
Abstract
Congenital myasthenic syndromes (CMS) are a heterogeneous group of disorders characterized by compromised neuromuscular signal transmission due to pathogenic germline variants in genes expressed at the neuromuscular junction (NMJ). A total of 40 genes have been reported in CMS (AGRN, ALG14, ALG2, CHAT, CHD8, CHRNA1, CHRNB1, CHRND, CHRNE, CHRNG, COL13A1, COLQ, DES, DOK7, DPAGT1, GFPT1, GMPPB, LAMA5, LAMB2, LRP4, MACF1, MUSK, MYO9A, PLEC, PREPL, PTPN11, PURA, RAPSN, RPH3A, SCN4A, SLC18A3, SLC25A1, SLC5A7, SNAP25, SYT2, TEFM, TOR1AIP1, UNC13A, UNC50 and VAMP1). The 40 genes are putatively classified into 13 subtypes by pathomechanical, clinical, and therapeutic features. A unique feature shared by recently identified genes is that CMS is concomitantly recognized in other mostly severer diseases. For example, four recently identified genes exhibit the following phenotypes: PURA-CMS, developmental delay; TEFM-CMS, mitochondrial disease; PTPN11-CMS, Noonan syndrome/Leopard syndrome; and DES-CMS, desmin myopathy. Conversely, these diseases are not always associated with CMS, although genetic and/or environmental factors that determine the involvement of the NMJ remain to be identified. In this review, particular emphasis will be placed on five recently identified genes (MACF1, TEFM, PTPN11, DES and UNC50).
Similar content being viewed by others

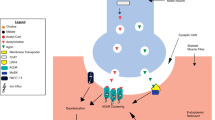

Introduction
CMS are caused by pathogenic germline variants in genes expressed at the neuromuscular junction (NMJ), and are characterized by defective neuromuscular signal transduction [1, 2]. Pathogenic variants have been identified in 40 genes (AGRN, ALG14, ALG2, CHAT, CHD8, CHRNA1, CHRNB1, CHRND, CHRNE, CHRNG, COL13A1, COLQ, DES, DOK7, DPAGT1, GFPT1, GMPPB, LAMA5, LAMB2, LRP4, MACF1, MUSK, MYO9A, PLEC, PREPL, PTPN11, PURA, RAPSN, RPH3A, SCN4A, SLC18A3, SLC25A1, SLC5A7, SNAP25, SYT2, TEFM, TOR1AIP1, UNC13A, UNC50 and VAMP1) (Fig. 1). The causative genes have been classified into three categories of presynaptic, synaptic, and postsynaptic CMS. To delineate clinical, pathomechanical, and therapeutic features of CMS, we classified the 40 genes into 13 subtypes (Tables 1 and 2). We extensively reviewed 35 genes causing CMS in 2023 [1]. In addition to the epidemiology, inheritance, and therapeutic features of CMS, five recently identified genes in CMS (MACF1, TEFM, PTPN11, DES and UNC50) will be introduced in detail in this review.

Schematic of 40 genes (red letters) causing congenital myasthenic syndromes. Five representative groups of gene products that cooperatively work at the NMJ and are compromised in CMS are explained below. First, adult AChR is comprised of α, β, δ and ε subunits encoded by CHRNA1, CHRNB1, CHRND and CHRNE, respectively. Gene products of RAPSN, CHD8 and MACF1 make subsynaptic structural network on which AChRs are clustered. UNC50 (UNC50) is essential for trafficking AChR. Defects in these genes cause endplate AChR deficiency (subtype 1). Second, agrin (AGRN) released from the motor nerve terminal binds to LRP4 (LRP4) at the motor endplate, and triggers MuSK (MUSK) phosphorylation, which is enhanced by cytoplasmic adaptor protein DOK7 (DOK7). Defects in these genes compromise AChR clustering (subtype 6). Third, choline generated by hydrolysis of acetylcholine by acetylcholinesterase in the synaptic space is taken up by high-affinity choline transporter (CHT1. SLC5A7) at the motor nerve terminal. Acetylcholine is resynthesized from choline by choline acetyl transferase (CHAT) and is incorporated into the synaptic vesicle by vesicular acetylcholine transporter (vAChT, SLC18A3). Defects in these genes compromise recycling of acetylcholine (subtype 8). Fourth, the action potential that reached the motor nerve terminal opens P/Q- and N-type calcium channels. Ca2+ ions entered in the nerve terminal bind to synaptotagmin 2 (SYT2) and activate the SNARE complex comprised of synaptobrevin/VAMP (VAMP1), SNAP25 (SNAP25), and syntaxin. Rabphilin 3a (RPH3A), α5 laminin (LAMA5), and Munc13-1 (UNC13A) are SNARE-associated proteins that play pivotal roles in the release of the synaptic vesicles. Defects in these genes cause LEMS-like CMS (subtype 9). Fifth, GFPT1 (GFPT1) is the rate limiting enzyme to generate UDP-GlcNAc that is required for N- and O-linked glycosylation of glycoproteins, as well as for making glycosaminoglycans and glycolipids. DPAGT1 (DPAGT1), ALG14 (ALG14) and ALG2 (ALG2) are enzymes in the N-glycosylation pathway. GMPPB (GMPPB) generates GDP-mannose, a major mannosyl donor for mannose-containing polymers. Defects in these genes cause glycosylation-deficient CMS (subtype 10)
Epidemiology
The prevalences of CMS per million in total population are 1.8 (18/10,000,000) in Brazil [3], 1.8 (64/35,500,000) in Spain [4], 3.1 (28/9,000,000) in Austria [5], and 3.2 (37/11,900,000) in Belgium [6], which gives rise to the weighted average of 2.2 per million in total population. Similarly, the prevalences of CMS per million under age 18 years are 9.2 (123/13,900,000) in UK [7], 22.2 (8/360,000) in Slovenia [8] and 10.5 (28/2,670,000) in Austria [5], and the weighted average becomes 9.7 per million under age 18 years.
