
Abstract
PARP1 initiates DNA repair pathways including single-strand break repair (SSBR) by recruiting multiple DNA repair factors via poly ADP-ribosylation (PARylation) of target proteins. However, how PARP1 is stabilized and activated to promote DNA damage repair remains unclear. Here we report that DNA damage generates a ROS signal, which triggers USP10 to interact with and stabilize PARP1 by deubiquitinating the K418 site in an ATM-dependent manner. In turn, PARP1 mediates PARylation of USP10 at amino acid residues D634, D645, and E648, which further promotes the deubiquitination activity of USP10 and DNA damage response to form a positive feedback loop. PARP1 is highly expressed in breast cancer tissues and positively correlates with USP10 protein levels. Moreover, breast cancer cells treated with a USP10 inhibitor show increased sensitivity to PARP1 inhibitor both in vivo and in vitro. Overall, our results unravel that the deubiquitination-PARylation positive feedback loop of the USP10-PARP1 axis promotes DNA damage repair, which might contribute to the improvement of PARP1 inhibitor efficacy in breast cancer treatment.
Similar content being viewed by others


Introduction
Poly(ADP-ribose) polymerase 1 (PARP1) is an essential sensor of DNA damage, rapidly binding to lesion sites and recruiting multiple DNA-repair factors. The activation of PARP1 initiates various DNA repair pathways, particularly single-strand break repair (SSBR), via its enzymatic activity of poly(ADP)ribosylation (PARylation) [1]. During PARylation, PARP1 catalyzes the attachment of PAR chains onto itself or other target proteins to facilitate DNA damage repair [2,3,4]. Importantly, impairing PARP1-dependent SSBR could elicit DNA double-strand breaks (DSBs), particularly in cells with faulty homologous recombination (HR)-dependent DSB repair due to mutations in breast cancer 1/2 genes (BRCA1/2), which is described as synthetic lethality [5, 6]. To date, several PARP1 inhibitors have been approved by the United States (US) Food and Drug Administration (FDA) for clinical use in BRCA1/2 mutant-driven breast cancer [7,8,9]. However, the biological heterogeneity and high PARP1 expression in some breast cancers limits the therapeutic benefit [10,11,12]. Hence, elucidating the molecular mechanisms of PARP1 stabilization upon DNA damage might contribute to the development of novel therapeutic strategies.
PARP1 is a target of ubiquitination, which is an important post-translational modification resulting from the covalent attachment of ubiquitin (Ub) molecules. Ubiquitination of target proteins can regulate their stability or functional activity [13]. This process is sequentially catalyzed by three groups of enzymes: ubiquitin-activating enzymes (E1s), ubiquitin-conjugating enzymes (E2s), and ubiquitin ligases (E3s) [14]. The addition of polymeric ubiquitin chains (poly-ubiquitination) mainly serves to guide the target protein to the proteasome degradation pathway [15, 16]. Protein ubiquitination is a reversible process that can be regulated by another group of enzymes with ubiquitin-cleavage activity, namely deubiquitinating enzymes (DUBs). Until now, many E3 ubiquitin ligases that target PARP1 have been identified including Smurf2, WWP2, TRIP12, RNF4, RNF144A, UHRF1, HECTD3 [17,18,19,20,21,22,23]. Although the deubiquitination of PARP1 have been mentioned [12, 24], how DUBs are activated to regulate PARP1 and the effect of their interactions on efficacy of PARP1 inhibitor remain largely unknown.
Ubiquitin-specific peptidase 10 (USP10) is a highly conserved DUB involved in diverse cellular processes and, importantly, is associated with a broad spectrum of cancer types [25, 26]. However, the roles that USP10 plays in tumorigenesis are varied and are determined by the tumor types and the substrates it modulates. The most important target proteins of USP10 include p53, PTEN, CFTR, AMPKα, SIRT6, FLT3 and (NF-κB)-essential modulator (NEMO), which regulates various cellular processes including metabolism, homeostasis and tumorigenesis [27,28,29,30,31,32,33].
In this study, we demonstrate that DNA damage generates reactive oxygen species (ROS), which promotes USP10 to interact with and stabilize PARP1 by deubiquitinating the K418 site in an ATM-dependent manner. In turn, PARP1 mediates PARylation of USP10 at the D634, D645, E648 sites, which further promotes the deubiquitination activity of USP10, thereby establishing a positive feedback loop. PARP1 is highly expressed in breast cancer tissues and positively correlates with USP10 protein level. Importantly, USP10 inhibitor sensitizes breast cancer cells to treatment with PARP1 inhibitor both in vivo and in vitro. Overall, we found that the deubiquitination-PARylation positive feedback loop of the USP10-PARP1 axis promotes DNA damage repair, which might contribute to the improvement of PARP1 inhibitor efficacy in breast cancer treatment.
Materials and methods
Cell culture
MCF7, MDA-MB-231, HEK293, HCT116, and H1299 cells were purchased from the Cell Bank in the Chinese Academy of Sciences Shanghai. MCF7, MDA-231, and HEK293 cells were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM); HCT116 cells were cultured in McCoy’s 5 A medium; H1299 cells were cultured in RPMI 1640 medium. All media described above were supplemented with 10% fetal bovine serum (FBS) (CLARK, Australia), penicillin (100U), and streptomycin (100 g/ml) before its use for culturing cells.
Antibodies and reagents
The following antibodies were used for Western blotting: anti-PARP1 (#9532, CST), anti-USP10 (#8501, CST), anti-PAR(#83732, CST), anti-Flag (SG4110-16, Shanghai Genomics Technology), anti-Myc (SG4110-18, Shanghai Genomics Technology), anti-HA (#3724, CST), anti-Ubiquitin (#3933, CST), anti-ATM (#2873, CST), anti-Phospho-ATM Ser1981 (#13050, CST), anti-DDB1 (#5428, CST), anti-XRCC1 (#2735, CST), anti-MRE11 (#4895, CST), anti-p53 (sc-126, Santa Cruz Biotechnology), anti-SIRT6 (#2590, CST), anti-Axin1 (#2087, CST), anti-α-Tubulin (AC012, ABclonal), anti-β-actin (AC004, ABclonal). Additionally, anti-USP10 (sc-365828, Santa Cruz Biotechnology) and anti-PARP1 (#9532, CST) were used for Immunofluorescent analysis and PLA assay. Anti-USP10 (#8501, CST) and anti-PARP1(13371-1-AP, proteintech) were used for Immunohistochemistry.
