
Abstract
Sodium is one of the most abundant elements on Earth and a sustainable alternative to less sustainable metals such as lithium, which is becoming increasingly depleted and expensive. Traditionally, however, organosodium reagents have been considered highly reactive, engaging in uncontrollable reactions, and as a result, they have been scarcely used in organic synthesis, especially in combination with transition-metal catalysis. Here we report the use of organosodium compounds as C(sp2)–Na nucleophilic partners in iron-catalysed oxidative homocoupling and cross-coupling with alkyl halides. Mechanistic investigations based on the preparation and characterization of putative organoiron intermediates reveal that a bidentate additive coordinates both sodium and the iron centre, exerting control over the catalytic reactivity. This combination of two abundant and non-toxic metals, powered by molecular-level mechanistic understanding, is expected to open new avenues for the use of sustainable organometallic reagents in organic synthesis.

Similar content being viewed by others
Main
Sodium is the most abundant alkali metal, and the resources of sodium on Earth (23,600 ppm in the crust, together with a large amount in the ocean) are substantially greater than those of lithium (20 ppm in the crust; Fig. 1a)1. Moreover, sodium is largely non-toxic: whereas the amount of permissible daily exposure to lithium in drugs is specified (560 μg per day), sodium is not subjected to such regulations2. Thus, sodium is an ideal metal for sustainable and environmentally benign organic synthesis. For over half a century, transition-metal-catalysed coupling reactions have revolutionized organic synthesis, enabling the precise and efficient creation of carbon–carbon bonds3. Notably, the historical beginnings of coupling reactions did not require the use of a precious transion metal and relied on sodium-mediated approaches such as homodimerization of alkyl halides (1855, the Wurtz reaction)4,5 or aryl halides (1862, the Fittig reaction)6 in the presence of a superstoichiometric amount of sodium metal (Fig. 1b) and cross-coupling of alkyl iodides with aryl bromides mediated by an excess amount of sodium to produce alkylarenes (the Wurtz–Fittig reaction; Fig. 1c)7,8. Despite these initial discoveries and the merits of sodium, the subsequent explosive development of transition-metal-catalysed coupling reactions has relied on various other organometallic reagents9, and the utilization of organosodium compounds has been largely neglected in modern organic synthesis10. This situation has been caused mainly by the higher ionic character of the C–Na bond as compared with other metals such as boron, zinc, magnesium and lithium, inducing uncontrollably high reactivity. As a result, the high reactivity of organosodium leads to non-selective reactions generating many undesired by-products. In addition, their high ionic character leads to the formation of highly aggregated structures, making organosodium compounds sparingly soluble in non-polar solvents. This, combined with their instability in ethereal solvents, makes their manipulation as well as the isolation of key reaction intermediates and mechanistic analysis particularly difficult.

a, The abundance and toxicity of sodium1,2. b, Sodium-mediated carbon–carbon bond formation through homodimerization4,5,6. c, Wurtz–Fittig reaction of aryl bromides with alkyl iodides using sodium metal7,8. d, This work: merger of organosodium chemistry and iron catalysis for oxidative homocoupling and cross-coupling with alkyl halides. PDE, permissible daily exposure; Ar, aryl.
Another reason for the slow development of organosodium chemistry has been the lack of efficient synthetic methods. Recently, there has been a surge of interest in the preparation of organosodium compounds11,12,13,14, alleviating the long-standing prejudice that organosodium compounds cannot be of synthetic utility, but applications have still been limited to a reduced number of organic transformations. Nevertheless, a handful of reports have shown that organosodium reagents are amenable to transition-metal catalysis. Thus, a few studies have reported the direct use of (hetero)arylsodiums in Pd- and Ni-catalysed C(sp2)–C(sp2) cross-coupling for the construction of biaryls15,16 and polythiophenes17,18, respectively, although these approaches may suffer from issues associated with sustainability or toxicity of the catalysts. Meanwhile, the use of organosodium reagents in iron-catalysed C–C bond formation processes remains unexplored. Breaking new ground in this evolving area of research, we report here the integration of organosodium chemistry with sustainable transition-metal catalysis, specifically the direct coupling of aryl and alkenyl organosodium compounds using abundant, non-toxic and environmentally benign iron as a catalyst (Fig. 1d)19,20,21,22. Furthermore, mechanistic studies based on the preparation and characterization of organoiron complexes and radical-clock experiments provided important insights into the dual role of the key bidentate donor additive, and into the catalytic cycle. Iron catalysis has also been notoriously difficult to control, and the present study demonstrates that two capricious metals can be tamed in a productive way for organic synthesis.
Results and discussion
As described in the introduction, the chemistry of organosodium is appealing for the development of sustainable synthesis, but the high reactivity of these compounds is difficult to control. A common way to control reactivity and selectivity in coupling reactions is the use of a transition-metal catalyst; however, organosodium compounds often uncontrollably reduce transition metals to unproductive mixtures of low-valent species. This is especially true for iron, which is notoriously difficult to control even for the reaction of organomagnesium or organolithium compounds23,24,25,26.
To develop practical coupling reactions with organosodium compounds, we decided to divide the reactions into two steps: the preparation of the organosodium compound and the catalytic C–C bond formation step, thereby suppressing the premature decomposition of organosodium (Fig. 1d). We have been exploring organosodium chemistry for some time15,16,27,28,29,30, and we expected that our recently developed efficient methods for generating organosodiums in hydrocarbon solvents under mild conditions, followed by judicious choice of oxidant and additive for the subsequent iron catalysis step, could suppress side reactions and achieve selective and efficient homo- and cross-coupling reactions.