Inheritance
All except for the following five forms of CMS are caused by loss-of-function variants with autosomal recessive inheritance (Table 2). Autosomal dominant inheritance or hemiallelic de novo variant is observed in slow-channel CMS (SCCMS) [1], PURA-CMS [9], PTPN11-CMS [10], SNAP25-CMS [11, 12] and some [13,14,15] but not all [16,17,18] patients of SYT2-CMS. SCCMS is caused by a missense variant that prolongs the channel openings of acetylcholine receptors (AChRs), and 50% abnormal AChRs are sufficient to cause SCCMS [1]. PURA-CMS and PTPN11-CMS are associated with developmental delay and are likely to be caused by loss-of-function of PURA [9] and gain-of-function of PTPN11 [10], respectively. Both SNAP25-CMS [11, 12] and SYT2-CMS [13,14,15,16,17,18] show LEMS-like CMS, and are likely to be caused by dominant negative effects.
Therapeutic features
Therapeutic agents for CMS include cholinesterase inhibitors (ChEIs) (pyridostigmine and neostigmine), β-adrenergic agents (ephedrine, salbutamol, and albuterol), amifampridine (3,4-diaminopyridine), quinidine, fluoxetine, and acetazolamide (Table 2) [19, 20].
ChEIs block both acetylcholinesterase and butyrylcholinesterase, and prolong the dwell time of acetylcholine released from the nerve terminal, thereby make acetylcholine receptor open for a prolonged time. ChEIs are effective in most but not all forms of CMS. ChEIs are contraindicated for COLQ-CMS [21,22,23] and LAMB2-CMS [24], because respiratory arrest may occur in some patients. Similarly, ChEIs are ineffective or worsen symptoms in patients with SCCMS, AGRN-CMS, LRP4-CMS and MUSK-CMS [25,26,27]. The reason for the lack of the effects of ChEIs in CMS associated with defective AChR clustering (AGRN, LRP4, MUSK and DOK7) remains unknown.
The sympathetic nerve directly innervate the NMJ, and sympathomimetics ameliorates electrophysiological and morphological deficits of the NMJ induced by sympathectomy in a mouse model of CMS [28]. Similarly, adrenaline, but not noradrenaline, increases the action potential-elicited Ca2+ entry into the motor nerve terminal and increases both spontaneous and evoked acetylcholine release [29]. The positive effect of natural agonist adrenaline is reproduced only by β2-adranergic agonist, fenoterol, but not by α1-, α2-, or β1-adranergic agonist [29]. In contrast to ChEIs, β-adrenergic agents are effective in most forms of CMS including SCCMS and COLQ-CMS, in which excessive openings of AChRs compromise the NMJ signal transmission [26]. Although some patients do not respond to β-adrenergic agents, no patients worsened with β-adrenergic agents. Especially, β-adrenergic agents are effective for CMS associated with defective AChR clustering (AGRN, LRP4, MUSK and DOK7). Amifampridine, a blocker of the voltage-gated potassium channel at the nerve terminal, is another commonly used drug for CMS. Amifampridine is also effective in many forms of CMS, but worsening of symptoms is observed in some patients with AGRN-CMS and DOK7-CMS [26]. Quinidine [30, 31] and fluoxetine [32] block AChR openings and ameliorate SCCMS. A marked effect of fluoxetine was reported in a case of COLQ-CMS [33]. Acetazolamide was effective in two patients of SCN4A-CMS [34, 35], but was not in another SCN4A-CMS [36].
MACF1-CMS in the subtype of ‘endplate acetylcholine receptor deficiency’
Screening for rapsyn-dependent AChR-binding molecules detected MACF1 (microtubule-actin cross-linking factor 1) that carries binding sites for microtubules and action [37]. MACF1 links rapsyn to microtubule-associated proteins including end-binding protein 1 (EB1) and microtubule-associated protein 1b (MAP1b), as well as to actin-associated protein, vinculin [37]. MACF1 is essential for the structural integrity, functional maturation, and long-term maintenance of the NMJ.
In two CMS patients in Serbia and India, recessive missense variants were identified in MACF1 [37]. Both patients showed decremental response to repetitive nerve stimulation. The Indian patient showed late-onset fatigable limb-girdle muscle weakness without ophthalmoparesis, and responded well to cholinesterase inhibitors and salbutamol [37]. In 197 pedigrees with CMS in India in another report, a patient with late-onset limb-girdle CMS was homozygous for a missense variant in MACF1 [27].
