
Abstract
The rapid adoption of machine learning in various scientific domains calls for the development of best practices and community agreed-upon benchmarking tasks and metrics. We present Matbench Discovery as an example evaluation framework for machine learning energy models, here applied as pre-filters to first-principles computed data in a high-throughput search for stable inorganic crystals. We address the disconnect between (1) thermodynamic stability and formation energy and (2) retrospective and prospective benchmarking for materials discovery. Alongside this paper, we publish a Python package to aid with future model submissions and a growing online leaderboard with adaptive user-defined weighting of various performance metrics allowing researchers to prioritize the metrics they value most. To answer the question of which machine learning methodology performs best at materials discovery, our initial release includes random forests, graph neural networks, one-shot predictors, iterative Bayesian optimizers and universal interatomic potentials. We highlight a misalignment between commonly used regression metrics and more task-relevant classification metrics for materials discovery. Accurate regressors are susceptible to unexpectedly high false-positive rates if those accurate predictions lie close to the decision boundary at 0 eV per atom above the convex hull. The benchmark results demonstrate that universal interatomic potentials have advanced sufficiently to effectively and cheaply pre-screen thermodynamic stable hypothetical materials in future expansions of high-throughput materials databases.
Similar content being viewed by others

Main
The challenge of evaluating, benchmarking and then applying the rapid evolution of machine learning (ML) models is common across scientific domains. Specifically, the lack of agreed-upon tasks and datasets can obscure the performance of the model, making comparisons difficult. Materials science is one such ___domain, where in the last decade, the numbers of ML publications and associated models have increased dramatically. Similar to other domains, such as drug discovery and protein design, the ultimate success is often associated with the discovery of a new material with specific functionality. In the combinatorial sense, materials science can be viewed as an optimization problem of mixing and arranging different atoms with a merit function that captures the complex range of properties that emerge. To date, ~105 combinations have been tested experimentally1,2, ~107 have been simulated3,4,5,6,7 and upwards of ~1010 possible quaternary materials are allowed by electronegativity and charge-balancing rules8. The space of quinternaries and higher is even less explored, leaving vast numbers of potentially useful materials to be discovered. The discovery of new materials is a key driver of technological progress and lies on the path to more efficient solar cells, lighter and longer-lived batteries, and smaller and more efficient transistor gates, just to name a few. In light of our sustainability goals, these advances cannot come fast enough. Any speed-up new discovery methods might yield should be leveraged to the fullest extent.
Computational materials discovery continues to present notable challenges despite advances in theory and methodology. The process typically requires performing extensive high-throughput calculations, which can be computationally intensive and time-consuming. Moreover, the complex relationship between structure and properties means that finding materials with desired characteristics often remains more art than science. ML approaches offer promising alternatives by efficiently identifying patterns within large datasets. These methods excel at handling multidimensional data, balancing multiple optimization objectives9, quantifying prediction uncertainty10,11,12 and extracting meaningful information from sparse or noisy data13,14. These capabilities make ML particularly valuable as a complementary tool to traditional computational methods in materials science.
In particular, we focus on the role of ML to accelerate the use of Kohn–Sham density functional theory (DFT) in the materials discovery pipeline. In comparison with other simulation frameworks, DFT offers a compelling compromise between fidelity and cost that has seen it adopted as a workhorse method by the computational materials science community. The great strengths of DFT as a methodology have led it to demand up to 45% of core hours at the UK-based Archer2 Tier 1 supercomputer15 and over 70% allocation time in the materials science sector at the National Energy Research Scientific Computing Center16,17. This heavy resource requirement drives demand for ways to reduce or alleviate its computational burden, such as efficiency improvements or substitution from ML approaches.
While typically exhibiting lower accuracy and reliability, ML models produce results notably faster—by orders of magnitude—than ab initio simulations. This speed advantage positions them ideally for high-throughput screening campaigns, where they can act as efficient pre-filters for computationally demanding, higher-fidelity methods such as DFT. The pioneering work of Behler and Parrinello18 demonstrated the use of neural networks to learn the DFT potential energy surface (PES). This breakthrough spurred rapid advancements and extensive efforts to train increasingly sophisticated ML models on available PES data. Early applications often involved deploying these models as interatomic potentials (or force fields) focused on specific materials, a process necessitating the creation of bespoke training datasets for each system under investigation19,20. As larger and more diverse datasets have emerged from initiatives such as the Materials Project (MP)3, AFLOW5 or the Open Quantum Materials Database4, researchers have begun to train so-called universal models that cover 90 or more of the most application-relevant elements in the periodic table. This opens up the prospect of ML-guided materials discovery to increase the hit rate of stable crystals and speed up DFT- and expert-driven searches.
Progress in ML for materials is often measured according to performance on standard benchmark datasets. As ML models have grown in complexity and applicability, benchmark datasets need to grow with them to accurately measure their usefulness. However, due to the rapid pace of the field and the variety of possible approaches for framing the discovery problem, no large-scale benchmark yet exists for measuring the ability of ML to accelerate materials discovery. As a result, it is unclear which methodologies or models are best suited for this task. The materials community has explored several approaches for computational discovery, including coordinate-free predictors that operate without requiring precise atomic positions11, sequential optimization methods based on Bayesian principles21 and physics-informed interatomic potentials with universal element coverage22,23,24. While each approach has demonstrated success in specific contexts, systematic comparison across methodologies has been lacking, preventing clear identification of optimal approaches for materials discovery at scale. Our work aims to identify the state-of-the-art model by proposing an evaluation framework that closely simulates a real-world discovery campaign guided by ML models. Our analysis reveals that universal interatomic potentials (UIPs) surpass all other methodologies we evaluated in terms of both accuracy and robustness.
We hope that creation of benchmarks following this framework creates a pathway through which interdisciplinary researchers with limited materials science backgrounds can contribute usefully to model architecture and methodology development on a relevant task and thereby aid progress in materials science. This work expands on initial research conducted in J.R.’s PhD thesis25.
Evaluation framework for materials discovery
This work proposes a benchmark task designed to address four fundamental challenges that we believe are essential to justify the effort of experimentally validating ML predictions:
-
(1)
Prospective benchmarking: Idealized and overly simplified benchmarks may not adequately capture the challenges encountered in real-world applications. This disconnect can arise from selecting inappropriate targets14 or using unrepresentative data splits26,27. For small datasets of materials properties, ‘Leave-Out’ data splitting strategies are often used to assess model performance28,29,30. However, in our target ___domain large quantities of diverse data (~105) are available and hence retrospective splitting strategies predicated on clustering can end up testing artificial or unrepresentative use cases. This encourages using new sources of prospectively generated test data to understand application performance. Adopting this principle, the intended discovery workflow should be used to generate the test data, leading to a substantial but realistic covariate shift between the training and test distributions that gives a much better indicator of likely performance on additional application of the same discovery workflow.
-
(2)
Relevant targets: For materials discovery, high-throughput DFT formation energies are widely used as regression targets but do not directly indicate thermodynamic stability or synthesizability. The true stability of a material depends on its energetic competition with other phases in the same chemical system, quantified by the distance to the convex hull phase diagram. This distance serves as the primary indicator of (meta-)stability under standard conditions31, making it a more suitable target despite other factors such as kinetic and entropic stabilization that influence real-world stability but are more expensive to simulate, especially at scale. Additionally, ML models that require relaxed structures as input create a circular dependency with the DFT calculations they are meant to accelerate, reducing their practical utility for discovery.
-
(3)
Informative metrics: Global metrics such as mean absolute error (MAE), root mean squared error (RMSE) and R2 may provide practitioners with misleading confidence regarding model reliability. Even models with strong regression performance can produce unexpectedly high rates of false-positive predictions when nominally accurate estimates fall near decision boundaries, resulting in substantial opportunity costs through wasted laboratory resources and time. Consequently, models should be evaluated based on their ability to facilitate correct decision-making patterns rather than regression accuracy alone. One effective approach is to define selection criteria and assess regression models primarily by their classification performance.
