identification of candidate genes and indel marker development
The WGRS was used to sequence the parental genotypes (Pusa Baisakhi and PMR-1) and sequence information was used to identify the variants within the QTLs identified through mapping. The raw reads were trimmed, aligned and resulting variants were functionally annotated using SnpEff ver. 5.1d65. The annotated information was used to identify the candidate genes exhibiting variations between the parents and potentially affected expression. For the development of markers, WGRS data was aligned to the reference genome and variant calling was performed and those InDels that are monomorphic or of < 15 bp size were excluded. Then 200 bp sequence flanking each nearest InDel to the QTL (100 bp on either side) was used for primer designing via primer3 web tool66. Primers with a length of 18–24 bp, melting temperature (Tm) of 53–62 °C and GC content of 40–60% were idealised. While, primers forming secondary structures (e.g., hairpins) and primer-dimers were avoided.
Digital gene expression analysis of identified candidate genes
The digital gene expression was done to validate the candidate genes using well-characterized Arabidopsis orthologs, by analyzing their localized expression patterns through Expression Angler, an ePlant web browser (https://bar.utoronto.ca/eplant/)47,67. The Arabidopsis orthologs were retrieved using OrthoDB V12.068.
Data availability
All data generated or analysed during this study are included in this article and its supplementary information files. Sequence data that support the findings of this study have been deposited in the NCBI with the primary accession codes NCBI (1) BioProject: PRJNA914895, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA914895; (2) NCBI BioProject: PRJNA1191361, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1191361; (3) NCBI BioProject: PRJNA1191375, https://www.ncbi.nlm.nih.gov/sra/PRJNA1191375.
References
Dasgupta, U. et al. Comparative RNA-Seq analysis unfolds a complex regulatory network imparting yellow mosaic disease resistance in Mungbean [Vigna radiata (L.) R. Wilczek]. Plos One 16(1), e0244593 (2021).
Dikshit, H.K., Mishra, G.P., Somta, P., Shwe, T., Alam, A.K.M.M. & Nair, R.M. Classical genetics and traditional breeding in mungbean. In (Nair, R., Schafleitner, R., Lee, S.H. eds.) The Mungbean Genome. Compendium of Plant Genomes. 43–54. 10.1007/978-3-030-20008-4_4. ISBN: 978-303020008-4 (Springer, 2020).
Mishra, G.P., Priti, Dikshit, H.K., Aski, M., Sangwan, S., Stobdan, T. & Praveen, S. Microgreens: A novel food for nutritional security. In Conceptualizing Plant-Based Nutrition: Bioresources, Nutrients Repertoire and Bioavailability. 123–156. (Springer, 2022).
Priti, Mishra, G.P., Dikshit, H.K., Vinutha, T., Mechiya, T., Stobdan, T. & Praveen, S. Diversity in phytochemical composition, antioxidant capacities, and nutrient contents among mungbean and lentil microgreens when grown at plain-altitude region (Delhi)and high-altitude region (Leh-Ladakh), India. Front. Plant Sci. 12, 710812 (2021).
Manjunatha, P.B., Aski, M.S., Mishra, G.P., Gupta, S., Devate, N.B., Singh, A. & Dikshit, H.K. Genome-wide association studies for phenological and agronomic traits in mungbean (Vigna radiata L. Wilczek). Front. Plant Sci. 14, 1209288 (2023).
Agricultural Land Use Area in India - Percentage of Land Used for Agriculture, Arable Land Statistics in India 1950–2018. https://avrdc.org/intl-mungbean-network/. International Mungbean Improvement Network - World Vegetable Center.
AICRP on Kharif Pulses. Indian Institute of Pulses Research, Kanpur, Uttar Pradesh, India. https://iipr.icar.gov.in/aicrp/ (2023).
Gupta, S., Aski, M., Mishra, G.P., Yadav, P.S., Tripathi, K., Lal, S.K. & Dikshit, H.K. Genetic variation for tolerance to pre-harvest sprouting in mungbean (Vigna radiata) genotypes. PeerJ 12, e17609 (2024).
Kumari, N., Aski, M.S., Mishra, G.P., Roy, A., Dikshit, H.K., Saxena, S. & Nair,R.M. Development of infectious clones of mungbean yellow mosaic India virus (MYMIV, Begomovirus Vigna radiataindiaense) infecting mungbean [Vigna radiata (L.) R. Wilczek] and evaluation of a RIL population for MYMIV resistance. Plos One. 10.1371/journal.pone.0310003 (2024).
Mathivathana, M. K. et al. Detection of QTLs associated with Mungbean yellow mosaic virus (MYMV) resistance using the interspecific cross of Vigna radiata × Vigna umbellata. J. Appl. Genet. 60 (3), 255–268 (2019).
