
Abstract
With the area of perovskite films and devices increases, the performance tends to deteriorate, which can be attributed to the accumulation of defects at the bottom surface without proper passivation. Here, we introduce the 1-(4-Fluorophenyl)−2-pyrrolidone molecule (FPP) as additive in large-area blade-coating perovskite films. During the top-down crystallization process, the FPP molecule form an intermediate phase with the perovskite components and subsequently self-deposit at the bottom surface. Consequently, the crystallization kinetics of the large-area thin films are regulated, and the bottom surface is effectively and uniformly passivated in one single-step processing. By employing this self-deposited passivation method (SDP), the efficiency decrease caused by the expansion of the device area has been significantly suppressed and the devices yield power conversion efficiency of 23.41% (0.09 cm2), 22.43% (1 cm2), and 20.75% (24 cm2). The method is compatible for commercial manufacturing with scaling up solar cell area and holding high efficiency.
Similar content being viewed by others

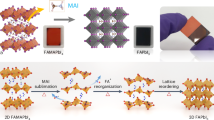

Introduction
Perovskite solar cells (PSCs) have achieved a notable power conversion efficiency (PCE) of 26.14%1, demonstrating the significant potential for commercial applications. However, the current state-of-the-art PSCs with high efficiencies are typically produced by the spin-coating method, which has limitations in terms of the active area, usually less than 0.1 cm2 (see refs. 2,3). Moreover, extra interface engineering methods are often required, making them incompatible with the cost-effective and scalable manufacturing processes needed for commercialization4. To address this challenge, researchers have explored various scalable techniques such as blade-coating, slot-die coating, bar coating, roll-to-toll coating, screen-coating, and spray-coating methods to expand the area of perovskite films5. Despite the significant progress made in depositing perovskite films using these scalable methods, the PCE of the resulting devices experiences a rapid decline as the device area increases. As depicted in Fig. 1a and Supplementary Fig. 1, which illustrates the PCEs and PCEs retention rate of PSCs obtained from recent reports, the efficiency typically decays below 88% when the device area from less than 1 cm2 to exceed 10 cm2, and the trend of efficiency drop is more noticeable as the area increases6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24.

a Normalized PCE of PSCs as a function of the area obtained from recent reports. b Illustration of blade-coating technology and the structure of FPP with its ESP plot. c PL statistical results of 25 pieces from 6.5 × 6.5 cm2 target perovskite film. d PCE of 81 dot cells on a 6.5 × 6.5 cm2 target substrate. e Champion J–V curves of control and target devices in forward scan and reverse scan under AM 1.5 G simulated solar illumination. The active area of PSCs is 0.09 cm2. f J–V curves of the 1 cm2 device and 24 cm2 module. The inset photograph shows a perovskite solar module.
There are many reasons responsible for the PCE loss, including the attainment of uniformity in film formation, heterogeneous rates of perovskite crystal growth across distinct regions, heightened occurrence of defects within the perovskite layer and at interfacial boundaries, and elevated module resistance25. Among them, the significant performance drop observed in large-area perovskite films is primarily attributed to the rapid growth of defects within the polycrystalline perovskite film, which is inherent to the ionic nature of perovskite materials26. Previous reports have indicated that these defects are not uniformly distributed throughout the perovskite film, with the highest defect concentration observed at the bottom surface in comparison to the top surface and bulk in the perovskite layer2,13,27 Unlike fabricating a nanometer-thick passivation layer in small-area perovskite films by spin-coating, it is not feasible to passivate the entire bottom surface effectively by simply using blade-coating or slot-die methods. Consequently, the bottom surface of the perovskite film remains inadequately passivized in large-area devices.
In response to the challenges associated with the area increase in films and devices, numerous efforts have been undertaken to mitigate the impact of the area amplification effect. Chen et al. introduced an in situ polymer framework to suppress the coffee ring effect and improve film uniformity, achieving efficiency of 23.94% (0.04 cm2) and 17.53% (25 cm2)28. Qi et al. introduced potassium sulfamate (H2KNO3S) as the passivation layer to both passivate the uncoordinated Pb2+ and mitigate the oxygen loss issue of the SnO2 electron transport layer, yielding a champion PCE of 21.98% (0.09 cm2) and 16.16% (22.4 cm2) of mini-modules29. These works have shown promise in addressing several aspects problem of the fabrication of large-area PSCs, yet few focus on the issue of bottom surface defects in large-scale films. In addition, the extra fabrication steps will also increase the process complexity.
In this work, we proposed a self-deposited passivation method (SDP) employing FPP into the perovskite solution and fabricated perovskite films by blade-coating in the open air (under room temperature and 40–60% relative humidity, RH). During the annealing process, as the residual solvent evaporates from the film’s top surface, the FPP molecules undergo self-deposited onto the bottom interface. As the fluorine atoms in FPP can form hydrogen bonding with FA+ and the C = O group in FPP coordinates with the Pb2+, FPP molecular forms the coordinate phase with perovskite components. This interaction reduces the reaction rate between organic salt and PbI2, resulting in the retardation of perovskite film crystallization. As a consequence, grains grow sufficiently in various positions across the large-area thin film, enabling a more controllable and homogeneous crystallization process, ultimately leading to the formation of uniform large-area perovskite films. In addition, FPP molecules will move downwards as the intermediate phase transitions to the perovskite phase and gather at the bottom surface, acting as a passivator. The champion efficiency reaches 23.41% with an active area of 0.09 cm2. Moreover, the FPP treatment brings about high efficiencies of 22.43% and 20.75% for 1 cm2 devices and 24 cm2 modules, respectively. In comparison, without FPP treatment, the PCE was 21.52% (0.09 cm2), 18.11% (1 cm2), and 15.62% (24 cm2). The efficiency loss decreased from 15.9 to 4.2% (0.09–1 cm2) and from 27.5 to 11.4% (0.09–24 cm2) by SDP strategy. The advantage of the method is its ability to effectively address both the crystal growth stage and the bottom surface defect, irrespective of the film’s size, which renders it particularly suitable for the production of large-area devices.
