
Abstract
Limited prognostic factors have been associated with overall survival (OS) post-relapse in childhood Acute Lymphoblastic Leukemia (ALL). Patients enrolled on 12 Children’s Oncology Group frontline ALL trials (1996–2014) were analyzed to assess for additional prognostic factors associated with OS post-relapse. Among 16,115 patients, 2053 (12.7%) relapsed. Relapse rates were similar for B-ALL (12.5%) and T-ALL (11.2%) while higher for infants (34.2%). Approximately 50% of B-ALL relapses occurred late (≥36 months) and 72.5% involved the marrow. Conversely, 64.8% of T-ALL relapses occurred early (<18 months) and 47.1% involved the central nervous system. The 5-year OS post-relapse for the entire cohort was 48.9 ± 1.2%; B-ALL:52.5 ± 1.3%, T-ALL:35.5 ± 3.3%, and infant ALL:21.5 ± 3.9%. OS varied by early, intermediate and late time-to-relapse; 25.8 ± 2.4%, 49.5 ± 2.2%, and 66.4 ± 1.8% respectively for B-ALL and 29.8 ± 3.9%, 33.3 ± 7.6%, 58 ± 9.8% for T-ALL. Patients with ETV6::RUNX1 or Trisomy 4 + 10 had median time-to-relapse of 43 months and higher OS post-relapse 74.4 ± 3.1% and 70.2 ± 3.6%, respectively. Patients with hypodiploidy, KMT2A-rearrangement, and TCF3::PBX1 had short median time-to-relapse (12.5-18 months) and poor OS post-relapse (14.2 ± 6.1%, 31.9 ± 7.7%, 36.8 ± 6.6%). Site-of-relapse varied by cytogenetic subtype. This large dataset provided the opportunity to identify risk factors for OS post-relapse to inform trial design and highlight populations with dismal outcomes post-relapse.
Similar content being viewed by others
Introduction
Acute lymphoblastic leukemia (ALL) is the most common childhood malignancy. With improved understanding of leukemia biology and the application of risk-directed therapy, 5-year event-free survival (EFS) for children in developed countries with newly diagnosed ALL is currently 85–90% [1,2,3,4,5,6]. However, for patients who relapse, outcome remains poor with limited progress made over the past three decades [7, 8].
During initial therapy, several well-established prognostic factors such as age, presenting white blood cell (WBC) count, central nervous system (CNS) involvement, cytogenetic and molecular subtype, and end-induction measurable residual disease (MRD) inform risk-directed therapy [9]. However, other than immunophenotype (B versus T), time-to-relapse, site of relapse, and MRD post re-induction, similar prognostic indicators have not been established for post-relapse outcomes [7, 10,11,12,13,14]. Smaller relapse studies suggest that patients with unfavorable cytogenetics or older age have worse outcomes but their role in risk stratification post-relapse is limited [11].
Children’s Oncology Group (COG) investigators previously reported overall survival (OS) after first relapse in children with ALL enrolled on an older group of non-overlapping frontline clinical trials between 1988 and 2002 [7]. Of 9585 patients on ten trials during this time period, 1961 (20.5%) relapsed. Five-year OS post-relapse for the entire cohort was 36%, similar to reports from other pediatric groups [10, 11]. In addition to time-to and site-of relapse, age at initial diagnosis, sex, and presence of CNS disease at diagnosis significantly influenced outcomes. However, due to limited immunophenotypic and cytogenetic data, detailed subgroup analyses were not possible.
In this report, we review the largest cohort to date of infant, pediatric, adolescent, and young adult patients with ALL enrolled on more contemporary COG trials. We identified additional clinical, cytogenetic, and biological risk factors that could enhance trial design and improve risk stratification algorithms for relapsed ALL.
Patients and methods
Patients and clinical trials
Patients with newly diagnosed B- or T-ALL enrolled on 12 frontline COG clinical trials from June 1996 - July 2014 were included in this study (n = 16,115; Table 1). Patients or guardians provided informed consent for trial participation. Patients <22 years were eligible for trials before 2004 after which eligibility expanded to patients <31 years. Trial details and 5-year EFS rates are listed in Table 1. For analysis of post-relapse survival, only patients with relapse as a first event were included. Patients with induction failure as defined by individual protocols (1.6%), death as first event (2.6%) and second malignant neoplasm as first event (0.96%) of 16,115 were excluded from the analysis. Data collected at first relapse included time from date of initial diagnosis to date of relapse (time-to-relapse), relapse site, and survival information. Data on subsequent therapy post-relapse were limited and therefore not addressed. Sex assigned at birth, race and ethnicity were per individual institutional submission.
The COG risk stratification algorithms used for patient eligibility are described in each trial publication [15,16,17,18,19,20,21,22,23,24,25]. Infants <1 year of age with B-ALL, treated on dedicated infant ALL trials, define the “Infant ALL” cohort in this report. Patients with B-ALL ≥ 1 year at diagnosis define the “B-ALL” cohort. Patients with T-ALL ≥ 1 year at diagnosis define the “T-ALL” cohort. All but 93 patients ≥1 year at diagnosis with T-ALL were treated on dedicated T-ALL trials. Outcomes post relapse in patients with Down Syndrome were reported separately [26].
