
Abstract
Head and neck cancer (HNC) is the seventh most common cancer worldwide. Currently-approved systemic therapies include chemotherapy, anti-EGFR antibodies, and PD-1 immunotherapy, with few genomic-based targeted therapies. Gene fusions involving cancer-driving kinase genes such as FGFR, NTRK, and ALK are clinically targetable in other solid tumors; however, there is limited knowledge about their prevalence in HNC. Here, we describe the genomic landscape and the biological impact of oncogenic fusions in a combined dataset of over 13,000 HNC tumors (excluding salivary gland tumors). We identified 66 cases (2.8%) harboring oncogenic fusions, including previously-reported FGFR3 fusions (n = 19) and gain-of-function EGFR fusions (n = 6). Fusion-positive HNC had significantly higher gene expression and higher prevalence of human papillomavirus than fusion-negative HNC (p < 0.001). Tumors with FGFR alterations were associated with enriched cell proliferation and higher abundance of NK cells and CD8+ T cells compared to wildtype. Our results provide expanded therapeutic opportunities for patients with HNCs.
Similar content being viewed by others

Introduction
Head and neck cancer (HNC) is the 7th most common cancer diagnosed worldwide with a 5-year survival rate of ~50%. This aggressive disease arises in various anatomical sites, including oropharynx, oral cavity, larynx, sinus, salivary gland, and nasal cavity. Environmental factors that play a significant role in the development of HNC include tobacco, alcohol, and viral infection of human papillomavirus (HPV), mainly HPV161,2. These factors are typically mutually exclusive from one another. Treating HNC remains a challenge, due to its heterogeneity and frequent late-stage diagnosis. Currently approved systemic therapies include radiotherapy, concurrent with either cisplatin chemotherapy or anti-EGFR antibody (cetuximab), administered as first-line therapy or after surgery3,4. However, response rates remain limited, ranging from 13 to 45%5,6. PD-1 immunotherapy has also emerged as an alternate form of therapy for recurrent and metastatic HNC patients7, although overall response rate remains at 16–20%8,9. One study showed that addition of the PD-1 inhibitor nivolumab with cetuximab improved the 1-year overall survival rate to 66% and median survival of 20.2 months5. Another study reported a 6-month overall response rate of 45% in a trial combining pembrolizumab and cetuximab10, and another identified a response rate of 39% with a median 8.6-month duration of response in a trial combining cetuximab with durvalumab11. However, combination therapy with anti-EGFR antibodies and immunotherapies is administered to advanced HNC patients in an unselected manner, irrespective of EGFR mutation or PD-L1 immunohistochemistry status. Therefore, there is a need to establish additional genomic biomarkers within HNC to expand eligibility for targeted therapy and personalize treatment options for early-stage patients.
While tumor genomic alterations in HNC have been described, personalized treatment based on molecular subsets has not significantly advanced in this disease. Next Generation Sequencing (NGS) has improved our understanding of HNC. These tumors often harbor alterations in tumor suppressor genes NOTCH112, PTEN13,14, and in oncogenic pathways including the JAK/STAT pathway15,16. In addition, the clinically actionable gene fusion FGFR3—TACC3 has been observed in HNC16,17,18, as well as copy number alterations in oncogenes including PIK3CA19, FGFR120, and EGFR21. In spite of this, few targeted therapies have been successfully developed for HNC patients, in part due to the perception that there are no clinically actionable targets. Currently, only patients harboring point mutations in HRAS (3–4%) are eligible for personalized therapy with tipifarnib22. This contrasts with other cancer types, in which precision oncology approaches are more widely used, such as lung adenocarcinoma and colorectal cancer, where molecular targets can guide therapy selection23,24. Here, we sought to identify genomic alterations that may be amenable to personalized targeted therapies in HNC.
Gene fusions are clinically actionable across many different cancer types, but have not been thoroughly studied in HNC. Many gene fusions involving kinase genes, including ALK25, ROS126, FGFR127, FGFR228,29, FGFR327,29, RET30, NTRK1, NTRK2, and NTRK331, can be targeted with clinically approved tyrosine kinase inhibitors (TKIs). Other oncogenic fusions involving EGFR32, BRAF33, and NRG134 show potential for targeted treatment with TKIs. Compared to other alteration types, including copy number alterations and point mutations, gene fusions are not well described in this cancer type. Fusions from case studies and small cohorts have been observed in genes involved in cancer proliferation or gene transcription, such as BRAF, ROS1, FGFR, and CD27417,35,36,37,38,39,40,41,42,43. Despite these reports, there is limited knowledge on clinically relevant gene fusions in HNC and their biological impact for two reasons: (1) HNC studies are small, often focusing on a single anatomical site, and are, therefore, insufficient for comprehensive analysis for rare alterations, such as gene fusions and (2) The few studies that leveraged large data (n > 200 samples) applied now out-of-date bioinformatics methods for gene fusion analysis. For example, TCGA published their multi-omic, comprehensive head and neck squamous cell carcinoma dataset (n = 279 patients) using MapSplice44 to detect gene fusions45. Using this method, which was designed to identify splice variation, they uncovered two fusion-positive cases, both harboring an FGFR3-TACC3 fusion45. Stransky et al46. analyzed RNA sequencing for kinase fusions using the updated TCGA cohort from the original study, totaling 411 head and neck cancer patients. They identified the same two FGFR3-TACC3 fusion-positive cases described by the TCGA investigators45 and two additional fusion-positive cases, one harboring an NTRK2 fusion and the other harboring an NTRK3 fusion46. Since these studies were published, several gene fusion software packages have been developed and optimized to improve gene fusion detection. In a study that compared various gene fusion software, it demonstrated that STAR-Fusion47 and Arriba48 were among the best gene fusion tools when balancing sensitivity and specificity47. In fact, following this publication, investigators leveraged these tools to reanalyze the entire TCGA dataset for gene fusions49 and is now available on the Genomic Data Commons (GDC) portal. However, there remains a need to define gene fusions that represent gain-of-function events and are potential targets for therapy. Here, we address this through a study of the largest cohort of HNC (n = 13,655 tumors) in which we sought to describe the prevalence of gain-of-function fusions, their biological impact, and determine whether HNC patients harboring recurring alterations involving EGFR and FGFR are sensitive to tyrosine kinase inhibitor therapy.
To discover and characterize cohorts of HNC patients who may benefit from gene fusion targeted therapy, we leveraged datasets with paired RNA sequencing, whole exome sequencing, and clinical data. We leveraged multiple data sources to create a cohort of over 13,000 HNC cases of all anatomical sites, excluding salivary gland, and all histology typesto identify and describe the prevalence of oncogenic gene fusions in HNC, as well as their biological and clinical impact. We demonstrate that tumors harboring clinically relevant gene fusions are prevalent in HNC, have increased gene expression, have higher prevalence of HPV positivity, and display distinct immune phenotypes. These findings can expand personalized therapy options for HNC harboring gene fusions.