Results and discussion
To reduce the perovskite band gap and get a broader light absorption spectrum, it is necessary to incorporate a high ratio of FA cations and iodine anions in the perovskite composition. Here, we employed (FAPbI3)0.95(MAPbBr3)0.05 compound as the perovskite absorber, and low-toxic 2Me was selected as the perovskite precursor solvent30. Figure 1b shows the illustration of blade-coating technology, and details of the fabrication process are given in “Methods”. Supplementary Fig. 2 shows a photograph of blade-coating perovskite film with a substrate area of 6.5 × 6.5 cm2 with 2.5 mg ml−1 FPP (Target, 2.5FPP), which has a smooth and reflective surface. The inset picture in Fig. 1b shows the Electrostatic potential (ESP) image of FPP, where a fluorine ion is connected to the benzene ring and contains a functional group of C = O. The red and blue regions are the maximum and minimum ESP values, corresponding to the nucleophilic and electrophilic sites, meaning that O and F atoms are electrophilic sites.
Large-area perovskite films (6.5 × 6.5 cm2) are divided into 25 pieces (Fig. 1c and Supplementary Fig. 3) to evaluate the perovskite coating uniformity via photoluminescence measurement (PL). A stronger and more indistinguishable PL intensity is observed in the target film compared with the control film. Then, we attempted to fabricate dot cells on 6.5 × 6.5 cm2 substrates before fabricating perovskite modules (Fig. 1d and Supplementary Fig. 4). The PSCs were fabricated with the typical n-i-p planar device architecture of ITO glass/SnO2/Perovskite/Spiro-OMeTAD/Au. The difference between the maximum and minimum efficiency in 81 dot target cells is 2.10% with an average efficiency of 22.79%, while the difference between control cells is 5.08% with an average efficiency of 19.27%. The uniform distribution of PL intensity and the dot cell efficiencies indicated that the uniformity of blade-coating perovskite films was improved by introducing FPP.
Figure 1e depicts the current density–voltage (J–V) curves of the control and target devices under simulated AM 1.5 G with detailed photovoltaic parameters. The control device with an active area of 0.09 cm2 exhibits a PCE of 21.52% with a VOC of 1.12 V, a JSC of 24.54 mA cm−2, and an FF of 77.63%. For the target devices, the best PCE reaches 23.41% with a VOC of 1.15 V, a JSC of 25.36 mA cm−2, and an FF of 80.23%. The JSC is further confirmed by External Quantum Efficiency (EQE) in Supplementary Fig. 5. We fabricated devices with an up-scaling area of 1 cm2 and the efficiency was improved from 18.11 to 22.43% (Fig. 1f). We further studied laser processing and fabricated perovskite modules (Supplementary Figs. 6 and 7). The best solar module with an active area of 24 cm2 displayed a PCE of 20.75% with a VOC of 4.47 V, a JSC of 6.14 mA cm2, and an FF of 75.45%, while the efficiency of the control module is 15.62%.
To study the effect of FPP on crystal formation, the XRD test of temporal evolution was conducted. For the control film (Supplementary Fig. 8), the diffraction peaks located at ≈13.97° are assigned to the (001) crystal planes of the α-FAPbI3 perovskite phase. With an annealing time of 600 s, the full width at half maximum value (FWHM) of the diffraction peak corresponding to the (001) crystal plane is 0.114°. The inset image in Fig. 2a is an XRD pattern of FPP powder with a high-intensity peak located at 9.01°. For the target film with an annealing time of 0 s, there is a new diffraction peak located at 8.92°. With the annealing time prolonged, the new diffraction peak intensity gradually decreases and finally disappears with an annealing time of 120 s. Therefore, we speculate that the phase near 9° in XRD patterns of the target film is an intermediate phase, which will adjust the crystallization rate and increase the grain size31. In consequence, the FWHM value of the diffraction peak corresponding to the (001) crystal plane decreases to 0.094° after annealing for 600 s. We also compared the XRD characterization of perovskite films with different concentrations of FPP (0, 1, 2.5, and 5 mg ml−1, Supplementary Fig. 9), no shift of the (001) crystal plane is observed, which means that the crystal structure of perovskite has not been changed by FPP. In addition, we compared the ultraviolet-visible absorption (UV–vis) and obtained the Tauc plot spectra (Supplementary Fig. 10). The target film showed stronger absorption over all wavelengths compared with the control film, which is consistent with the enhanced crystallinity in XRD results and is beneficial for increasing the JSC of devices. The Tauc plot spectra implied that FPP did not induce a band gap change, and both perovskite films showed a band gap of 1.53 eV. As aforementioned showed that FPP may not exist within the lattice of the perovskite crystal after annealing for 600 s.