Definition of relapse
Patients who achieved a morphologic remission with <5% blasts and clearance of extramedullary disease with frontline therapy who then developed disease recurrence were defined as having a relapse. Bone marrow relapse was defined by ≥25% morphologic blasts or ≥5% blasts with concomitant extramedullary relapse, CNS relapse by CNS 3 status (≥5 WBC/microliter cerebrospinal fluid, with blasts on cytospin) or clinical signs of CNS leukemia. Isolated extramedullary recurrence had to be proven by biopsy. Relapses that occurred <18 months from diagnosis were considered “early”, 18- <36 months “intermediate”, and ≥36 months “late”.
MRD determination and definitions
MRD was determined by centralized COG flow cytometry laboratories at the end of the 4-week Induction (EOI) therapy. On the CCG and POG trials MRD was done on a research basis and did not affect risk stratification but on the COG (AALL) trials, MRD was used in risk stratification and treatment decisions and are described in each trial publication [20,21,22,23,24,25]). For the majority of patients sensitivity was determined to 10−4, but in 87 patients data was only available to 10−3.
Cytogenetics and molecular subtypes
From 1996 to 2003, karyotype data from 12 reference cytogenetics laboratories (Pediatric Oncology Group) or from more than 100 institutional cytogenetic laboratories (Children’s Cancer Group) was collected and reviewed by members of their central cytogenetics committees. The requirement for fluorescence in situ hybridization (FISH) studies for chromosomes 4, 10, and 17 was added for patients on COG trials in 2003 and was initially performed in COG central or approved local laboratories (2003 to 2006) and later (2007 to 2014) exclusively in COG approved local cytogenetic laboratories. Testing for ETV6::RUNX1, BCR::ABL1, TCF3::PBX1 and KMT2A rearrangements was done by reverse transcriptase polymerase chain reaction (RT-PCR) or FISH in one of two central reference laboratories (2003 to 2006) or by FISH in COG approved local laboratories (2007 to 2014). Ascertainment of iAMP21 may have been incomplete prior to 2007. Standard karyotype analysis was performed in COG-approved local laboratories. All FISH and karyotype data from local laboratories were centrally reviewed. Patients found to have a BCR::ABL1 translocation during Induction therapy would be removed from protocol therapy on the eligible trials in this analysis and, if available, offered enrollment in dedicated Ph+ ALL treatment trials. Data on relapse and outcomes post relapse were available on these patients.
Statistical analyses
Among the eligible patients enrolled on 12 frontline studies, EFS from date of initial diagnosis was calculated for each study, with events defined as induction failure, relapse, second malignant neoplasm, or death of any cause, whichever occurred first. Patient and disease characteristics at initial diagnosis were compared between patients who relapsed and those who did not using Pearson’s chi-squared tests. Among patients who relapsed, OS post-relapse was defined as the time between date of first relapse and date of death; patients alive at last follow-up were censored. Data cutoff date was June 30, 2021. The EFS or OS probabilities were calculated using the Kaplan–Meier method with Greenwood standard errors. Analyses of OS post-relapse in relation to patient and disease characteristics were based on the logrank tests and univariate and multivariable Cox regression models [27, 28]. Analyses were performed for B-ALL, T-ALL, and infant ALL separately. All p-values reported are two-sided. A p ≤ 0.05 was considered statistically significant. Statistical analyses were performed using STATA software [29].
Results
Between June 1996 and July 2014, 16,115 patients enrolled on the 12 frontline clinical trials and 2053 (12.7%) relapsed (Table 1). Five-year EFS rates ranged from 45% to 91% depending upon the study population within each trial. For the entire relapse cohort, the median time-to-relapse was 31.0 months. Median follow-up from initial diagnosis was 121.7 months, and from first relapse, 70.5 months.
Most patients (13,771, 85.5%) had B-ALL, of which 12.5% relapsed (n = 1715) (Table 2). A significantly higher percentage of patients with National Cancer Institute (NCI) high-risk (HR) B-ALL relapsed (754/3974; 19%), compared to patients with NCI standard-risk (SR) B-ALL (961/9797; 9.8%; p < 0.0001). Among 2019 patients with T-ALL, 227 relapsed (11.2%) and of 325 infants, 111 relapsed (34.2%).
Relapse characteristics
Characteristics of all patients with and without relapse are presented in Table 2. By univariate analysis relapse rate was significantly higher for males, as well as infants and patients aged ≥10 years at initial diagnosis, with incremental increased risk in patients ≥16 years. Patients of Hispanic ethnicity of all races, and those with WBC ≥ 50,000/uL or CNS 2/3 status at initial diagnosis also had significantly increased risk of relapse. The distribution of relapse site and time differed by immunophenotype and is detailed in Fig. 1 and Supplementary Table 1. Isolated testicular relapse was exceedingly rare (n = 66; 0.7%).