Results
Gene fusions occur across diverse anatomical sites in head and neck cancers
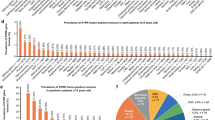
We leveraged various data sources to create a substantial research cohort of 2564 and a clinical database of 11,091 HNC cases, totaling in 13,655 HNC cases. These sources include ORIEN (n = 1441), TCGA (n = 528), CPTAC (n = 110), and 17 published datasets from dbGaP and ArrayExpress (n = 485) (Supplementary Table 1). STAR-Fusion and Arriba were used to analyze the RNA sequencing data from these cases for gene fusions. Applying this method, we identified 66 cases (2.57% prevalence) harboring gene fusions with a clinically relevant gene partner (Fig. 1A and Supplementary Table 2). Most gene fusion-positive cases derived from squamous cell carcinomas (64/66), with one deriving from adenocarcinoma and another from adenosquamous carcinoma (Fig. 1B). We uncovered gene fusions involving oncogenic genes that have been shown to be clinically targetable, including FGFR3 (n = 19), EGFR (n = 6), FGFR2 (n = 6), and NRG1 (n = 5). (Fig. 2A). The anatomical site with the most gene fusions was the oropharynx (n = 25), followed by oral cavity (n = 20) and larynx (n = 17) (Fig. 2B). Leveraging the OncoKB database, we found that 48/66 (72.7%) of gene fusions characterized as “Oncogenic” or “Likely Oncogenic” (Supplementary Fig. 1). Additionally, investigating the RNA sequencing data from 11,091 samples from the Caris Life Sciences clinical dataset, we observed 54 clinically relevant gene fusions, including alterations involving FGFR3 (n = 43), FGFR2 (n = 2), and EGFR (ERBB1) (n = 2) (Supplementary Fig. 2A). Notably, this clinical dataset has stringent criteria for reporting previously observed gene fusions. Nonetheless, the overall results were similar compared to the discovery group (Fig. 2), the anatomical site harboring the most gene fusions was the oropharynx (Supplementary Fig. 2B).

A Flow chart of data curation process and sources. From these sources, we curated a total of 13,655 head and neck cancer cases. ORIEN oncology research information exchange network, TCGA the cancer genome atlas, CPTAC clinical proteomic tumor analysis consortium B Pie chart of histology types in the fusion-positive cohort.

A Count plot of the number of samples harboring a gene fusion involving a clinically relevant gene (y-axis) in a given anatomical site (x-axis). B Distribution of clinically relevant gene fusions present in each anatomical site. Left (orange) denotes the percentage of gene fusions present in the given anatomical site and right (blue) denotes the total number of cases harboring cancer in the given anatomical site.
Gain-of-function EGFR fusions are observed in head and neck cancers
To our surprise, we identified 6 EGFR fusions in our combined cohort. While our study focused on squamous cell carcinomas, we did also observe an EGFR fusion in one patient with salivary gland cancer (ORIEN cohort) wherein transcription factor and NTRK fusions are better known. We found diverse gene fusion partners associated with EGFR (Fig. 3), most of which involved EGFR as the 5′ partner (n = 5) (Fig. 3A). Interestingly, we also observed fusions with EGFR as the 3′ partner, which maintained an intact kinase ___domain (n = 2) (Fig. 3B). Four of these cases harbored one of two specific breakpoints that have previously shown to be gain-of-function and/or have demonstrated clinical responses in patients receiving EGFR TKI. Through a literature evaluation of EGFR fusions, 17 patients received EGFR TKI, 15 had a partial response, one had a complete response, and one had stable disease (Supplementary Table 3). Most of these cases had lung adenocarcinoma (n = 16/17), and one case had colorectal adenocarcinoma (n = 1/17). Consistent with the more common EGFR mutations observed in lung adenocarcinoma, nearly half of these fusion cases (8/17) were non-smokers. The most common EGFR fusion breakpoint was at exon 24 (encompassing exons 1–24), where the kinase ___domain ends, with EGFR as the 5′ partner (n = 8/17). We also observed gene fusion breakpoints at exon 18 (encompassing exons 18-28), where the kinase starts (n = 2/17). We compared these cases to our EGFR fusion-positive cohort and found 4/6 cases that harbored the same EGFR fusion constructs observed among the responders: 3 with an EGFR breakpoint at exon 24 (5′ partner) and one with a breakpoint at exon 18 (3′ partner). From both our fusion data analysis and the curated literature, the most common EGFR breakpoint occurred at the end of exon 24, the final exon encoding the kinase ___domain. Intrigued by this, we leveraged our pan-cancer cohorts from TCGA (n = 10,533) and ORIEN (n = 11,489) and found 66 total cases with a fusion breakpoint at EGFR exon 24 from 12 unique cancer types (Supplementary Fig. 3).

Gene fusion constructs of EGFR. A Schematic of 5′ EGFR fusions; breakpoints occurred in intron 24 (top 3) and exon 28 (bottom). B Schematic of 3’ EGFR fusions; breakpoints occurred in intron 17 (top) and exon 9 (bottom).
Clinically relevant gene fusions demonstrate upregulated transcript expression
Next, we analyzed the biological impact associated with gene fusion-positive HNC. We leveraged RNAseq data from our two large cohorts, TCGA (n = 528 samples) and ORIEN (n = 1618 samples), and our combined cohort of 488 curated RNAseq samples from the literature (Supplementary Table 1). We found that fusion-positive cases displayed upregulated expression of their respective clinically relevant gene fusion partner (Supplementary Fig. 4). To analyze for an association between gene fusion-positive cancer and upregulated gene expression, we normalized the RNA counts by converting the counts to z scores. We found that fusion-positive cases had significantly higher z scores in their given gene partner compared to fusion-negative cases (p < 0.001) for each of the separate datasets described previously (Fig. 4A–C).

Violin plots of the z scores for each given gene (denoted in the y-axis) for samples in (A) ORIEN (B) TCGA and (C) curated studies (dbGaP and Array Express). Violin plots of tumor mutation burden (TMB) comparing fusion-positive versus fusion wildtype cases in (D) ORIEN and (E) TCGA. E, F stacked bar charts of the proportion of samples in each group positive for human papillomavirus (HPV) in (F) ORIEN and (G) TCGA.
Clinically relevant gene fusions do not have a unique co-mutation pattern
Because oncogenic gene fusions are associated with low tumor mutational burden, we investigated mutation status in these tumors. Out of 42 fusion-positive samples from ORIEN, 31 had whole exome sequencing data available for mutation analysis. Intriguingly, when we compared our fusion-positive cohort in ORIEN (n = 31 to our fusion-negative group (n = 1579), we found no significant difference (p > 0.5) in tumor mutational burden (Fig. 4D). These results were consistent when we analyzed TMB between fusion-positive samples (n = 12) and fusion-negative samples in TCGA (n = 516) (Fig. 4E). Additionally, we investigated oncogenic co-mutations associated with fusion-positive cases. In ORIEN, 6/31 fusion-positive cases harbored functional mutations in PIK3CA (n = 4), NOTCH1 (n = 1), HRAS (n = 1), and CDK2NA (n = 1), with one case harboring a mutation in both PIK3CA and HRAS. In TCGA, 3/12 cases harbored oncogenic mutations in PIK3CA (n = 2) and TP53 (n = 1). Notably, these fusion-positive cases were mutually exclusive from functional mutations in FGFR2, FGFR3, PTEN, and NRAS.