a XRD characterization of target film under different annealing time at 150 °C. The inset image shows the XRD characterization of FPP powder. b H NMR spectra of FAI and FAI·FPP. The inset shows the interaction between FAI and FPP. c FTIR spectrogram of FPP·2Me solution, FPP·FAI·2Me solution, and FPP·FAI·PbI2·2Me solution. d The 3D maps showed the distribution of FPP in the target film deposited on ITO glass. e XPS of spectra of F 1s in target-top and target-bottom surface. XPS of spectra of N 1s (f) and Pb 4f (g) of control-bottom and target-bottom surface. h Schematic illustration of the self-deposited passivation model.
In addition, semi-in-situ SEM images and UV–vis absorption spectra for the control and target perovskite films with different annealing time were performed. As the annealing time is 20 s, the control perovskite grain becomes relatively large. After annealing for 600 s, the average grain size is 0.95 μm. However, the grain size of the target gradually changes, and the final average grain size is 1.22 μm (Supplementary Figs. 11–13). This may be ascribed to the formation of the intermediate phase, which will adjust the growth rate of crystallization. The larger grains with less grain boundary are beneficial for suppressing charge trapping and improving charge mobility. Supplementary Fig. 14 shows the semi-in-situ UV–vis absorption spectra and the control film exhibited a strong red-shift in the region between 550 and 800 nm in a short period of 60 s. However, the target film displays gentle absorption changes. The variation of absorbance intensity at 650 nm is summarized in Supplementary Fig. 14c, the target film rises more slowly during the growth process compared to the control film, proving that the formation of perovskite crystals was slowed down by FPP treatment32.
The liquid-state nuclear magnetic resonance spectra (NMR) and liquid-state Fourier transform infrared spectroscopy measurement (FTIR) were carried out to reveal the interaction between FPP and perovskite in the solution environment. Figure 2b shows an obvious signal at 7.84 ppm, which is assigned to the H-bond with nitrogen atoms in cationic FA+33. After introducing FPP into the FAI-DMSO-d6 solution, the signal of ammonium protons upshifted to 7.88 ppm. Subsequently, the interaction between fluorine in FPP and FAI is explored (Supplementary Fig. 15), demonstrating that the chemical shifts of F shifted by 0.03 ppm when FAI was added to the FPP-DMSO-d6 solution. Thus, we speculate that the F in FPP can form hydrogen bonding with FA+34. In the FTIR spectrogram (Fig. 2c), FPP was added into 2Me solution showing a C = O absorption peak appeared at 1680 cm−1. When FAI and FPP were added into the 2Me solution, the C = O absorption peak did not change35. In contrast, the C = O absorption peak shifted to 1672 cm−1 after the introduction of PbI2 in FPP·FAI·2Me solution. The shifted stretching vibration frequency of C = O is mainly attributed to the formation of the coordinate bond between FPP with PbI236.
To reveal the distribution of perovskite components and FPP in target perovskite film, the time-of-flight secondary ion mass spectrometry (TOF-SIMS) was conducted (Fig. 2d and Supplementary Fig. 16). The 3D maps show the distribution of the PbI3− and FPP signals in the target perovskite film. The line of PbI3− is horizontal in Supplementary Fig. 16a, indicating that the ions are uniformly distributed in the film. Notably, the FPP signal distributes throughout the surveyed depth and sharply increases in the area where the Sn intensity increases, indicating that FPP was mainly gathering at the perovskite film bottom37. The 3D maps of target perovskite film show the same distribution trend as the TOF-SIMS depth profiles.
As FPP is gathered at the bottom surface in the target films, direct and visualized investigation on the bottom surface is a favor for understanding the mechanism of the FPP. Thus, we peeled off the perovskite film and exposed the perovskite bottom surface (Supplementary Fig. 17)38. The bottom-view SEM images of the control perovskite film show that there are some holes, but disappeared in the target film (Supplementary Fig. 18). X-ray photoelectron spectroscopy (XPS) was performed to confirm the interaction between FPP and perovskite compounds. Figure 2e depicts the XPS spectra of F 1 s of the target-top and target-bottom surface. Notably, there are no signals of F ions on the top surface, but exist on the bottom surface. Thus, we rationally believe that FPP mainly exists at the bottom of perovskite films, which is consistent with the TOF-SIMS test. Figure 2f shows the N 1 s characteristic peaks shifted from 399.85 (for the control-bottom surface) to 400.30 eV (for the target-bottom surface), which is relative to the H-bond. As exhibited in Fig. 2g, the XPS characteristic peaks of Pb 4f 5/2 shifted from 142.55 eV (for the control-bottom surface) to 143.00 eV (for the target-bottom surface). The chemical shift of the corresponding energy-binding peaks in the XPS spectrum is relative to the electron density around an atom. The uncoordinated Pb2+ can accept electrons, making it act as a Lewis acid, while the C = O could serve as the Lewis base, leading to the change of binding energy of Pb 4f 39,40.