-
(4)
Scalability: Future materials discovery efforts are likely to target broad chemical spaces and large data regimes. Small benchmarks can lack chemical diversity, and obfuscate poor scaling relations or weak out-of-distribution performance. For instance, random forests achieve excellent performance on small datasets but are typically outperformed by neural networks on large datasets due to the benefits of representation learning32. While we propose that large training sets are necessary to adequately differentiate the ability of models to learn in the larger data regime, given the enormous size of the configurational space of materials yet to be explored, we also propose that it is important to construct a task where the test set is larger than the training set to mimic true deployment at scale. No other inorganic materials benchmarks test the prospects of large-scale deployment in this manner.
We highlight two specific benchmarking efforts that have partially addressed the above challenges: Matbench33 and the Open Catalyst Project (OCP)34. Other valuable efforts such as MatSciML35 and JARVIS-Leaderboard36 aggregate a wide variety of materials science-related benchmark tasks, including from Matbench and OCP, but do not introduce distinct benchmarking design patterns to those seen in Matbench or the OCP.
By providing a standardized collection of 13 datasets ranging in size from ~300 to ~132,000 samples from both DFT and experimental sources, Matbench addresses the scalability challenge, highlighting how model performance changes as a function of data regime. Matbench helped focus the field of ML for materials, increase comparability across papers and provide a quantitative measure of progress in the field. Importantly, all tasks were exclusively concerned with the properties of known materials. We believe a task that simulates a materials discovery campaign by requiring materials stability prediction from unrelaxed structures to be a missing piece here.
OCP is a large-scale initiative aimed at discovering substrate–adsorbate combinations that can catalyse critical industrial reactions, transforming these adsorbates into more useful products. The OCP has released two datasets thus far, OCP20 (ref. 34) and OCP22 (ref. 37), for training and benchmarking ML models. OCP certainly addressed challenge 1 of closely mimicking a real-world problem by recently showing that despite not reaching their target accuracy to entirely replace DFT, using ML in conjunction with confirmatory DFT calculations dramatically speeds up their combinatorial screening workflow38. The team behind the OCP has a second initiative targeting materials for direct air capture called OpenDAC that has shared the ODAC23 dataset39. The OpenDAC benchmark is set up identically to the OCP.
We believe that addressing these four challenges will result in benchmarks that enable future ML-guided discovery efforts to confidently select appropriate models and methodologies for the expansion of computational materials databases. Figure 1 provides an overview of how data is used in our proposed Matbench Discovery framework.
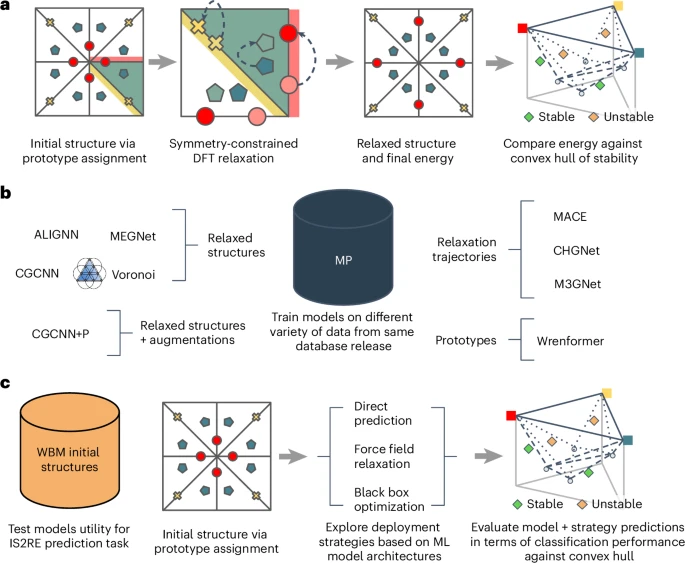
a, A conventional prototype-based discovery workflow where different elemental assignments to the sites in a known prototype are used to create a candidate structure. This candidate is relaxed using DFT to arrive at a relaxed structure that can be compared against a reference convex hull. This sort of workflow was used to construct the WBM dataset. b, Databases such as the MP provide a rich set of data that different academic groups have used to explore different types of models. While earlier work tended to focus on individual modalities, our framework enables consistent model comparisons across modalities. c, The proposed test evaluation framework where the end user takes an ML model and uses it to predict a relaxed energy given an initial structure (IS2RE). This energy is then used to make a prediction as to whether the material will be stable or unstable with respect to a reference convex hull. From an applications perspective, this classification performance is better aligned with intended use cases in screening workflows.
Results
Table 1 shows performance metrics for all models included in the initial release of Matbench Discovery reported on the unique protostructure subset. EquiformerV2 + DeNS achieved the highest performance in ML-guided materials discovery, surpassing all other models across the nine reported metrics. When computing metrics in the presence of missing values or obviously pathological predictions (error of 5 eV per atom or greater), we assign the dummy regression values and a negative classification prediction to these points. The discovery acceleration factor (DAF) quantifies how many times more effective a model is at finding stable structures compared with random selection from the test set. Formally, the DAF is the ratio of the precision to the prevalence. The maximum possible DAF is the inverse of the prevalence, which on our dataset is (33,000/215,000)−1 ≈ 6.5. Thus, the current state-of-the-art of 5.04 achieved by EquiformerV2 + DeNS leaves room for improvement. However, evaluating each model on the subset of the 10,000 materials that each model ranks as being most stable (Supplementary Table 2), we see an impressive DAF of 6.33 for EquiformerV2 + DeNS, which is approaching optimal performance for this task.
A notable performance gap emerges between models predicting energy directly from unrelaxed inputs (the MatErials Graph Network (MEGNet), Wrenformer, the Crystal Graph Convolutional Neural Network (CGCNN), CGCNN+P, the Atomistic Line Graph Neural Network (ALIGNN), Voronoi RF) and UIPs, which leverage force and stress data to emulate DFT relaxation for final energy prediction. While the energy-only models exhibit surprisingly strong classification metrics (F1, DAF), their regression performance (R2, RMSE) is considerably poorer. Notably, only ALIGNN, BOWSR and CGCNN+P among the energy-only models achieve a positive coefficient of determination (R2). Negative R2 means model predictions explain the observed variation in the data less than simply predicting the test set mean. In other words, these models are not predictive in a global sense (across the full dataset range). Nevertheless, models with negative R2 may still show predictive capability for materials far from the stability threshold (that is, in the distribution tails). Their performance suffers most near the 0 eV per atom stability threshold, the region with the highest concentration of materials. This illustrates a limitation of using R2 alone to evaluate models for classification tasks such as stability prediction.
The reason CGCNN+P achieves better regression metrics than CGCNN but is still worse as a classifier becomes apparent from Supplementary Fig. 5 by noting that the CGCNN+P histogram is more sharply peaked at the 0 hull distance stability threshold. This causes even small errors in the predicted convex hull distance to be large enough to invert a classification. Again, this is evidence to choose carefully which metrics to optimize. Regression metrics are far more prevalent when evaluating energy predictions. However, our benchmark treats energy predictions as merely means to an end to classify compound stability. Improvements in regression accuracy are of limited use to materials discovery in their own right unless they also improve classification accuracy. Our results demonstrate that this is not a given.