De novo heterozygous variants in MACF1 were previously reported in nine patients with lissencephaly 9 with complex brainstem malformation (LIS9) (OMIM #618325) [38]. In LIS9, heterozygous pathogenic missense or inframe variants were observed the GAR ___domain, and a dominant negative effect was speculated [38]. In contrast, in three patients with MACF1-CMS, homozygous or compound homozygous pathogenic variants were observed either in the plakin ___domain or the spectrin repeats [27, 37]. Thus, affected domains may determine the phenotype and heredity.
Although lack of ophthalmoparesis is unusual in the subtype of ‘endplate AChR deficiency’, MACF1-CMS is classified into ‘endplate AChR deficiency’, in which pathogenic variants are also present in CHRNA1, CHRNB1, CHRND, CHRNE, RAPSN and CHD8 encoding chromodomain helicase DNA-binding protein 8 (Tables 1 and 2).
TEFM-CMS in the subtype of ‘CMS caused by defective nerve terminal formation’
TEFM (transcription elongation factor, mitochondrial) is essential for mitochondrial DNA transcription by mitochondrial RNA polymerase [39]. Knockout of Tefm in zebrafish showed defective NMJ structures and defective mitochondrial transcription [40]. Especially, synaptic vesicles at the nerve terminal were markedly decreased in Tefm-knockout zebrafish.
In seven patients in five families with mitochondrial myopathy, eight recessive pathogenic variants were identified in TEFM [40]. Three patients showed fatigable muscle weakness, and one patient showed decremental response to repetitive nerve stimulation. Two patients were treated with salbutamol with favorable responses. The other patients variably showed lactic acidosis, epilepsy, developmental delay, and motor ataxia, which are characteristic of mitochondrial diseases. In biopsied skeletal muscle and primary skin fibroblasts, marked reduction of the transcription of the H and L strands of mitochondrial DNA was observed [40]. The protein levels of mitochondrial electron transport complex proteins were markedly decreased, but mitochondrial outer membrane protein TOMM20 as well as the number of mitochondria were preserved. Another report showed that two siblings were homozygous for a missense variant in TEFM [27]. They showed fatigable limb and ocular muscle weakness along with epilepsy and ataxia, and abnormal decrement on repetitive nerve stimulation. They also showed elevated lactate and decreased mitochondrial electron transport chain complexes I and IV in muscle biopsy. Pathogenic variants in TEFM have not been reported in any other diseases.
TEFM-CMS is classified into ‘CMS caused by defective nerve terminal formation’, in which pathogenic variants are also observed in MYO9A encoding myosin 9A and SLC25A1 encoding mitochondrial tricarboxylate transporter [41] (Tables 1 and 2). The gene product of SLC25A1 shuttles citrate and malate between mitochondria and cytoplasm. The nerve terminal is rich in mitochondria due to high energy demand required for the recycling of acetylcholine and the repeated releases of synaptic vesicles. Based on the presence of TEFM-CMS and SLC25A1-CMS, mitochondrial CMS was proposed [42]. Mitochondrial CMS is an attractive idea because of the essential roles of mitochondrial energy production at the NMJ. However, most patients with mitochondrial disease do not exhibit defects in the NMJ signal transmission, and genetic and mechanistic factors that affect the NMJ signal transmission remain unelucidated. In addition, MYO9A-CMS that shows defective nerve terminal formation fits well with SLC25A1-CMS. We thus classified MYO9A-CMS, SLC25A1-CMS and TEFM-CMS into the subtype of ‘defective nerve terminal formation’.
PTPN11-CMS in the subtype of ‘CMS associated with developmental disorders’
PTPN11 (protein-tyrosine phosphatase, nonreceptor type 11) encodes SHP-2 (src homology region 2-___domain phosphatase-2). SHP-2 is a ubiquitously expressed signaling molecule especially in the RAS/MAPK pathway, and plays essential roles in cell proliferation, differentiation, migration, and apoptosis [43].
In four patients with Noonan/Leopard syndrome with muscle weakness, heterozygous PTPN11 variants were observed [10]. Three showed fatigability and bulbar signs. One showed delayed motor milestones. Two out of three who were examined for repetitive nerve stimulation showed decremental responses. Pyridostigmine was effective in a patient but not in two other patients. Salbutamol was effective in a single patient. Patients with Noonan and Leopard syndrome show variable degrees of muscle weakness, but the ratio of the patients exhibiting CMS symptoms remains unclear.
Pathogenic variants in PTPN11 have been reported in Noonan syndrome [43] and Leopard syndrome [44]. Noonan syndrome is an autosomal dominant disorder characterized by short stature, hypertelorism, mild mental retardation, skeletal malformation, and congenital heart defects [45]. Sixteen causative genes have been reported in Noonan syndrome (OMIM PS163950), and heterozygous PTPN11 variants are responsible for ~50% of the patients. Clinical features of Leopard syndrome are overlapping with those of Noonan syndrome. Leopard is an acronym for Lentigines, ECG conduction abnormalities, Ocular hypertelorism, Pulmonary stenosis, Abnormal genitalia, Retardation of growth, and sensorineural Deafness [46]. Leopard syndrome is also called ‘Noonan syndrome with multiple lentigines’. Three causative genes (PTPN11, RAF1, and BRAF) have been reported in Leopard syndrome (OMIM PS151100), and PTPN11 variants constitute most of them. Factors that drive the expression of CMS phenotypes remain unknown.