Clinically relevant gene fusions are more common in HPV-positive HNCs
Because HPV is associated with some HNC, we assessed the prevalence of HPV in fusion-positive cases. Overall, the fusion-positive cohort had a higher proportion of samples that were also HPV+ compared to the fusion-negative cohort in ORIEN and TCGA (Fig. 4F, G). In the ORIEN cohort, 18% (250/1376) of fusion-negative patients were positive for HPV16, compared to the fusion-positive cohort that had a higher prevalence of 31% (12/37) (p = 0.05, odds ratio (OR) = 2.0) (Fig. 4F). These cases derived mostly from oropharynx tumors (n = 11), followed by oral cavity (n = 1) which made up 28.9% and 2.6% of the fusion-positive cohort, respectively (Fig. 4F and Supplementary Fig. 5A). Notably, among the 15 fusion-positive oropharynx tumors, 73% (n = 11/15) were HPV16 positive while fusion negative tumors had a slightly lower HPV16 prevalence of 62% (n = 244/391). In particular, we observed a significant enrichment of HPV16 among FGFR3 fusion-positive patients (p = 0.015, OR = 5.44) (data not shown). We identified 10 different HPV strains present in RNA samples from head and neck tumors; however, in the fusion-positive group, the strains observed were exclusively HPV16 (n = 12), and HPV18 (n = 1). Similarly, in TCGA, 42% (5/12) fusion-positive cases were positive for HPV compared to 14% (74/509) of the wildtype group (p = 0.024, OR = 4.19). Consistent with our results from the ORIEN cohort, most of these cases derived from oropharynx tumors (n = 3) followed by oral cavity (n = 1) and hypopharynx (n = 1) and represented 25%, 8.3%, and 8.3%, respectively, of the fusion-positive cohort (Fig. 4G and Supplementary Fig. 5B). We also note that HPV16 was present in 2/2 FGFR3 fusion-positive cases, showing a similar pattern that observed in the ORIEN dataset.
FGFR-altered HNC patients respond well to FGFR TKI therapies
Because FGFR fusions showed the highest prevalence in our cohort, we sought to further this subset of HNC including fusions as well as gain-of-function point mutations. While the most common fusion observed in our cohort was FGFR3-TACC3 (n = 15), we also observed additional FGFR3 fusions with the same breakpoint, but with different fusion gene partners (n = 4) (Fig. 5A). Additionally, we identified six fusions involving FGFR2 with five unique fusion partners (Fig. 5B). Five of these cases harbor breakpoints in FGFR2 that were observed in clinical cases of FGFR2 fusion and who demonstrated response to FGFR kinase inhibitor therapies50. To look for additional activating mutations in FGFR1/2/3, we utilized whole exome data available from ORIEN (n = 1610), TCGA (n = 528), and CPTAC (n = 110) (Supplementary Table 4). We identified 17 FGFR3 point mutations from ORIEN and 11 from TCGA, totaling 28 mutations observed in 24 unique tumors. These mutation events were mutually exclusive from the gene fusion-positive cases. The point mutation with the highest prevalence, as expected, was FGFR3 S249C (n = 16). Other gain-of-function mutations we observed included FGFR3 G380R (n = 4), R248C (n = 4), R669G (n = 2), V50I (n = 1), and D786N (n = 1) (Fig. 5C). We did not find gain-of-function point mutations in the CPTAC cohort (n = 0/110) and no mutations from FGFR1 or FGFR2 in any of the datasets. Altogether, from our combined cohort we identified 49 cases harboring alterations in FGFR2 or FGFR3 with a prevalence of 1.84% (n = 49/2564).

A Unique gene fusion constructs involving FGFR2. All 5′ FGFR breakpoints occurred at intron 17 and the 3’ breakpoint occurred at intron 4. B Unique gene fusion construct of FGFR3 with canonical breakpoints. Two unique 5′ FGFR3 breakpoints occurred in intron 14 (top), and the end of exon 15 (middle). The 3’ FGFR3 breakpoint occurred at exon 9. C Lollipop plot of gain of function mutations observed in FGFR3 in ORIEN, TCGA, and CPTAC.
Due to the success of FGFR kinase inhibitors in other cancer types harboring FGFR alterations (cholangiocarcinoma and bladder cancer), we sought to determine how FGFR-altered head and neck cancers respond to FGFR inhibitor therapies. We performed a comprehensive literature review to find reports and past clinical trials where head and neck cancer cases harboring FGFR alterations were treated with FGFR inhibitors. We found 19 cases from eight separate studies that showed durable responses to FGFR inhibitor therapy (Table 1). Overall, 11/19 (58%) achieved at least a partial response, two of which had a complete response. The average duration of therapy was 10 months. Various therapies were administered, including FGFR selective kinase inhibitors futibatinib, erdafitinib, and pemigatinib, as well as drugs that target multiple tyrosine kinases in addition to FGFR, including ninetedanib, lenvatinib, and pazopanib.
FGFR-altered HNCs display a distinct tumor immune phenotype
Previous studies show that FGFR signaling alters the tumor microenvironment51,52 and there is evidence that immune checkpoint inhibitor therapy combined with FGFR kinase inhibitor therapy could enhance the anti-tumor activity in FGFR-mutated cancers53,54,55. Therefore, we sought to describe the impact of FGFR-mutated HNC on the tumor immune microenvironment. We leveraged CIBERSORT, Quantiseqr, and ImSig to analyze biological signatures and immune infiltration in the ORIEN RNAseq dataset, where RNAseq samples were available. We divided the FGFR-altered cases by mutation type: FGFR fusions (n = 15), which included fusions in FGFR2 (n = 5) and FGFR3 (n = 10), and FGFR3 point mutations (n = 12). Although there was no difference in cytotoxic T cells (CD8+ T cell) or CD4-activated T cell abundance in the FGFR fusion-positive group compared to the FGFR wildtype group, we observed significant upregulation of both these cell types in the FGFR3 point mutation group (Fig. 6A, B). Additionally, we observed lower levels of CD4 resting T cells in the FGFR3-mutated group compared to wildtype (Fig. 6C). Quantiseqr analysis showed higher levels of natural killer (NK) cells in the FGFR3-mutated group compared to wildtype (Fig. 6D). Interestingly, we observed significant upregulation of ImSig-defined cell proliferation signature in both the FGFR fusion and FGFR-mutated groups (Fig. 6E).

Violin plots of immune cell abundance estimates as measured by CIBERSORT assessing (A) CD8 T cells, (B) Activated CD4 T cells, and (C) Resting CD4 T cells. D Violin plot of NK cell abundance measured by Quantiseqr. E Violin plot of cell proliferation signature measured by ImSig.