Patterns of first relapse differed between (A) B-ALL, (B) T-ALL, (C) Infant ALL. iBM isolated bone marrow, BM bone marrow, CNS central nervous system, iCNS isolated central nervous system.
B-ALL
Patients with B-ALL had a median time-to-relapse of 34.6 months (range 1.7–186); significantly longer for NCI SR B-ALL compared to HR B-ALL (p < 0.0001) (37 months vs 31.6 months, Table 2). Approximately 16% of B-ALL relapses occurred >5 years from diagnosis and 1.7% >10 years (Fig. 1A). Patients with SR B-ALL accounted for over half of the B-ALL relapse cohort (961/1715). Patients with B-ALL most commonly experienced isolated bone marrow (iBM) relapses (58.7%); with any bone marrow (BM) involvement 72.5%, isolated CNS (iCNS) relapses 21.7%; and overall CNS involvement at relapse 32.9%. CNS relapses occurred earlier than BM relapses (Fig. 1A, Supplementary Table 1). Of 371 iCNS relapses, 113 occurred <18 months (30.5%), 205 (55.3%) 18- < 36 months and only 53 (14.3%) occurred ≥36 months. Patients with end-induction MRD ≥ 0.01% had increased risk of relapse (p < 0.0001).
Karyotype and FISH data for seven common cytogenetic subtypes were available at diagnosis for 10,094 of 13,771 patients with B-ALL (73.3%); 6440 patients were positive for one subtype; 3654 patients were negative for all seven subtypes, classified here as B-ALL, other. Complete cytogenetic data were not available for 3677 patients. Time-to-relapse differed among cytogenetic subtypes (Table 3). Patients with favorable cytogenetics: Trisomy 4 + 10 and ETV6::RUNX1 relapsed later, with a median time-to-relapse of 43 months for both subtypes. They comprised 52% of newly diagnosed patients and 30% (391/1307) of the B-ALL relapse cohort. Patients with iAMP21 also had a long median time-to-relapse (43.6 months). Those with unfavorable subtypes, i.e., hypodiploid and KMT2A-rearranged (KMT2A-R), had very short time-to-relapse (12.5 and 18 months respectively). Patients with TCF3::PBX1 also relapsed early, with median time-to-relapse 15.8 months.
Sites of relapse relative to cytogenetic subtype are detailed in Table 3. The proportion of patients with iBM relapse was highest for those with hypodiploidy (91.7%) and BCR::ABL1 (80%), while CNS involvement at relapse, either isolated or combined, was most frequent in patients with TCF3::PBX1 (43.9%).
T-ALL
In stark contrast to B-ALL, patients with T-ALL had a median time-to-relapse of 13.8 months (Table 2), the majority of whom (64.8%) relapsed <18 months from diagnosis and 82% relapsed <36 months from diagnosis. Relapse >5 years from diagnosis was less frequent than B-ALL (8.8%). The rate of iBM relapse in patients with T-ALL was 40.8%, any BM involvement 56.5%, iCNS relapse 31.4% and any CNS involvement at relapse 47.1% (Fig. 1B, Supplementary Table 1). Over 80% of iCNS relapses occurred <18 months from diagnosis. iCNS relapses accounted for 67.9%, 38.6%, and 21.1% of relapses that occurred in patients with CNS3, CNS2 and CNS1 at initial diagnosis respectively, demonstrating that burden of CNS disease at initial diagnosis was associated with iCNS site at relapse (Supplementary Table 2).
Infant ALL
Similar to patients with T-ALL, infants relapsed early with median time-to-relapse of 13.9 months (Table 2). Only 3.6% (4/111) infants who relapsed did so more than 36 months from diagnosis (Fig. 1C, Supplementary Table 1). BM was the most common site involved at relapse (77.8%) while 23.1% had any CNS involvement. Of 111 infants who relapsed, 98 were KMT2A-R and 13 were KMT2A-germline. Median time-to-relapse was 13.1 months (3.4–57.5) for KMT2A-R vs. 16.4 months (13.3–45.4) for KMT2A-germline infants (p = 0.009).
Survival post-relapse
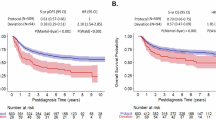
Post-relapse 5-year OS was 48.9 ± 1.2% for all patients, 52.5 ± 1.3% for B-ALL, 35.5 ± 3.3% for T-ALL, and 21.5 ± 3.9% for infants (p < 0.001, Table 2, Fig. 2A). Enrollment on earlier era clinical trials (POG/CCG 1996–2006) did not show significantly different OS post-relapse (49.2 ± 1.8%) when compared to more contemporary COG trials conducted 2004–2014 (48.7 ± 1.5%) (Table 2).

Kaplan–Meier estimates of overall survival rates post relapse based on (A) mmunophenotype, (B) B-ALL time to relapse, (C) B-ALL site of relapse, (D) B-ALL cytogenetic subtype, (E) T-ALL time to relapse, (F) T-ALL site of relapse, (G) Infant ALL cytogenetic subtype, (H) Infant ALL time to relapse, (I) Infant ALL site of relapse.