Olaparib (SC9118), Spautin-1 (SC5498) and NAC (S0077) were purchased from Beyotime. Hydroxyurea (HY-B0313) was purchased from MedChemExpress. CHX (S7418), PDD00017273 (PARG inhibitor, S8862) and KU-55933 (ATM inhibitor, S1092) were purchased from Selleck.
Plasmid constructions, transfection and lentivirus infection
PARP1 and USP10 overexpression plasmids were constructed in our previous studies. Plasmids expressing protein truncations were purchased from MiaoLingBio., Wuhan, China. Mutations in PARP1 (K131R, K249, K418R, K548R, K579R, K633R) and mutations in USP10 (K620A, E621A, D634A, D645A, E648A, E654A, K687A, E692A, K693A and K699A) were generated via site-directed mutagenesis. Specific siRNAs targeting PARP1 or USP10 were purchased from RiboBio Co., Ltd., Guangzhou, China. Plasmid transfection was performed with jetPrime Transfection reagent and the corresponding jet buffer (Polyplus PT-114-15). Cells were harvested 48 hours after transfection. To construct stable knockdown cell lines, cells were infected with control shRNA (shNC) lentivirus, shRNA against USP10 (shUSP10) lentivirus, shRNA against PARP1 (shPARP1) lentivirus, or shRNA against ATM (shATM) lentivirus purchased from Shanghai GeneChem Company for 72 h. Subsequently, the cells were subjected to a five-day puromycin (1ug/ml) selection to screen for successfully infected cells.
Mass spectrometry
To analyze PARP1 interacting proteins and ubiquitination sites of PARP1, HEK293 cells were transfected with Myc-PARP1 plasmids for 48 h and subjected to co-IP assays with anti-Myc magnetic agarose. After washing three times with PBS buffer, samples were boiled in 2 × SDS loading buffer, resolved in SDS-PAGE. The immune complex proteins were excised in gel digested with trypsin and subjected to liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. MS experiments were performed on a nanoscale UHPLC system (EASY-Nlc1000 from Proxeon Biosystems, Odense, Denmark) connected to an Orbitrap Q-Exactive equipped with a nanoelectros pray source (Thermo Fisher Scientific, Bremen, Germany). The raw data were processed using Proteome Discoverer (PD, version 2.1), and MS/MS spectra were searched against the reviewed Swiss-Prot human proteome database. Only peptides with at least six amino acids in length were considered. The peptide and protein identifications were filtered by PD to control the false discovery rate (FDR) < 1%. At least one unique peptide was required for protein identification. The dataset relative to the mass spectrometry analysis of PARP1-associated proteins was uploaded to the National Genomics Data Center (https://ngdc.cncb.ac.cn/omix/release/OMIX006504).
Western blot analysis and Co-immunoprecipitation assay
To prepare the samples, cells were lysed for 30 min on ice using IP lysis buffer (0.25% Sodium deoxycholate, 50 mM Tris-HCL, PH7.4, 1 mM EDTA, 1% TritonX-100, 1% NP40 and 150 mM NaCl) containing protease inhibitor cocktails. When necessary, phosphorylation protease inhibitor was also added. The lysates were then centrifuged at 13,500 rpm for 20 min at 4 °C to collect the lysed protein. The protein concentration was then assessed using G250 and 40 μg of protein was used for sample preparation.
To perform co-immunoprecipitation, cells were lysed using IP lysis buffer, which was prepared in a similar manner as for the western blot analysis. Primary antibody was then added to the cell lysates and mixed by rotation for at least an hour. Next, protein A/G beads (Santa Cruz) were introduced to the mixture, which was then incubated at 4 °C overnight. The protein samples were washed with PBS at 4 °C three times and eluted with 2×loading. The protein samples were subjected to 8%, 10%, 12% or 15% SDS-PAGE and then transferred to PVDF membrane (Millipore, IPVH00010) for 120 min to 150 min depending on the target molecular weight range. After blocking in 5% BSA in TBST for an hour at room temperature, the membranes were probed with specific primary antibodies and incubated at 4 °C overnight. Following this, the membranes were washed with TBST three times and incubated at room temperature for an hour with HRP-conjugated secondary antibody. After three washes, bands were detected by using the enhanced chemiluminescence detection kit (Thermo Fisher Scientific, 32106) and visualized via the DNR western blot detection system (Azure Biosystems C600).
GST-pull down assay
The bacterial expression constructs (pGEX-4T-1) containing the target gene USP10 were transformed into BL21-competent cells (Takara). Cells were induced to overexpress the GST-fusion protein by 1 mM IPTG for 3.5 h while shaking at 30 °C. Cells were resuspended in bacterial lysis buffer (PBS containing 1 mM PMSF, 5 mM β-mercaptoethanol, 0.5% TritonX-100 and 2 mM EDTA), followed by ultrasonication. The proteins were purified in a single step using glutathione beads according to the manufacturer’s protocol (Promega Science). In vitro transcription and translation of Myc-PARP1 were performed using T7-TNT Kits (Promega, L1170) in accordance with the manufacturer’s instructions.