Iron-catalysed oxidative homocoupling of organosodium
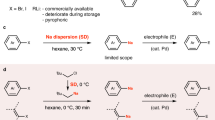
We began our investigation with identifying the key parameters for the iron-catalysed oxidative homocoupling of organosodiums, the catalytic version of the Fittig-type coupling. We generated 4-(tert-butyl)phenylsodium (8Na) in situ by reductive sodiation of the corresponding aryl chloride (8′) with sodium dispersion13,15, and we used it as a model substrate for iron-catalysed oxidative homocoupling in one pot (Fig. 2 and Supplementary Table 1). Screening of iron sources and other parameters identified commercially available iron (III) acetylacetonate (acac) as the optimal catalyst, whereas iron fluoride, chloride, bromide, acetate and triflate resulted in low yields (<5%), which we speculate is partly due to the low solubility of these iron salts in hydrocarbon solvent. Methylcyclohexane (MCH) was a better solvent than hexane, and di-tert-butyl peroxide (DTBP)31 was a much more efficient oxidant than 1,2-dichloroethane32.

a, The preparation of organosodium compounds 1Na–30Na by reductive sodiation of aryl chlorides or halogen–sodium exchange with aryl- and alkenylbromides. b, Iron-catalysed oxidative homocoupling of aryl- and alkenylsodium compounds. Yields were determined by isolation of the homocoupled products. aThe homocoupling was conducted at 50 °C. bFe(acac)3 (10 mol%) was used. Ar, aryl.
A variety of aryl and alkenyl organosodium compounds 1Na–30Na were prepared in situ through reductive sodiation of aryl chlorides15,33 or halogen–sodium exchange of aryl bromides16,33,34, and the scope of the iron-catalysed oxidative homocoupling was explored for the construction of biaryl derivatives 1–30 (Fig. 2a,b). A key factor in minimizing the formation of undesired side products is the efficiency of the sodiation protocol; the starting materials (1′–30′) were converted to the corresponding organosodium compounds (1Na–30Na) with high yields, avoiding unproductive reactions of unreacted aryl halides in the subsequent step. One-pot homocoupling of phenylsodium (1Na) prepared by reductive sodiation of chlorobenzene (1′) went smoothly. This reaction was also applicable to mono- and dialkylated arylsodium compounds, to give the corresponding biaryl products (2–8, 15 and 16) in moderate-to-good yields. Other arylsodium compounds bearing silyl (9Na and 10Na), alkoxy (11Na and 12Na) and amino (13Na and 14Na) groups also reacted well. The synthesis of fully deuterated biphenyl (17) was achieved using readily available chlorobenzene-d5 (17′), and oligoarylene derivatives (18 and 19) could be conveniently synthesized through this Fittig-type coupling. For compounds 21–30, the organosodiums were generated through halogen–sodium exchange of the corresponding aryl bromides with neopentylsodium and used for one-pot iron-catalysed oxidative homocoupling under the same conditions. Conjugated molecules were synthesized by the reaction of π-extended aryl- and alkenylsodium nucleophiles (21Na, 22Na and 27Na–29Na). Despite the strong reducing and basic conditions, a silyl protecting group on phenol (23 and 24) and alcohol (25) and a carbazolyl group (30) were well tolerated. The iron-catalysed homocoupling of organosodium was successfully applied to the synthesis of functional organic molecules such as TPB ((20, NN,N,N′,N′-tetraphenylbenzidine), DPVBi (29, 4,4’-bis(2,2-diphenylvinyl)-1,1’-biphenyl), and CBP (30,4,4’-di(9H-carbazol-9-yl)-1,1’-biphenyl), demonstrating the potential of iron/sodium chemistry for synthetic applications in materials science.
Investigation of additives for the development of cross-coupling
Building upon the success in controlling the catalytic reactivity for the oxidative homocoupling using organosodium reagents and an iron catalyst, we subsequently investigated the C(sp2)–C(sp3) cross-coupling reaction of arylsodiums with alkyl halides (Wurtz–Fittig-type coupling). The initial attempt at iron-catalysed cross-coupling of 4-(tert-butyl)phenylsodium (8Na) with a secondary alkyl bromide such as bromocyclohexane gave only a trace amount of the desired cross-coupled product 31, and the main compound obtained was the homocoupled product 8 (Fig. 3 and Supplementary Fig. 1). Thus, we decided to investigate the reaction parameters in order to switch the selectivity towards cross-coupling.

a, The effect of polar and Lewis donor additives. b, Selective cross-coupling reaction under the optimized reaction conditions with Fe(acac)3 (5 mol%) and TMEDA as a cosolvent (see the details in the Supplementary Information). Yields were determined by gas chromatography analysis using dodecane as an internal standard, after calibration.
Highly ionic organosodium compounds typically aggregate to form complex polymeric structures in solution and solid state35,36. Therefore, to control the aggregation state of the organosodium compounds in MCH, we screened various additives capable of disrupting their aggregation. Thus, the iron-catalysed cross-coupling of arylsodium 8Na with bromocyclohexane was conducted in the presence of various polar and Lewis donor additives (Fig. 3a). Although the use of polar additives, such as N-methylpyrrolidone (NMP)37,38, N,N'-dimethylethyleneurea (DMI)39 and N,N′-dimethylpropyleneurea (DMPU)40 is known to enhance the iron-catalysed coupling of Grignard reagents, the amide and urea moieties are incompatible with the highly nucleophilic organosodium, and both cross- and homocoupling products were obtained in low yield. The use of ethereal compounds improved the yield of the cross-coupling product41 but also promoted homocoupling, where the haloalkane acted as an oxidant rather than as an electrophile. The results obtained with mono-, di- and higher dentate N-donor additives42,43 mirrored those observed when O-donor additives were used. Intriguingly, only N,N,N′,N′-tetramethylethylenediamine (TMEDA)44,45 inverted the selectivity of the reaction, resulting in cross-selective coupling (cross/homo = 76:24). With TMEDA as a cosolvent46, increasing the amount of organosodium nucleophile to 1.7 equivalent resulted in a much improved yield of cross-coupled product with high cross-selectivity (Fig. 3b). A control experiment confirmed that, in the absence of the iron catalyst, the cross-coupling product was not obtained at all (Supplementary Table 3). Notably, the use of other transition-metal catalysts such as palladium, nickel and copper did not promote the cross-coupling reaction and produced exclusively the homocoupled product (Supplementary Table 3). Thus, the metallic impurities contained in the iron salt are considered to exert a minimal influence on the cross-coupling reaction47, and the unique efficiency of iron as a catalyst illustrates the importance of appropriate pairing of the two metals to control their reactivity.