Figure 2 shows models ranking materials by model-predicted hull distance from most to least stable: materials farthest below the known hull at the top, materials right on the hull at the bottom. For each model, we iterate through that list and calculate at each step the precision and recall of correctly identified stable materials. This simulates exactly how these models would be used in a prospective materials discovery campaign and reveals how a model’s performance changes as a function of the discovery campaign length. As a practitioner, you typically have a certain amount of resources available to validate model predictions. These curves allow you to read off the best model given these constraints. For instance, plotting the results in this manner shows that CHGNet initially achieves higher precision than models such as EquiformerV2 + DeNS, ORB MPtrj, SevenNet and MACE, which report higher precision across the whole test set.

A typical discovery campaign will rank hypothetical materials by model-predicted hull distance from most to least stable and validate the most stable predictions first. A higher fraction of correct stable predictions corresponds to higher precision and fewer stable materials overlooked corresponds to higher recall. Precision is calculated based only on the selected materials up to that point, while the cumulative recall depends on knowing the total number of positives upfront. Models such as eqV2 S DeNS and Orb MPtrj perform better for exhaustive discovery campaigns (screening a higher share of the candidate pool); others such as CHGNet do better when validating a smaller percentage of the materials predicted to be most stable. UIPs offer notably improved precision on shorter campaigns of ~20,000 or fewer materials validated, as they are less prone to false-positive predictions among highly stable materials.
In Fig. 2 each line terminates when the model believes there are no more materials in the Wang-Botti-Marques (WBM) test set below the MP convex hull. The dashed vertical line shows the actual number of stable structures in our test set. All models are biased towards stability to some degree as they all overestimate this number, most of all BOWSR by 133%. This overestimation primarily affects exhaustive discovery campaigns aiming to validate all materials predicted as stable. In practice, campaigns are often resource-limited (for example, to 10,000 DFT relaxations). By ranking candidates by predicted stability and validating only the top fraction dictated by the budget, the higher concentration of false positives typically found among less stable predictions is avoided without diminishing the campaign’s effective discovery rate (see Supplementary Table 2 where even the DAF of the worst performing model benchmarked, Voronoi RF, jumps from 1.58 to 2.49).
The diagonal ‘Optimal Recall’ line on the recall plot in Fig. 2 would be achieved if a model never made a false negative prediction and stopped predicting stable crystals exactly when the true number of stable materials was reached. Examining the UIP models, we find that they all achieve similar recall values, ranging from approximately 0.75 to 0.86. This is substantially smaller than the variation we see in the precision for the same models, ~0.44–0.77. Inspecting the overlap, we find that the intersection of the models’ correct agreements accounts for a precision of only 0.57 within the ~0.75–0.86 range, with just 0.04 of the examples where all models are wrong simultaneously. These results indicate that the models are making meaningfully different predictions.
Examining the precision plot in Fig. 2, we observe that the energy-only models exhibit a much more pronounced drop in their precision early on, falling to 0.6 or less in the first 5,000 screened materials. Many of these models (all except BOWSR, Wrenformer and Voronoi RF) display an interesting hook shape in their cumulative precision, recovering again slightly in the middle of the simulated campaign between 5,000 and up to 30,000 before dropping again until the end.
Figure 3 provides a visual representation of the reliability of different models as a function of a material’s DFT distance to the MP convex hull. The lines show the rolling MAE of model-predicted hull distances versus DFT. The red-shaded area, which we coin the ‘triangle of peril’, emphasizes the zone where the average model error surpasses the distance to the stability threshold at 0 eV. As long as the rolling MAE remains within this triangle, the model is highly susceptible to misclassifying structures. The average error in this region is larger than the distance to the classification threshold at 0, and consequently in cases where the error points towards the stability threshold it would be large enough to flip a correct classification into an incorrect one. Inside this region, the average error magnitude surpasses the distance to the classification threshold at 0 eV. Consequently, when errors point toward the stability boundary, they are sufficiently large to potentially reverse a correct classification. The faster a model’s error curve exits the triangle on the left side (representing negative DFT hull distances), the lower its tendency to mistakenly classify stable structures as unstable, thereby reducing false negatives. Exiting promptly on the right side (positive DFT hull distances) correlates with a decreased probability of predicting unstable structures as stable, resulting in fewer false positives.

The lines represent rolling MAE on the WBM test set as a function of distance to the MP training set convex hull. The red ‘triangle of peril’ indicates regions where the mean error exceeds the distance to the stability threshold (0 eV). Within this triangle, models are more likely to misclassify materials as the errors can flip classifications. Earlier exit from the triangle correlates with fewer false positives (right side) or false negatives (left side). The width of the ‘rolling window’ indicates the range over which prediction errors were averaged.
Models generally exhibit lower rolling errors towards the left edge of the plot compared with the right edge. This imbalance indicates a greater inclination towards false-positive predictions than false negative ones. Put differently, all models are less prone to predicting a material at −0.2 eV per atom DFT hull distance as unstable than they are to predicting a material at +0.2 eV per atom DFT hull distance as stable. From a practical perspective, this is undesirable because the opportunity cost associated with validating an incorrectly predicted stable material (a false positive) is typically much higher than that of missing a genuinely stable one (a false negative). We hypothesize that this error asymmetry arises from the MP training set’s uncharacteristically high proportion of stable materials, causing statistical models trained on it to be biased towards assigning low energies even to high-energy atomic arrangements. Training on datasets with more high-energy structures, such as Alexandria7 and OMat24 (ref. 40), would be expected to improve performance by balancing out this source of bias.
Discussion
We have demonstrated the effectiveness of ML-based triage in high-throughput materials discovery and posit that the benefits of including ML in discovery workflows now clearly outweigh the costs. Table 1 shows in a realistic benchmark scenario that several models achieve a discovery acceleration greater than 2.5 across the whole dataset and up to 6 when considering only the 10,000 most stable predictions from each model (Supplementary Table 2). Initially, the most promising ML methodology for accelerating high-throughput discovery was uncertain. Our results reveal a distinct advantage for UIPs regarding both accuracy and extrapolation performance. Incorporating force information allows UIPs to better simulate the relaxation pathway towards the DFT-relaxed structure, enabling a more accurate final energy determination.
Ranked best-to-worst by their test set F1 score on thermodynamic stability prediction, we find EquiformerV2 + DeNS > Orb > SevenNet > MACE > CHGNet > M3GNet > ALIGNN > MEGNet > CGCNN > CGCNN+P > Wrenformer > BOWSR > Voronoi fingerprint random forest. The top models are UIPs which we establish to be the best methodology for ML-guided materials discovery, achieving F1 scores of 0.57–0.82 for crystal stability classification and DAFs of up to 6× on the first 10,000 most stable predictions compared with dummy selection.
As the convex hull becomes more comprehensively sampled through future discoveries, the fraction of unknown stable structures will naturally decline. This will lead to less enriched test sets and, consequently, more challenging and discriminative discovery benchmarks. However, the discovery task framed here addresses only a limited subset of potential UIP applications. We believe that additional benchmarks are essential to effectively guide UIP development. These efforts should prioritize task-based evaluation frameworks that address the four critical challenges we identify for narrowing the deployment gap: adopting prospective rather than retrospective benchmarking, tackling relevant targets, using informative metrics and scalability.
Looking ahead, the consistently linear log–log learning curves observed in related literature41 suggest that further decreases in the error of UIPs can be readily unlocked with increased training data. This has been borne out in the scaling results of GNoME42, MatterSim43, Alexandria7 and OMat24 (ref. 40), which all show improvements in performance when training on much larger datasets. To realize the full potential of scaling these models, future efforts should deploy their resources to generate large quantities of higher-than-PBE fidelity training data. The quality of a UIP model is circumscribed by the quality and level of theory of its training data.