In vitro de-ubiquitination assay
To determine the direct effects of USP10 on PARP1 polyubiquitination, Myc-PARP1, Flag-USP10 and HA-Smurf2 plasmids were transfected into HEK293 cells for 48 h, followed by purification with anti-Myc beads, anti-Flag beads or anti-HA beads, respectively. These purified proteins were added to the reaction mixture containing Mg-ATP, Ubiquitin, Ubiquitin-Activating Enzyme(E1), UbcH5a (E2) and Ub E3 Ligase Buffer which all provided by the E3 Ligase Auto-Ubiquitination Assay Kit (Abcam, ab139469). After incubation at 37 °C for 1 h, the reaction was then terminated with 2× SDS loading buffer and subjected to western blot with anti-ubiquitin antibody.
In vitro Poly (ADP-ribose) (PAR) binding assay
The PAR-binding feature of recombinant protein was determined by GST pull down assay. Briefly, recombinant GST fusions proteins or GST proteins immobilized using Glutathione agarose beads were incubated with 20pmol biotin-PAR (Trevigen, 4336-100-01) diluted in NETN buffer [20 mM Tris–HCl (pH8.0), 0.15 M NaCl, 1 mM EDTA, 0.5%NP-40 and a protease inhibitor cocktail] at 4 °C overnight. The beads were washed with NETN buffer 5 times, and bound proteins were released by adding SDS sample buffer followed by heating at 90 °C for 10 min. The reaction was analyzed by dot-blotting onto PVDF membranes and immunoblotting with an anti-PAR antibody.
Immunofluorescent analysis
To prepare for immunofluorescence in cultured cells, the culture medium was removed, and the cells were washed three times with PBS. Afterward, the cells were treated with tissue fixative for 20 min. After washing three times with PBS, the cells were treated with 0.25% TritonX-100 for 15 min. The samples were then washed three times with PBS, blocked with 5% BSA for an hour, and incubated with anti-USP10 (#8501S, 1:200, CST) and anti-PARP1 (#9532S,1:800, CST) antibodies at 4 °C overnight. After three washes with PBS, the corresponding fluorescent secondary antibody (1:400) was incubated at room temperature for an hour in the dark. After another three washes with PBS, the samples were stained with DAPI for 5 min and mounted upside down on a glass slide to protect the sample from light and to dry for observation under a laser confocal microscope.
PLA assays
PLA assays were carried out according to the Duolink® In Situ – Fluorescence manual. The cells on the glass slides were fixed with 4% PFA for 15 min. Subsequently, the glass slides were permeabilized with Triton X-100 for 15 min. Blocking Solution was added to the glass slide, and slides were incubated in a pre-heated humidity chamber for 30 min at 37 °C. The glass slides with diluted primary antibody were incubated overnight at 4 °C. The primary antibody solution was tapped off and the slides were washed in 1x Wash Buffer A. The PLA probe solution was added and slides were incubated in a pre-heated humidity chamber for an hour at 37 °C. The PLA probe solution was tapped off and the slides were washed in 1x Wash Buffer A. The Ligation solution with Ligase was added and slides were incubated in a pre-heated humidity chamber for 30 min at 37 °C. The Ligation solution was tapped off and the slides were washed in 1x Wash Buffer A. The Amplification-Polymerase solution was added to each sample followed by incubation in a pre-heated humidity chamber for 100 min at 37 °C. Ultimately, The Amplification-Polymerase solution was tapped off and slides were washed in 1x Wash Buffer B and then 0.01x Wash Buffer B. Duolink Slides were mounted using In Situ Mounting Medium with DAPI (4’,6-diamidino-2-phenylindole) with a cover slip. The cells were imaged using a fluorescence microscope (Olympus).
Comet assays
Comet assays were performed following protocols provided by KeyGen Biotech (Nanjing, China). Briefly, cells transfected by wild-type USP10 or USP10-3A mutant were washed with ice-cold 1 × PBS, then harvested and resuspended in ice-cold 1 × PBS at 1 × 106 cells/ml. The slides were first precoated with a layer of 1% normal melting point agarose in PBS. Next, a second layer of the cell–agarose solution, consisting of a mixture of resuspended cells and 0.7% low-melting-point agarose in PBS at a ratio of 1:7.5 (v/v), was added. Subsequently, the slide was laid flat in the dark and maintained for 30 min at 4 °C to enhance attachment. Next, the slides were coated with 0.7% low-melting-point agarose in PBS and stored at 4 °C until solidification. The cells on the slides were lysed in ice-cold lysis buffer at 4 °C for 1 h, and then, the slides were immersed in alkaline unwinding solution (300 mM NaOH and 1 mM EDTA) for 1 h at room temperature. Electrophoresis was carried out at 25 V for 30 min in a horizontal electrophoresis apparatus. After rinsing 3 times with Tris-HCl (0.4 mM, pH7.5), the slides were stained with PI for 10 min at 4 °C in the dark. Images were acquired using a fluorescence microscope (Olympus, Tokyo, Japan).
Cell proliferation assay
Cells were seeded in triplicate at a density of 1 × 104 cells per well in 96-well plates. After incubation in complete DMEM with 10% FBS for 24 h, cells were treated with corresponding reagents at the indicated concentrations. To measure cell survival, we added DMEM medium and CCK8 (Cell Counting Kit-8) staining solution to each well with cells and left them for 2 h at 37 °C. We used an absorbance reader (TECAN, Switzerland) to measure the absorbance at 450 nm on a daily basis. Finally, we calculated the percentage of cell survival.
Cell colony formation assay
Cells were seeded in six-well plates at a density of 5 × 103 cells per well and were cultured under 5% CO2 at 37 °C. Cells were treated with corresponding reagents at the indicated concentrations. All the colonies were fixed in methyl alcohol for 15 min and stained with Coomassie brilliant blue R250 for 10 min. Colonies were then counted and photographed.
Flow cytometry analysis
To measure cell apoptosis, cells were treated with different combinations of Spautin-1 and PARP1 inhibitor for 24 h, followed by incubation with APC-Annexin V and 7-AAD (KeyGEN BioTECH, KGA1026). The manufacturer’s protocol was followed to measure the percentage of apoptotic cells.