PTPN11-CMS is classified into ‘CMS associated with developmental disorders’, in which pathogenic variants also are observed in PURA encoding purine-rich element-binding protein A (Tables 1 and 2).
DES-CMS in the subtype of 'CMS caused by defective structural molecule at the NMJ'
Desmin encoded by DES is a muscle-specific intermediate filament protein [47]. Desmin forms a cytoplasmic scaffold in skeletal, cardiac, and smooth muscles.
In three unrelated patients with fatigable muscle weakness, ptosis, and ophthalmoparesis without cardiomyopathy, a homozygous intronic variant in DES (NM_001927.4: c.1023 + 5 G > A) was observed [27, 48]. The patients showed decremental responses to repetitive nerve stimulation. Pyridostigmine was effective in two patients, and salbutamol was effective in one patient. The intronic variant caused leaky retention of complete intron 5 of DES, and the protein levels of desmin in skeletal muscle were reduced to 60–75% of normal. There was another report of DES-CMS in 2016, in which two cousins with homozygous truncating DES variants showed fatigable limb and facial muscle weakness, ptosis, and severe ophthalmoparesis [49]. Decremental responses to repetitive nerve stimulation were observed in both patients. Salbutamol was effective in both patients.
Pathogenic variants in DES have been reported in desmin-related myopathy (DRM) that is characterized by myofibrillar degeneration with desmin-positive aggregates [50]. DRM is also referred to as myofibrillar myopathy [51]. Heterozygous missense variants in DES in DRD show dominant negative effects and generate abnormal desmin aggregates [50]. Homozygous DES variants are rare and are reported in 19 DRM patients [52]. Fifteen patients carried truncating DES variants and four were homozygous for missense variants. In all the patients, loss-of-function mechanisms were speculated. Lack of dominant negative effect and mild degrees of desmin reduction in patients with DES-CMS may account for the CMS phenotype.
DES-CMS is classified into ‘CMS caused by defective structural molecule at the NMJ’, in which pathogenic variants are also observed in PLEC encoding plectin (Tables 1 and 2).
UNC50-CMS in the subtype of ‘CMS with arthrogryposis multiplex congenita (AMC)’
UNC50 (Unc-50 inner nuclear membrane RNA-binding protein) is a transmembrane protein in the Golgi apparatus. UNC50 was identified by screening for a mammalian homolog of unc-50 in C. elegans, and found to be an essential molecule for trafficking AChR [53, 54].
In a stillborn baby with arthrogryposis multiplex congenita (AMC), a homozygous frameshift variant was detected in UNC50 [55]. C. elegans carrying an orthologous mutant showed a marked decrease in the surface expression of muscle AChR [55]. Similarly, in five babies in two families, three resulted in stillbirth or neonatal death, one pregnancy was terminated, and one baby died in early infancy [56]. Two genetically identified babies were homozygous for an intronic deletion in the polypyrimidine tract that activated a cryptic 3’ splice site in UNC50 causing a frameshift. Although electrophysiological or morphological studies of the NMJ were not available due to early neonatal death, the babies were reported to be a subtype of CMS [56].
AMC is a key symptom of multiple pterygium syndrome (MPS), which is characterized by pterygia across multiple joints. MPS is divided into the lethal variant (LMPS, OMIM #253290) and the nonlethal variant (Escobar syndrome, OMIM #265000). Similarly, AMC is a key feature of fetal akinesia deformation sequence (FADS, OMIM #618388, #618389, #618393), which is characterized by multiple deformities including pulmonary hypoplasia, craniofacial anomalies, and hypoplastic dermal ridges. AMC/LMPS/Escovar/FADS are caused by reduced fetal movements. Although more than 320 causative genes have been identified in AMC/LMPS/Escobar/FADS, AMC/LMPS/Escobar/FADS are frequently caused by pathogenic variants affecting the NMJ signal transmission. Escobar syndrome is exclusively caused by pathogenic variants in CHRNG [57,58,59,60]. CHRNG encodes the fetal γ subunit of AChR, and is expressed only in embryos. Defects in AChR γ subunit affect fetal movements, but not neonatal or later movements. Thus, Escovar syndrome is mostly benign and nonprogressive.
AMC/LMPS/FADS are also caused by pathogenic variants in AGRN [56, 61], CHRNA1 [62, 63], CHRNB1 [64,65,66], CHRND [62, 67], DOK7 [68, 69], MUSK [70,71,72,73], MYO9A [74], RAPSN [62, 75, 76], SLC18A3 [77], SLC5A7 [78] and SNAP25 [11] (Tables 1 and 2). These genes also cause other subtypes of CMS, but pathogenic variants in AMC/LMPS/FADS are more deleterious than those in the other subtypes of CMS.
Conclusions
CMS are caused by pathogenic germline variants in 40 genes, which are classified into 13 subtypes according to clinical, mechanical, and therapeutic features. Concomitant recognition of myasthenic features in other mostly severer diseases facilitated the identification of novel genes in recent years. Prevalences of CMS in total population and under 18 years of age are 2.2 and 9.7 per million, respectively. Five forms of CMS (SCCMS, PURA-CMS, PTPN11-CMS, SNAP25-CMS and SYT2-CMS) are caused by autosomal dominant variant, and the others are by autosomal recessive variants.