Scope of the iron-catalysed cross-coupling of organosodium
With the optimized conditions, we examined the substrate scope for the iron-catalysed cross-coupling (Fig. 4). First, several arylsodium compounds were evaluated for the cross-coupling with bromocyclohexane, and the corresponding products were obtained in moderate-to-good yield. Methyl (32, 33 and 37), ethyl (34 and 35), butyl (31 and 36), silyl (38), methoxy (39) and amino (40, 41 and 42) groups on the para-, meta- and ortho-positions of the nucleophilic aromatic ring were tolerated. We next explored the scope of secondary and primary alkyl halides. The use of cyclohexyl iodide or chloride instead of bromide gave the cross-coupled product (31) in a lower yield. Cycloalkyl bromides with different ring sizes (43 and 44) and those containing oxygen (45), nitrogen (46) and fluorine (47) reacted well. An acyclic secondary alkyl bromide gave product 48 in good yield. Primary alkyl bromides substituted with a β-alkyl group gave the Wurtz–Fittig-type products (49–53) in good-to-excellent yields; the iron-free coupling reaction of arylsodium in TMEDA cosolvent did not proceed under these conditions (49 and 50). Finally, we applied the cross-coupling protocol for the selective functionalization of a bioactive molecule; the arylation of cholesteryl bromide (54) proceeded smoothly with the retention of the stereochemistry, forming 55 as a single stereoisomer48,49.

Yields were determined by isolation of the cross-coupled products. aFe(acac)3 (10 mol%) was used. bFe(acac)3 (20 mol%) was used and the cross-coupling was conducted at 30 °C. cYields were determined by gas chromatography analysis using dodecane as an internal standard. Ar, aryl.
Mechanistic investigations
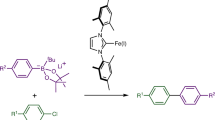
To better understand the selective formation of the cross-coupled product with alkyl bromides, we first examined the effect of varying equivalents of TMEDA on the composition of the formed arylsodiums. To that end, we prepared salt- and donor-free 8Na and 12Na and studied their solution behaviour upon addition of the Lewis donor TMEDA. We observed that, upon addition of stoichiometric amounts of the donor, 8Na remained insoluble in cyclohexane, whereas 12Na was solubilized. 1H diffusion-ordered spectroscopy nuclear magnetic resonance (1H DOSY NMR) spectroscopic studies in deuterated cyclohexane of the latter revealed the formation of an aggregate with an estimated molecular weight of 816 g mol−1, which would fit with a tetrameric structure of the form [(ArNa)4(TMEDA)2]. Crystallization at −30 °C provided single crystals that confirmed the suspected tetrameric structure suggested by 1H DOSY NMR spectroscopic studies, [(C6H4OMeNa)4(TMEDA)2] (Fig. 5a). This compound exhibits a cubane structure, with Na and C at alternate corners. Interestingly, two different sodium environments are present; Na1/Na2 are stabilized by forming Na–O interactions with two neighbouring OMe groups, whereas Na3/Na4 achieve coordinative saturation by bonding to the two N atoms of a TMEDA molecule (Fig. 5a). Further addition of an excess of TMEDA into the highly insoluble 8Na (30 equiv.) and 12Na (2 equiv.) in deuterated cyclohexane delivered more soluble aggregates, with estimated molecular weights of 709 g mol−1 and 609 g mol−1. The calculated molecular weights of the aggregates present in an excess of TMEDA suggest that a dimeric motif (dimer, 545 g mol−1) is present in the solution for 8Na and that an aggregate with a motif between the tetrameric structure observed by crystallography ([(C6H4OMeNa)4(TMEDA)2], 753 g mol−1) and a dimer (dimer, 493 g mol−1) is present in the solution for 12Na. The results of our 1H DOSY NMR spectroscopic studies demonstrate how the addition of TMEDA is able to not only solubilize these arylsodiums but also decrease their level of aggregation, increasing their kinetic reactivity. We can correlate these observations with the key role of TMEDA for an efficient cross-coupling, being able to form in this case less aggregated and more soluble arylsodium reagents. The role of other common Lewis donors in deaggregating 12Na was also probed by 1H DOSY NMR spectroscopic studies. A tetrameric structure was observed in solution upon the addition of 1 equiv. of PMDETA, and the addition of 4 equiv. of tetrahydrofuran (THF) to 12Na led to a trisolvated dimeric structure, whereas the addition of the crown ether 15-crown-5 led to a rapid decomposition of the donor as monitored by NMR spectroscopy. Although the deaggregation of 12Na was observed with other donors, it was also noticed that different yields of the cross-coupled product were obtained when using different donors, suggesting that the donor may play an additional role in the catalysis beyond that of solvating the arylsodium intermediates.

a, TMEDA deaggregation of organosodium reagents. b, The direct synthesis and characterization of organoiron species. c, Stoichiometric reaction with organoiron complexes. d, Radical-clock experiments. e, Plausible mechanism of iron-catalysed direct coupling of organosodium compounds. Ar, aryl; r.t., room temperature; n.d., not detected.