Discussion
In this study, we describe the landscape of potentially targetable gene fusions in head and neck cancer (HNC). While several cancer types have various established genomic biomarkers for targeted therapy, squamous cell carcinoma of the head and neck has only one: point mutations in HRAS, which are present in only 3–4% of HNC22. Therefore, we sought to uncover additional targetable alterations in HNC and expand potential therapy opportunities. Overall, we identified 66 clinically relevant gene fusions, with a prevalence of 2.73% (66/2564). This gene fusion prevalence we observed in HNC tumors is comparable with other solid tumor cancers that have been described in literature. Across solid tumors, clinically relevant gene fusions are prevalent in about 1–2% of cancers. Gene fusions that have an approved therapy, including NTRK56, RET57, and FGFR58 are prevalent in 0.3%, 0.8–1%, and 0.5–0.9% of solid tumors, respectively. Specific cancer types like colorectal cancer59, pancreatic cancer60, and non-small lung cancer61 harbor clinically targetable fusions at a rate of ~1%. Additionally, our analysis uncovered EGFR fusions that have not been previously described in HNC. These EGFR fusions had canonical breakpoints previously observed in lung cancer patients who responded to EGFR TKI therapies. Finally, we provide evidence that HNC patients harboring FGFR fusions respond to FGFR TKI inhibitors. Together, these results provide evidence for expanded targeted treatment eligibility in HNC. Previous work has underestimated the prevalence and impact of gene fusions in HNC; however, we show that leveraging large datasets and optimal bioinformatics tools for gene fusion analysis revealed alterations that are biologically impactful and more common than once thought.
We postulate that small cohorts and older bioinformatics tools for gene fusion analysis contributed to the limited knowledge of clinically relevant gene fusions in HNC. Previous studies consisted of less than 100 cases and this population size is insufficient for comprehensive analysis of gene fusions, which have a pan-cancer prevalence of about 1%49. To date, reports of gene fusions in HNC are limited to case studies. There have been some larger genomic studies of HNC; for instance, TCGA investigators performed analysis of HNC (n = 279 tumors) in 2015. Leveraging then contemporary bioinformatics software Mapsplice, they identified only 2 kinase fusions, both involving FGFR343. Mapsplice can identify gene fusions by detecting “splice junctions”, essentially treating the alteration as a splice event. In contrast, newer software designed to detect gene fusion events, such as STAR-Fusion and Arriba, utilize both junction reads as well as discordant read pairs, improving sensitivity to detect additional gene fusions. Discordant read pairs occur where mated reads are mapped to different genes, which may also indicate a gene fusion event. Incorporating both types of reads significantly improves the algorithm’s performance47. Gao et al. analyzed the TCGA cohort using both these and other fusion detection tools for comprehensive fusion analysis49. Out of 9 fusions we observed in TCGA, 6 were observed from this prior study. Following that publication, TCGA re-analyzed the RNA sequencing data for gene fusions using STAR-Fusion and Arriba, from which we observed 3 additional clinically relevant gene fusions in TCGA that have not yet been reported. Given the identification of additional fusions, we sought to curate an even larger cohort of HNCs. We combined the updated TCGA analysis along with large datasets from ORIEN, CPTAC, and Caris Life Sciences and data from 17 published studies to create a combined cohort of 13,655 head and neck cancer cases. To our knowledge, aside from salivary gland cancer studies, this is the largest cohort of HNC cases reported. We applied optimal bioinformatics tools STAR-Fusion and Arriba to this large dataset of RNA samples creating an optimal setting to comprehensively identify and describe clinically targetable gene fusions in HNC. Our methods satisfy the unmet needs of a larger cohort and improved bioinformatics methods for comprehensive gene fusion detection in head and neck cancer cases.
Unexpectedly, our study uncovered 6 novel gene fusions involving EGFR in HNC. EGFR fusions are a rare alteration that occurs in 0.4% of cancer cases62 and has mainly been observed in glioblastoma and non-small lung cancer (NSCLC). Most of these HNC cases harbored breakpoints in the EGFR gene partner that have been observed in responders on EGFR TKI therapy. To our knowledge, this is the first report of potentially targetable EGFR gene fusions in HNC. While EGFR fusions have recently been reported in head and neck cancer, these studies do not distinguish between gain-of-function vs passenger fusion events for the observed breakpoints or structures63,64. For example, recurring EGFR fusion in which only exon 1 is retained49 is likely not functional nor clinically actionable. Our literature curation showed that patients with other cancer types harboring EGFR fusions responded well to EGFR TKI therapies, including one colorectal cancer case65, which has not previously been described in that cancer type. All cases showed benefit from EGFR TKI therapy. Given this and the fact that many EGFR fusion-positive HNC cases in our cohort harbored identical EGFR fusion breakpoints and structures observed in these responders, we suggest that EGFR fusion-positive HNC cases may benefit from EGFR TKI therapies. 5/6 HNC EGFR fusion cases were from the ORIEN database, which includes medication history. As expected, none (0/5) of these patients received EGFR TKI therapy; however, surprisingly, none of them received EGFR therapy cetuximab, a routine treatment for HNC. We speculate that there may be exceptional responders to cetuximab who may have an underlying EGFR gene fusion. In the future, we hope to investigate the response of HNC patients harboring EGFR fusions to cetuximab and EGFR TKI inhibitors. Intrigued by the prevalence of EGFR fusions in HNC, we leveraged our pan-cancer cohort and found additional EGFR fusions across several tumor types in which EGFR fusions have not been reported or not well described (Supplementary Fig. 3). These observations demonstrate that patients with cancers other than brain and lung cancer harbor EGFR fusions that may be clinically targetable.
We found that FGFR alterations are prevalent in 1.8% of our HNC cohort (70 out of 2564). FGFR alterations, consisting of gene amplifications, point mutations, and gene fusions, are one of the most frequently altered receptor tyrosine kinase genes in HNC, making it a promising target for FGFR inhibitor therapies45,66. FGFR fusions had the highest prevalence (16/66) in our fusion-positive HNC cohort (Fig. 1A). This is consistent with the literature, which has reported on FGFR fusion-positive cases in HNC, most frequently FGFR3 fusions45,67,68,69; however, FGFR2 fusions have also been observed in clinical case reports17,70,71. We curated clinical trial studies and case studies that reported FGFR-altered HNC patients who received FGFR TKI therapy27,72,73,74,75,76. Importantly, most FGFR-altered cases achieved at least a partial response on FGFR TKI therapy (Table 1), demonstrating that these alterations could become an effective predictive biomarker for this disease. The patient with the best response to FGFR inhibitor therapy was a 38-year-old female with nasal cavity cancer who harbored a FGFR1 frameshift deletion; she was on nintedanib, a multi-TKI, for 36 months, achieved a tumor shrinkage of 81.4% and a partial response74. Our results show that there is a higher-than-expected prevalence of clinically actionable FGFR fusions in HNC. More work is needed to establish FGFR fusions as a genomic target in HNC, including the arrival of second-generation FGFR inhibitors in clinical trials.