Beyond simply predicting thermodynamic stability at 0 K, future models will need to understand and predict material properties under varying environmental conditions, such as finite temperature and pressure, to aid in materials discovery. In this context, temperature-dependent dynamical properties constitute an area ripe for interatomic potentials. Another key open question is how effectively these models can contribute to the computational prediction of synthesis pathways. Many current methods for predicting reaction pathways employ heuristic rules to manage the considerable complexity introduced by metastability, in addition to relying on conventional ground-state ab initio data44,45,46. These algorithms will massively benefit from more efficient estimates of reaction energy barriers47 and non-crystalline, out-of-equilibrium materials48, opening up a whole new field to ML-accelerated inquiry.
Methods
Matbench Discovery framework
As first presented in J.R.ʼs PhD thesis25, we propose an evaluation framework that places no constraints on the type of data a model is trained on as long as it would be available to a practitioner conducting a real materials discovery campaign. This means that for the high-throughput DFT data considered, any subset of the energies, forces, stresses or any other properties that can be routinely extracted from DFT calculations, such as magnetic moments, are valid training targets. All of these would be available to a practitioner performing a real materials discovery campaign and hence are permitted for training any model submission. We enforce only that at test time, all models must make predictions on the convex hull distance of the relaxed structure with only the unrelaxed structure as input. This setup avoids circularity in the discovery process, as unrelaxed structures can be cheaply enumerated through elemental substitution methodologies and do not contain information inaccessible in a prospective discovery campaign. Figure 1 provides a visual overview of design choices.
The convex hull distance of a relaxed structure is chosen as the measure of its thermodynamic stability, rather than the formation energy, as it informs the decision on whether to pursue a potential candidate crystal. This decision was also motivated by ref. 14, which found that even composition-only models are capable of predicting DFT formation energies with useful accuracy. However, when tasking those same models with predicting decomposition enthalpy, performance deteriorated sharply. This insight exposes how ML models are much less useful than DFT for discovering new inorganic solids than would be expected given their low prediction errors for formation energies due to the impact of random as opposed to systematic errors.
Standard practice in ML benchmarks is to hold all variables fixed—most importantly the training data—and vary only the model architecture to isolate architectural effects on the performance. We deliberately deviate from this practice due to diverging objectives from common ML benchmarks. Our goal is to identify the best methodology for accelerating materials discovery. What kind of training data a model can ingest is part of its methodology. Unlike energy-only models, UIPs benefit from the additional training data provided by the forces and stresses recorded in DFT relaxations. This allows them to learn a fundamentally higher-fidelity model of the physical interactions between ions. That is a genuine advantage of the architecture and something any benchmark aiming to identify the optimal methodology for materials discovery must reflect. In light of this utilitarian perspective, our benchmark contains models trained on varying datasets, and any model that can intake more physical modalities from DFT calculations is a valid model for materials discovery.
We define the MP3 v.2022.10.28 database release as the maximum allowed training set for any compliant model submission. Models may train on the complete set of relaxation frames, or any subset thereof such as the final relaxed structures. Any subsets of the energies, forces and stresses are valid training targets. In addition, any auxiliary tasks such as predicting electron densities, magnetic moments, site-partitioned charges and so on that can be extracted from the output of the DFT calculations are allowed for multi-task learning49. Our test set consists of the unrelaxed structures in the WBM dataset50. Their target values are the PBE formation energies of the corresponding DFT-relaxed structures.
Limitations of this framework
While the framework proposed here directly mimics a common computational materials discovery workflow, it is worth highlighting that there still exist notable limitations to these traditional computational workflows that can prevent the material candidates suggested by such a workflow from being able to be synthesized in practice. For example, high-throughput DFT calculations often use small unit cells which can lead to artificial orderings of atoms. The corresponding real material may be disordered due to entropic effects that cannot be captured in the 0-K thermodynamic convex hull approximated by DFT51.
Another issue is that, when considering small unit cells, the DFT relaxations may get trapped at dynamically unstable saddle points in the true PES. This failure can be detected by calculating the phonon spectra for materials predicted to be stable. However, the cost of doing so with DFT is often deemed prohibitive for high-throughput screening. The lack of information about the dynamic stability of nominally stable materials in the WBM test set prevents this work from considering this important criterion as an additional screening filter. However, recent progress in the development of UIPs suggests that ML approaches will soon provide sufficiently cheap approximations of these terms for high-throughput searches52,53. As the task presented here begins to saturate, we believe that future discovery benchmarks should extend upon the framework proposed here to also incorporate criteria based on dynamic stability.
When training UIP models there is competition between how well given models can fit the energies, forces and stresses simultaneously. The metrics in the Matbench Discovery leaderboard are skewed towards energies and consequently UIP models trained with higher weighting on energies can achieve better metrics. We caution that optimizing hyperparameters purely to improve performance on this benchmark may have unintended consequences for models intended for general purpose use. Practitioners should also consider other involved evaluation frameworks that explore orthogonal use cases when developing model architectures. We highlight work from Póta et al.54 on thermal conductivity benchmarking, Fu et al.55 on MD stability for molecular simulation and Chiang et al.56 on modelling reactivity (hydrogen combustion) and asymptotic correctness (homonuclear diatomic energy curves) as complementary evaluation tasks for assessing the performance of UIP models.
We design the benchmark considering a positive label for classification as being on or below the convex hull of the MP training set. An alternative formulation would be to say that materials in WBM that are below the MP convex hull but do not sit on the combined MP + WBM convex hull are negatives. The issue with such a design is that it involves unstable evaluation metrics. If we consider the performance against the final combined convex hull rather than the initial MP convex hull, then each additional sample considered can retroactively change whether or not a previous candidate would be labelled as a success as it may no-longer sit on the hull. Since constructing the convex hull is computationally expensive, this path dependence makes it impractical to evaluate cumulative precision metrics (Fig. 2). The chosen setup does increase the number of positive labels and could consequently be interpreted as overestimating the performance. This overestimation decreases as the convex hull becomes better sampled. Future benchmarks building on this work could make use of the combination of MP + WBM to control this artefact. An alternative framework could report metrics for each WBM batch in turn and retrain between batches; this approach was undesirable here as it increases the cost of submission fivefold and introduces many complexities, for example, should each model only retrain on candidates it believed to be positive, that would make fair comparison harder.
Datasets
MP training set
The MP is a widely used database of inorganic materials properties that have been calculated using high-throughput ab initio methods. At the time of writing, the MP database3 has grown to ~154,000 crystals, covering diverse chemistries and providing relaxed and initial structures as well as the relaxation trajectory for every entry.
Our benchmark defines the training set as all data available from the v.2022.10.28 MP release. We recorded a snapshot of energies, forces, stresses and magnetic moments for all MP ionic steps on 15 March 2023 as the canonical training set for Matbench Discovery, and provide convenience functions through our Python package for easily feeding those data into future model submissions to our benchmark.
Flexibility in specifying the dataset allows authors to experiment with and fully exploit the available data. This choice is motivated by two factors. First, it may seem that models trained on multiple snapshots containing energies, forces and stresses receive more training data than models trained only on the energies of relaxed structures. However, the critical factor is that all these additional data were generated as a byproduct of the workflow to produce relaxed structures. Consequently, all models are being trained using data acquired at the same overall cost. If some architectures or approaches can leverage more of these byproduct data to make improved predictions this is a fair comparison between the two models. This approach diverges philosophically from other benchmarks such as the OCP and Matbench where it has been more common to subcategorize different models and look at multiple alternative tasks (for example, composition-only versus structure available in Matbench or IS2RS, IS2RE, S2EF in OCP) and which do not make direct comparisons of this manner. Second, recent work in the space from refs. 57,58 has claimed that much of the data in large databases such as MP are redundant and that models can be trained more effectively by taking a subset of these large data pools. From a systems-level perspective, identifying innovative cleaning or active-learning strategies to make better use of available data may be as crucial as architectural improvements, as both can similarly enhance performance, especially given the prevalence of errors in high-throughput DFT. Consequently, such strategies where they lead to improved performance should be able to be recognized within the benchmark. We encourage submissions to submit ablation studies showing how different system-level choices affect performance. Another example of a system-level choice that may impact performance is the choice of optimizer, for example, FIRE59 versus L-BFGS, in the relaxation when using UIP models.