Figure 2h shows the schematic illustration of the self-deposited passivation model. During the annealing time of perovskite films, with the evaporation of residual solvent from the top surface and the intermediate phases are converted to perovskite grains, the perovskite polycrystalline film follows the downward growth from the intermediate phase. From the previous discussion, it could form the coordinate phase (FPP·FAI·PbI2) as F in FPP can form hydrogen bonding with FA+ and the C = O group in FPP can coordinate with the Pb2+. Thus, in the annealing process, the particular molecules exhibit bi-functional properties: on the one hand, they autonomously interact with the perovskite, aiding in slowing down the crystallization process; on the other hand, FPP not exist within the lattice of the perovskite crystal after annealing. Thus, with the upper part of the film transformed from the FPP-related intermediate phase to the perovskite phase, FPP will be separated from the intermediate phase and move downward. The process is repeated until perovskite layers are completely formed. Finally, it results in a small amount of FPP present at the grain boundaries in the film bulk and most of the FPP will accumulate at the bottom of the film, acting as passivator41,42,43.
To determine the potential difference between the top and bottom surfaces of the perovskite film in the devices, as well as the average electric field intensity in the perovskite film regions, cross-section atomic force microscope (AFM, Fig. 3a–d) and cross-section Kelvin probe force microscopy measurement (KPFM, Supplementary Fig. 19e–h) was conducted. The device for this measurement has a structure of glass/ITO/SnO2/Perovskite/Spiro-OMeTAD/Au. The potential difference between the top and the bottom surface is 52, 91, 153, and 157 mV, for the control, 1FPP, 2.5FPP, and 5FPP device, respectively, indicating the potential difference between the top and bottom surface increases after adding FPP. Then we took the derivative of the potential data to obtain the average electric field of the perovskite thin film (Fig. 3e–h). The average electric field intensity in perovskite film regions of devices is 73.15, 104.9, 176.2, and 244.4 mV μm−1, for the control, 1FPP, 2.5FPP, and 5FPP devices, respectively. The larger electric field intensity is beneficial for enhancing carrier mobility and reducing reverse charge accumulation44,45,46.

a–d Cross-section AFM images of the devices with different FPP. e–h The potential difference and the average electric field intensity were obtained from the cross-section KPFM images. i, j UPS spectra of the secondary electron cutoff region of the control top, control bottom, target top, and target-bottom surface. Energy level diagrams of the (k) control and (l) target devices.
Ultraviolet photoelectron spectroscopy (UPS) was performed to evaluate the energy level alignment between the perovskite surface and the charge transport layer (Fig. 3i, j). The Fermi energy level is estimated for different surfaces (control-top surface, control-bottom surface, target-top surface, and target-bottom surface) by subtracting the binding energy of the secondary edge (17.05, 16.99, 17.00, 16.90 eV) from the excitation energy of the UV photons (21.22 eV, He I UPS) spectra47. The estimated Fermi energy levels are −4.17, −4.23, −4.22, and −4.32 eV, respectively. The difference in Fermi energy levels between the bottom and top of the control and target is 60 and 100 mV, respectively. Figure 3k, l shows the energy level diagrams of the control and target devices. After doping with FPP, the CBM of the bottom surface shifts downward for 0.03 eV, causing a smaller energy gap with the CBM of SnO2. The VBM of the TOP surface shifts upward for 0.02 eV, causing a smaller energy gap with the VBM of Spiro-OMeTAD. This is beneficial for reducing energy loss at the perovskite interface of devices.
The photoelectric characteristics of films and devices were systematically studied to further evaluate the effect of FPP. Under the PL test, the incident light from the bottom side generates excessive carriers mainly on the bottom surface, and the incident light from the top side generates excessive carriers mainly on the top surface48. Regardless of the direction of the incident light, the PL intensity of the target film is higher than the control film, suggesting suppressed non-radiative recombination at the top surface and bottom surface by introducing FPP (Fig. 4a)49. Figure 4b shows the time-resolved photoluminescence measurement (TRPL), and the average carrier lifetime was calculated by Eq. (1):

a PL spectra. The films were deposited on glass and the light incident from the top and bottom side. b TRPL spectra of control and target films. c SCLC measurement of control and target electron-only devices. d SCLC measurement of control and target hole-only devices. e EIS spectra of control and target devices. The inset shows the equivalent circuit diagram. f VOC versus light intensity for control and target devices.
The carrier lifetime was 387.86 μs for the control film and 780.36 μs for the target film. The significantly increasing carrier extraction lifetime in the target perovskite agrees well with its higher steady-state PL intensity50.
Furthermore, the trap density of the perovskite films is evaluated by the space-charge limited current measurements (SCLC). The dark I–V curves with the configurations of electron-only devices (ITO glass/SnO2/Perovskite/PCBM/Au) are shown in Fig. 4c. The trap-filled limited voltage (VTFL) is the transition voltage between the Ohmic and trap-filling regions, and the VTFL for the target device (0.40 V) is smaller than the control devices (0.48 V). The defects concentration value (Nt) can be determined by the Eq. (2):
where ε0 is the vacuum dielectric constant, εr is the relative dielectric constant of perovskite, q is the elemental charge, and L is the thickness of perovskite film. According to the equation, the target device demonstrates a Nt of 3.90 × 1015 cm−3, which is smaller than that of the control device (5.92 × 1015 cm−3). The dark I–V curves with the configurations of hole-only devices (ITO glass/MeO-2PACZ/Perovskite/Spiro-OMeTAD/Au) are shown in Fig. 4d. The hole trap density (Nt) declined from 8.89 × 1015 (control) to 3.70 × 1015 cm−3 (target). These confirm the better charge extraction and transport at the SnO2/perovskite interface and perovskite/Spiro-OMeTAD interface51.