Manjunatha, P.B., Revanth Ragul, A., Kohli, M., Chowdhury, S., Shivaprasad, K.M.,Shashidhar, B.R. & Dikshit, H.K. Genome-wide association to identify the genetic loci associated with various agro-economical traits in mungbean (Vigna radiata L. Wilczek). Int. J. Environ. Clim. Change 14(3), 325–341 (2024).
Kumari, N., Mishra, G.P., Dikshit, H.K., Gupta, S., Roy, A., Sinha, S.K. & Aski, M. Identification of quantitative trait loci (QTLs) regulating leaf SPAD value and trichome density in mungbean (Vigna radiata L.) using genotyping-by-sequencing (GBS) approach. Peer J. 12, e16722 (2024).
Kajonphol, T., Sangsiri, C., Somta, P., Toojinda, T. & Srinives, P. SSR map construction and quantitative trait loci (QTL) identification of major agronomic traits in Mungbean (Vigna radiata (L.) Wilczek). SABRAO J. Breed. Genet. 44(1) (2012).
Rohilla, V. et al. Association mapping for yield attributing traits and yellow mosaic disease resistance in mung bean [Vigna radiata (L.) Wilczek]. Front. Plant. Sci. 12, 749439 (2022).
Singh, N., Mallick, J., Sagolsem, D., Mandal, N. & Bhattacharyya, S. Mapping of molecular markers linked with MYMIV and yield attributing traits in Mungbean. Indian J. Genet. Plant. Breed. 78 (01), 118–126 (2018).
Chen, T. et al. Construction of high-density genetic map and identification of a bruchid resistance locus in mung bean (Vigna radiata L). Front. Genet. 13 (2022).
Kohli, M., Bansal, H., Mishra, G.P., Dikshit, H.K., Reddappa, S.B., Roy, A. & Aski, M. Genome-wide association studies for earliness, MYMIV resistance, and other associated traits in mungbean (Vigna radiata L. Wilczek) using genotyping by sequencing approach. Peer J. 12, e16653 (2024).
Mishra, G.P., Dikshit, H.K., Tripathi, K., Aski, M., Pratap, A., Dasgupta, U. & Gupta, S. Mungbean breeding. In ( Yadava, D.K. et al. eds.) Fundamentals of Field Crop Breeding. ISBN: 978-9811692567. 10.1007/978-981-16-9257-4_22 (2022).
Young, N. D. et al. RFLP mapping of a major bruchid resistance gene in Mungbean (Vigna radiata, L. Wilczek). Theor. Appl. Genet. 84, 839–844 (1992).
Somta, P., Laosatit, K., Yuan, X. & Chen, X. Thirty years of Mungbean genome research: where do we stand and what have we learned? Front. Plant. Sci. 13, 944721 (2022).
Verhoeven, K. J., Macel, M., Wolfe, L. M. & Biere, A. Population admixture, biological invasions and the balance between local adaptation and inbreeding depression. Proc. Royal Soc. B: Biol. Sci. 278 (1702), 2–8 (2011).
Mather, K. & Jinks, J. L. Biometrical Genetics: the Study of Continuous Variation (Springer, 1982).
Latta, R. G., Gardner, K. M. & Johansen-Morris, A. D. Hybridization, recombination, and the genetic basis of fitness variation across environments in Avena barbata. Genetica 129, 167–177 (2007).
Umani, K., Zhang, C., McGee, R. J., Vandemark, G. J. & Sankaran, S. A pulse crop dataset of agronomic traits and multispectral images from multiple environments. Data Brief. 53, 110013 (2024).
Sousa Silva, A. et al. Growth and production of soybean in response to Spatial arrangement in Savannah climate. Aust J. Crop Sci. 13 (11), 1901–1907 (2019).
Chiteri, K. O. et al. Dissecting the genetic architecture of leaf morphology traits in Mungbean (Vigna radiata (L.) Wizcek) using genome-wide association study. Plant. Phenome J. 6(1), e20062 (2023).
Boerma, H. R. & Ashley, D. A. Canopy photosynthesis and seed-fill duration in recently developed soybean cultivars and selected plant introductions. Crop Sci. 28 (1), 137–140 (1988).
Digrado, A. et al. Cowpea leaf width correlates with above ground biomass across diverse environments. Legume Sci. 4 (4), e144 (2022).
Baisakh, B., Swain, S. C., Panigrahi, K. K., Das, T. R. & Mohanty, A. Estimation of genetic variability and character association in micro mutant lines of Greengram [Vigna radiata (L.) Wilczek] for yield attributes and cold tolerance. Legume Genomics Gene 7(2), (2016).
Ye, W. et al. InDel marker development and QTL analysis of agronomic traits in mung bean [Vigna radiate (L.)Wilczek]. Mol. Plant. Breed. 41, 1–14 (2021).