Thus, we expected that the addition of TMEDA would also have an impact on the iron catalytic cycle. For iron-catalysed cross-coupling reactions, a mechanistic understanding of the catalytic cycle and identification of the reactive species is critically important for successful reaction development, as iron is known to exhibit a rich redox chemistry with formal oxidation states typically from Fe(−II) to Fe(+III), and a variety of reactivity patterns50,51,52. To gain insights into the mechanism of iron-catalysed coupling of organosodiums, we first investigated the reactivity of arylsodium reagents with an iron complex. We started with (TMEDA)FeBr2, because we speculated that Fe(II) bromide species might form during the catalytic cycle of the cross-coupling with alkyl bromides in the presence of TMEDA53. Upon addition of 1 or 2 equiv. of 12Na in the presence of TMEDA, we obtained the corresponding [(TMEDA)FeBr(C6H4OMe)] Fe-1 and [(TMEDA)Fe(C6H4OMe)2] Fe-2 (Fig. 5b). These complexes decompose rapidly in a few hours at room temperature, forming variable amounts of 2,2′-dimethoxybiphenyl (12) and other unidentified products. Crystallographic studies confirmed the identity of these species, showing their monomeric structure, analogous to the structures reported bearing a phenyl group by Neidig and coworkers, which were prepared by salt metathesis with PhMgBr in THF53. Similarly, the reaction of in situ formed Fe-2 with cyclohexyl bromide delivered the corresponding cross-coupled product in 52% yield (104% yield versus Fe-2), whereas Fe-1 delivered the cross-coupled product in just 10% yield (Fig. 5c). These observations are consistent with the radical mechanism proposed for the coupling of aryl Grignard reagents catalysed by iron in the presence of TMEDA53, based on extensive analysis of the reaction mixtures by Mössbauer spectroscopy and the isolation of iron intermediates. The feasibility of the proposed mechanism was also supported by DFT calculations54, showing that the Fe(II)/Fe(III)/Fe(I) is the most plausible mechanism. Because we access similar iron species in solution using organosodium nucleophiles instead of organomagnesium, we propose that the formed Fe-2 is able to react with alkyl bromides in a similar way to generate an alkyl radical species and provide a higher yield of cross-coupled product 39, whereas Fe-1 is unable to generate the alkyl radical efficiently and results in a poor yield of the cross-coupled product. Control experiments reacting Fe(acac)3 with the tetrameric form of 12Na, [(C6H4OMeNa)4(TMEDA)2], in the presence or absence of additional TMEDA in C6D6 show the formation of Fe-2 in solution (Supplemantary Information section 8 and Supplementary Figs. 6 and 7), suggesting that the same TMEDA-bound iron species are formed in catalytic conditions where Fe(acac)3 is used instead of FeBr2.
To confirm the formation of radical intermediates in solution, we performed radical-clock experiments using electrophile probes such as (bromomethyl)cyclopropane and 6-bromohex-1-ene, in parallel with control experiments in the absence of any iron catalyst (Fig. 5d). The reaction with (bromomethyl)cyclopropane resulted in the exclusive formation of the ring-opened product (57), with no cyclic product (56) observed. When 6-bromohex-1-ene was used as a substrate, the ring-closed product (52) was obtained as the main product, together with some uncyclized product (58). These results indicated that an alkyl radical species is generated by homolytic cleavage of the C–Br bond facilitated by the active iron species.
On the basis of these experimental results and the consideration of literature reports31,53,55,56, we herein propose a plausible mechanism in iron-catalysed coupling reactions of organosodium compounds (Fig. 5e). The Fe(III) precursor is reduced by arylsodium to generate reduced Fe(I) or Fe(II) species, with the release of the biaryl compound, in the absence or presence of TMEDA. For the oxidative homocoupling of arylsodium reagents, the Fe(I) complex is reoxidized by DTBP and the subsequent transmetallation with arylsodium followed by reductive elimination affords the biaryl product to close the catalytic cycle. In the case of cross-coupling in the presence of excess TMEDA, an arylsodium complex solvated by TMEDA can transmetallate to the Fe(II) centre to form (TMEDA)FeArX (Fe-Ar1) or (TMEDA)FeAr2 (Fe-Ar2), the latter of which can react with alkyl bromide to generate an alkyl radical. The recombination of Fe-Ar1 with the alkyl radical yields the corresponding Fe(III) intermediate, which reductively eliminates the cross-coupled product. The resulting reduced (TMEDA)Fe(I)X complex can react more efficiently with the alkyl bromide to generate the Fe(II) species and release an alkyl radical that will recombine with Fe-Ar1 as in the first cycle and propagate the catalytic cycle. The occasional formation of Fe-Ar2 can explain the radical initiation pathway of the catalytic cycle as well as the formation of a small amount of homocoupled product, which is observed in experimental conditions. The low solubility of arylsodium reagents (even in the presence of excess TMEDA) would limit the amount of diaryliron species formed in solution, favouring the formation of cross-coupling product and avoiding the need for slow addition of the organometallic reagent, which is typically required when using other polar organometallics such as ArMgX or ArLi (refs. 44,57,58,59). Nevertheless, we cannot completely rule out the formation of sodium ferrates in solution (such as NaFeAr3). Previous studies have shown that ferrate complexes can react with alkyl bromides to give the corresponding cross-coupled products; Bedford showed that the [FeMes3]− intermediate generated from a bulky mesitylmagnesium bromide is more reactive than (TMEDA)FeMes2(ref. 60), although more recent studies by Neidig assessing the reactivity of ArMgBr in these reactions have shown that ferrates with less sterically hindered substituents are thermally less stable and react less efficiently with an electrophile than the relevant (TMEDA)FeAr2 species53,61. Multiple attempts to isolate sodium tris(aryl)ferrate by reacting (TMEDA)FeBr2 with an excess of 12Na resulted in the isolation of iron bis(aryl) Fe-2. We next attempted the synthesis of a heteroleptic sodium ferrate via co-complexation of a bis(alkyl) complex, (TMEDA)Fe(CH2SiMe3)2, with 12Na. The Fe(II) alkyl complex has been shown to be thermally robust62 and can form stable ferrates with sodium alkyl reagents63. Interestingly, the stoichiometric reaction of the in situ formed ferrate (TMEDA)2NaFe(CH2SiMe3)2(Ar) (I) with cyclohexyl bromide led to the formation of the cross-coupled product in a reasonable yield of 56%. However, attempts to isolate ferrate (I) gave (TMEDA)2NaFe(CH2SiMe3)3 as a crystalline solid63. This is consistent with ligand redistribution in I, and concomitant formation of (TMEDA)2NaFe(Ar)3, which can ultimately lead to the reactive (TMEDA)FeAr2 (Fe-2) complex, possibly explaining the formation of the cross-coupled product in this stoichiometric experiment.