Electrochemical impedance spectroscopy (EIS) was measured to further evaluate interfacial carrier transfer and recombination52. Nyquist plots were obtained from the solar cells under dark conditions. The fitted equivalent circuit model is shown in the inset picture in Fig. 4e. From the plots, two semicircle arcs were observed. The semicircle arc in the high-frequency region was attributed to the charge transfer resistance (Rct), and the semicircle arc in the low-frequency region was ascribed to the charge recombination resistance (Rrec). After the introduction of FPP, the Rct value was reduced from 2049 to 1960 Ω and the Rrec value was increased from 474.8 to 504.1 Ω, indicating that interfacial charge transfer was promoted and interfacial charge recombination was suppressed. From the light intensity-dependent VOC measurement in Fig. 4f, the target device shows a smaller slope (1.08 kT/q) as compared to the control device (1.23 kT/q). Since the deviation of the slope from unity kT/q (where k is the Boltzmann constant, T is the absolute temperature, and q is the elementary charge) reflects trap-assisted non-radiative recombination in PSCs. The lower slope value reflected that the carrier-trap-assisted recombination was greatly suppressed in the target film53.
Figure 5a shows the normalized PCEs of PSCs as a function of the area in this work. As the area of devices expanded from 0.09 to 1 cm2 and 0.09 to 24 cm2, the PCE retention rate of the control devices was 84.1% and 72.5%, respectively. In comparison, the target device is 95.8% and 88.6%, respectively, indicating that the efficiency decrease caused by the expansion of the device area has been significantly suppressed. We believe this is mainly due to a more uniform film and less defect concentration by introducing FPP.

a Normalized PCEs of PSCs as a function of the area in this work. b MPP tracking of JSC at continuous Xenon lamp light irradiation of control and target unencapsulated devices. c Thermal stability of control and target unencapsulated devices. d Long-term stability of control and target unencapsulated devices.
The stability of perovskite films and devices was systematically studied under different conditions. Supplementary Fig. 20 shows the images of water droplet contact angle measurements of the control and target films. The target film had a contact angle of 80.36° with that of the control film of 70.51°, indicating that the perovskite films containing the FPP were more hydrophobic. This excellent hydrophobicity results in no significant changes in the target perovskite film after 60 days of aging under high humidity conditions (50–80% RH, Supplementary Fig. 21). However, on the 30th day of aging, the control film showed a yellow phase (δ-FAPbI3) and on the 60th day, almost all of the control film turned yellow54. The stability of the unencapsulated devices under continuous illumination was tested at the maximum power point (MPP, Fig. 5b). One hundred percent of the initial current density was maintained within 1800 s for the target device. However, the initial and end current densities for the control device are 24.01 and 23.08 mA cm−2, which decay to 96.12%. Figure 5c shows the thermal stability of PCE under 85 °C for the unencapsulated control and target devices. The unencapsulated target device maintains 86.89% of its initial PCE after aging for 240 h, which is higher than control devices (below 80%). The long-term stability of devices (Fig. 5d) demonstrates the target device retained 86.11% of its initial PCE after over 12,400 h of aging under <20% RH at 25 °C, while the control maintained 79.73% within 2280 h of aging.
The improved stability of the film and device can be attributed to the following reasons. Moisture, heat, and light-driven degradation are initiated from the defect states at the grain boundary and surfaces and then gradually propagate to the grain bulks in polycrystalline thin films55. After adding FPP, the fewer defects in the perovskite film cause less degradation sites in the film. Therefore, the target perovskite films and devices became less vulnerable to environmental stress during the aging period.
Conclusions
In this article, we presented a self-passivation technique tailored for large-area perovskite solar cells to mitigate the efficiency decline. We fabricated large-area perovskite films using the blade-coating method and introduced FPP to improve perovskite film quality. The FPP self-deposited downward and finally gathered at the perovskite film bottom, passivating the defects in the bottom surface. Consequently, grains grow sufficiently in various positions across the large-area thin film, enabling a more controllable and homogeneous crystallization process, ultimately forming uniform large-area perovskite films. As a result, the phenomenon of efficiency decline caused by device area amplification is suppressed. This strategy achieved a PCE of 23.41% for single-junction devices and 20.75% for solar modules with excellent stability, which provides a simple and effective method for scalable perovskite solar cells.
Methods
Materials
SnO2 (tin (IV) oxide, 15 wt% in H2O colloidal dispersion liquid) was purchased from Alfa Aesar. Purified water was purchased from Wahaha. Potassium chloride (KCl, GR) was purchased from Sinopharm. ITO glass, formamidinium iodide (FAI, 99.9%), and methylamine chloride (MACl, 99.9%) were purchased from Advanced Election Technology Co., Ltd, Lead (II) iodide (PbI2, 98%), methylamine bromine (MABr, 98%), and 2-methoxyethanol (2Me, 99%) were purchased from TCI. Lead (II) bromide (PbBr2, 99.999%) and 1-cyclohexyl-2-pyrrolidinon (CHP, 99%) were purchased from Aladdin. 2-methylthio-2-imidazoline hydroiodide (95%) was purchased from Bidepharm. Chlorobenzene (CB, 99%), 2-propanol (IPA, 99.9%), and acetonitrile (ACN, 99.8%) were purchased from Sigma-Aldrich. Spiro-OMeTAD (99.8%), 4-tert-butylpyridine (tBP, >96%), FK 209 Co (III) TFSI salt, and Li-TFSI salt were purchased from Xi’an Polymer Light Technology Corporation. 1-(4-Fluorophenyl)−2-pyrrolidone (FPP, 99.58%) was purchased from Leyan. Au (99.999%) was purchased from ZhongNuo Advanced Material Technology. All purchased materials were used without further purification.