References
Ohno K, Ohkawara B, Shen XM, Selcen D, Engel AG. Clinical and pathologic features of congenital myasthenic syndromes caused by 35 genes-a comprehensive review. Int J Mol Sci. 2023;24:3730.
Ramdas S, Beeson D, Dong YY. Congenital myasthenic syndromes: increasingly complex. Curr Opin Neurol. 2024;37:493–501.
Mihaylova V, Scola RH, Gervini B, Lorenzoni PJ, Kay CK, Werneck LC, et al. Molecular characterisation of congenital myasthenic syndromes in Southern Brazil. J Neurol Neurosurg Psychiatry. 2010;81:973–7.
Natera-de Benito D, Topf A, Vilchez JJ, Gonzalez-Quereda L, Dominguez-Carral J, Diaz-Manera J, et al. Molecular characterization of congenital myasthenic syndromes in Spain. Neuromuscul Disord. 2017;27:1087–98.
Krenn M, Sener M, Rath J, Zulehner G, Keritam O, Wagner M, et al. The clinical and molecular landscape of congenital myasthenic syndromes in Austria: a nationwide study. J Neurol. 2023;270:909–16.
Smeets N, Gheldof A, Dequeker B, Poleur M, Maldonado Slootjes S, Van Parijs V, et al. Congenital myasthenic syndromes in Belgium: genetic and clinical characterization of pediatric and adult patients. Pediatr Neurol. 2024;158:57–65.
Parr JR, Andrew MJ, Finnis M, Beeson D, Vincent A, Jayawant S. How common is childhood myasthenia? The UK incidence and prevalence of autoimmune and congenital myasthenia. Arch Dis Child. 2014;99:539–42.
Troha Gergeli A, Neubauer D, Golli T, Butenko T, Loboda T, Maver A, et al. Prevalence and genetic subtypes of congenital myasthenic syndromes in the pediatric population of Slovenia. Eur J Paediatr Neurol. 2020;26:34–8.
Qashqari H, McNiven V, Gonorazky H, Mendoza-Londono R, Hassan A, Kulkarni T, et al. PURA syndrome: neuromuscular junction manifestations with potential therapeutic implications. Neuromuscul Disord. 2022;32:842–4.
Pugliese A, Della Marina A, de Paula Estephan E, Zanoteli E, Roos A, Schara-Schmidt U, et al. Mutations in PTPN11 could lead to a congenital myasthenic syndrome phenotype: a Noonan syndrome case series. J Neurol. 2024;271:1331–41.
Shen XM, Selcen D, Brengman J, Engel AG. Mutant SNAP25B causes myasthenia, cortical hyperexcitability, ataxia, and intellectual disability. Neurology. 2014;83:2247–55.
Reynolds HM, Wen T, Farrell A, Mao R, Moore B, Boyden SE, et al. Rapid genome sequencing identifies a novel de novo SNAP25 variant for neonatal congenital myasthenic syndrome. Cold Spring Harb Mol Case Stud. 2022;8:a006242.
Herrmann DN, Horvath R, Sowden JE, Gonzalez M, Sanchez-Mejias A, Guan Z, et al. Synaptotagmin 2 mutations cause an autosomal-dominant form of Lambert-Eaton myasthenic syndrome and nonprogressive motor neuropathy. Am J Hum Genet. 2014;95:332–9.
Whittaker RG, Herrmann DN, Bansagi B, Hasan BA, Lofra RM, Logigian EL, et al. Electrophysiologic features of SYT2 mutations causing a treatable neuromuscular syndrome. Neurology. 2015;85:1964–71.
Montes-Chinea NI, Guan Z, Coutts M, Vidal C, Courel S, Rebelo AP, et al. Identification of a new SYT2 variant validates an unusual distal motor neuropathy phenotype. Neurol Genet. 2018;4:e282.
Donkervoort S, Mohassel P, Laugwitz L, Zaki MS, Kamsteeg EJ, Maroofian R, et al. Biallelic loss of function variants in SYT2 cause a treatable congenital onset presynaptic myasthenic syndrome. Am J Med Genet A. 2020;182:2272–83.
Maselli RA, van der Linden H Jr, Ferns M. Recessive congenital myasthenic syndrome caused by a homozygous mutation in SYT2 altering a highly conserved C-terminal amino acid sequence. Am J Med Genet A. 2020;182:1744–9.
Maselli RA, Wei DT, Hodgson TS, Sampson JB, Vazquez J, Smith HL, et al. Dominant and recessive congenital myasthenic syndromes caused by SYT2 mutations. Muscle Nerve. 2021;64:219–24.
Schara U, Lochmuller H. Therapeutic strategies in congenital myasthenic syndromes. Neurotherapeutics. 2008;5:542–7.
Thompson R, Bonne G, Missier P, Lochmuller H. Targeted therapies for congenital myasthenic syndromes: systematic review and steps towards a treatabolome. Emerg Top Life Sci. 2019;3:19–37.
Wargon I, Richard P, Kuntzer T, Sternberg D, Nafissi S, Gaudon K, et al. Long-term follow-up of patients with congenital myasthenic syndrome caused by COLQ mutations. Neuromuscul Disord. 2012;22:318–24.