Conclusions
We have developed an iron-catalysed homo- and cross-coupling of organosodium compounds, which enables the formation of C(sp2)–C(sp2) and C(sp2)–C(sp3) bonds in a more sustainable manner. We applied this reaction to the synthesis of various biaryl and alkylarene molecules, including π-conjugated molecules and a cholesterol derivative. A plausible catalytic cycle involving the generation of organoiron(II) species, which reacts with an alkyl halide through single-electron transfer to initiate the radical process, was proposed and supported by mechanistic studies, paralleling the proposed mechanism for the iron-catalysed cross-coupling of organomagnesium compounds. Iron-catalysed couplings have focused primarily on the utilization of organomagnesium, organozinc and organoboron reagents and, recently, organolithiums59,64. When reactive compounds such as organomagnesium and organolithium are used, the slow addition of the organometallic reagent and/or cryogenic conditions are generally required for controlling the catalytic reactivity of iron; in this study, iron catalysis has met organosodium chemistry, and notably, the reactions proceeded under mild conditions and without slow addition of reagents.
The use of sustainable metals such as iron for catalysis has become an important area of modern research65; we believe that the present demonstration of the direct use of organosodium compounds in catalysis will lay the foundations for a concept of sustainable organometallic reagents66. There has been a renaissance of interest in the development of synthetic reactions using organosodium chemistry67,68,69,70,71, some of which use more sustainable approaches such as green solvents33, flow chemistry34,72,73,74 and mechanochemistry75. We, thus, believe that the coupling reactions described here will further stimulate the development of organosodium chemistry for sustainable synthesis.
Methods
General procedure for iron-catalysed oxidative homocoupling of organosodium compounds
In a dry Schlenk tube equipped with a glass-coated stirring bar, aryl chloride (0.50 mmol, if liquid) was added to a mixture of MCH (1.0 ml) and sodium dispersion (ca. 26 wt%, ca. 92.8 mg, 1.05 mmol, 210 mol%) under nitrogen. If the starting material was solid, the sodium dispersion was added to a mixture of aryl chloride (0.50 mmol) and MCH (1.0 ml). After stirring at 30 °C for 1 h to generate the corresponding arylsodium, Fe(acac)3 (8.8 mg, 25 μmol, 5 mol%), benzene (0.50 ml) and DTBP (46 μl, 0.25 mmol, 0.5 equiv.) were added to the suspension of the arylsodium in MCH at 0 °C, and the reaction mixture was stirred at 30 °C for 30 min. The reaction was quenched with D2O (0.1 ml) and then with a saturated aqueous solution of NH4Cl (1 ml) at 0 °C. After extraction with ethyl acetate (EtOAc), diethyl ether (Et2O) or dichloromethane (CH2Cl2) three times, the combined organic layers were passed through a plug of silica gel with EtOAc, Et2O or CH2Cl2 and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel to afford the desired compound.
General procedure for iron-catalysed cross-coupling of organosodium compounds with alkyl halides
The organosodium compound was prepared using the same procedure as above. Fe(acac)3 (5.3 mg, 15 μmol, 5 mol%), TMEDA (0.50 ml) and alkyl halide (0.30 mmol, 1.0 equiv.) were added to the suspension of the arylsodium in MCH (1.0 ml) at 0 °C, and the reaction mixture was stirred at 0 °C for 1 h. The reaction was quenched with D2O (0.1 ml) and then with a saturated aqueous solution of NH4Cl (1 ml) at 0 °C. After extraction with EtOAc, Et2O or CH2Cl2 three times, the combined organic layers were passed through a plug of silica gel with EtOAc, Et2O or CH2Cl2 and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel to afford the desired compound.
Data availability
The data that support the findings of this study are available within the Article and its Supplementary Information. Detailed conditions for each reaction and compound characterization data are provided in the Supplementary Methods, Supplementary Fig. 1 and Supplementary Tables 1–5. NMR and electron paramagnetic resonance (EPR) spectra are available in Supplementary Figs. 2–14 and 18–136. Crystal structure data have been deposited at the Cambridge Crystallographic Data Centre (CCDC nos. 2375562 (Fe-2), 2375563 ([(C6H4OMeNa)4(TMEDA)2]) and 2375564 (Fe-1)), and crystallographic data are provided in the Supplementary Methods, Supplementary Figs. 15–17 and Supplementary Table 6. These data can be obtained free of charge via The Cambridge Crystallographic Data Centre at www.ccdc.cam.ac.uk/structures/.
References
Haynes, W. M. CRC Handbook of Chemistry and Physics 105th edn. (CRC Press, 2024).
ICH Harmonised Guideline, Guideline for Elemental Impurities, Q3D(R2) (International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, 2022).
de Meijere, A., Bräse, S. & Oestreich, M. Metal-Catalyzed Cross-Coupling Reactions and More Vols. 1–3 (Wiley, 2014).
Wurtz, A. Sur une nouvelle classe de radicaux organiques. Ann. Chim. Phys. 44, 275–312 (1855).