Precursor solution preparation
Electron transport layer precursor solution: 1 g SnO2 colloidal solution and 15 mg KCl were dissolved in 4 ml purified water to form the SnO2 solution. The SnO2 solution was stirred at least 6 h before use and should be used within 14 days.
Perovskite precursor solution: 1.1 M (FAPbI3)0.95(MAPbBr3)0.05 perovskite solution: 179.7 mg FAI, 481.7 mg PbI2, 6.1 mg MABr, 20.1 mg PbBr2, 25.9 mg MACl (35% mol compared to Pb), and 0.1 ml CHP were dissolved in 1 ml 2Me solution (as control perovskite solution). FPP with different concentrations was added to the control solution (1 mg ml−1, 2.5 mg ml−1 (target solution), and 5 mg ml−1, compared with 2Me). The solution was stirred at least 2 hours before use and should be used within 24 h.
Passivation layer precursor solution: 5 mg 2-methylthio-2-imidazoline hydroiodide solution was dissolved in 1 ml IPA as passivation solution for the perovskite top surface. The solution was stirred at least 2 h before use and should be used within 24 h.
Hole transport layer precursor solution: Spiro-OMeTAD solution was prepared by mixing 73.5 mg Spiro-OMeTAD in 1 ml CB with 39.5 μl tBP, 23 μl Li-TFSI salt solution (520 mg ml−1 in ACN), and 10 μl Co (III)-TFSI salt solution (375 mg ml−1 in ACN). The solution was stirred at least 2 hours before use and should be used within 24 h.
Single-junction PSCs fabrication
ITO glass substrates (7 Ω square−1, 6.5 × 6.5 cm2) were washed sequentially by detergent, deionized water three times, and ethanol in an ultrasonic bath system. Each step takes 15 min. Then, the ITO glasses were dried with nitrogen gas or cold dry air. Before use, the ITO glass was treated with plasma for 2 min to improve the glass wettability. The electron transfer layer was deposited on the precleaned 6.5 × 6.5 cm2 ITO glass with 1 ml SnO2 solution by spin-coating at 5000 rpm for 25 s (2000 rpm s−1 of acceleration), followed by annealing at 90 °C for 60 min in open air. Before depositing the perovskite film, the SnO2 layer was treated by plasma for 2 min to improve the wettability. The perovskite film was deposited by blade-coating at room temperature under 40–60% RH. A pipette gun was used to inject perovskite solution droplets into the space between the blade and substrate. The total amount of solution is 24 μl with 6 μl applied each time. The gap between the blade coater and substrate is 160 μm (the thickness of ITO glass is 1100 μm, and the height of the blade coater is 1270 μm). The speed of the blade is 25 mm s−1. The N2 knife followed the blade coater with a pressure of 0.4 MPa. Then, the perovskite wet film was annealed at 150 °C for 10 minutes under a 40–60% RH ambient environment. Then the 6.5 × 6.5 cm2 glass was cut into 1.5 × 2 cm2 small glass. In all, 60 μl 2-methylthio-2-imidazoline hydroiodide solution was spin-coated on the perovskite layer at 3000 rpm for 20 s (2000 rpm s−1 of acceleration) and heated at 100 °C for 10 min in an air glove box. 45 μl Spiro-OMeTAD solution was spin-coated at 3000 rpm for 20 s (2000 rpm s−1 of acceleration) in an air glove box. Finally, a 100-nm-thick Au electrode was deposited via the thermal evaporation method. A non-reflective shadow mask (0.09 cm2, 1 cm2) was used to define the active area of the PSCs.
Modules fabrication
For the 6.5 × 6.5 cm2 module, 600 μl of 2-methylthio-2-imidazoline hydroiodide solution and 600 μl of Spiro-OMeTAD solution were used. Four laser-patterning lines, P1, P2, P3, and P4 were accomplished by the 355 nm laser beam with a power of 3 W. Using a single laser can reduce the operating process and improve the accuracy of line spacing.
Before cleaning the ITO glass, P1 was used to remove the ITO layer from the substrate, with a laser beam of 500 mm s−1 laser scribing speed, 30 kHz scribing frequency, and 8 μs pulse width. Before depositing Au, P2 is used to remove all the deposited layers with a laser beam of 500 mm s−1 laser scribing speed, 30 kHz scribing frequency, and 2 μs pulse width. Notably, we use the low-energy laser twice or more times to remove the functional layer and reduce the damage to ITO. After depositing Au, P3 removed all the deposited layers with a laser beam of 500 mm s−1 scribing speed, 30 kHz scribing frequency, and 3 μs pulse width. We used the low-energy laser twice in P3 to remove the functional layer and reduce the damage to ITO. P4 removes all the deposited layers with a laser beam of 500 mm s−1 scribing speed, 30 kHz scribing frequency, and 5 μs pulse width. The line widths of P1, P2, P3, and P4 are about 50, 100, 100, and 100 μm, respectively. The line spacing between P1 and P2, P2 and P3 is about 100 μm.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the study are available in the supplementary material of this article.
References
NREL. Best research-cell effciencies, https://www.nrel.gov/pv/cell-efficiency.html (2024).