B-ALL
Five-year post-relapse OS rates by patient and disease characteristics are presented in Supplementary Table 3, with univariate and multivariable analyses in Table 4. Variables that were significant by univariate analysis included time-to-relapse, relapse site, age and WBC at initial diagnosis, race/ethnicity, end-induction MRD at frontline therapy and age at relapse (all p ≤ 0.003). Longer time-to-relapse was associated with better OS post-relapse; 66.4 ± 1.8% for late relapse, 49.5 ± 2.2% for intermediate, and 25.8 ± 2.4% for early relapses (Fig. 2B, Supplementary Table 3). For relapse site, OS post-relapse was 45.1 ± 1.7% for iBM relapse, 58.7 ± 3.4% for combined BM relapse and 65.7 ± 2.6% for iCNS relapse (Fig. 2C, Supplementary Table 3). End-induction MRD ≥0.1% after frontline therapy was associated with inferior OS post-relapse: 58.2 ± 1.6% for patients with end-induction MRD < 0.1%, 37.7 ± 2.6% for those with MRD ≥ 0.1%; p < 0.005). All clinical factors remained significant in multivariable analysis, except for WBC at initial diagnosis and race/ethnicity as a group, although OS post-relapse for Non-Hispanic White patients remained significantly better than Hispanics of all races (p = 0.006).
Post-relapse OS was significantly associated with cytogenetic subtype (p < 0.001, Table 3, Fig. 2D). Similar to de novo B-ALL, cytogenetic subtypes with the best OS post-relapse were ETV6::RUNX1 and Trisomy 4 + 10 with 5-year OS of 74.4 ± 3.1% and 70.2 ± 3.6% respectively, while those with the worst outcomes were hypodiploid (14.2 ± 6.1%), and KMT2A-R (31.9 ± 7.7%). For the other subtypes, 5-year OS post-relapse ranged from 36.8–48.2%. All cytogenetic features except iAMP21 were significantly associated with OS post-relapse in B-ALL in univariate analysis. Only Trisomy 4 + 10 (p = 0.004), ETV6::RUNX1 (p = 0.001), KMT2A-R (p = 0.050) and hypodiploid (p = 0.002) remained significant after adjusting for time-to-relapse, relapse site(s) and other patient and disease characteristics (Table 4).
T-ALL
For patients with T-ALL, time-to-relapse, relapse site, and age at initial diagnosis were significantly associated with OS post-relapse in univariate and multivariable analyses (all p ≤ 0.002; Table 4). Unlike B-ALL, race and ethnicity were not significant for survival post-relapse (Table 4, Supplementary Table 3). Five-year OS post-relapse was highest for patients with late relapse (58 ± 9.8%) but was equally poor for T-ALL patients with intermediate (33 ± 7.6%) or early (29.8 ± 3.9%) relapse (Fig. 2E). For site of relapse, iBM and combined BM relapse had similar 5-year OS post-relapse (23.7 ± 4.7% and 22.7 ± 7.4%, respectively) compared to 52.4 ± 6% for iCNS (Fig. 2F).
Infant ALL
Time-to-relapse (p < 0.001) and relapse site (p = 0.002) were significantly associated with OS post-relapse in multivariable analysis (Table 4). In addition, female infants had higher risk of death compared to males (p = 0.023). Infants with early relapses <18 months rarely survived (9.0 ± 3.2% 5-year OS post-relapse), but OS post-relapse was 51.1 ± 8.8% for infants with relapses ≥18 months (Fig. 2H). Infants with iBM involvement at relapse had significantly worse OS than those with combined BM or iCNS relapses (Fig. 2I). No infant with age at relapse <1 year survived (Supplementary Table 3).
Five-year OS post-relapse was 18.3 ± 3.9% for KMT2A-R versus 46.2 ± 13.8% for KMT2A-germline (p = 0.012, Table 3, Supplementary Table 3), but KMT2A-R was not significantly associated with OS post-relapse after adjusting for time-to-relapse, relapse site and other characteristics (p = 0.62).
Discussion
We analyzed the largest dataset to date of pediatric, adolescent and young adult patients with B-ALL, T-ALL, and infant ALL and describe clinical features at first relapse, as well as prognostic indicators for survival after relapse. In comparison to the previous COG report [7], relapse rates decreased from 20.5% to 12.7%. Patients with relapsed B-ALL had significant improvement in 5-year OS post-relapse (37.2 ± 21% prior vs. 52.5 ± 1.3% current, p < 0.001); more apparent in NCI HR-ALL (22.6 ± 2.1% prior vs. 43.5 ± 1.9% current, p < 0.001) compared to SR-ALL (50.4 ± 2.4% prior vs. 59.5 ± 1.7% current, p < 0.01). Patients with T-ALL had improved OS post-relapse of 35.5 ± 3.3%, compared to 23.0 ± 4.0% in the prior COG report (p < 0.05), while OS post-relapse for infants remained dismal at approximately 20% in both cohorts.