Given the association of FGFR alterations and altered tumor immune microenvironments in other cancers such as bladder cancer, we further characterized these FGFR-altered HNCs. Although evidence was inconclusive for FGFR fusion cases alone due to low sample size, there was a distinct biological difference between FGFR-altered tumors compared to wild type. FGFR-altered tumors display increased cell proliferation according to ImSig’s RNA signature. FGFR-mutant HNC was also associated with lower proportions of resting CD4+ T cells and higher levels of NK cells, CD4+ activated T cells, and CD8+ T cells according to Quantiseqr. NK cells play a significant role in anti-tumor response77. Studies suggest that PD-1 may serve as a checkpoint for NK cells and that PD-1 blockade increases NK cell activity78. CD8+ T cells are a well-established marker for inflammation; there is a positive correlation between CD8+ T cell abundance and progression-free survival on immunotherapy79. Intriguingly, FGFR mutation appears to impact the immune microenvironment differently across cancers. FGFR3 mutated bladder cancer displays an immune suppressive phenotype compared to wildtype measured by lower levels of immune infiltration and increased T cell exhaustion51,80. On the other hand, Zhang et al. showed that FGFR-mutated melanoma is associated with higher levels of CD4+ T cells, consistent with our findings81. Despite this inconsistency, there is evidence in preclinical studies showing that FGFR TKI can enhance tumor immunogenicity in multiple cancers, demonstrating the potential to combine PD-1 immunotherapy with FGFR TKI in therapy strategies82,83,84. We postulate that HNC patients with FGFR mutations may benefit from combined therapy options.85. We acknowledge that this sample size is limited (n = 28); therefore, an expanded cohort of FGFR-mutant HNC is needed to further understand the unique associated biological phenotypes.
Although our study identified clinically relevant gene fusions, there were some important limitations. Our results about the distribution of gene fusions in head and neck cancer derived from a combined discovery dataset (n = 2564) and the Caris Life Sciences dataset (n = 11,091). The distributions of cohorts vary due to their differing purposes. The discovery cohort of combined data was assembled and analyzed with the intent of uncovering clinically relevant gene fusions in head and neck cancer. Caris’s bioinformatics pipeline was developed to identify and confirm the presence of established clinically targetable alterations as part of a tumor diagnostic report; thus, the filtering methods are more stringent to ensure true positive results.
Another limitation of our study was the sampling bias of our cohort. Treatment protocols differ for each anatomical site in head and neck cancer; tumors in sites that are more often surgically resectable, such as the oral cavity, are more frequently analyzed for genomic sequencing than sites less accessible including the larynx and nasal cavity, leading to an over-representation of surgically resectable cancer types. One study estimated that the most common anatomical site of HNC was oropharynx (31.1%) followed by the oral cavity (29.1%), then larynx (28.6%)86. In contrast, in our HNC cohort, the most common anatomical site was oral cavity (40.4%), followed by oropharynx (30.0%) and larynx (18.0%), showing an over-representation of oral cavity tumors. It is evident that there is a need for more routine genomic sequencing for all head and neck tumors to comprehensively understand the prevalence of targetable alterations in this cancer type. Another limitation was the sample specimens analyzed, specifically from the ORIEN cohort. For FGFR alteration immune phenotyping analysis, we leveraged RNA sequencing data, 15.6% of which was derived from lymph nodes. Because lymph nodes are the source of immune cells, resulting in higher levels of immune infiltration than other sample sites, this may influence our observed results. However, there was an even distribution of lymph node-derived samples between the mutated and wild-type groups. We uncovered targetable alterations, and gene fusions, leveraging RNA sequencing data that are novel to head and neck cancer. DNA sequencing can be analyzed for somatic mutations, copy number alterations, and other aberrations, and is applied more often than RNA sequencing data for tumor genomic profiling. Although there are bioinformatics algorithms that can detect genomic structural variations that may include gene fusions, RNA sequencing is more optimal for gene fusion detection than DNA sequencing methods and DNA-based methods often miss gene fusion events detected in RNA sequencing87. Our results show that RNA sequencing is an important genomic analysis that should be incorporated with DNA sequencing methods for more complete tumor profiling to detect targetable alterations that would otherwise be missed.
Our findings from this study provide future directions to study targetable alterations in head and neck cancer. Further characterization of the immune microenvironment is warranted in FGFR-altered head and neck cancer to assess the potential benefit of combination therapy including PD-1 immunotherapy and FGFR TKI’s. Finally, given the frequent EGFR fusions with canonical, we observed in our pan-cancer cohorts, we propose a pan-cancer clinical trial treating EGFR fusion-positive cancers with EGFR TKI therapy to help establish this alteration as a biomarker for targeted therapy. Together these pursuits will expand our knowledge of the targetable alterations prevalent in head and neck cancer and help establish additional biomarkers for precision therapies. In conclusion, these results comprise the largest comprehensive evaluation of gene fusions in HNC. Although these alterations are rare, they occur at a prevalence comparable to clinically targetable HRAS alterations, demonstrating that this study may help contribute toward providing expanded therapeutic opportunities for HNC patients.
Methods
Data acquisition
We leveraged various data sources to create an expanded cohort of 2564 HNC cases. These sources include ORIEN (n = 1538), TCGA (n = 528), CPTAC (n = 110), and 17 published datasets from Database of Genotypes and Phenotypes (dbGaP) (https://www.ncbi.nlm.nih.gov/gap/) and ArrayExpress (https://www.ebi.ac.uk/biostudies/arrayexpress) (n = 485) (Supplementary Table 1). Samples from these datasets were derived from primary sites and metastatic sites where primary samples weren’t available. We included tumor samples from head and neck anatomical sites including oral cavity, oropharynx, larynx, sinus, hypopharynx, nasal cavity, and nasopharynx. Additionally, we included cases of all histological types, including squamous cell carcinoma, adenocarcinoma, spindle cell neoplasm, and basaloid carcinoma. For the purpose of this study, we excluded salivary gland cancer cases. We annotated sample histology type, including keratinizing status, according to how it was reported in the data source, where that information was available. ORIEN, we obtained 1619 RNA sequencing data and 1470 whole exome sequencing data from head and neck cancer patients from 1538 patients. Additionally, in collaboration with Caris Life Sciences, we evaluated de-identified real-world data on gene fusions from whole transcriptome sequencing in a cohort of 11,091 head and neck cancer patients. For fusion calling, BAM files from STAR aligner were processed for RNA variants using a proprietary custom detection pipeline; fusions were classified as pathogenic or likely pathogenic by board-certified molecular geneticists according to ACMG criteria.
Gene fusion calling
We downloaded fusion output from STAR-Fusion (v1.6.0) and Arriba (v1.1.0) from cases from TCGA and CPTAC from the Genomic Data Commons (GDC) (https://portal.gdc.cancer.gov/). STAR-Fusion (v1.8.0) and Arriba (v1.1.0) were used to analyze RNA sequencing samples from ORIEN for gene fusions. We applied the following filters to identify high-quality fusion calls. First, we kept fusion calls that were detected by both tools. For calls only detected by STAR-Fusion, we selected for calls with a STAR-Fusion calculated fusion fragments per million total RNA-seq fragments (FFPM) score ≥ 0.1, as recommended by the authors. For calls only detected by Arriba, we selected calls that had a confidence score of “medium” or “high”. Finally, for fusions involving a kinase gene, we selected candidates with a fully retained kinase ___domain.