Wurtz, A. Ueber eine neue Klasse organischer Radicale. Ann. Chem. Pharm. 96, 364–375 (1855).
Fittig, R. Ueber das Monobrombenzol. Justus Liebigs Ann. Chem. 121, 361–365 (1862).
Tollens, B. & Fittig, R. Ueber die Synthese der Kohlenwasserstoffe der Benzolreihe. Justus Liebigs Ann. Chem. 131, 303–323 (1864).
Tollens, B. & Fittig, R. Vorläufige Notiz über gemischte Alkoholradicale der Aethyl-und Phenylreihe. Justus Liebigs Ann. Chem. 129, 369–370 (1864).
Johansson Seechurn, C. C. C., Kitching, M. O., Colacot, T. J. & Snieckus, V. Palladium-catalyzed cross-coupling: a historical contextual perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed. 51, 5062–5085 (2012).
Seyferth, D. Alkyl and aryl derivatives of the alkali metals: useful synthetic reagents as strong bases and potent nucleophiles. 1. Conversion of organic halides to organoalkali-metal compounds. Organometallics 25, 2–24 (2006).
Mulvey, R. E. & Robertson, S. D. Synthetically important alkali-metal utility amides: lithium, sodium, and potassium hexamethyldisilazides, diisopropylamides, and tetramethylpiperidides. Angew. Chem. Int. Ed. 52, 11470–11487 (2013).
Woltornist, R. A. et al. Structure, reactivity, and synthetic applications of sodium diisopropylamide. Synthesis 52, 1478–1488 (2020).
De, P. B., Asako, S. & Ilies, L. Recent advances in the use of sodium dispersion for organic synthesis. Synthesis 53, 3180–3192 (2021).
Anderson, D. E., Tortajada, A. & Hevia, E. New frontiers in organosodium chemistry as sustainable alternatives to organolithium reagents. Angew. Chem. Int. Ed. 63, e202313556 (2024).
Asako, S., Nakajima, H. & Takai, K. Organosodium compounds for catalytic cross-coupling. Nat. Catal. 2, 297–303 (2019).
Asako, S., Takahashi, I., Nakajima, H., Ilies, L. & Takai, K. Halogen–sodium exchange enables efficient access to organosodium compounds. Commun. Chem. 4, 76 (2021).
Inoue, T. et al. Cross-coupling polymerization of organosodium for polythiophene synthesis. Organometallics 40, 3506–3510 (2021).
Inoue, T., Kuwayama, A., Okano, K., Horie, M. & Mori, A. Generation of sodium–thiophene species with metal amide-free approach toward polythiophene synthesis by cross-coupling polymerization. Asian J. Org. Chem. 11, e202200253 (2022).
Bolm, C., Legros, J., Le Paih, J. & Zani, L. Iron-catalyzed reactions in organic synthesis. Chem. Rev. 104, 6217–6254 (2004).
Bauer, I. & Knölker, H.-J. Iron catalysis in organic synthesis. Chem. Rev. 115, 3170–3387 (2015).
Piontek, A., Bisz, E. & Szostak, M. Iron-catalyzed cross-couplings in the synthesis of pharmaceuticals: in pursuit of sustainability. Angew. Chem. Int. Ed. 57, 11116–11128 (2018).
Rana, S., Biswas, J. P., Paul, S., Paik, A. & Maiti, D. Organic synthesis with the most abundant transition metal–iron: from rust to multitasking catalysts. Chem. Soc. Rev. 50, 243–472 (2021).
Bedford, R. B. How low does iron go? Chasing the active species in Fe-catalyzed cross-coupling reactions. Acc. Chem. Res. 48, 1485–1493 (2015).
Parchomyk, T. & Koszinowski, K. Iron-catalyzed cross-coupling: mechanistic insight for rational applications in synthesis. Synthesis 49, 3269–3280 (2017).
Bakas, N. J. & Neidig, M. L. Additive and counterion effects in iron-catalyzed reactions relevant to C–C bond formation. ACS Catal. 11, 8493–8503 (2021).
Wowk, V. & Lefèvre, G. The crucial and multifaceted roles of main-group cations and their salts in iron-mediated cross-couplings. Dalton Trans. 51, 10674–10680 (2022).
Asako, S., Kodera, M., Nakajima, H. & Takai, K. Lithium-free synthesis of sodium 2,2,6,6-tetramethylpiperidide and its synthetic applications. Adv. Synth. Catal. 361, 3120–3123 (2019).
Bole, L. J., Tortajada, A. & Hevia, E. Enhancing metalating efficiency of the sodium amide NaTMP in arene borylation applications. Angew. Chem. Int. Ed. 61, e202204262 (2022).
Tortajada, A., Anderson, D. E. & Hevia, E. Gram-scale synthesis, isolation and characterisation of sodium organometallics: nBuNa and NaTMP. Helv. Chim. Acta 105, e202200060 (2022).
Anderson, D. E., Tortajada, A. & Hevia, E. Highly reactive hydrocarbon soluble alkylsodium reagents for benzylic aroylation of toluenes using Weinreb amides. Angew. Chem. Int. Ed. 62, e202218498 (2023).
Wang, Z.-Y., Peng, X.-S. & Wong, H. N. C. Ligand-free iron-catalyzed homo-coupling of aryllithium reagents. Asian J. Org. Chem. 9, 1834–1840 (2020).
Nagano, T. & Hayashi, T. Iron-catalyzed oxidative homo-coupling of aryl Grignard reagents. Org. Lett. 7, 491–493 (2005).
Dilauro, G. et al. Introducing water and deep eutectic solvents in organosodium chemistry: chemoselective nucleophilic functionalizations in air. Angew. Chem. Int. Ed. 62, e202304720 (2023).