Min, H. et al. Perovskite solar cells with atomically coherent interlayers on SnO2 electrodes. Nature 598, 444–450 (2021).
Park, J. et al. Controlled growth of perovskite layers with volatile alkylammonium chlorides. Nature 616, 724–730 (2023).
Gao, Z. W., Wang, Y. & Choy, W. C. Buried interface modification in perovskite solar cells: a materials perspective. Adv. Energy Mater. 12, 2104030 (2022).
Saki, Z., Byranvand, M. M., Taghavinia, N., Kedia, M. & Saliba, M. Solution-processed perovskite thin-films: the journey from lab-to large-scale solar cells. Energy Environ. Sci. 14, 5690–5722 (2021).
Yoo, J. W. et al. R4N+ and Cl− stabilized α-formamidinium lead triiodide and efficient bar-coated mini-modules. Joule 7, 797–809 (2023).
Chung, J. et al. Engineering perovskite precursor inks for scalable production of high‐efficiency perovskite photovoltaic modules. Adv. Energy Mater. 13, 2300595 (2023).
Sangale, S. S., Kwon, S. N., Patil, P., Lee, H. J. & Na, S. I. Locally supersaturated inks for a slot‐die process to enable highly efficient and robust perovskite solar cells. Adv. Energy Mater. 13, 2300537 (2023).
Bu, T. et al. Lead halide–templated crystallization of methylamine-free perovskite for efficient photovoltaic modules. Science 372, 1327–1332 (2021).
Uddin, M. A. et al. Blading of conformal electron‐transport layers in p–i–n perovskite solar cells. Adv. Mater. 34, 2202954 (2022).
Liang, Q. et al. Manipulating crystallization kinetics in high‐performance blade‐coated perovskite solar cells via cosolvent‐assisted phase transition. Adv. Mater. 34, 2200276 (2022).
Yin, J., Shi, X., Wang, L., Yan, H. & Chen, S. High‐performance inverted perovskite solar devices enabled by a polyfullerene electron transporting material. Angew. Chem. Int. Ed. 61, e202210610 (2022).
Chen, S. et al. Stabilizing perovskite-substrate interfaces for high-performance perovskite modules. Science 373, 902–907 (2021).
Chen, C. et al. Perovskite solar cells based on screen-printed thin films. Nature 612, 266–271 (2022).
Chen, S., Xiao, X., Gu, H. & Huang, J. Iodine reduction for reproducible and high-performance perovskite solar cells and modules. Sci. Adv. 7, eabe8130 (2021).
Deng, Y. et al. Tailoring solvent coordination for high-speed, room-temperature blading of perovskite photovoltaic films. Sci. Adv. 5, eaax7537 (2019).
Li, J. et al. Ink design enabling slot‐die coated perovskite solar cells with> 22% power conversion efficiency, micro‐modules, and 1 year of outdoor performance evaluation. Adv. Energy Mater. 13, 2203898 (2023).
Hu, J. et al. Spontaneously self‐assembly of a 2D/3D heterostructure enhances the efficiency and stability in printed perovskite solar cells. Adv. Energy Mater. 10, 2000173 (2020).
Hu, J. et al. Overcoming photovoltage deficit via natural amino acid passivation for efficient perovskite solar cells and modules. J. Mater. Chem. A 9, 5857–5865 (2021).
Deng, Y. et al. Surfactant-controlled ink drying enables high-speed deposition of perovskite films for efficient photovoltaic modules. Nat. Energy 3, 560–566 (2018).
Yang, M. et al. Perovskite ink with wide processing window for scalable high-efficiency solar cells. Nat. Energy 2, 1–9 (2017).
Li, J. et al. A regularity‐based fullerene interfacial layer for efficient and stable perovskite solar cells via blade‐coating. Adv. Funct. Mater. 32, 2105917 (2022).
Xu, Z. et al. Reducing energy barrier of δ-to-α phase transition for printed formamidinium lead iodide photovoltaic devices. Nano Energy 91, 106658 (2022).
Zhang, J. et al. Two-step sequential blade-coating of high quality perovskite layers for efficient solar cells and modules. J. Mater. Chem. A 8, 8447–8454 (2020).
Niu, X., Li, N., Chen, Q. & Zhou, H. Insights into large‐scale fabrication methods in perovskite photovoltaics. Adv. Energy Sust. Res. 2, 2000046 (2021).
Chen, B., Rudd, P. N., Yang, S., Yuan, Y. & Huang, J. Imperfections and their passivation in halide perovskite solar cells. Chem. Soc. Rev. 48, 3842–3867 (2019).
Ni, Z. et al. Resolving spatial and energetic distributions of trap states in metal halide perovskite solar cells. Science 367, 1352–1358 (2020).
Li, L. et al. In‐situ polymer framework strategy enabling printable and efficient perovskite solar cells by mitigating “coffee ring” effect. Adv. Mater. 36, 2310752 (2024).
Tong, G. et al. Holistic strategies lead to enhanced efficiency and stability of hybrid chemical vapor deposition based perovskite solar cells and modules. Adv. Energy Mater. 13, 2300153 (2023).
Yoo, J. W. et al. Efficient perovskite solar mini-modules fabricated via bar-coating using 2-methoxyethanol-based formamidinium lead tri-iodide precursor solution. Joule 5, 2420–2436 (2021).
Xiang, W. et al. Intermediate phase engineering of halide perovskites for photovoltaics. Joule 6, 315–339 (2022).