Vu, T. T. H., Kilian, A., Carling, J. & Lawn, R. J. Consensus genetic map and QTLs for morphological and agronomical traits in Mungbean using dart markers. Physiol. Mol. Biol. Plants. 28 (6), 1277–1295 (2022).
Mishra, P. & Panigrahi, K. C. GIGANTEA–an emerging story. Front. Plant. Sci. 6, 108096 (2015).
Laha, N. P. et al. INOSITOL (1, 3, 4) TRIPHOSPHATE 5/6 KINASE1-dependent inositol polyphosphates regulate auxin responses in Arabidopsis. Plant. Physiol. 190 (4), 2722–2738 (2022).
Nishimura, N., Sarkeshik, A., Nito, K., Park, S. Y., Wang, A., Carvalho, P. C., …Schroeder, J. I. PYR/PYL/RCAR family members are major in-vivo ABI1 protein phosphatase 2 C‐interacting proteins in Arabidopsis. Plant J., 61(2), 290–299 (2010).
Kishor, P. B. K., Tiozon, R. N., Fernie, A. R. & Sreenivasulu, N. Abscisic acid and its role in the modulation of plant growth, development, and yield stability. Trends Plant. Sci. 27 (12), 1283–1295 (2022).
Isemura, T., Kaga, A., Tabata, S., Somta, P., Srinives, P., Shimizu, T., … Tomooka,N. Construction of a genetic linkage map and genetic analysis of domestication related traits in mungbean (Vigna radiata). PLoS One. 7(8), e41304 (2012).
Liu, C. et al. Quantitative trait locus mapping under irrigated and drought treatments based on a novel genetic linkage map in Mungbean (Vigna radiata L). Theor. Appl. Genet. 130 (11), 2375–2393 (2017).
Yu, K. et al. High-density QTL mapping of leaf-related traits and chlorophyll content in three soybean RIL populations. BMC Plant Biol. 20, 1–14 (2020).
Wang, L., Conteh, B., Fang, L., Xia, Q. & Nian, H. QTL mapping for soybean (Glycine max L.) leaf chlorophyll-content traits in a genotyped RIL population by using RAD-seq based high-density linkage map. BMC Genomics 21, 1–18 (2020).
Yao, X., Nie, J., Bai, R. & Sui, X. Amino acid transporters in plants: identification and function. Plants 9 (8), 972 (2020).
Khattak, G. S., Ashraf, M., Elahi, T. & Abbas, G. Selection for large seed size at the seedling stage in Mungbean (Vigna radiata (L.) Wilczek). Breed. Sci. 53 (2), 141–143 (2003).
Sompong, U., Somta, P., Raboy, V. & Srinives, P. Mapping of quantitative trait loci for phytic acid and phosphorus contents in seed and seedling of Mungbean (Vigna radiata (L.) Wilczek). Breed. Sci. 62 (1), 87–92 (2012).
Chen, H. M. et al. The major quantitative trait locus for Mungbean yellow mosaic Indian virus resistance is tightly linked in repulsion phase to the major bruchid resistance locus in a cross between Mungbean [Vigna radiata (L.) Wilczek] and its wild relative Vigna radiata ssp. sublobata. Euphytica 192, 205–216 (2013).
Alam, A. M., Somta, P., Muktadir, M. A. & Srinives, P. Quantitative trait loci associated with seed weight in Mungbean (Vigna radiata (L.) Wilczek). Agric. Nat. Resour. 48 (2), 197–204 (2014).
Humphry, M.E., Lambrides, C.J., Chapman, S.C., Aitken, E.A.B., Imrie, B.C., Lawn,R.J. & Liu, C.J. Relationships between hard-seededness and seed weight in mungbean (Vigna radiata) assessed by QTL analysis. Plant Breed. 124(3), 292–298 (2005).
He, L., Li, M., Qiu, Z., Chen, D., Zhang, G., Wang, X. & Zhu, L. Primary leaf-type ferredoxin 1 participates in photosynthetic electron transport and carbon assimilation in rice. Plant J. 104(1), 44–58 (2020).
Austin, R. S., Hiu, S., Waese, J., Ierullo, M., Pasha, A., Wang, T. T. & Provart,N. J. New BAR tools for mining expression data and exploring Cis-elements in Arabidopsis thaliana. Plant J. 88(3), 490–504 (2016).
Kato, Y. & Sakamoto, W. FtsH protease in the thylakoid membrane: physiological functions and the regulation of protease activity. Front. Plant. Sci. 9, 387734 (2018).
Xiao, W., Hu, S., Zou, X., Cai, R., Liao, R., Lin, X. & Guo, X. Lectin receptor-like kinase LecRK-VIII. 2 is a missing link in MAPK signaling-mediated yield control. Plant Physiol. 187(1), 303–320 (2021).
Saidou, M. & Zhang, Z. The l-type lectin-like receptor kinase gene TaLecRK-IV. 1 regulates the plant height in wheat. Int. J. Mol. Sci. 23 (15), 8208 (2022).