Harenberg, J. H. et al. (2-Ethylhexyl)sodium: a hexane-soluble reagent for Br/Na-exchanges and directed metalations in continuous flow. Angew. Chem. Int. Ed. 60, 14296–14301 (2021).
Weiss, E. Structures of organo alkali metal complexes and related compounds. Angew. Chem. Int. Ed. 32, 1501–1523 (1993).
Klett, J. Structural motifs of alkali metal superbases in non-coordinating solvents. Chem. Eur. J. 27, 888–904 (2021).
Cahiez, G. & Marquais, S. Cu-catalyzed alkylation and Fe-catalyzed alkenylation of organomanganese reagents. Pure Appl. Chem. 68, 53–60 (1996).
Fürstner, A. & Leitner, A. Iron-catalyzed croos-coupling reactions of alkyl-Grignard reagents with aryl chlorides, tosylates, and triflates. Angew. Chem. Int. Ed. 41, 609–612 (2002).
Bisz, E. & Szostak, M. Cyclic ureas (DMI, DMPU) as efficient, sustainable ligands in iron-catalyzed C(sp2)–C(sp3) coupling of aryl chlorides and tosylates. Green Chem. 19, 5361–5366 (2017).
Cahiez, G., Gager, O. & Habiak, V. Iron-catalyzed alkenylation of Grignard reagents by enol phosphates. Synthesis https://doi.org/10.1055/s-2008-1067194 (2008).
Nagano, T. & Hayashi, T. Iron-catalyzed Grignard cross-coupling with alkyl halides possessing β-hydrogens. Org. Lett. 6, 1297–1299 (2004).
Bedford, R. B., Bruce, D. W., Frost, R. M. & Hird, M. Simple iron-amine catalysts for the cross-coupling of aryl Grignards with alkyl halides bearing β-hydrogens. Chem. Commun. https://doi.org/10.1039/B507133J (2005).
Cahiez, G., Habiak, V., Duplais, C. & Moyeux, A. Iron-catalyzed alkylations of aromatic Grignard reagents. Angew. Chem. Int. Ed. 46, 4364–4366 (2007).
Nakamura, M., Matsuo, K., Ito, S. & Nakamura, E. Iron-catalyzed cross-coupling of primary and secondary alkyl halides with aryl Grignard reagents. J. Am. Chem. Soc. 126, 3686–3687 (2004).
Martin, R. & Fürstner, A. Cross-coupling of alkyl halides with aryl Grignard reagents catalyzed by a low-valent iron complex. Angew. Chem. Int. Ed. 43, 3955–3957 (2004).
Trimitsis, G. B., Tuncay, A. & Beyer, R. D. Metalations of dimethylarenes with organosodium reagents. The catalytic effect of certain tertiary amines. J. Am. Chem. Soc. 94, 2152–2153 (1972).
Bedford, R. B. et al. Iron-catalysed Suzuki coupling? A cautionary tale. Tetrahedron Lett. 50, 6110–6111 (2009).
Nakamura, M., Ito, S., Matsuo, K. & Nakamura, E. Iron-catalyzed chemoselective cross-coupling of primary and secondary alkyl halides with arylzinc reagents. J. Am. Chem. Soc. https://doi.org/10.1021/ja049744t (2005).
Bauer, G., Cheung, C. W. & Hu, X. Cross-coupling of nonactivated primary and secondary alkyl halides with aryl Grignard reagents catalyzed by chiral iron pincer complexes. Synthesis 47, 1726–1732 (2015).
Fürstner, A. et al. Preparation, structure, and reactivity of nonstabilized organoiron compounds. Implications for iron-catalyzed cross coupling reactions. J. Am. Chem. Soc. 130, 8773–8787 (2008).
Guérinot, A. & Cossy, J. Iron-catalyzed C–C cross-couplings using organometallics. Top. Curr. Chem. 374, 49 (2016).
Zhou, E. et al. Mechanistic facets of the competition between cross-coupling and homocoupling in supporting ligand-free iron-mediated aryl–aryl bond formations. ACS Org. Inorg. Au 2, 359–369 (2022).
Bakas, N. J., Sears, J. D., Brennessel, W. W. & Neidig, M. L. A TMEDA–iron adduct reaction manifold in iron-catalyzed C(sp2)–C(sp3) cross-coupling reactions. Angew. Chem. Int. Ed. 61, e202114986 (2022).
Wan, X., Zhao, T., Ren, Q. & Zhou, G. Role of TMEDA in iron-catalyzed C(sp2)–C(sp3) cross-coupling reactions: a mechanistic study using DFT calculations. Organometallics 43, 449–456 (2024).
Sharma, A. K. et al. DFT and AFIR study on the mechanism and the origin of enantioselectivity in iron-catalyzed cross-coupling reactions. J. Am. Chem. Soc. 139, 16117–16125 (2017).
Lee, W., Zhou, J. & Gutierrez, O. Mechanism of Nakamura’s bisphosphine-iron-catalyzed asymmetric C(sp2)–C(sp3) cross-coupling reaction: the role of spin in controlling arylation pathways. J. Am. Chem. Soc. 139, 16126–16133 (2017).
Guérinot, A., Reymond, S. & Cossy, J. Iron-catalyzed cross-coupling of alkyl halides with alkenyl Grignard reagents. Angew. Chem. Int. Ed. 46, 6521–6524 (2007).
Jin, M., Adak, L. & Nakamura, M. Iron-catalyzed enantioselective cross-coupling reactions of α-chloroesters with aryl Grignard reagents. J. Am. Chem. Soc. 137, 7128–7134 (2015).
Jia, Z., Liu, Q., Peng, X.-S. & Wong, H. N. C. Iron-catalysed cross-coupling of organolithium compounds with organic halides. Nat. Commun. 7, 10614 (2016).