Liu, K. et al. Moisture-triggered fast crystallization enables efficient and stable perovskite solar cells. Nat. Commun. 13, 4891 (2022).
Huang, Y. et al. Finite perovskite hierarchical structures via ligand confinement leading to efficient inverted perovskite solar cells. Energy Environ. Sci. 16, 557–564 (2023).
Yang, Y. et al. Improving photovoltaic performance and stability of perovskite solar cells via molecular bridge strategy. Adv. Opt. Mater. 11, 2300684 (2023).
Yang, Z. et al. Slot-die coating large-area formamidinium-cesium perovskite film for efficient and stable parallel solar module. Sci. Adv. 7, eabg3749 (2021).
Lee, D. K., Lim, K. S., Lee, J. W. & Park, N. G. Scalable perovskite coating via anti-solvent-free lewis acid-base adduct engineering for efficient perovskite solar module. J. Mater. Chem. A 9, 3018–3028 (2021).
Liang, Z. et al. Homogenizing out-of-plane cation composition in perovskite solar cells. Nature 624, 557–563 (2023).
Luo, C. et al. Engineering the buried interface in perovskite solar cells via lattice-matched electron transport layer. Nat. Photonics 17, 856–864 (2023).
Wang, R. et al. Constructive molecular configurations for surface-defect passivation of perovskite photovoltaics. Science 366, 1509–1513 (2019).
Li, C., Zhang, N. & Gao, P. Lessons learned: how to report XPS data incorrectly about lead-halide perovskites. Mater. Chem. Front. 7, 3797–3802 (2023).
Chen, S. et al. Crystallization in one-step solution deposition of perovskite films: upward or downward? Sci. Adv. 7, eabb2412 (2021).
Tan, Q. et al. Inverted perovskite solar cells using dimethylacridine-based dopants. Nature 620, 545–551 (2023).
Tao, Y. et al. Bidirectional anions gathering strategy afford efficient mixed Pb- Sn perovskite solar cells. Small 19, 2207480 (2023).
Xu, H. et al. Constructing robust heterointerfaces for carrier viaduct via interfacial molecular bridges enables efficient and stable inverted perovskite solar cells. Energy Environ. Sci. 16, 5792–5804 (2023).
Wu, Y. et al. Intrinsic dipole arrangement to coordinate energy levels for efficient and stable perovskite solar cells. Adv. Mater. 35, 2300174 (2023).
Tan, S. et al. Stability-limiting heterointerfaces of perovskite photovoltaics. Nature 605, 268–273 (2022).
Liang, Z. et al. A selective targeting anchor strategy affords efficient and stable ideal‐bandgap perovskite solar cells. Adv. Mater. 34, 2110241 (2022).
Wang, H. et al. In situ dual‐interface passivation strategy enables the efficiency of formamidinium perovskite solar cells over 25%. Adv. Mater. 36, 2307855 (2024).
Fei, C. et al. Lead-chelating hole-transport layers for efficient and stable perovskite minimodules. Science 380, 823–829 (2023).
Zhu, P. et al. Simultaneous contact and grain‐boundary passivation in planar perovskite solar cells using SnO2‐KCl composite electron transport layer. Adv. Energy Mater. 10, 1903083 (2020).
Liu, G. et al. Mixed-phase low-dimensional perovskite-assisted interfacial lead directional management for stable perovskite solar cells with efficiency over 24%. ACS Energy Lett. 6, 4395–4404 (2021).
Hu, M. et al. Regulating the surface passivation and residual strain in pure tin perovskite films. ACS Energy Lett. 6, 3555–3562 (2021).
Du, M. et al. High‐pressure nitrogen‐extraction and effective passivation to attain highest large‐area perovskite solar module efficiency. Adv. Mater. 32, 2004979 (2020).
Wang, Z. et al. Supple formamidinium-based low-dimension perovskite derivative for sensitive and ultrastable X-ray detection. ACS Nano 17, 13638–13647 (2023).
Fu, L. et al. Defect passivation strategies in perovskites for an enhanced photovoltaic performance. Energy Environ. Sci. 13, 4017–4056 (2020).
Acknowledgements
The authors acknowledge financial support from National Natural Science Foundation of China (Grant No. U22A20142, No. 52302324, and No. 52272252), the National Key R&D Program of China (No. 2021YFB3800102), Key Lab of Photovoltaic and Energy Conservation Materials, Chinese Academy of Sciences (Grant No. PECL2021QN002) and CASHIPS Director’s Fund (YZJJ202304-CX, GGZX-GTCX-2023-10). The authors would like to thank Liying Zhang, Du Du, Yanan Song, Boyuan Liu, and Tianle Guo for the experiment support and the mechanism analysis.
Author information
Authors and Affiliations
Contributions
Yuli Tao, Zheng Liang, and Jiajiu Ye conceived the idea. Yuli Tao and Zheng Liang fabricated the perovskite devices. Shendong Xu and Huifen Xu analyzed the experiment data of this manuscript. Hui Zhang, Chentai Cao and Quan Yang assisted in SEM, PL, and XRD measurements. Zameer Abbas, Guozhen Liu, and Xu Pan revised the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Materials thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: John Plummer.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tao, Y., Liang, Z., Ye, J. et al. Self-deposited passivation for decreasing scalable processing loss of perovskite solar cells. Commun Mater 5, 166 (2024). https://doi.org/10.1038/s43246-024-00585-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43246-024-00585-2