Yis U, Becker K, Kurul SH, Uyanik G, Bayram E, Haliloglu G, et al. Genetic landscape of congenital myasthenic syndromes from Turkey: novel mutations and clinical insights. J Child Neurol. 2017;32:759–65.
Durmus H, Shen XM, Serdaroglu-Oflazer P, Kara B, Parman-Gulsen Y, Ozdemir C, et al. Congenital myasthenic syndromes in Turkey: clinical clues and prognosis with long term follow-up. Neuromuscul Disord. 2018;28:315–22.
Maselli RA, Ng JJ, Anderson JA, Cagney O, Arredondo J, Williams C, et al. Mutations in LAMB2 causing a severe form of synaptic congenital myasthenic syndrome. J Med Genet. 2009;46:203–8.
Ohkawara B, Cabrera-Serrano M, Nakata T, Milone M, Asai N, Ito K, et al. LRP4 third beta-propeller ___domain mutations cause novel congenital myasthenia by compromising agrin-mediated MuSK signaling in a position-specific manner. Hum Mol Genet. 2014;23:1856–68.
Theuriet J, Masingue M, Behin A, Ferreiro A, Bassez G, Jaubert P, et al. Congenital myasthenic syndromes in adults: clinical features, diagnosis and long-term prognosis. Brain. 2024;147:3849–62.
Polavarapu K, Sunitha B, Topf A, Preethish-Kumar V, Thompson R, Vengalil S, et al. Clinical and genetic characterisation of a large Indian congenital myasthenic syndrome cohort. Brain. 2024;147:281–96.
Khan MM, Lustrino D, Silveira WA, Wild F, Straka T, Issop Y, et al. Sympathetic innervation controls homeostasis of neuromuscular junctions in health and disease. Proc Natl Acad Sci USA. 2016;113:746–50.
Arkhipov A, Zhilyakov N, Sibgatullina G, Nevsky E, Bukharaeva EA, Petrov AM. Adrenergic modulation of acetylcholine release at the mouse neuromuscular junctions of fast-twitch skeletal muscle. Neurochem Res. 2025;50:162.
Fukudome T, Ohno K, Brengman JM, Engel AG. Quinidine normalizes the open duration of slow-channel mutants of the acetylcholine receptor. Neuroreport. 1998;9:1907–11.
Harper CM, Engel AG. Quinidine sulfate therapy for the slow-channel congenital myasthenic syndrome. Ann Neurol. 1998;43:480–4.
Harper CM, Fukodome T, Engel AG. Treatment of slow-channel congenital myasthenic syndrome with fluoxetine. Neurology. 2003;60:1710–3.
Vidanagamage A, Gooneratne IK, Nandasiri S, Gunaratne K, Fernando A, Maxwell S, et al. A rare mutation in the COLQ gene causing congenital myasthenic syndrome with remarkable improvement to fluoxetine: A case report. Neuromuscul Disord. 2021;31:246–8.
Tsujino A, Maertens C, Ohno K, Shen XM, Fukuda T, Harper CM, et al. Myasthenic syndrome caused by mutation of the SCN4A sodium channel. Proc Natl Acad Sci USA. 2003;100:7377–82.
Berghold VM, Koko M, Berutti R, Plecko B. Case report: novel SCN4A variant associated with a severe congenital myasthenic syndrome/myopathy phenotype. Front Pediatr. 2022;10:944784.
Habbout K, Poulin H, Rivier F, Giuliano S, Sternberg D, Fontaine B, et al. A recessive Nav1.4 mutation underlies congenital myasthenic syndrome with periodic paralysis. Neurology. 2016;86:161–9.
Oury J, Liu Y, Topf A, Todorovic S, Hoedt E, Preethish-Kumar V, et al. MACF1 links Rapsyn to microtubule- and actin-binding proteins to maintain neuromuscular synapses. J Cell Biol. 2019;218:1686–705.
Dobyns WB, Aldinger KA, Ishak GE, Mirzaa GM, Timms AE, Grout ME, et al. MACF1 mutations encoding highly conserved zinc-binding residues of the GAR ___domain cause defects in neuronal migration and axon guidance. Am J Hum Genet. 2018;103:1009–21.
Hillen HS, Temiakov D, Cramer P. Structural basis of mitochondrial transcription. Nat Struct Mol Biol. 2018;25:754–65.
Van Haute L, O’Connor E, Diaz-Maldonado H, Munro B, Polavarapu K, Hock DH, et al. TEFM variants impair mitochondrial transcription causing childhood-onset neurological disease. Nat Commun. 2023;14:1009.
O’Connor E, Topf A, Muller JS, Cox D, Evangelista T, Colomer J, et al. Identification of mutations in the MYO9A gene in patients with congenital myasthenic syndrome. Brain. 2016;139:2143–53.
O’Connor K, Spendiff S, Lochmuller H, Horvath R. Mitochondrial mutations can alter neuromuscular transmission in congenital myasthenic syndrome and mitochondrial disease. Int J Mol Sci. 2023;24:8505.
Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29:465–8.
Digilio MC, Conti E, Sarkozy A, Mingarelli R, Dottorini T, Marino B, et al. Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am J Hum Genet. 2002;71:389–94.
Mendez HM, Opitz JM. Noonan syndrome: a review. Am J Med Genet. 1985;21:493–506.