Immunohistochemistry
The microarrays of breast cancer tissue (HBreD180Bc01-1) were purchased from Shanghai Outdo Biotech Company, China. After they were deparaffinized in xylene and rehydrated in graded ethanol, tissue microarrays were immersed in citrate buffer or EDTA buffer for antigen retrieval. Endogenous peroxidase was quenched using 3% hydrogen peroxide for 30 min. To decrease the nonspecific staining, 10% normal goat serum was subsequently used to block tissue collagen for 30 min Tissue sections were then incubated with antibody anti-USP10 (#8501S, 1:100, CST) or anti-PARP1 (13371-1-AP, 1:3000, proteintech) at 4 °C overnight. After that, biotinylated secondary antibody and streptavidin-biotin peroxidase were added to tissue sections for 10 min each in turn. Slides were stained with DAB chromogenic reagent for 1 min, and afterward counterstained with hematoxylin. UltraSensitiveTM SPIHC Kit (KIT-9720, Maixin Inc., Fujian, China) was used in this experiment.
Two investigators that were unaware of the clinical information independently reviewed and scored the stained sections. We used a semi-quantitative scoring method to evaluate the expression of USP10 and PARP1. Staining intensity was categorized as 0 (no staining), 1 (weak staining), 2 (moderate), or 3 (strong), while the percentage of stained cells was classified as 0 (0–5%), 1 (6–25%), 2 (26–50%), 3 (51–75%), or 4 (76–100%). The final scores were obtained by multiplying the staining intensity by the percentage of cells.
Tumor xenografts
A total of 20 female nude mice (BALB/cA-nu Mice) at 4 weeks of age were subcutaneously inoculated with 5 × 106 MCF7 cells. After one week, the mice were randomly divided into Control, Spautin-1, Olaparib, Spautin-1+ Olaparib groups. Spautin-1 and Olaparib were administered twice a week at a concentration of 10 mg/kg and 40 mg/kg, respectively. The tumor size was measured daily. The tumor volume was calculated (length × width2). The mice were sacrificed after 25 days, and the tumors were removed for subsequent analyses. All animal experiments were approved by Institutional Animal Care and Use Committee of China Medical University.
Statistical analysis
SPSS version 20.0 (SPSS Inc., Chicago IL, USA) was used for statistical analyses. Two-tailed Student’s t test was used for continuous variables. Spearman’s rank correlation test was used to evaluate the correlation between USP10 and PARP1 expression. Survival outcomes were assessed by the Kaplan–Meier method, and the log-rank test was used to compare the differences among the groups. In addition, patients with breast cancer who received Olaparib treatment from The Cancer Genome Atlas (TCGA) data were analyzed. The Cox proportional hazards model was used to investigate the differences in prognosis among different groups. Results were considered statistically significant when P < 0.05 (*, P < 0.05, **, P < 0.01, and ***, P < 0.001).
Results
USP10 interacts with PARP1
Efficient DNA damage repair activity relies on the precise regulation of proteins by various post-translational modifications (PTMs). In recent years, PARP1 has been reported to be a target of the ubiquitination modification [17,18,19,20,21,22,23], however, the factors that perform deubiquitination of PARP1 have remained elusive. To search for candidate DUB factors, we performed mass spectrometry analysis in HEK293 cells to find PARP1-associated proteins. From this analysis, we identified USP10, a member of the DUB family, as a potential interacting partner of PARP1 (Fig. 1A). Glutathione-S-transferase (GST) pull-down analysis confirmed that USP10 directly binds to PARP1 in vitro (Fig. 1B). Subsequent co-immunoprecipitation (co-IP) assays also suggested the interaction between USP10 and PARP1 in HEK293 (Fig. 1C), MCF7 (Fig. 1D), HCT116 (Supplementary Fig. 1A) and MDA-MB-231 (Supplementary Fig. 1B) cells.

A HEK293 cells transfected with Myc-PARP1 plasmids for 48 h were lysed and subjected to co-immunoprecipitation and SDS-PAGE. The immune complex was excised in gel digested with trypsin and subjected to liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. The ubiquitin-specific peptidase 10 (USP10), denoted in red, was identified as a potential PARP1 interacting partner. B GST in vitro pull-down assay to test the interaction between USP10 and PARP1. Recombinant human myc-PARP1 was incubated with bacterially expressed GST-USP10. Co-IP analysis of USP10 and PARP1 in HEK293 cells (C) and MCF7 cells (D). Lysates were immunoprecipitated with IgG control or an anti-USP10 antibody or an anti-PARP1 antibody and then immunoblotted with the indicated antibodies. E Schematic showing the truncated PARP1 constructs that lack various domains, including amino acids 1–203, 1–476, 484–1014 and 203–1014. F Co-IP analysis to determine the interaction between endogenous USP10 and the functional domains of PARP1. Lysates were prepared from HEK293 cells that were transfected with plasmids encoding the indicated Myc-PARP1 truncations. G Schematic of the USP10 truncations lacking various domains including amino acids 1–205, 206–414, 415–600 and 601–798. H The PARP1-binding region of USP10 was determined by co-IP analysis of HEK293 cells transfected with plasmids expressing full-length Flag-USP10 or truncations as indicated.
To clarify the specific ___domain in PARP1 to which USP10 binds, we constructed Myc-PARP1 truncations and tested them in co-IP assays (Fig. 1E). PARP1 consists of three main domains: an amino-terminal DNA-binding ___domain (DBD) that contains zinc finger motifs, a central auto-modification ___domain that contains a BRCA1 C-terminal (BRCT) ___domain, and a highly conserved carboxy-terminal nicotinamide adenine dinucleotide (NAD)-binding catalytic ___domain [3]. Our results revealed that USP10 interacts with the carboxy-terminal catalytic ___domain of PARP1 (Fig. 1F). Next, to map the PARP1-binding region on USP10, we constructed four Flag-tagged truncated versions of USP10 (Fig. 1G) and overexpressed them in HEK293 cells. Co-IP analysis showed that PARP1 binds to the carboxy-terminal region (aa601-aa798) of USP10 (Fig. 1H).