Bedford, R. B. et al. TMEDA in iron-catalyzed Kumada coupling: amine adduct versus homoleptic “ate” complex formation. Angew. Chem. Int. Ed. 53, 1804–1808 (2014).
Carpenter, S. H., Baker, T. M., Muñoz, S. B. III, Brennessel, W. W. & Neidig, M. L. Multinuclear iron–phenyl species in reactions of simple iron salts with PhMgBr: identification of Fe4(μ-Ph)6(THF)4 as a key reactive species for cross-coupling catalysis. Chem. Sci. 9, 7931–7939 (2018).
Bart, S. C., Hawrelak, E. J., Schmisseur, A. K., Lobkovsky, E. & Chirik, P. J. Synthesis, reactivity, and solid state structures of four-coordinate iron(II) and manganese(II) alkyl complexes. Organometallics 23, 237–246 (2004).
Tortajada, A. & Hevia, E. Room-temperature intermolecular hydroamination of vinylarenes catalyzed by alkali-metal ferrate complexes. ACS Org. Inorg. Au 5, 62–68 (2025).
Zhong, Z., Peng, X.-S. & Wong, H. N. C. Recent advances on iron-catalyzed coupling reactions involving organolithium reagents. Chin. Chem. Lett. 30, 1463–1467 (2019).
Nakamura, E. & Sato, K. Managing the scarcity of chemical elements. Nat. Mater. 10, 158–161 (2011).
Nguyen, T. Sustainable organosodium compounds step up in cross-coupling reactions. C&EN News https://cen.acs.org/synthesis/reagents/Sustainable-organosodium-compounds-step-cross/97/i12 (2019).
Huang, Y., Chan, G. H. & Chiba, S. Amide-directed C–H sodiation by a sodium hydride/iodide composite. Angew. Chem. Int. Ed. 56, 6544–6547 (2017).
Zhang, J.-Q. et al. Conversion of triphenylphosphine oxide to organophosphorus via selective cleavage of C–P, O–P, and C–H bonds with sodium. Commun. Chem. 3, 1 (2020).
Bole, L. J. & Hevia, E. Activation of polar organometallic reagents with alkali-metal alkoxides. Nat. Synth. 1, 195–202 (2022).
Davison, N. et al. Li vs Na: divergent reaction patterns between organolithium and organosodium complexes and ligand-catalyzed ketone/aldehyde methylenation. J. Am. Chem. Soc. 145, 6562–6576 (2023).
Takahashi, F., Kurogi, T. & Yorimitsu, H. Synthesis of trans-1,2-dimetalloalkenes through reductive anti-dimagnesiation and dialumination of alkynes. Nat. Synth. 2, 162–171 (2023).
Harenberg, J. H., Annapureddy, R. R., Karaghiosoff, K. & Knochel, P. Continuous flow preparation of benzylic sodium organometallics. Angew. Chem. Int. Ed. 61, e202203807 (2022).
Weidmann, N., Ketels, M. & Knochel, P. Sodiation of arenes and heteroarenes in continuous flow. Angew. Chem. Int. Ed. 57, 10748–10751 (2018).
Jiang, Y., Kurogi, T. & Yorimitsu, H. Reductive stereo- and regiocontrolled boryllithiation and borylsodiation of arylacetylenes using flow microreactors. Nat. Synth. 3, 192–201 (2024).
Kondo, K., Kubota, K. & Ito, H. Mechanochemistry enabling highly efficient Birch reduction using sodium lumps and d-(+)-glucose. Chem. Sci. 15, 4452–4457 (2024).
Acknowledgements
This work was financially supported by JSPS KAKENHI Grant-in-Aid for Scientific Research (B) no. JP22H02125 (S.A.), Grant-in-Aid for Young Scientists nos. JP22K14719 and JP24K17716 (I.T.), MEXT KAKENHI Grant-in-Aid for Transformative Research Areas (A) no. JP24H01874 (Green Catalysis Science) (S.A.), Fukuoka Naohiko Memorial Foundation (S.A.), The Hattori Hokokai Foundation (S.A.) and The Foundation for the Promotion of Ion Engineering (S.A.). I.T. is grateful to the SPDR Program in RIKEN. We thank Z. Hou and M. Takimoto (RIKEN) for generously allowing us to use the mass spectrometer. The generous support of the Swiss National Science Foundation (SNF) (project numbers 219318 (E.H.) and 210608 (A.T.)) is also acknowledged, and we thank the X-ray crystal structure service at the University of Bern for measuring, solving, refining and summarizing the structure of compounds Fe-1, Fe-2 and [(C6H4OMeNa)4(TMEDA)2].
Funding
Open access funding provided by University of Bern.
Author information
Authors and Affiliations
Contributions
L.I., E.H. and S.A. conceived and directed the project. I.T. developed the method and performed the synthetic and part of the mechanistic experiments. A.T. and D.E.A. performed the mechanistic studies. All authors contributed to the writing of the manuscript. All authors analysed the data and contributed to discussions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Synthesis thanks Guillaume Lefèvre and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Thomas West, in collaboration with the Nature Synthesis team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Experimental procedures, Characterization data, Mechanistic studies, Crystallographic details, Supplementary Figs. 1–136, Supplementary Tables 1–6, Supplementary Methods and Supplementary References.
Supplementary Data 1
Crystallographic data for Fe-1, CCDC 2375564.
Supplementary Data 2
Crystallographic data for Fe-2, CCDC 2375562.
Supplementary Data 3
Crystallographic data for [(C6H4OMeNa)4(TMEDA)2], CCDC 2375563.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Takahashi, I., Tortajada, A., Anderson, D.E. et al. Iron-catalysed direct coupling of organosodium compounds. Nat. Synth (2025). https://doi.org/10.1038/s44160-025-00771-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s44160-025-00771-1
This article is cited by
-
Coupling reactions using iron–sodium pairs
Nature Synthesis (2025)