When assessed by immunophenotype, time and site of relapse are distinct. Half of B-ALL relapses occur ≥36 months post-diagnosis and over 70% involve the bone marrow, whereas 82% of T-ALL relapses occur within 36 months and the CNS is involved in almost half of cases. The majority (67.9%) of patients with iCNS relapse of T-ALL had CNS3 disease at initial diagnosis, despite receiving therapeutic cranial irradiation on frontline regimens suggesting inadequate clearance of this sanctuary site. Though CNS relapses occur earlier than BM/combined relapses in both B and T-ALL they had significantly better OS post-relapse. Early relapse of B-ALL and T-ALL have significantly worse outcomes but T-ALL patients with intermediate relapses did not have the survival advantage seen with B-ALL, possibly accounting for the worse OS for T-ALL post-relapse.
Beyond known risk factors, i.e., time-to-relapse, site of relapse, immunophenotype and age <1 year or ≥10 years [7, 10, 11], we identified additional risk factors that independently predict for worse outcomes in relapsed B-ALL. End-induction MRD ≥ 0.01% during frontline therapy increased risk of relapse, but a higher MRD threshold of ≥0.1% at end-induction was associated with worse OS post-relapse on multivariable analyses. End-induction MRD ≥ 1% had an even shorter time to relapse and 5-year OS post-relapse. We also demonstrate that cytogenetic subtype in B-ALL is an important prognostic factor at relapse and is closely linked to time-to-relapse, substantiating that biology drives clinical behavior. Interestingly, patients with TCF3::PBX1, considered cytogenetically neutral at initial diagnosis, had short time-to-relapse (median 15.8 months) and inferior OS post-relapse (36.8±6.6%) while patients with iAMP21, traditionally considered a high-risk subtype at initial diagnosis, had one of the longest time-to-relapse (median 43.6 months) with a 48.2 ± 7.7% OS post-relapse. These findings indicate that tumor biology associated with cytogenetic subtypes alters clonal dynamics and the emergence of subclones with varying responses to retrieval therapy [30,31,32]. Further investigation by next generation testing including single cell sequencing of diagnosis, end-induction MRD and relapse blasts will likely provide insights into the dynamics of resistance and relapse [33]. In multivariable analysis, Trisomy 4 + 10, ETV6::RUNX1, KMT2A-R and hypodiploidy retained their independent significant impact on outcome after adjusting for time-to-relapse and other variables. It remains to be seen if risk stratification algorithms at relapse can be refined by incorporating cytogenetic variables similar to initial diagnosis. More recently described cytogenetic subtypes with prognostic impact, such as Ph-like ALL, or secondary lesions such as TP53 and RAS mutations were not tested in this cohort but are being considered for new risk stratification algorithms [10, 34].
Our analysis by race and ethnicity was limited by the use of individual institutional submission but did show that Hispanic patients with B-ALL of all races were more likely to have worse outcomes post-relapse than non-Hispanic White patients. In addition to race and ethnicity, it is imperative going forward that clinical trials collect comprehensive data on non-medical social determinants of health which also likely influences outcomes. Intervention in these domains in populations at risk will be required to improve survival post relapse.
Infant ALL remains one of the greatest challenges; sequential studies have failed to demonstrate any improvement in survival post-relapse [17, 23]. Infants who relapse <18 months have a dismal 9.0 ± 3.2% post-relapse OS and no infant who relapsed while still <1 year of age survived. With our small sample size of KMT2A-germline patients (n = 13), the analysis assessing independent contribution of KMT2A-R in post-relapse outcomes in infants lacked statistical power. We were able to identify some infant ALL populations who are more salvageable, including those who relapse ≥18 months from diagnosis, infants with iCNS relapse and infants without KMT2A-R (5-yr OS 51.1 ± 8.8%, 46.7 ± 12.9% and 46.2 ± 13.8%, respectively). Further intensification of therapy for infants is challenging due to toxicity and targeted therapies to date have not showed benefit [23]. Early data combining blinatumomab with chemotherapy is promising for newly diagnosed KMT2A-R infants [35].
The significance of some of the findings from this study could be altered by more modern frontline therapy; including changes in CNS-directed therapy which evolved during the study period [18]. To assess this concern, we performed an analysis by “era treated” comparing the earlier CCG/POG trials (1996–2006) versus more contemporary COG trials (2004–2014) and did not detect outcome differences seen between the two groups post-relapse. A similar result was noted in the prior COG analysis wherein historical era of initial treatment, as a surrogate marker of therapy intensity, did not predict for OS post-relapse [2]. In addition, the CCG-1961 trial randomizing patients with HR B-ALL and T-ALL to intensified versus standard therapy post-Induction, showed no difference in post-relapse survival on the more intensive therapy arm [36].
A major limitation to these data is the lack of information about relapse therapy received, including transplant utilization; as well as MRD response to relapse re-induction therapy. We also lack additional information on post-relapse events such as subsequent relapse or death due to toxicity during chemotherapy or transplant. It is presumed most patients would have received intensive first relapse chemotherapy ± radiation to involved extramedullary sites, with or without a consolidative stem cell transplant. Another limitation is that our cohort only includes patients who were enrolled on front line pediatric therapy trials during time periods they were open. However, the wide catchment area of the over 200 COG treatment centers and the size of our cohort provides a broad representation of all children with ALL.