USP10 stabilizes PARP1 and positively correlates with PARP1 expression in breast cancer
Since USP10 can stabilize its substrates via deubiquitination, we explored whether USP10 indeed regulates the stability of the PARP1 protein. As the results showed, overexpression of Flag-USP10 in HEK293 cells resulted in an increase in endogenous PARP1, and similar results were obtained in HCT116 cells (Fig. 2A, Supplementary Fig. 2A). Overexpression of Flag-USP10 also increased exogenous Myc-PARP1 in MCF7 cells (Supplementary Fig. 2B). By contrast, transfection of MCF7 cells with siRNA fragments that target USP10 resulted in a decrease in PARP1 protein level (Fig. 2B). Similar results were obtained in USP10 stable knockdown cells (Fig. 2C). Consistently, 24 h treatment of MCF7 cells with Spautin-1, a specific USP10 inhibitor, obviously decreased PARP1 levels in a concentration-dependent manner (Fig. 2D). In addition, enzymatic inactive mutant of USP10 (C424A) failed to increase PARP1 protein level as wild-type USP10 did (Fig. 2E). By determining PARP1 protein levels after 0 h, 4 h, 8 h treatment of the protein synthesis inhibitor CHX, we found that overexpression of wild-type USP10 prolonged the half-life of PARP1 protein, while enzymatic inactive mutant of USP10 (C424A) could not (Fig. 2F). Furthermore, we detected PARP1 level in cells with or without stable USP10 knockdown after treatment with CHX for 0 h, 4 h, 8 h and found that shUSP10 was associated with a shortened PARP1 half-life (Fig. 2G). Thus, these results indicate that USP10 likely directly regulates PARP1 levels through increasing its stability.

A HEK293 cells overexpressing Flag-USP10 were subjected to western blot analysis to determine PARP1 expression levels. B PARP1 levels were determined in MCF7 cells that were transfected with three different siRNA-USP10 target sequences for 48 h. C PARP1 levels were determined by western blot analysis of cell lines with or without USP10 knockdown. D PARP1 levels were determined in MCF7 cells treated with or without USP10 inhibitor (Spautin-1) for 24 h at the indicated concentrations. E HEK293 cells were co-transfected with Flag-PARP1 and Myc vector, Myc-USP10 or Myc-USP10 C424A plasmids for 48 h, followed by western blot analysis with anti-Flag or anti-Myc antibodies. F Flag-PARP1 protein levels were assessed at different time points following CHX (20 μM) treatment of HEK293 cells co-transfected with Flag-PARP1 and Myc vector, Myc-USP10 or Myc-USP10 C424A plasmids for 48 h. G PARP1 protein levels were assessed at different time points following CHX (20 μM) treatment of HEK293 cells with or without USP10 shRNA-mediated knockdown. H USP10 and PARP1 protein levels were assessed in seven representative cancer specimens. ‘N’ represents normal tissue, and ‘T’ represents tumor tissue. I Representative sections and semiquantitative analyses of USP10 expression in breast cancer tissue (n = 30). Data are expressed as mean ± SEM. **P < 0.01 (Mann–Whitney test). J Representative sections and semiquantitative analyses of PARP1 expression in breast cancer tissue (n = 30). Data are expressed as mean ± SEM. ***P < 0.001 (Mann–Whitney test). K Expression of USP10 and PARP1 are positively correlated in consecutive sections of breast cancer tissue. The results of Spearman correlation analyses of P value and correlation coefficient are shown.
Importantly, examination of tumor tissues revealed that relatively high levels of PARP1 were accompanied by high USP10 expression (Fig. 2H). In order to determine this ulteriorly, we conducted an immunohistochemistry analysis of tissues from 150 patients diagnosed with breast cancer (Supplementary Fig. 2C). Indeed, USP10 and PARP1 were highly expressed in carcinomas compared with paired peri-tumor tissues (Fig. 2I, J). Moreover, a significant positive correlation between USP10 and PARP1 expression was identified in breast cancer tissues (P < 0.001, r = 0.332) (Fig. 2K).
USP10 deubiquitinates PARP1 at the K418 site
We next explored whether USP10 regulates the ubiquitination level of PARP1. Overexpression of Flag-USP10 significantly reduced the ubiquitination of endogenous PARP1 in HEK293 and HCT116 cells, and similar results were obtained in HEK293 cells transfected with exogenous HA-Ub, Myc-USP10 and Flag-PARP1 expression plasmids (Fig. 3A, Supplementary Fig. 3A, B). Moreover, in vitro de-ubiquitination assay further confirmed that USP10 deubiquitinates PARP1 (Fig. 3B). By contrast, transfection with siRNA targeting USP10 markedly increased the level of ubiquitinated PARP1 (Fig. 3C, Supplementary Fig. 3C). Similar results were obtained in cells which USP10 was stably silenced (Fig. 3D). In addition, treatment with the USP10 inhibitor Spautin-1 significantly increased the ubiquitination level of PARP1 protein in MCF7 and HEK293 cells (Fig. 3E, Supplementary Fig. 3D).