Augur, C., Stiefel, V., Darvill, A., Albersheim, P. & Puigdomenech, P. Molecular cloning and pattern of expression of an α-L-Fucosidase gene from pea seedlings. J. Biol. Chem. 270 (42), 24839–24843 (1995).
Gu, Q., Ferrándiz, C., Yanofsky, M. F. & Martienssen, R. The FRUITFULL MADS-box gene mediates cell differentiation during Arabidopsis fruit development. Development 125 (8), 1509–1517 (1998).
Chen, X. et al. SQUAMOSA promoter-binding protein‐like transcription factors: star players for plant growth and development. J. Integr. Plant. Biol. 52 (11), 946–951 (2010).
Al-Saharin, R., Hellmann, H. & Mooney, S. Plant E3 ligases and their role in abiotic stress response. Cells 11 (5), 890 (2022).
Mendiburu, F. & Yaseen, M. Agricolae: Statistical Procedures for Agricultural Research. R Package Version 1.4.0 (R Foundation for Statistical Computing, 2020).
Popat, R., Patel, R. & Parmar, D. Variability: Genetic Variability Analysis for Plant Breeding Research. R Package Version 0.1.0. (2020).
Campagne, F., Digan, W. E. & Simi, M. MetaR: Simple, high-level languages for data analysis with the R ecosystem. bioRxiv 030254 (2015).
R Core Team. A Language and Environment for Statistical Computing. (R Foundation for Statistical Computing, 2013). https://www.R-project.org/
Racine, J. S. RStudio: A platform-independent IDE for R and Sweave. (2012).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30 (15), 2114–2120 (2014).
Kang, Y.J., Kim, S.K., Kim, M.Y., Lestari, P., Kim, K.H., Ha, B.K. & Lee, S.H. Genome sequence of mungbean and insights into evolution within Vigna species. Nat. Commun. 5(1), 5443 (2014).
Bradbury, P. J. et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23 (19), 2633–2635 (2007).
Broman, K. W. & Sen, S. A. Guide To QTL Mapping with R/qtl. Vol. 46 (Springer, 2009).
Schurack, S., Beuch, S., Cowan, S., Griffiths, I., Lunzer, M., Morales, L. & Herrmann, M.H. Genetic mapping of the powdery mildew resistance gene Pm13 on oat chromosome 1D. bioRxiv 2024-01 (2024).
Cingolani, P., Platts, A., Wang, L.L., Coon, M., Nguyen, T., Wang, L. & Ruden, D.M.A program for annotating and predicting the effects of single nucleotide polymorphisms,SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6(2), 80–92 (2012).
Untergasser, A. et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 40 (15), e115–e115 (2012).
Fucile, G., Di Biase, D., Nahal, H., La, G., Khodabandeh, S., Chen, Y. & Provart,N. J. ePlant and the 3D data display initiative: integrative systems biology on the world wide web. PLoS One 6, e15237 (2011).
Waterhouse, R. M., Tegenfeldt, F., Li, J., Zdobnov, E. M. & Kriventseva, E. V. OrthoDB: a hierarchical catalog of animal, fungal and bacterial orthologs. Nucleic Acids Res. 41, 358–365 (2013).
Acknowledgements
The technical support received by Mr. Dilip Kumar (Indian Agricultural Research Institute, New Delhi) is duly acknowledged. First author also acknowledges the support received from The Graduated School, IARI, New Delhi during his PhD programme.
Funding
This research work was funded by SERB (Science and Engineering Research Board), New Delhi (CRG/2019/002024), and the Indian Council of Agricultural Research (ICAR), New Delhi. Ramakrishnan M. Nair acknowledges the long-term strategic donors to the World Vegetable Center: Taiwan, the United States Agency for International Development (USAID), the UK Government’s Foreign, Commonwealth & Development Office (FCDO), Australian Centre for International Agricultural Research (ACIAR), Germany, Thailand, Philippines, Korea, Japan, and funding from ACIAR Project on International Mungbean Improvement Network (CROP/2019/144). The funders had no role in the study design, data collection, analysis, the decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
GPM, HKD, and RMN: Conceptualization, resources, writing—original draft; writing—review and editing; SBR, MSA, SG, and KMS: methodology, formal analysis, validation, investigation, writing—original draft; SKS, RRK, DCM, SD: methodology, formal analysis; SBR, MSA, AR, PAMP: investigation, data analysis, validation; GPM, HKD, RMN: Funding acquisition, supervision.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Reddappa, S.B., Aski, M.S., Mishra, G.P. et al. QTL mapping for yield contributing traits in mungbean (Vigna radiata L.) using a RIL population. Sci Rep 15, 20795 (2025). https://doi.org/10.1038/s41598-025-99687-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-99687-1