Mutation analysis
We downloaded mutation analysis data in the form of mafs from TCGA and CPTAC from the GDC. Specifically, we selected the maf files that were generated using an ensemble approach from mutation software: Mutect288, VarScan289, Pindel90, and MuSE91. Similarly, we performed mutation analysis for whole exome samples from ORIEN using Mutect2 (generated by using GATK v4.3.0.0), VarScan2 (v2.3.5), and MuSE (v2.0), and using software SomaticCombiner (v1.02)92 to combine the three outputs. Tumor mutational burden was calculated by taking the number of somatic mutations divided by the capture region for each sample. To assess statistically significant mutational associations with gene fusion status, we leveraged OncoKB93 to select for mutations that were considered “Oncogenic” or “Likely Oncogenic”.
RNAseq analysis
For each dataset (ORIEN, TCGA, and dbGaP/ArrayExpress studies), we performed batch correction on the RNA expression outputs using CombatSeq and institution of origin for batch94. For the data from dbGaP and Array Express (n = 488), we performed batch correction on the combined cohort and treated each individual study as a batch. To determine fusion-positive samples that were outliers in gene expression of the oncogenic fusion partner, we converted the batch-adjusted counts to transcript per million (TPM). Samples were considered overexpressed if the expression value of the oncogenic gene partner was above the third percentile in the entire cohort (ORIEN: N = 1618 samples, TCGA: N = 527 samples). Samples were considered outliers in expression if they exceeded the value above the threshold using Tukey’s rule for outliers (1.5 Inner Quartile Range + 3rd Quartile). Z scores were calculated by applying the Python package z score, which takes expression counts and determines the z score relative to the cohort mean and standard deviation.
To assess for overexpression across all fusion cases, we converted the expression values to z scores for each gene. Next, we took the z scores from fusion-positive cases of their clinically relevant gene fusion partner and compared that to the rest of all the z scores. We used a Mann–Whitney U-test to assess significance.
We leveraged RNA-based software VirTect95 to assess for the presence of human papillomavirus (HPV) in tumors.
We leveraged various software packages to assess the immune phenotypes associated with fusion-positive samples. To estimate immune cell-type proportions and signatures, we used CIBERSORT96, Quantiseqr96, and ImSig97.
Code availability
The code used for analysis and to generate figures is available upon request to the corresponding author.
References
Hashibe, M. et al. Alcohol drinking in never users of tobacco, cigarette smoking in never drinkers, and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. J. Natl. Cancer Inst. 99, 777–789 (2007).
Proia, N. K. et al. Smoking and smokeless tobacco-associated human buccal cell mutations and their association with oral cancer-a review. Cancer Epidemiol. Biomark. Prev. 15, 1061–1077 (2006).
Oosting, S. F. & Haddad, R. I. Best practice in systemic therapy for head and neck squamous cell carcinoma. Front. Oncol. 9, 815 (2019).
Krishnamurthy, S. et al. The dogma of Cetuximab and Radiotherapy in head and neck cancer - A dawn to dusk journey. Clin. Transl. Radiat. Oncol. 34, 75–81 (2022).
Chung, C. H. et al. Phase II multi-institutional clinical trial result of concurrent cetuximab and nivolumab in recurrent and/or metastatic head and neck squamous cell carcinoma. Clin. Cancer Res. 28, 2329–2338 (2022).
Bonner, J. A. et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 11, 21–28 (2010).
Botticelli, A. et al. Anti-PD-1 and anti-PD-L1 in head and neck cancer: a network meta-analysis. Front. Immunol. 12, 705096 (2021).
Bauml, J. M., Aggarwal, C. & Cohen, R. B. Immunotherapy for head and neck cancer: where are we now and where are we going?. Ann. Transl. Med. 7, S75 (2019).
Hashemi-Sadraei, N., Sikora, A. G. & Brizel, D. M. Immunotherapy and checkpoint inhibitors in recurrent and metastatic head and neck cancer. Am. Soc. Clin. Oncol. Educ. Book 35, e277–e282 (2016).
Sacco, A. G. et al. Pembrolizumab plus cetuximab in patients with recurrent or metastatic head and neck squamous cell carcinoma: an open-label, multi-arm, non-randomised, multicentre, phase 2 trial. Lancet Oncol. 22, 883–892 (2021).
Gulati, S. et al. Durvalumab plus cetuximab in patients with recurrent or metastatic head and neck squamous cell carcinoma: an open-label, nonrandomized, phase II clinical trial. Clin. Cancer Res. 29, 1906–1915 (2023).
Shah, P. A., et al. NOTCH1 signaling in head and neck squamous cell carcinoma. Cells 9, 2677 (2020).
Shao, X. et al. Mutational analysis of the PTEN gene in head and neck squamous cell carcinoma. Int. J. Cancer 77, 684–688 (1998).
Poetsch, M., Lorenz, G. & Kleist, B. Detection of new PTEN/MMAC1 mutations in head and neck squamous cell carcinomas with loss of chromosome 10. Cancer Genet. Cytogenet. 132, 20–24 (2002).
Lui, V. W. et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 3, 761–769 (2013).
Sen, M. et al. JAK kinase inhibition abrogates STAT3 activation and head and neck squamous cell carcinoma tumor growth. Neoplasia 17, 256–264 (2015).
Chu, Y. H. et al. FGFR1/2/3-rearranged carcinoma of the head and neck: expanded histological spectrum crossing path with high-risk HPV in the sinonasal tract. Histopathology 84, 589–600 (2024).
Yuan, L. et al. Recurrent FGFR3-TACC3 fusion gene in nasopharyngeal carcinoma. Cancer Biol. Ther. 15, 1613–1621 (2014).
Brauswetter, D. et al. Copy number gain of PIK3CA and MET is associated with poor prognosis in head and neck squamous cell carcinoma. Virchows Arch. 468, 579–587 (2016).
Seiwert, T. Y. et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin. Cancer Res. 21, 632–641 (2015).
Dou, S. et al. EGFR mutation and 11q13 amplification are potential predictive biomarkers for immunotherapy in head and neck squamous cell carcinoma. Front. Immunol. 13, 813732 (2022).
Coleman, N. et al. HRAS mutations define a distinct subgroup in head and neck squamous cell carcinoma. JCO Precis Oncol. 7, e2200211 (2023).
Friedlaender, A. et al. Oncogenic alterations in advanced NSCLC: a molecular super-highway. Biomark. Res. 12, 24 (2024).
Haslam, A., Kim, M. S. & Prasad, V. Updated estimates of eligibility for and response to genome-targeted oncology drugs among US cancer patients, 2006-2020. Ann. Oncol. 32, 926–932 (2021).
Koivunen, J. P. et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin. Cancer Res. 14, 4275–4283 (2008).
Drilon, A. et al. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: integrated analysis of three phase 1-2 trials. Lancet Oncol. 21, 261–270 (2020).
Meric-Bernstam, F. et al. Futibatinib, an irreversible FGFR1-4 inhibitor, in patients with advanced solid tumors harboring FGF/FGFR aberrations: a phase I dose-expansion study. Cancer Discov. 12, 402–415 (2022).
Abou-Alfa, G. K. et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 21, 671–684 (2020).