A PARP1 ubiquitination level determined by immunoblotting analysis. Lysates prepared from HEK293 cells transfected with or without Flag-USP10 for 48 h were immunoprecipitated with anti-PARP1 antibody, followed by immunoblotting with anti-ubiquitin antibody. B In vitro de-ubiquitination assay to determine the direct effect of USP10 on PARP1 polyubiquitination. Myc-PARP1, Flag-USP10 and HA-Smurf2 were purified from HEK293 cells. These purified proteins were incubated with Mg-ATP, Ubiquitin, E1 and E2 at 37 °C for 1 h. After termination, the reaction was subjected to immunoblotting with anti-ubiquitin antibody. C PARP1 ubiquitination in HEK293 cells transfected with or without USP10 siRNA for 48 h. Lysates were immunoprecipitated with anti-PARP1 antibody, followed by immunoblotting with anti-ubiquitin antibody. D PARP1 ubiquitination in HCT116 cells with or without USP10 knockdown. Lysates were immunoprecipitated with anti-PARP1 antibody, followed by immunoblotting with anti-ubiquitin antibody. E PARP1 ubiquitination in MCF7 cells treated with or without Spautin-1 (30 μM) for 24 h. Lysates were immunoprecipitated with anti-PARP1 antibody, followed by immunoblotting with anti-ubiquitin antibody. F The potential ubiquitination sites of PARP1 protein identified by mass spectrometry. G Western blot analysis of lysates prepared from HEK293 cells co-transfected with wild-type or mutant Flag-PARP1 (K418R) and control (Myc) or Myc-USP10 expression plasmids, as indicated, for 48 h. Data are presented as the mean ± SEM from three independent experiments using the Student’s t test. **P < 0.01, N.S. not significant. H–M Co-IP analysis of PARP1 wild-type and mutant proteins. HEK293 cells were co-transfected with HA-ubiquitin, Myc-USP10, and Flag-PARP1 wild-type, Flag-PARP1 K418R, Flag-PARP1 K131R, Flag-PARP1 K249R, Flag-PARP1 K548R, Flag-PARP1 K579R or Flag-PARP1 K633R plasmids for 48 h. Lysates were immunoprecipitated with anti-Flag antibody, followed by immunoblotting with anti-HA antibody.
We next looked for the specific USP10-mediated deubiquitination sites on the PARP1 protein. Mass spectrometry analysis was used to screen the potential ubiquitination modification sites of PARP1 (Fig. 3F). Each of the six identified putative acetylation sites was mutated to arginine (R) through site-directed mutagenesis, thus producing mutants that cannot be ubiquitinated. Overexpression of USP10 showed little to no effect on PARP1 mutated lysine (K) 418 to R, while significantly increased the stability of PARP1 WT and the other PARP1 mutants (K131R/K249R/K548R/K579R/ K633R) (Fig. 3G, Supplementary Fig. 3E–I). Furthermore, co-immunoprecipitation assay results showed that the deubiquitination function of USP10 on PARP1 was lost when lysine (K) 418 of PARP1 was substituted by arginine (K418R) (Fig. 3H). In contrast, USP10 still mediated PARP1 deubiquitination after the mutation of other PARP1 residues (K131R/K249R/K548R/K579R/K633R) (Fig. 3I–M). Collectively, these results indicate that PARP1 K418 is the specific site that is deubiquitinated by USP10.
PARP1 mediates PARylation of USP10 at D634, D645, E648 site
PARylation is a PTM required for the initiation of the DNA damage response (DDR). PARP1 is among 17 members of the (ADP-ribosyl)transferase family in mammals and is the key enzyme for catalyzing PARylation [34]. Given that USP10 is a PARP1 interacting partner, we asked if PARP1 covalently modifies USP10 by PARylation. Co-IP of endogenous proteins in HEK293 (Fig. 4A, B) and MCF7 cells (Supplementary Fig. 4A, B) suggested that USP10 can bind to poly-ADP-ribose (PAR). Moreover, we co-incubated bacterially produced recombinant GST-USP10 with PAR polymers and performed in vitro PAR-binding assay followed by dot blot analyses. PAR polymers were present in the GST-USP10 pulldown complex, but not in the negative control (GST alone), which confirms that USP10 directly binds to PAR polymers (Fig. 4C).

A PARylation of USP10 determined by immunoprecipitation analysis in HEK293 cells. Lysates were immunoprecipitated with control IgG or anti-USP10 antibody, followed by immunoblotting with the indicated antibodies. B HEK293 cell lysates were immunoprecipitated with control IgG or anti-PAR antibody and then immunoblotted with the indicated antibodies. C In vitro Poly (ADP-ribose) (PAR) binding assay of USP10. Bacterially produced re-combinant GST-USP10 was used to pull down Biotin PAR-polymer (20 pmol) followed by dot blot analyses. USP10 PARylation level was determined in HEK293 cells that were transfected with Myc or Myc-PARP1 plasmids for 48 h (D), treated with or without PARP1-siRNA for 48 h (E), treated with or without 10 mM Olaparib for 24 h (F), or treated with or without 10 mM PARG inhibitor for 24 h (G). Lysates were immunoprecipitated with anti-USP10 antibody, followed by immunoblotting with anti-PAR antibody. H The PARylation level of the USP10 truncations was determined in HEK293 cells transfected with the indicated Flag-USP10 expression plasmids. I HEK293 cells were transfected with Flag-USP10 wild-type, Flag-USP10 (K620A, E621A, D634A, D645A, E648A, E654A, K687A, E692A, K693A and K699A) plasmids for 48 h. Lysates were immunoprecipitated with anti-Flag antibody, and the level of PARylation was determined by immunoblotting with an anti-PAR antibody.
To further determine whether the PARylation of USP10 is mediated by PARP1, we overexpressed PARP1 and looked for any effect on USP10. Indeed, USP10 showed increased PARylation in HEK293 cells upon overexpression of Myc-PARP1 (Fig. 4D). By contrast, transient silencing of PARP1 with siRNA (Fig. 4E), or inhibition of PARP1 activity with the PARP1 inhibitor Olaparib (Fig. 4F), led to a significant reduction of USP10 PARylation. Treatment of cells with an inhibitor that targets PARG, which is the main PAR-degrading enzyme [35], enhanced USP10 PARylation level (Fig. 4G).