Sarkozy A, Digilio MC, Dallapiccola B. Leopard syndrome. Orphanet J Rare Dis. 2008;3:13.
Clemen CS, Herrmann H, Strelkov SV, Schroder R. Desminopathies: pathology and mechanisms. Acta Neuropathol. 2013;125:47–75.
Polavarapu K, O’Neil D, Thompson R, Spendiff S, Nandeesh B, Vengalil S, et al. Partial loss of desmin expression due to a leaky splice site variant in the human DES gene is associated with neuromuscular transmission defects. Neuromuscul Disord. 2024;39:10–8.
Durmus H, Ayhan O, Cirak S, Deymeer F, Parman Y, Franke A, et al. Neuromuscular endplate pathology in recessive desminopathies: Lessons from man and mice. Neurology. 2016;87:799–805.
Capetanaki Y, Papathanasiou S, Diokmetzidou A, Vatsellas G, Tsikitis M. Desmin related disease: a matter of cell survival failure. Curr Opin Cell Biol. 2015;32:113–20.
Ohno SelcenD, Engel K. AG. Myofibrillar myopathy: clinical, morphological and genetic studies in 63 patients. Brain. 2004;127:439–51.
Onore ME, Savarese M, Picillo E, Passamano L, Nigro V, Politano L. Bi-Allelic DES gene variants causing autosomal recessive myofibrillar myopathies affecting both skeletal muscles and cardiac function. Int J Mol Sci. 2022;23:15906.
Fitzgerald J, Kennedy D, Viseshakul N, Cohen BN, Mattick J, Bateman JF, et al. UNCL, the mammalian homologue of UNC-50, is an inner nuclear membrane RNA-binding protein. Brain Res. 2000;877:110–23.
Eimer S, Gottschalk A, Hengartner M, Horvitz HR, Richmond J, Schafer WR, et al. Regulation of nicotinic receptor trafficking by the transmembrane Golgi protein UNC-50. EMBO J. 2007;26:4313–23.
Abiusi E, D’Alessandro M, Dieterich K, Quevarec L, Turczynski S, Valfort AC, et al. Biallelic mutation of UNC50, encoding a protein involved in AChR trafficking, is responsible for arthrogryposis. Hum Mol Genet. 2017;26:3989–94.
Shravya MS, Purushothama G, Radhakrishnan P, Hebbar M, Guruvare S, Mathew M, et al. Biallelic variant, c.644-13_644-9del in UNC50 is associated with congenital myasthenia syndrome. Am J Med Genet A. 2025:e64086. https://doi.org/10.1002/ajmg.a.64086. Online ahead of print.
Morgan NV, Brueton LA, Cox P, Greally MT, Tolmie J, Pasha S, et al. Mutations in the embryonal subunit of the acetylcholine receptor (CHRNG) cause lethal and Escobar variants of multiple pterygium syndrome. Am J Hum Genet. 2006;79:390–5.
Hoffmann K, Muller JS, Stricker S, Megarbane A, Rajab A, Lindner TH, et al. Escobar syndrome is a prenatal myasthenia caused by disruption of the acetylcholine receptor fetal gamma subunit. Am J Hum Genet. 2006;79:303–12.
Seo J, Choi IH, Lee JS, Yoo Y, Kim NK, Choi M, et al. Rare cases of congenital arthrogryposis multiplex caused by novel recurrent CHRNG mutations. J Hum Genet. 2015;60:213–5.
Shen XM, Nakata T, Mizuno S, Imoto I, Selcen D, Ohno K, et al. Impaired gating of gamma- and epsilon-AChR respectively causes Escobar syndrome and fast-channel myasthenia. Ann Clin Transl Neurol. 2023;10:732–43.
Geremek M, Dudarewicz L, Obersztyn E, Paczkowska M, Smyk M, Sobecka K, et al. Null variants in AGRN cause lethal fetal akinesia deformation sequence. Clin Genet. 2020;97:634–8.
Michalk A, Stricker S, Becker J, Rupps R, Pantzar T, Miertus J, et al. Acetylcholine receptor pathway mutations explain various fetal akinesia deformation sequence disorders. Am J Hum Genet. 2008;82:464–76.
Shamseldin HE, Swaid A, Alkuraya FS. Lifting the lid on unborn lethal Mendelian phenotypes through exome sequencing. Genet Med. 2013;15:307–9.
Agerholm JS, McEvoy FJ, Menzi F, Jagannathan V, Drogemuller C. A CHRNB1 frameshift mutation is associated with familial arthrogryposis multiplex congenita in Red dairy cattle. BMC Genom. 2016;17:479.
Freed AS, Schwarz AC, Brei BK, Clowes Candadai SV, Thies J, Mah JK, et al. CHRNB1-associated congenital myasthenia syndrome: expanding the clinical spectrum. Am J Med Genet A. 2021;185:827–35.
Ravenscroft G, Clayton JS, Faiz F, Sivadorai P, Milnes D, Cincotta R, et al. Neurogenetic fetal akinesia and arthrogryposis: genetics, expanding genotype-phenotypes and functional genomics. J Med Genet. 2021;58:609–18.
Chen C, Han J, Xue J, Li R, Chen G, Yang X, et al. Case Report: Early diagnosis of lethal multiple pterygium syndrome with micrognathia: two novel mutations in the CHRND gene. Front Genet. 2023;14:1005624.