We highlight several example datasets that are valid within the rules of the benchmark that take advantage of these freedoms. The first is the MP-crystals-2019.4.1 dataset60, which is a subset of 133,420 crystals and their formation energies that form a subset of the v.2021.02.08 MP release. The MP-crystals-2022.10.28 dataset is introduced with this work comprising a set of 154,719 structures and their formation energies drawn from the v.2021.02.08 MP release. The next is the MPF.2021.2.8 dataset22 curated to train the M3GNet model, which takes a subset of 62,783 materials from the v.2021.02.08 MP release. The curators of the MPF.2021.2.8 dataset down-sampled the v.2021.02.08 release notably to select a subset of calculations that they believed to be most self-consistent. Rather than taking every ionic step from the relaxation trajectory, this dataset opts to select only the initial, final and one intermediate structure for each material to avoid biasing the dataset towards examples where more ionic steps were needed to relax the structure. Consequently the dataset consists of 188,349 structures. The MPF.2021.2.8 is a proper subset of the training data as no materials were deprecated between the v.2021.02.08 and v.2022.10.28 database releases. The final dataset we highlight, with which several of the UIP models have been trained, is the MPtrj dataset23. This dataset was curated from the earlier v.2021.11.10 MP release. The MPtrj dataset is a proper subset of the allowed training data but several potentially anomalous examples from within MP were cleaned out of the dataset before the frames were subsampled to remove redundant frames. It is worth noting that the v.2022.10.28 release contains a small number of additional Perovskite structures not found in MPtrj that could be added to the training set within the scope of the benchmark.
We note that the v.2023.11.1 deprecated a large number of calculations so data queried from subsequent database releases are not considered valid for this benchmark.
WBM test set
The WBM dataset50 consists of 257,487 structures generated via chemical similarity-based elemental substitution of MP source structures followed by DFT relaxation and calculating each crystal’s convex hull distance. The element substitutions applied to a given source structure were determined by random sampling according to the weights in a chemical similarity matrix data-mined from the ICSD61.
The WBM authors performed five iterations of this substitution process (we refer to these steps as batches). After each step, the newly generated structures found to be thermodynamically stable after DFT relaxation flow back into the source pool to partake in the next round of substitution. This split of the data into batches of increasing substitution count is a unique and compelling feature of the test set as it allows out-of-distribution testing by examining whether model performance degrades for later batches. A higher number of elemental substitutions on average carries the structure farther away from the region of material space covered by the MP training set (see Supplementary Fig. 6 for details). While this batch information makes the WBM dataset an exceptionally useful data source for examining the extrapolation performance of ML models, we look primarily at metrics that consider all batches as a single test set.
To control for the potential adverse effects of leakage between the MP training set and the WBM test set, we cleaned the WBM test set based on protostructure matching. We refer to the combination of a materials prototype and the elemental assignment of its wyckoff positions as a protostructure following ref. 62. First we removed 524 pathological structures in WBM based on formation energies being larger than 5 eV per atom or smaller than −5 eV per atom. We then removed from the WBM test set all examples where the final protostructure of a WBM material matched the final protostructure of an MP material. In total, 11,175 materials were cleaned using this filter. We further removed all duplicated protostructures within WBM, keeping the lowest energy structure in each instance, leaving 215,488 structures in the unique prototype test set.
Throughout this work, we define stability as being on or below the convex hull of the MP training set (EMP hull dist ≤ 0). In total, 32,942 of 215,488 materials in the WBM unique prototype test set satisfy this criterion. Of these, ~33,000 are unique prototypes, meaning they have no matching structure prototype in MP nor another higher-energy duplicate prototype in WBM. Our code treats the stability threshold as a dynamic parameter, allowing for future model comparisons at different thresholds. For initial analysis in this direction, see Supplementary Fig. 1.
As WBM explores regions of materials space not well sampled by MP, many of the discovered materials that lie below MP’s convex hull are not stable relative to each other. Of the ~33,000 that lie below the MP convex hull less than half, or around ~20,000, remain on the joint MP + WBM convex hull. This observation suggests that many WBM structures are repeated samples in the same new chemical spaces. It also highlights a critical aspect of this benchmark in that we knowingly operate on an incomplete convex hull. Only current knowledge of competing points on the PES is accessible to a real discovery campaign and our metrics are designed to reflect this.
Models
To test a wide variety of methodologies proposed for learning the potential energy landscape, our initial benchmark release includes 13 models. Next to each model’s name we give the training targets that were used: E, energy; F, forces; S, stresses; and M, magnetic moments. The subscripts G and D refer to whether gradient-based or direct prediction methods were used to obtain force and stress predictions.
-
(1)
EquiformerV2 + DeNS63,64 (EFSD): EquiformerV2 builds on the first Equiformer model65 by replacing the SO(3) convolutions with equivariant Spherical Channel Network convolutions66 as well as a range of additional tweaks to make better use of the ability to scale to higher \({L}_{\max }\) using equivariant Spherical Channel Network convolutions. EquiformerV2 uses direct force prediction rather than taking the forces as the derivative of the energy predictions for computational efficiency. Here we take the pre-trained ‘eqV2 S DeNS’40 trained on the MPtrj dataset. This model in addition to supervised training on energies, forces and stresses makes use of an auxiliary training task based on de-noising non-equilibrium structures64. We refer to this model as ‘EquiformerV2 + DeNS’ in the text and ‘eqV2 S DeNS’ in plots.
-
(2)
Orb67 (EFSD): Orb is a lightweight model architecture developed to scale well for the simulation of large systems such as metal organic frameworks. Rather than constructing an architecture that is equivariant by default, Orb instead makes use of data augmentation during training to achieve approximate equivariance. This simplifies the architecture, allowing for faster inference. We report results for the ‘orb-mptrj-only-v2’ model, which was pre-trained using a diffusion-based task on MPtrj before supervised training on the energies, forces and stresses in MPtrj. For simplicity we refer to this model as ‘ORB MPtrj’.
-
(3)
SevenNet68 (EFSG): SevenNet emerged from an effort to improve the performance of message-passing neural networks69 when used to conduct large-scale simulations involving that benefit from parallelism via spatial decomposition. Here we use the pre-trained ‘SevenNet-0_11July2024’ trained on the MPtrj dataset. The SevenNet-0 model is an equivariant architecture based on a NequIP70 architecture that mostly adopts the GNoME42 hyperparameters. SevenNet-0 differs from NequIP and GNoME by replacing the tensor product in the self-connection layer with a linear layer applied directly to the node features, and this reduces the number of parameters from 16.24 million in GNoME to 0.84 million for SevenNet-0. For simplicity we refer to this model as ‘SevenNet’.
-
(4)
MACE24 (EFSG): MACE builds upon the recent advances70,71 in equivariant neural network architectures by proposing an approach to computing high-body-order features efficiently via Atomic Cluster Expansion72. Unlike the other UIP models considered, MACE was primarily developed for molecular dynamics of single material systems and not the universal use case studied here. The authors trained MACE on the MPtrj dataset; these models have been shared under the name ‘MACE-MP-0’ (ref. 52) and we report results for the ‘2023-12-03’ version commonly called ‘MACE-MP-0 (medium)’. For simplicity we refer to this model as ‘MACE’.