In addition, we observed that the PARylation level in the carboxy-terminal region (aa601-aa798) of USP10 was significantly higher than in the other three regions (Fig. 4H). PARylation most frequently occurs on aspartate (D), glutamate (E) and lysine (K) residues of target proteins [36]. An analysis of the conserved sequence alignments of USP10 in different species revealed ten residues (K620, E621, D634, D645, E648, E654, K687, E692, K693 and K699) as potential PARylated sites. Individual substitution of the ten residues with alanine (A) revealed that mutation of D634, D645 or E648 site correlated with a notable reduction in USP10 PARylation (Fig. 4I). Then, we created a combination mutant in which D634, D645, and E648 were all substituted with alanine (A) (3 A) of USP10. We found the 3 A mutant showed nearly no detectable PARylation compared with the WT and the three single mutants (Supplementary Fig. 4C). In addition, the mutations did not change the overall structural features or complex formations with PARP1, which validates the ADP-ribose acceptor site of USP10 (Supplementary Fig. 4D, E). The protein-protein docking was performed by the HDOCK web server [37], and the visualization of molecular docking was conducted using the Chimera software.
DNA damage generates a ROS signal that promotes ATM-dependent stabilization of PARP1 via USP10
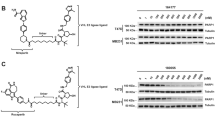
Given the critical role that PARP1 plays in the DDR pathway, we set out to investigate if USP10 is required for the stabilization and activation of PARP1 in response to DNA damage. First, we treated cells with Hydroxyurea (HU), a nonalkylating antineoplastic agent that could induce DNA damage by inhibiting ribonucleotide reductase. After HU treatment, we observed a significant increase in PARP1 protein levels in both HEK293 (Fig. 5A) and MCF7 (Supplementary Fig. 5A) cells. Consistent with previous reports that HU treatment elevated intracellular reactive oxygen species (ROS) levels [38, 39], H2O2 treatment also resulted in an increase in PARP1 level in HEK293 cells (Supplementary Fig. 5B). Treatment with N-acetylcysteine (NAC), a scavenger of free radicals, significantly inhibited the PARP1 increase induced by HU treatment in both MCF7 (Fig. 5B) and HEK293 (Supplementary Fig. 5C) cells. Consistently, we found that H2O2 or HU treatment resulted in PARP1 deubiquitination (Fig. 5C, D), while NAC could significantly inhibit the deubiquitination of PARP1 induced by HU (Fig. 5E). These results confirm the involvement of ROS in the mediation of PARP1 deubiquitination.

A Western blot analysis of PARP1 protein levels in HEK293 cells treated with HU (5 mM) for the indicated time. B Western blot analysis of PARP1 in MCF7 cells pretreated with or without NAC (3 mM) and then treated with or without HU (10 mM) for 4 h. PARP1 ubiquitination level was determined in HEK293 cells treated with or without H2O2 (500 μM) for 5 h (C), treated with or without HU (10 mM) for 7 h (D). Lysates were immunoprecipitated with IgG control or anti-PARP1 antibody, followed by immunoblotting with anti-ubiquitin antibody. E HEK293 cells pretreated with or without NAC (3 mM) and then treated with or without HU (5 mM) for 4 h. Lysates were immunoprecipitated with anti-PARP1 antibody, followed by immunoblotting with anti-ubiquitin antibody. F HEK293 cells transfected with Flag-USP10 treated with or without H2O2 (500 μM) for 4 h. Lysates were immunoprecipitated with anti-Flag antibody, followed by immunoblotting with anti-ATM and anti-PARP1 antibody. G, H HEK293 cells transfected with Flag-USP10 pretreated with or without NAC (3 mM) and then treated with or without HU (15 mM) for 6 h. Lysates were immunoprecipitated with anti-Flag antibody, followed by immunoblotting with anti-ATM antibody and anti-PARP1 antibody. I Immunofluorescence analysis of USP10 and PARP1 in HEK293 cells treated with or without H2O2 (100 μM) for 1 h. Cells were stained with anti-USP10, anti-PARP1, and DAPI. Scale bar, 10 μm. Pearson’s coefficient was calculated to quantify the co-localization of USP10 and PARP1. Data are presented as mean ± SEM from six independent experiments, ***P < 0.001 (Student’s t test). J Proximity ligation assay (PLA) passes were performed to determine the interaction between PARP1 and USP10. Red indicates the presence of interaction, and blue (DAPI) indicates nuclear staining. Scale bar, 10 μm. K H1299 control and shRNA ATM knockdown cells were treated with or without H2O2 (500 μM) for 1.5 h, and lysates were immunoblotted with the indicated antibodies. L H1299 control and shRNA ATM knockdown cells were treated with or without HU (20 mM) for 4 h. Lysates were immunoprecipitated with anti-PARP1 antibody, followed by immunoblotting with anti-ubiquitin antibody. M H1299 control and shRNA ATM knockdown cells were transfected with Flag-USP10 plasmids for 48 h, and then treated with or without H2O2 (500 μM) for 4 h. Lysates were immunoprecipitated with anti-Flag antibody, followed by immunoblotting with anti-phospho-Ser/Thr antibody. N Analysis of PARP1 ubiquitination in HEK293 cells transfected with Flag-USP10 wild-type or Flag-USP10 T42A/S337A plasmids, treated with or without H2O2 (500 μM) for 4 h. Lysates were immunoprecipitated with anti-PARP1 antibody, followed by immunoblotting with anti-ubiquitin antibody. O Western blot analysis of PARP1 in HEK293 cells pretreated with or without Spautin-1 (20 μM) for 24 h and then treated with or without HU (10 mM) for 6 h. P PARP1 ubiquitination in MCF7 cells pretreated with or without Spautin-1 (30 μM) for 24 h and then treated with or without HU (30 mM) for 7 h. Lysates were immunoprecipitated with anti-PARP1 antibody, followed by immunoblotting with anti-ubiquitin antibody.
Our results suggest that genotoxic stress triggers a transduction signal, potentially by activating a mechanism involving USP10 to increase the stability of PARP1. Upon DNA damage, USP10 is phosphorylated by ATM at Thr42 and Ser337 to deubiquitinate p53 [27]. Indeed, we found that both ATM-USP10 and USP10-PARP1 interactions were enhanced after H2O2 treatment (Fig. 5F) or HU treatment (Fig. 5G). NAC significantly inhibited ATM-USP10 and USP10-PARP1 interactions induced by HU (Fig. 5H). Moreover, both immunofluorescence analysis and PLA assay revealed co-localization of USP10 with PARP1 upon H2O2 treatment (Fig. 5I, J).