Hoy, S. M. Pemigatinib: first approval. Drugs 80, 923–929 (2020).
Cascetta, P. et al. RET inhibitors in non-small-cell lung cancer. Cancers 13, 4415 (2021).
O’Haire, S. et al. Systematic review of NTRK 1/2/3 fusion prevalence pan-cancer and across solid tumours. Sci. Rep. 13, 4116 (2023).
Di Federico, A. et al. EGFR-RAD51 gene fusion NSCLC responsiveness to different generation EGFR-TKIs: two cases and review of the literature. Transl. Lung Cancer Res.11, 497–503 (2022).
Ross, J. S. et al. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int. J. Cancer 138, 881–890 (2016).
Liu, S. V. et al. Real-world outcomes associated with afatinib use in patients with solid tumors harboring NRG1 gene fusions. Lung Cancer 188, 107469 (2024).
Fang, S. G. et al. BCL6-SPECC1L: a novel fusion gene in nasopharyngeal carcinoma. Technol. Cancer Res. Treat. 21, 15330338221139981 (2022).
Valouev, A. et al. Discovery of recurrent structural variants in nasopharyngeal carcinoma. Genome. Res. 24, 300–309 (2014).
Chung, G. T. et al. Identification of a recurrent transforming UBR5-ZNF423 fusion gene in EBV-associated nasopharyngeal carcinoma. J. Pathol. 231, 158–167 (2013).
Kuo, Y. J. et al. DEK-AFF2 fusion-associated papillary squamous cell carcinoma of the sinonasal tract: clinicopathologic characterization of seven cases with deceptively bland morphology. Mod. Pathol. 34, 1820–1830 (2021).
Wang, J. et al. Nasopharyngeal papillary adenocarcinoma harboring a fusion of ROS1 with GOPC: a case report. Medecine100, e24377 (2021).
Wang, J. et al. Interfering expression of chimeric transcript SEPT7P2-PSPH promotes cell proliferation in patients with nasopharyngeal carcinoma. J. Oncol. 2019, 1654724 (2019).
Zhong, Q. et al. The RARS-MAD1L1 fusion gene induces cancer stem cell-like properties and therapeutic resistance in nasopharyngeal carcinoma. Clin. Cancer Res. 24, 659–673 (2018).
Bubola, J. et al. A poorly differentiated non-keratinizing sinonasal squamous cell carcinoma with a novel ETV6-TNFRSF8 fusion gene. Head. Neck Pathol. 15, 1284–1288 (2021).
Lawrence, M. S. et al. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517, 576–582 (2015).
Wang, K. et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 38, e178 (2010).
Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517, 576–582 (2015).
Stransky, N. et al. The landscape of kinase fusions in cancer. Nat. Commun. 5, 4846 (2014).
Haas, B. J. et al. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 20, 213 (2019).
Keller, H. et al. Arriba: effects of an educational intervention on prescribing behaviour in prevention of CVD in general practice. Eur. J. Prev. Cardiol. 19, 322–329 (2012).
Gao, Q. et al. Driver fusions and their implications in the development and treatment of human cancers. Cell Rep. 23, 227–238.e3 (2018).
Krook, M. A., et al. Tumor heterogeneity and acquired drug resistance in FGFR2-fusion-positive cholangiocarcinoma through rapid research autopsy. Cold Spring Harb. Mol. Case Stud. 5, a004002 (2019).
Robinson, B. D. et al. Upper tract urothelial carcinoma has a luminal-papillary T-cell depleted contexture and activated FGFR3 signaling. Nat. Commun. 10, 2977 (2019).
Yang, C. et al. Comprehensive analysis of the prognostic value and immune infiltration of FGFR family members in gastric cancer. Front. Oncol. 12, 936952 (2022).
Palakurthi, S. et al. The combined effect of FGFR inhibition and PD-1 blockade promotes tumor-intrinsic induction of antitumor immunity. Cancer Immunol. Res. 7, 1457–1471 (2019).
Lee, H. W. & Seo, H. K. Fibroblast growth factor inhibitors for treating locally advanced/metastatic bladder urothelial carcinomas via dual targeting of tumor-specific oncogenic signaling and the tumor immune microenvironment. Int. J. Mol. Sci. 22, 9526 (2021).
Ruan, R. et al. Unleashing the potential of combining FGFR inhibitor and immune checkpoint blockade for FGF/FGFR signaling in tumor microenvironment. Mol. Cancer 22, 60 (2023).
Westphalen, C. B. et al. Genomic context of NTRK1/2/3 fusion-positive tumours from a large real-world population. NPJ Precis. Oncol. 5, 69 (2021).
Parimi, V. et al. Genomic landscape of 891 RET fusions detected across diverse solid tumor types. NPJ Precis. Oncol. 7, 10 (2023).
Murugesan, K. et al. Pan-tumor landscape of fibroblast growth factor receptor 1-4 genomic alterations. ESMO Open 7, 100641 (2022).
Pagani, F. et al. The landscape of actionable gene fusions in colorectal cancer. Int. J. Mol. Sci. 20, 5319 (2019).
Gkountakos, A. et al. Fusion genes in pancreatic tumors. Trends Cancer 10, 430–443 (2024).
Seker-Cin, H. et al. Analysis of rare fusions in NSCLC: genomic architecture and clinical implications. Lung Cancer 184, 107317 (2023).
Schubert, L. et al. ERBB family fusions are recurrent and actionable oncogenic targets across cancer types. Front. Oncol. 13, 1115405 (2023).
Nair, S., J. A. Bonner, & Bredel, M. EGFR mutations in head and neck squamous cell carcinoma. Int. J. Mol. Sci. 23, 3818 (2022).
Zhang, H. et al. Distribution of EGFR fusions in 35,023 Chinese patients with solid tumors-the frequency, fusion partners and clinical outcome. World J. Surg. Oncol. 22, 194 (2024).
Li, Y. et al. A rare EGFR-SEPT14 fusion in a patient with colorectal adenocarcinoma responding to erlotinib. Oncologist 25, 203–207 (2020).
Wang, Z. & Anderson, K. S. Therapeutic targeting of FGFR signaling in head and neck cancer. Cancer J. 28, 354–362 (2022).
Pant, S. et al. Erdafitinib in patients with advanced solid tumours with FGFR alterations (RAGNAR): an international, single-arm, phase 2 study. Lancet Oncol. 24, 925–935 (2023).
Rodón, J. et al. Pemigatinib in previously treated solid tumors with activating FGFR1-FGFR3 alterations: phase 2 FIGHT-207 basket trial. Nat. Med. 30, 1645–1654 (2024).
Subbiah, V. et al. FIGHT-101, a first-in-human study of potent and selective FGFR 1-3 inhibitor pemigatinib in pan-cancer patients with FGF/FGFR alterations and advanced malignancies. Ann. Oncol. 33, 522–533 (2022).
Rodón, J. et al. Publisher correction: pemigatinib in previously treated solid tumors with activating FGFR1-FGFR3 alterations: phase 2 FIGHT-207 basket trial. Nat. Med. 30, 2377 (2024).