-
(5)
CHGNet23 (EFSGM): CHGNet is a UIP for charge-informed atomistic modelling. Its distinguishing feature is that it was trained to predict magnetic moments on top of energies, forces and stresses in the MPtrj dataset (which was prepared for the purposes of training CHGNet). By modelling magnetic moments, CHGNet learns to accurately represent the orbital occupancy of electrons, which allows it to predict both atomic and electronic degrees of freedom. We make use of the pre-trained ‘v.0.3.0’ CHGNet model from ref. 23.
-
(6)
M3GNet (ref. 22) (EFSG): M3GNet is a graph neural network (GNN)-based UIP for materials trained on up to three-body interactions in the initial, middle and final frames of MP DFT relaxations. The model takes the unrelaxed input and emulates structure relaxation before predicting energy for the pseudo-relaxed structure. We make use of the pre-trained ‘v.2022.9.20’ M3GNet model from ref. 22 trained on the compliant MPF.2021.2.8 dataset.
-
(7)
ALIGNN73 (E): ALIGNN is a message-passing GNN architecture that takes as input both the interatomic bond graph and a line graph corresponding to three-body bond angles. The ALIGNN architecture involves a global pooling operation, which means that it is ill-suited to force field applications. To address this the ALIGNN-FF model was later introduced without global pooling74. We trained ALIGNN on the MP-crystals-2022.10.28 dataset for this benchmark.
-
(8)
MEGNet60 (E): MEGNet is another GNN-based architecture that also updates a set of edge and global features (such as pressure and temperature) in its message-passing operation. This work showed that learned element embeddings encode periodic chemical trends and can be transfer-learned from large datasets (formation energies) to predictions on small data properties (band gaps, elastic moduli). We make use of the pre-trained ‘Eform_MP_2019’ MEGNet model trained on the compliant MP-crystals-2019.4.1 dataset.
-
(9)
CGCNN75 (E): CGCNN was the first neural network model to directly learn eight different DFT-computed material properties from a graph representing the atoms and bonds in a periodic crystal. CGCNN was among the first to show that just as in other areas of ML, given large enough training sets, neural networks can learn embeddings that outperform human-engineered structure features directly from the data. We trained an ensemble of 10 CGCNN models on the MP-crystals-2022.10.28 dataset for this benchmark.
-
(10)
CGCNN+P76 (E): This work proposes simple, physically motivated structure perturbations to augment stock CGCNN training data of relaxed structures with structures resembling unrelaxed ones but mapped to the same DFT final energy. Here we chose P = 5, meaning the training set is augmented with five random perturbations of each relaxed MP structure mapped to the same target energy. In contrast to all other structure-based GNNs considered in this benchmark, CGCNN+P is not attempting to learn the Born–Oppenheimer PES. The model is instead taught the PES as a step-function that maps each valley to its local minimum. The idea is that during testing on unrelaxed structures, the model will predict the energy of the nearest basin in the PES. The authors confirm this by demonstrating a lowering of the energy error on unrelaxed structures. We trained an ensemble of ten CGCNN+P models on the MP-crystals-2022.10.28 dataset for this benchmark.
-
(11)
Wrenformer (E): For this benchmark, we introduce Wrenformer, which is a variation on the coordinate-free Wren model11 constructed using standard QKV-self-attention blocks77 in place of message-passing layers. This architectural adaptation reduces the memory usage, allowing the architecture to scale to structures with greater than 16 Wyckoff positions. Similar to its predecessor, Wrenformer is a fast, coordinate-free model aimed at accelerating screening campaigns where even the unrelaxed structure is a priori unknown62. The key idea is that by training on the coordinate anonymized Wyckoff positions (symmetry-related positions in the crystal structure), the model learns to distinguish polymorphs while maintaining discrete and computationally enumerable inputs. The central methodological benefit of an enumerable input is that it allows users to predict the energy of all possible combinations of spacegroup and Wyckoff positions for a given composition and maximum unit cell size. The lowest-ranked protostructures can then be fed into downstream analysis or modelling. We trained an ensemble of ten Wrenformer models on the MP-crystals-2022.10.28 dataset for this benchmark.
-
(12)
BOWSR21 (E): BOWSR combines a symmetry-constrained Bayesian optimizer with a surrogate energy model to perform an iterative exploration–exploitation-based search of the potential energy landscape. Here we use the pre-trained ‘Eform_MP_2019’ MEGNet model60 for the energy model as proposed in the original work. The high sample count needed to explore the PES with a Bayesian optimizer makes this by far the most expensive model tested.
-
(13)
Voronoi RF78 (E): A random forest trained to map a combination of composition-based Magpie features79 and structure-based relaxation-robust Voronoi tessellation features (effective coordination numbers, structural heterogeneity, local environment properties, …) to DFT formation energies. This fingerprint-based model predates most deep learning for materials but notably improved over earlier fingerprint-based methods such as the Coulomb matrix80 and partial radial distribution function features81. It serves as a baseline model to see how much value the learned featurization of deep learning models can extract from the increasingly large corpus of available training data. We trained Voronoi RF on the MP-crystals-2022.10.28 dataset for this benchmark.
Data availability
The Matbench Discovery training set is the latest Materials Project (MP)3 database release (v.2022.10.28 at time of writing). The test set is the WBM dataset50, which is available via Figshare at https://figshare.com/articles/dataset/22715158 (ref. 82). A snapshot of every ionic step including energies, forces, stresses and magnetic moments in the MP database is available via Figshare at https://figshare.com/articles/dataset/23713842 (ref. 83). All other data files such as phase diagrams and structures in both ASE and pymatgen format are also available in the WBM dataset via Figshare at https://figshare.com/articles/dataset/22715158 (ref. 82).
Code availability
The Matbench Discovery framework, including benchmark implementation, evaluation code and model submission tools, is available as an open-source Python package and via GitHub at https://github.com/janosh/matbench-discovery, with a permanent version available via Zenodo at https://doi.org/10.5281/zenodo.13750664 (ref. 84). We welcome further model submissions via pull requests.
References
Bergerhoff, G., Hundt, R., Sievers, R. & Brown, I. D. The inorganic crystal structure data base. J. Chem. Inf. Comput. Sci. 23, 66–69 (1983).
Belsky, A., Hellenbrandt, M., Karen, V. L. & Luksch, P. New developments in the Inorganic Crystal Structure Database (ICSD): accessibility in support of materials research and design. Acta Crystallogr. B 58, 364–369 (2002).
Jain, A. et al. Commentary: the Materials Project: a materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
Saal, J. E., Kirklin, S., Aykol, M., Meredig, B. & Wolverton, C. Materials design and discovery with high-throughput density functional theory: the Open Quantum Materials Database (OQMD). JOM (1989) 65, 1501–1509 (2013).
Curtarolo, S. et al. AFLOW: an automatic framework for high-throughput materials discovery. Comput. Mater. Sci. 58, 218–226 (2012).
Draxl, C. & Scheffler, M. NOMAD: the FAIR concept for big data-driven materials science. MRS Bull. 43, 676–682 (2018).
Schmidt, J. et al. Improving machine-learning models in materials science through large datasets. Mater. Today Phys. 48, 101560 (2024).
Davies, D. W. et al. Computational screening of all stoichiometric inorganic materials. Chem 1, 617–627 (2016).
Riebesell, J. et al. Discovery of high-performance dielectric materials with machine-learning-guided search. Cell Rep. Phys. Sci. 5, 102241 (2024).
Borg, C. K. H. et al. Quantifying the performance of machine learning models in materials discovery. Digit. Discov. 2, 327–338 (2023).
Goodall, R. E. A., Parackal, A. S., Faber, F. A., Armiento, R. & Lee, A. A. Rapid discovery of stable materials by coordinate-free coarse graining. Sci. Adv. 8, eabn4117 (2022).