Vogt J, Morgan NV, Marton T, Maxwell S, Harrison BJ, Beeson D, et al. Germline mutation in DOK7 associated with fetal akinesia deformation sequence. J Med Genet. 2009;46:338–40.
Radhakrishnan P, Shukla A, Girisha KM, Nayak SS. Biallelic c.1263dupC in DOK7 results in fetal akinesia deformation sequence. Am J Med Genet A. 2020;182:804–7.
Wilbe M, Ekvall S, Eurenius K, Ericson K, Casar-Borota O, Klar J, et al. MuSK: a new target for lethal fetal akinesia deformation sequence (FADS). J Med Genet. 2015;52:195–202.
Tan-Sindhunata MB, Mathijssen IB, Smit M, Baas F, de Vries JI, van der Voorn JP, et al. Identification of a Dutch founder mutation in MUSK causing fetal akinesia deformation sequence. Eur J Hum Genet. 2015;23:1151–7.
Li N, Qiao C, Lv Y, Yang T, Liu H, Yu WQ, et al. Compound heterozygous mutation of MUSK causing fetal akinesia deformation sequence syndrome: a case report. World J Clin Cases. 2019;7:3655–61.
Tiwari AK, Srinivasan VM, Phadke SR, Saxena D. Variants in DOK7 results in fetal akinesia deformation sequence: a case report and review of literature. Clin Genet. 2024;105:226–7.
Bayram Y, Karaca E, Coban Akdemir Z, Yilmaz EO, Tayfun GA, Aydin H, et al. Molecular etiology of arthrogryposis in multiple families of mostly Turkish origin. J Clin Invest. 2016;126:762–78.
Vogt J, Harrison BJ, Spearman H, Cossins J, Vermeer S, ten Cate LN, et al. Mutation analysis of CHRNA1, CHRNB1, CHRND, and RAPSN genes in multiple pterygium syndrome/fetal akinesia patients. Am J Hum Genet. 2008;82:222–7.
Winters L, Van Hoof E, De Catte L, Van Den Bogaert K, de Ravel T, De Waele L, et al. Massive parallel sequencing identifies RAPSN and PDHA1 mutations causing fetal akinesia deformation sequence. Eur J Paediatr Neurol. 2017;21:745–53.
Hakonen AH, Polvi A, Saloranta C, Paetau A, Heikkila P, Almusa H, et al. SLC18A3 variants lead to fetal akinesia deformation sequence early in pregnancy. Am J Med Genet A. 2019;179:1362–5.
Bauche S, O’Regan S, Azuma Y, Laffargue F, McMacken G, Sternberg D, et al. Impaired presynaptic high-affinity choline transporter causes a congenital myasthenic syndrome with episodic apnea. Am J Hum Genet. 2016;99:753–61.
Chaouch A, Muller JS, Guergueltcheva V, Dusl M, Schara U, Rakocevic-Stojanovic V, et al. A retrospective clinical study of the treatment of slow-channel congenital myasthenic syndrome. J Neurol. 2012;259:474–81.
Finsterer J. Slow-channel congenital myasthenic syndrome due to the novel variant c.1396G_A in CHRNA1 that responds favorably to 3,4-diaminopyridine: a case report. Cureus. 2024;16:e73601.
Mihaylova V, Muller JS, Vilchez JJ, Salih MA, Kabiraj MM, D’Amico A, et al. Clinical and molecular genetic findings in COLQ-mutant congenital myasthenic syndromes. Brain. 2008;131:747–59.
Nishikawa A, Mori-Yoshimura M, Okamoto T, Oya Y, Nakata T, Ohno K, et al. Beneficial effects of 3,4-diaminopyridine in a 26-year-old woman with DOK7 congenital myasthenic syndrome who was originally diagnosed with facioscapulohumeral dystrophy]. Rinsho Shinkeigaku. 2014;54:561–4.
Santos M, Cruz S, Peres J, Santos L, Tavares P, Basto JP, et al. DOK7 myasthenic syndrome with subacute adult onset during pregnancy and partial response to fluoxetine. Neuromuscul Disord. 2018;28:278–82.
Mroczek M, Durmus H, Topf A, Parman Y, Straub V. Four individuals with a homozygous mutation in exon 1f of the PLEC gene and associated myasthenic features. Genes. 2020;11:716.
Wyrebek R, DiBartolomeo M, Brooks S, Geller T, Crenshaw M, Iyadurai S. Hypotonic infant with PURA syndrome-related channelopathy successfully treated with pyridostigmine. Neuromuscul Disord. 2022;32:166–9.
Funding
This study was supported by Grants-in-Aid from the Japan Agency for Medical Research and Development (JP23ek0109678), the Japan Society for the Promotion of Science (JP23H02794, JP23K18273, and JP23K06412); the Ministry of Health, Labour and Welfare of Japan (23FC1014); and the National Center of Neurology and Psychiatry (5–6). Open Access funding provided by Nagoya University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ohno, K., Ito, M. & Ohkawara, B. Review of 40 genes causing congenital myasthenic syndromes. J Hum Genet (2025). https://doi.org/10.1038/s10038-025-01355-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s10038-025-01355-9