Hammerman, P. S., Hayes, D. N. & Grandis, J. R. Therapeutic insights from genomic studies of head and neck squamous cell carcinomas. Cancer Discov. 5, 239–244 (2015).
Garmezy, B. et al. A phase I study of KIN-3248, an irreversible small-molecule pan-FGFR inhibitor, in patients with advanced FGFR2/3-driven solid tumors. Cancer Res. Commun. 4, 1165–1173 (2024).
Yang, J. et al. Case report lenvatinib is an effective treatment for refractory nasopharyngeal carcinoma harboring FGFR3-TACC fusion: a case report. In. J. Clin. Exp. Med. 16, 121–126 (2023).
Kim, K. H. et al. A phase II trial of nintedanib in patients with metastatic or recurrent head and neck squamous cell carcinoma: in-depth analysis of nintedanib arm from the KCSG HN 15-16 TRIUMPH trial. Cancer Res Treat. 56, 37–47 (2024).
Guo, Y. et al. Phase I results of gunagratinib (ICP-192), a highly selective irreversible FGFR 1-4 inhibitor in patients with head and neck cancer harboring FGF/FGFR gene aberrations. Am. Soc. Clin. Oncol. 39, 4092–4092 (2022).
Liao, R. G. et al. Inhibitor-sensitive FGFR2 and FGFR3 mutations in lung squamous cell carcinoma. Cancer Res. 73, 5195–5205 (2013).
Hsu, J. et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Investig. 128, 4654–4668 (2018).
Quatrini, L. et al. The immune checkpoint PD-1 in natural killer cells: expression, function and targeting in tumour immunotherapy. Cancers 12, 3285 (2020).
Rosenberg, J. & Huang, J. CD8(+) T cells and NK cells: parallel and complementary soldiers of immunotherapy. Curr. Opin. Chem. Eng. 19, 9–20 (2018).
Song, Y., et al., Fibroblast growth factor receptor 3 mutation attenuates response to immune checkpoint blockade in metastatic urothelial carcinoma by driving immunosuppressive microenvironment. J. Immunother Cancer 11, e006643 (2023).
Zhang, W. et al. Fibroblast growth factor receptor family mutations as a predictive biomarker for immune checkpoint inhibitors and its correlation with tumor immune microenvironment in melanoma. Front. Immunol. 13, 1030969 (2022).
Wu, Y. et al. FGFR blockade boosts T cell infiltration into triple-negative breast cancer by regulating cancer-associated fibroblasts. Theranostics 12, 4564–4580 (2022).
Kono, M. et al. Immunomodulation via FGFR inhibition augments FGFR1 targeting T-cell based antitumor immunotherapy for head and neck squamous cell carcinoma. Oncoimmunology 11, 2021619 (2022).
Adachi, Y. et al. Inhibition of FGFR reactivates IFNγ signaling in tumor cells to enhance the combined antitumor activity of lenvatinib with anti-PD-1 antibodies. Cancer Res. 82, 292–306 (2022).
Zhang, B. et al. Characteristics of the immune microenvironment and their clinical significance in non-small cell lung cancer patients with ALK-rearranged mutation. Front. Immunol. 13, 974581 (2022).
Fakhry, C. et al. Head and neck squamous cell cancers in the United States are rare and the risk now is higher among white individuals compared with black individuals. Cancer 124, 2125–2133 (2018).
Bruno, R. & Fontanini, G. Next generation sequencing for gene fusion analysis in lung cancer: a literature review. Diagnostics 10, 521 (2020).
Benjamin, D. et al. Calling somatic SNVs and indels with mutect2. bioRxiv, 861054 (2019).
Koboldt, D. C. et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome. Res. 22, 568–576 (2012).
Ye, K. et al. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871 (2009).
Fan, Y. et al. MuSE: accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data. Genome. Biol. 17, 178 (2016).
Wang, M. et al. SomaticCombiner: improving the performance of somatic variant calling based on evaluation tests and a consensus approach. Sci. Rep. 10, 12898 (2020).
Chakravarty, D. et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol. 2017, 00011 (2017).
Zhang, Y., Parmigiani, G. & Johnson, W. E. ComBat-seq: batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2, lqaa078 (2020).
Khan, A. et al. Detection of human papillomavirus in cases of head and neck squamous cell carcinoma by RNA-seq and VirTect. Mol. Oncol. 13, 829–839 (2019).
Finotello, F. et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome. Med. 11, 34 (2019).
Nirmal, A. J. et al. Immune cell gene signatures for profiling the microenvironment of solid tumors. Cancer Immunol. Res. 6, 1388–1400 (2018).
Acknowledgements
We would like to thank the Ohio Supercomputer Center (https://www.osc.edu/) for providing the Owens cluster on which this work was performed. We would like to thank Barb Hughes and Jenny Badillo for their administrative assistance.
Author information
Authors and Affiliations
Contributions
E.L.H.—conceptualization, data curation, data analysis, methods, project administration, software implementation, data validation, data visualization, manuscript writing; R.V.—conceptualization, data analysis, methods, software implementation, data validation, data visualization, manuscript writing, manuscript review, and editing; J.W.R.—conceptualization, project supervisor, data visualization, validation, manuscript writing; MRW- conceptualization, project supervisor, data visualization, validation, manuscript review, and editing; E.S.—data analysis, methods, software implementation, manuscript writing, manuscript review and editing; A.T.—data analysis, methods L.S.—data analysis, data visualization; E.B.—data curation; Z.A.R.—data analysis; L.Y.—conceptualization, supervision, methods; M.C.—resources; N.S.— resources; J.C.—resources; M.M.—resources; M.J.C.—resources; M.R. —resources; C.S.—resources; K.N.—resources; M.P.—resources; B.S. —resources; S.E.—resources; A.Z.—resources; resources—T.W.D.; M.K.W.—resources; S.R.—conceptualization, funding acquisition, methods, project administration, resources, supervision, data validation, manuscript review and editing.
Corresponding author
Ethics declarations
Competing interests
S.R. participated in Advisory Boards for Incyte Corporation (2017, 2023), AbbVie, Inc. (2017), QED Therapeutics (2018, 2019), Bayer (2020), Seagen (2023), Tyra Biosciences (2023), Taiho (2024). [Health care companies) (<10,000 USD). S.R. received honoraria from IDT Integrated DNA Technologies (2017), Illumina (2018). [Technology companies]. S.R. received consulting fees from QED Therapeutics (2018, 2019) and Merck (2019). [Health care companies] (<10,000 USD). S.R. received travel reimbursement from Incyte Corporation (2019)(Less than 999 USD). T.W.D. Consulting/Ad Board; Caris Life Sciences, NEED Inc, Merck & Co., EMD Serono, Adlai Nortye, Genmab, Replimmune; Stock: High EnrollClinical trial funding: Merck & Co., BMS, Janssen, AstraZeneca, Tesaro/GSK
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hoskins, E.L., Vella, R., Reeser, J.W. et al. Prevalence and biological impact of clinically relevant gene fusions in head and neck cancers. npj Precis. Onc. 9, 221 (2025). https://doi.org/10.1038/s41698-025-00889-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-025-00889-7