Zhu, A., Batzner, S., Musaelian, A. & Kozinsky, B. Fast uncertainty estimates in deep learning interatomic potentials. J. Chem. Phys. 158, 164111 (2023).
Depeweg, S., Hernandez-Lobato, J.-M., Doshi-Velez, F. & Udluft, S. Decomposition of uncertainty in Bayesian deep learning for efficient and risk-sensitive learning. In Proc. 35th International Conference on Machine Learning, 1184–1193 (PMLR, 2018).
Bartel, C. J. et al. A critical examination of compound stability predictions from machine-learned formation energies. NPJ Comput. Mater. 6, 1–11 (2020).
Montanari, B., Basak, S. & Elena, A. Goldilocks convergence tools and best practices for numerical approximations in density functional theory calculations (EDC, 2024); https://ukerc.rl.ac.uk/cgi-bin/ercri4.pl?GChoose=gdets&GRN=EP/Z530657/1
Griffin, S. M. Computational needs of quantum mechanical calculations of materials for high-energy physics. Preprint at https://arxiv.org/abs/2205.10699 (2022).
Austin, B. et al. NERSC 2018 Workload Analysis (Data from 2018) (2022); https://portal.nersc.gov/project/m888/nersc10/workload/N10_Workload_Analysis.latest.pdf
Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007).
Bartók, A. P., Kermode, J., Bernstein, N. & Csányi, G. Machine learning a general-purpose interatomic potential for silicon. Phys. Rev. X 8, 041048 (2018).
Deringer, V. L., Caro, M. A. & Csányi, G. A general-purpose machine-learning force field for bulk and nanostructured phosphorus. Nat. Commun. 11, 5461 (2020).
Zuo, Y. et al. Accelerating materials discovery with Bayesian optimization and graph deep learning. Mater. Today 51, 126–135 (2021).
Chen, C. & Ong, S. P. A universal graph deep learning interatomic potential for the periodic table. Nat. Comput. Sci. 2, 718–728 (2022).
Deng, B. et al. CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling. Nat. Mach. Intell. 5, 1031–1041 (2023).
Batatia, I., Kovács, D. P., Simm, G. N. C., Ortner, C. & Csányi, G. MACE: higher order equivariant message passing neural networks for fast and accurate force fields. Preprint at http://arxiv.org/abs/2206.07697 (2023).
Riebesell, J. Towards Machine Learning Foundation Models for Materials Chemistry. PhD Thesis, Univ. of Cambridge (2024); www.repository.cam.ac.uk/handle/1810/375689
Wu, Z. et al. MoleculeNet: a benchmark for molecular machine learning. Chem. Sci. 9, 513–530 (2018).
Kpanou, R., Osseni, M. A., Tossou, P., Laviolette, F. & Corbeil, J. On the robustness of generalization of drug-drug interaction models. BMC Bioinformatics 22, 477 (2021).
Meredig, B. et al. Can machine learning identify the next high-temperature superconductor? Examining extrapolation performance for materials discovery. Mol. Syst. Des. Eng. 3, 819–825 (2018).
Cubuk, E. D., Sendek, A. D. & Reed, E. J. Screening billions of candidates for solid lithium-ion conductors: a transfer learning approach for small data. J. Chem. Phys. 150, 214701 (2019).
Zahrt, A. F., Henle, J. J. & Denmark, S. E. Cautionary guidelines for machine learning studies with combinatorial datasets. ACS Comb. Sci. 22, 586–591 (2020).
Sun, W. et al. The thermodynamic scale of inorganic crystalline metastability. Sci. Adv. 2, e1600225 (2016).
Goodall, R. E. A. & Lee, A. A. Predicting materials properties without crystal structure: deep representation learning from stoichiometry. Nat. Commun. 11, 6280 (2020).
Dunn, A., Wang, Q., Ganose, A., Dopp, D. & Jain, A. Benchmarking materials property prediction methods: the Matbench test set and Automatminer reference algorithm. NPJ Comput. Mater. 6, 1–10 (2020).
Chanussot, L. et al. Open Catalyst 2020 (OC20) dataset and community challenges. ACS Catal. 11, 6059–6072 (2021).
Lee, K. L. K. et al. Matsciml: a broad, multi-task benchmark for solid-state materials modeling. Preprint at https://arxiv.org/abs/2309.05934 (2023).
Choudhary, K. et al. Jarvis-leaderboard: a large scale benchmark of materials design methods. NPJ Comput. Mater. 10, 93 (2024).
Tran, R. et al. The Open Catalyst 2022 (OC22) dataset and challenges for oxide electrocatalysts. ACS Catal. 13, 3066–3084 (2023).
Lan, J. et al. AdsorbML: a leap in efficiency for adsorption energy calculations using generalizable machine learning potentials. NPJ Comput. Mater. 9, 172 (2023).
Sriram, A. et al. The Open DAC 2023 dataset and challenges for sorbent discovery in direct air capture. ACS Cent. Sci. 10, 923–941 (2024).
Barroso-Luque, L. et al. Open materials 2024 (OMat24) inorganic materials dataset and models. Preprint at https://arxiv.org/abs/2410.12771 (2024).
Lilienfeld, O. A. V. & Burke, K. Retrospective on a decade of machine learning for chemical discovery. Nat. Commun. 11, 4895 (2020).
Merchant, A. et al. Scaling deep learning for materials discovery. Nature 624, 80–85 (2023).
Yang, H. et al. MatterSim: a deep learning atomistic model across elements, temperatures and pressures. Preprint at https://arxiv.org/abs/2405.04967 (2024).
McDermott, M. J., Dwaraknath, S. S. & Persson, K. A. A graph-based network for predicting chemical reaction pathways in solid-state materials synthesis. Nat. Commun. 12, 3097 (2021).
Aykol, M., Montoya, J. H. & Hummelshøj, J. Rational solid-state synthesis routes for inorganic materials. J. Am. Chem. Soc. 143, 9244–9259 (2021).
Wen, M. et al. Chemical reaction networks and opportunities for machine learning. Nat. Comput. Sci. 3, 12–24 (2023).
Yuan, E. C.-Y. et al. Analytical ab initio Hessian from a deep learning potential for transition state optimization. Nat. Commun. 15, 8865 (2024).
Aykol, M., Dwaraknath, S. S., Sun, W. & Persson, K. A. Thermodynamic limit for synthesis of metastable inorganic materials. Sci. Adv. 4, eaaq0148 (2018).
Shoghi, N. et al. From molecules to materials: pre-training large generalizable models for atomic property prediction. Preprint at https://arxiv.org/abs/2310.16802 (2023).
Wang, H.-C., Botti, S. & Marques, M. A. L. Predicting stable crystalline compounds using chemical similarity. NPJ Comput. Mater. 7, 1–9 (2021).
Cheetham, A. K. & Seshadri, R. Artificial intelligence driving materials discovery? Perspective on the article: scaling deep learning for materials discovery. Chem. Mater. 36, 3490–3495 (2024).
Batatia, I. et al. A foundation model for atomistic materials chemistry. Preprint at https://arxiv.org/abs/2401.00096 (2023).
Deng, B. et al. Systematic softening in universal machine learning interatomic potentials. NPJ Comput. Mater. 11, 9 (2025).
Póta, B., Ahlawat, P., Csányi, G. & Simoncelli, M. Thermal conductivity predictions with foundation atomistic models. Preprint at https://arxiv.org/abs/2408.00755 (2024).
Fu, X. et al. Forces are not enough: benchmark and critical evaluation for machine learning force fields with molecular simulations. Transact. Mach. Learn. Res. https://openreview.net/forum?id=A8pqQipwkt (2023).