Although overall survival post-relapse rates appear to be improved when compared to our prior analyses, our data are unlikely to reflect the current revolution in immunotherapy and targeted therapy [13, 14, 37]. Only a minority of patients in this cohort enrolled on COG first ALL relapse trials involving investigational immunotherapies and targeted therapies. The use of tyrosine kinase inhibitors (TKIs) for Ph+ ALL increased significantly during this time period with pediatric de novo ALL trials incorporating TKIs into therapy as of the early 2000’s [38, 39]. Post-induction, 33% of our Ph+ ALL cohort enrolled on TKI containing COG frontline therapy trials (AALL0031 and AALL0622) but due to small patient numbers it was not possible to decipher if TKI use upfront impacted OS rate post relapse. It remains to be seen if newer interventions for relapsed ALL, including CAR T-cell immunotherapy, will improve long-term survival. The results reported herein will be valuable as a benchmark to compare the impact of immunotherapy and small molecule targeted therapy in improving survival post relapse [40, 41].
For patients with truly dismal outcomes post-relapse: infants, adolescents ≥16 years, T-ALL relapses involving the BM, and hypodiploid ALL, it would be prudent to consider alternatives to conventional cytotoxic chemotherapy at relapse. In a separate analysis patients with Down Syndrome on frontline COG trials also fit into the dismal outcomes category with a post-relapse 5-year OS of 14.3 ± 8.9% [26]. The COG current first relapse ALL trial studying blinatumomab and nivolumab with chemotherapy, AALL1821 (NCT04546399), now stratifies patient therapy based upon age ≥18 years, regardless of time-to-relapse given their poor survival outcomes and known increased toxicity in this population. International collaborations, developing combined pediatric and adult trails for the adolescent and young adult population and partnerships with industry to design trials of synergistic combinations of targeted therapies may overcome some of the challenges associated with clinical trials in smaller patient populations.
Despite these limitations, this large dataset has provided the opportunity to study various patient cohorts based on time to and site of relapse that including immunophenotype, cytogenetics, and race/ethnicity, which should inform future trial design and highlight populations for testing more novel therapy options.
Data availability
De-identified data from each of the included Children’s Oncology Group trials are available upon request to the relevant trial committees. Requests for access to COG protocol research data should be sent to: [email protected].
References
Hunger SP, Lu X, Devidas M, Camitta BM, Gaynon PS, Winick NJ, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children’s oncology group. J Clin Oncol. 2012;30:1663–9.
Campbell M, Kiss C, Zimmermann M, Riccheri C, Kowalczyk J, Felice MS, et al. Childhood acute lymphoblastic leukemia: results of the randomized acute lymphoblastic leukemia Intercontinental-Berlin-Frankfurt-Munster 2009 trial. J Clin Oncol. 2023;41:3499–511.
Jeha S, Pei D, Choi J, Cheng C, Sandlund JT, Coustan-Smith E, et al. Improved CNS control of childhood acute lymphoblastic leukemia without cranial irradiation: St Jude Total Therapy Study 16. J Clin Oncol. 2019;37:3377–91.
Toft N, Birgens H, Abrahamsson J, Griskevicius L, Hallbook H, Heyman M, et al. Results of NOPHO ALL2008 treatment for patients aged 1-45 years with acute lymphoblastic leukemia. Leukemia. 2018;32:606–15.
Vora A, Goulden N, Mitchell C, Hancock J, Hough R, Rowntree C, et al. Augmented post-remission therapy for a minimal residual disease-defined high-risk subgroup of children and young people with clinical standard-risk and intermediate-risk acute lymphoblastic leukaemia (UKALL 2003): a randomised controlled trial. Lancet Oncol. 2014;15:809–18.
Vrooman LM, Blonquist TM, Harris MH, Stevenson KE, Place AE, Hunt SK, et al. Refining risk classification in childhood B acute lymphoblastic leukemia: results of DFCI ALL Consortium Protocol 05-001. Blood Adv. 2018;2:1449–58.
Nguyen K, Devidas M, Cheng SC, La M, Raetz EA, Carroll WL, et al. Factors influencing survival after relapse from acute lymphoblastic leukemia: a Children’s Oncology Group study. Leukemia. 2008;22:2142–50.
Bhojwani D, Pui CH. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 2013;14:e205–17.
Hunger SP, Mullighan CG. Redefining ALL classification: toward detecting high-risk ALL and implementing precision medicine. Blood. 2015;125:3977–87.
Eckert C, Parker C, Moorman AV, Irving JA, Kirschner-Schwabe R, Groeneveld-Krentz S, et al. Risk factors and outcomes in children with high-risk B-cell precursor and T-cell relapsed acute lymphoblastic leukaemia: combined analysis of ALLR3 and ALL-REZ BFM 2002 clinical trials. Eur J Cancer. 2021;151:175–89.
Oskarsson T, Soderhall S, Arvidson J, Forestier E, Montgomery S, Bottai M, et al. Relapsed childhood acute lymphoblastic leukemia in the Nordic countries: prognostic factors, treatment and outcome. Haematologica. 2016;101:68–76.