Chiang, Y. et al. MLIP arena: advancing fairness and transparency in machine learning interatomic potentials through an open and accessible benchmark platform. AI for Accelerated Materials Design - ICLR 2025 https://openreview.net/forum?id=ysKfIavYQE (2025).
Li, K., DeCost, B., Choudhary, K., Greenwood, M. & Hattrick-Simpers, J. A critical examination of robustness and generalizability of machine learning prediction of materials properties. NPJ Comput. Mater. 9, 55 (2023).
Li, K. et al. Exploiting redundancy in large materials datasets for efficient machine learning with less data. Nat. Commun. 14, 7283 (2023).
Bitzek, E., Koskinen, P., Gähler, F., Moseler, M. & Gumbsch, P. Structural relaxation made simple. Phys. Rev. Lett. 97, 170201 (2006).
Chen, C., Ye, W., Zuo, Y., Zheng, C. & Ong, S. P. Graph networks as a universal machine learning framework for molecules and crystals. Chem. Mater. 31, 3564–3572 (2019).
Glawe, H., Sanna, A., Gross, E. K. U. & Marques, M. A. L. The optimal one dimensional periodic table: a modified Pettifor chemical scale from data mining. New J. Phys. 18, 093011 (2016).
Parackal, A. S., Goodall, R. E., Faber, F. A. & Armiento, R. Identifying crystal structures beyond known prototypes from x-ray powder diffraction spectra. Phys. Rev. Mater. 8, 103801 (2024).
Liao, Y.-L., Wood, B., Das, A. & Smidt, T. EquiformerV2: improved equivariant transformer for scaling to higher-degree representations. International Conference on Learning Representations (ICLR) https://openreview.net/forum?id=mCOBKZmrzD (2024).
Liao, Y.-L., Smidt, T., Shuaibi, M. & Das, A. Generalizing denoising to non-equilibrium structures improves equivariant force fields. Preprint at https://arxiv.org/abs/2403.09549 (2024).
Liao, Y.-L. & Smidt, T. Equiformer: equivariant graph attention transformer for 3D atomistic graphs. International Conference on Learning Representations (ICLR) https://openreview.net/forum?id=KwmPfARgOTD (2023).
Passaro, S. & Zitnick, C. L. Reducing SO(3) convolutions to SO(2) for efficient equivariant GNNs. Preprint at https://arxiv.org/abs/2302.03655 (2023).
Neumann, M. et al. Orb: a fast, scalable neural network potential. Preprint at https://arxiv.org/abs/2410.22570 (2024).
Park, Y., Kim, J., Hwang, S. & Han, S. Scalable parallel algorithm for graph neural network interatomic potentials in molecular dynamics simulations. J. Chem. Theory Comput. 20, 4857–4868 (2024).
Gilmer, J., Schoenholz, S. S., Riley, P. F., Vinyals, O. & Dahl, G. E. Neural message passing for quantum chemistry. In International Conference on Machine Learning (eds Precup, D. & Teh, Y. W.) 1263–1272 (PMLR, 2017).
Batzner, S. et al. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nat. Commun. 13, 2453 (2022).
Thomas, N. et al. Tensor field networks: rotation- and translation-equivariant neural networks for 3D point clouds. Preprint at http://arxiv.org/abs/1802.08219 (2018).
Drautz, R. Atomic cluster expansion for accurate and transferable interatomic potentials. Phys. Rev. B 99, 014104 (2019).
Choudhary, K. & DeCost, B. Atomistic line graph neural network for improved materials property predictions. NPJ Comput. Mater. 7, 1–8 (2021).
Choudhary, K. et al. Unified graph neural network force-field for the periodic table: solid state applications. Digit. Discov. 2, 346–355 (2023).
Xie, T. & Grossman, J. C. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys. Rev. Lett. 120, 145301 (2018).
Gibson, J., Hire, A. & Hennig, R. G. Data-augmentation for graph neural network learning of the relaxed energies of unrelaxed structures. NPJ Comput. Mater. 8, 1–7 (2022).
Vaswani, A. et al. Attention is all you need. In Advances in Neural Information Processing Systems, Vol. 30 (Curran Associates, Inc., 2017); https://proceedings.neurips.cc/paper_files/paper/2017/hash/3f5ee243547dee91fbd053c1c4a845aa-Abstract.html
Ward, L. et al. Including crystal structure attributes in machine learning models of formation energies via Voronoi tessellations. Phys. Rev. B 96, 024104 (2017).
Ward, L., Agrawal, A., Choudhary, A. & Wolverton, C. A general-purpose machine learning framework for predicting properties of inorganic materials. NPJ Comput. Mater. 2, 1–7 (2016).
Rupp, M., Tkatchenko, A., Müller, K.-R. & Lilienfeld, O. A. V. Fast and accurate modeling of molecular atomization energies with machine learning. Phys. Rev. Lett. 108, 058301 (2012).
Schütt, K. T. et al. How to represent crystal structures for machine learning: towards fast prediction of electronic properties. Phys. Rev. B 89, 205118 (2014).
Riebesell, J. & Goodall, R. Matbench discovery: WBM dataset. Figshare https://figshare.com/articles/dataset/22715158 (2023).
Riebesell, J. & Goodall, R. Mp ionic step snapshots for matbench discovery. Figshare https://figshare.com/articles/dataset/23713842 (2023).
Riebesell, J. et al. janosh/matbench-discovery: v1.3.1. Zenodo https://doi.org/10.5281/zenodo.13750664 (2024).
Acknowledgements
J.R. acknowledges support from the German Academic Scholarship Foundation (Studienstiftung). A.A.L. acknowledges support from the Royal Society. A.J. and K.A.P. acknowledge the US Department of Energy, Office of Science, Office of Basic Energy Sciences, Materials Sciences and Engineering Division under contract no. DE-AC02-05-CH11231 (Materials Project programme KC23MP). This work used computational resources provided by the National Energy Research Scientific Computing Center (NERSC), a US Department of Energy Office of Science User Facility operated under contract no. DE-AC02-05-CH11231. We thank H.-C. Wang, S. Botti and M. A. L. Marques for their valuable contribution in crafting and freely sharing the WBM dataset. We thank R. Armiento, F. A. Faber and A. S. Parackal for helping develop the evaluation procedures for Wren upon which this work builds. We also thank R. Elijosius for assisting in the initial implementation of Wrenformer and M. Neumann, L. Barroso-Luque and Y. Park for submitting compliant models to the leaderboard. We thank J. Blake Gibson, S. Ping Ong, C. Chen, T. Xie, P. Zhong and E. Dogus Cubuk for helpful discussions.
Author information
Authors and Affiliations
Contributions
J.R. was responsible for the methodology, software, data curation, investigation (training: CGCNN, CGCCN+P, Wrenformer, Voronoi RF), validation, formal analysis and writing the original draft. R.E.A.G. was responsible for conceptualization, software, validation, formal analysis, writing the original draft, and reviewing and editing the manuscript. P.B. was responsible for software, investigation (training: ALIGNN, MACE) and writing the original draft. Y.C. was responsible for investigation (training: MACE), formal analysis and reviewing and editing the manuscript. B.D. was responsible for data curation (MPtrj), investigation (training: CHGNet), and reviewing and editing the manuscript. G.C. and M.A. were responsible for supervision and funding acquisition. A.A.L. and A.J. supervised the project. K.A.P. was responsible for supervision, reviewing and editing the manuscript, and funding acquisition.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Machine Intelligence thanks Ju Li, Logan Ward and Tian Xie for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–13, discussion and Tables 1 and 2.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Riebesell, J., Goodall, R.E.A., Benner, P. et al. A framework to evaluate machine learning crystal stability predictions. Nat Mach Intell 7, 836–847 (2025). https://doi.org/10.1038/s42256-025-01055-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42256-025-01055-1