Tallen G, Ratei R, Mann G, Kaspers G, Niggli F, Karachunsky A, et al. Long-term outcome in children with relapsed acute lymphoblastic leukemia after time-point and site-of-relapse stratification and intensified short-course multidrug chemotherapy: results of trial ALL-REZ BFM 90. J Clin Oncol. 2010;28:2339–47.
Brown PA, Ji L, Xu X, Devidas M, Hogan LE, Borowitz MJ, et al. Effect of postreinduction therapy consolidation with blinatumomab vs chemotherapy on disease-free survival in children, adolescents, and young adults with first relapse of B-cell acute lymphoblastic leukemia: a randomized clinical trial. JAMA. 2021;325:833–42.
Locatelli F, Zugmaier G, Rizzari C, Morris JD, Gruhn B, Klingebiel T, et al. Improved survival and MRD remission with blinatumomab vs. chemotherapy in children with first high-risk relapse B-ALL. Leukemia. 2023;37:222–5.
Matloub Y, Bostrom BC, Hunger SP, Stork LC, Angiolillo A, Sather H, et al. Escalating intravenous methotrexate improves event-free survival in children with standard-risk acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Blood. 2011;118:243–51.
Asselin BL, Devidas M, Wang C, Pullen J, Borowitz MJ, Hutchison R, et al. Effectiveness of high-dose methotrexate in T-cell lymphoblastic leukemia and advanced-stage lymphoblastic lymphoma: a randomized study by the Children’s Oncology Group (POG 9404). Blood. 2011;118:874–83.
Dreyer ZE, Hilden JM, Jones TL, Devidas M, Winick NJ, Willman CL, et al. Intensified chemotherapy without SCT in infant ALL: results from COG P9407 (Cohort 3). Pediatr Blood Cancer. 2015;62:419–26.
Winick N, Martin PL, Devidas M, Shuster J, Borowitz MJ, Paul Bowman W, et al. Randomized assessment of delayed intensification and two methods for parenteral methotrexate delivery in childhood B-ALL: Children’s Oncology Group Studies P9904 and P9905. Leukemia. 2020;34:1006–16.
Bowman WP, Larsen EL, Devidas M, Linda SB, Blach L, Carroll AJ, et al. Augmented therapy improves outcome for pediatric high risk acute lymphocytic leukemia: results of Children’s Oncology Group trial P9906. Pediatr Blood Cancer. 2011;57:569–77.
Larsen EC, Devidas M, Chen S, Salzer WL, Raetz EA, Loh ML, et al. Dexamethasone and high-dose methotrexate improve outcome for children and young adults with high-risk B-acute lymphoblastic leukemia: a report from Children’s Oncology Group Study AALL0232. J Clin Oncol. 2016;34:2380–8.
Maloney KW, Devidas M, Wang C, Mattano LA, Friedmann AM, Buckley P, et al. Outcome in children with standard-risk B-cell acute lymphoblastic leukemia: results of Children’s Oncology Group Trial AALL0331. J Clin Oncol. 2020;38:602–12.
Dunsmore KP, Winter SS, Devidas M, Wood BL, Esiashvili N, Chen Z, et al. Children’s Oncology Group AALL0434: a phase III randomized clinical trial testing nelarabine in newly diagnosed T-cell acute lymphoblastic leukemia. J Clin Oncol. 2020;38:3282–93.
Brown PA, Kairalla JA, Hilden JM, Dreyer ZE, Carroll AJ, Heerema NA, et al. FLT3 inhibitor lestaurtinib plus chemotherapy for newly diagnosed KMT2A-rearranged infant acute lymphoblastic leukemia: Children’s Oncology Group trial AALL0631. Leukemia. 2021;35:1279–90.
Angiolillo AL, Schore RJ, Devidas M, Borowitz MJ, Carroll AJ, Gastier-Foster JM, et al. Pharmacokinetic and pharmacodynamic properties of calaspargase pegol Escherichia coli L-asparaginase in the treatment of patients with acute lymphoblastic leukemia: results from Children’s Oncology Group Study AALL07P4. J Clin Oncol. 2014;32:3874–82.
Rodriguez V, Kairalla J, Salzer WL, Raetz EA, Loh ML, Carroll AJ, et al. A pilot study of intensified PEG-asparaginase in high-risk acute lymphoblastic leukemia: Children’s Oncology Group Study AALL08P1. J Pediatr Hematol Oncol. 2016;38:409–17.
Rabin KR, Devidas M, Chen Z, Ji L, Kairalla J, Hitzler JK, et al. Outcomes in children, adolescents, and young adults with down syndrome and ALL: a report from the Children’s Oncology Group. J Clin Oncol. 2024;42:218–27.
Kalbfleisch JD, Prentice RL. The statistical analysis of failure time data. 2nd ed. New York: John Wiley and Sons; 2002.
Cox DR, Oakes D. Analysis of survival data. London: Chapman and Hall; 1984.
StataCorp. 2021. Stata Statistical Software: Release 17. College Station, TX: StataCorp LLC.
Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322:1377–80.
Li B, Brady SW, Ma X, Shen S, Zhang Y, Li Y, et al. Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood. 2020;135:41–55.
van Delft FW, Horsley S, Colman S, Anderson K, Bateman C, Kempski H, et al. Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood. 2011;117:6247–54.
Zhang Y, Wang S, Zhang J, Liu C, Li X, Guo W, et al. Elucidating minimal residual disease of paediatric B-cell acute lymphoblastic leukaemia by single-cell analysis. Nat Cell Biol. 2022;24:242–52.
Irving JA, Enshaei A, Parker CA, Sutton R, Kuiper RP, Erhorn A, et al. Integration of genetic and clinical risk factors improves prognostication in relapsed childhood B-cell precursor acute lymphoblastic leukemia. Blood. 2016;128:911–22.
van der Sluis IM, de Lorenzo P, Kotecha RS, Attarbaschi A, Escherich G, Nysom K, et al. Blinatumomab added to chemotherapy in infant lymphoblastic leukemia. N Engl J Med. 2023;388:1572–81.
Freyer DR, Devidas M, La M, Carroll WL, Gaynon PS, Hunger SP, et al. Postrelapse survival in childhood acute lymphoblastic leukemia is independent of initial treatment intensity: a report from the Children’s Oncology Group. Blood. 2011;117:3010–5.
Hunger SP, Raetz EA. How I treat relapsed acute lymphoblastic leukemia in the pediatric population. Blood. 2020;136:1803–12.
Schultz KR, Carroll A, Heerema NA, Bowman WP, Aledo A, Slayton WB, et al. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children’s Oncology Group study AALL0031. Leukemia. 2014;28:1467–71.
den Boer ML, Cario G, Moorman AV, Boer JM, de Groot-Kruseman HA, Fiocco M, et al. Outcomes of paediatric patients with B-cell acute lymphocytic leukaemia with ABL-class fusion in the pre-tyrosine-kinase inhibitor era: a multicentre, retrospective, cohort study. Lancet Haematol. 2021;8:e55–66.
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–17.
Teachey DT, Hunger SP. Acute lymphoblastic leukaemia in 2017: immunotherapy for ALL takes the world by storm. Nat Rev Clin Oncol. 2018;15:69–70.
Funding
This study was supported by research funding from the National Cancer Institute of the National Institutes of Health grants under award numbers U10CA13539, U10CA29139, U10CA98543 and U10CA180886 (COG, CCG, POG Chair’s grants), U10CA98413 and U10CA180899 (COG Statistics and Data Center grants), U24CA114766 and U24CA196173 (COG Specimen Banking), and St Baldrick’s Foundation funding. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
SRR, DB, SPH, EAR, and MLL all conceived of the study concept and design, performed data analyses and data interpretation of the results. LJ, XX, and MD conducted the statistical analyses. JAK, MS, NAH, AJC, and HB conducted the cytogenetic analyses. MB and BLW conducted the minimal residual disease analyses. SRR and DB wrote the manuscript which was edited and approved by all authors. All authors had full access to the data and accept responsibility for submission. LG is the Ergen Family Chair in Pediatric Oncology at Children’s Hospital Colorado. EAR is a KiDS of NYU Foundation Professor at NYU Langone Health. SPH is the Jeffrey E. Perelman Distinguished Chair in Pediatrics at the Children’s Hospital of Philadelphia. MLL is the Aldarra Foundation, June and Bill Boeing, Founders, Endowed Chair of Pediatric Cancer Research at Seattle Children’s Hospital.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The trial protocols were approved by the National Cancer Institute and by the Pediatric Central Institutional Review Board and by each trial center Institutional Review Board. All patients or a parent/guardian provided written informed consent for clinical trial enrollment. The independent Children’s Oncology Group, and historic Children’s Cancer Group and Pediatric Oncology Group, data and safety monitoring committees met regularly to review trial safety and efficacy data according to its charter and standard operating procedures. All individual trial references which include NCI registration numbers are found in Table 1.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rheingold, S.R., Bhojwani, D., Ji, L. et al. Determinants of survival after first relapse of acute lymphoblastic leukemia: a Children’s Oncology Group study. Leukemia 38, 2382–2394 (2024). https://doi.org/10.1038/s41375-024-02395-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-024-02395-4
This article is cited by
-
Venetoclax and azacitidine in combination with homoharringtonine, cytarabine, and aclarubicin for salvage therapy of relapsed/refractory T cell acute lymphoblastic leukemia
International Journal of Hematology (2025)
-
Neo-antigen specific cancer vaccines for acute lymphoblastic leukemia—challenges, opportunities, and future directions
Cancer Immunology, Immunotherapy (2025)
-
Symptom distress among Chinese children with cancer: what factors influence parent–child concordance?
European Journal of Pediatrics (2025)
-
Late-onset epilepsy in survivors of childhood cancer outside the central nervous system: a study within the Adult Life after Childhood Cancer in Scandinavia (ALiCCS) study
Journal of Cancer Survivorship (2025)