Next, we asked if ATM and USP10 were necessary for PARP1 stability under genotoxic stress conditions. As the results showed, stable knockdown of ATM in cells diminished PARP1 increase upon H2O2 treatment (Fig. 5K) and prevented the reduction of PARP1 ubiquitination upon HU treatment (Fig. 5L). Silencing ATM inhibited the Ser/Thr phosphorylation of USP10 induced by H2O2, which is consistent with previous reports that ATM could phosphorylate USP10 at Thr42 and Ser337 to activate its deubiquitination function [27] (Fig. 5M). Furthermore, USP10 TS/AA mutant failed to deubiquitinate PARP1 protein upon H2O2 treatment as wild-type USP10 did (Fig. 5N). Likewise, treatment with the USP10 inhibitor Spautin-1 also suppressed the increase of PARP1 levels (Fig. 5O, Supplementary Fig. 5E) and decrease of PARP1 ubiquitination upon HU stimulation (Fig. 5P, Supplementary Fig. 5F). These results indicate that ATM-mediated phosphorylation of USP10 at Thr42 and Ser337 is required for USP10-mediated PARP1 stabilization. In other words, upon DNA damage, the ROS signal triggers USP10 to stabilize PARP1 in an ATM-dependent manner.
PARP1-mediated USP10 PARylation contributes to DNA damage response
We further investigate whether USP10 PARylation is stimulated by DNA damage. Notably, cells treated with HU showed a significant increase in USP10 PARylation (Fig. 6A). By contrast, stable knockdown of ATM (Fig. 6B), PARP1 (Fig. 6C) or inhibition of PARP1 activity by the inhibitor Olaparib (Fig. 6D) significantly suppressed the increase in USP10 PARylation upon HU treatment. Furthermore, overexpression of wild-type USP10 caused an increase of PARP1, but this effect was compromised in cells expressing PARylation-defective USP10-3A (D634A/D645A/E648A) mutant (Fig. 6E). Indeed, results showed that cells expressing wild-type USP10 reduced the ubiquitination level of PARP1, while expressing the USP10-3A (D634A/D645A/E648A) mutant did not (Fig. 6F). These results indicate that the PARP1-mediated USP10 PARylation is stimulated by DNA damage, which in turn promotes deubiquitination and stabilization of PARP1, forming a USP10-PARP1 positive feedback loop.

A PARylation of USP10 in HEK293 cells treated with or without HU (10 mM) for 24 h. Lysates were immunoprecipitated with IgG control or anti-USP10 antibody, followed by immunoblotting with anti-PAR antibody. Data are presented as the mean ± SEM from three independent experiments using the Student’s t test. ***P < 0.001. B PARylation of USP10 in H1299 control and shRNA ATM knockdown cells treated with or without HU (20 mM) for 8 h. Lysates were immunoprecipitated with anti-USP10 antibody, followed by immunoblotting with anti-PAR antibody. C PARylation of USP10 in HEK293 control and shRNA PARP1 knockdown cells treated with or without HU (20 mM) for 24 h. Lysates were immunoprecipitated with anti-USP10 antibody, followed by immunoblotting with anti-PAR antibody. D PARylation of USP10 in HEK293 cells treated with Olaparib (10 μM) and/or HU (20 mM) for 24 h as indicated. Lysates were immunoprecipitated with anti-USP10 antibody, followed by immunoblotting with anti-PAR antibody. E HEK293 cells transfected with Flag-vector, Flag-USP10 wild-type or Flag-USP10-3A (D634A/D645A/E648A) plasmids for 48 h were subjected to western blot analysis to determine PARP1 levels. F HEK293 cells were transfected with Flag-vector, Flag-USP10 wild-type or Flag-USP10-3A (D634A/D645A/E648A) plasmids for 48 h. Lysates were immunoprecipitated with an anti-PARP1 antibody, followed by immunoblotting with an anti-ubiquitin antibody. Representative images of comet assay and quantitative analysis of tail moment after exposure to HU for 6 h (G) or H2O2 for 24 h (H) in HEK293 cells transfected with Flag-USP10 wild-type or Flag-USP10-3A plasmids. All data are presented as mean ± SD (30 cells for each group). ***P < 0.001. I HEK293 cells expressing Flag-USP10 wild-type or Flag-USP10-3A were treated with or without 5 mM HU for 4 h. Then cell lysates were immunoprecipitated with an anti-PAR antibody and immunoblotted with the indicated antibodies. J In vitro Poly (ADP-ribose) (PAR) binding assay of XRCC1, DDB1 and MRE11. Bacterially produced re-combinant GST-XRCC1, GST-DDB1 or GST-MRE11 was used to pull down Biotin PAR-polymer (20 pmol) followed by dot blot analyses. K HEK293 cells expressing Flag-USP10 wild-type or Flag-USP10-3A were treated with or without increasing doses of HU for 12 h. Cell viability was assessed by CCK8 assay. L HEK293 cells expressing Flag-USP10 wild-type or Flag-USP10-3A were treated with or without increasing doses of H2O2 for 12 h. The representative images of cell colonies are shown.
Furthermore, we aim to examine whether the positive feedback loop is involved in DNA damage response. Comet assays were performed to measure DNA damage kinetics in cells transfected with wild-type USP10 or PARylation-defective USP10-3A mutant after HU treatment (Fig. 6G) or H2O2 treatment (Fig. 6H). The results showed that cells transfected with PARylation-defective USP10-3A mutant prolonged the tail moment compared with cells transfected with wild-type USP10, which further confirms the contribution of the feedback loop of USP10-PARP1 to DNA damage response. As is known, upon initial DNA damage, PARP1 PARylates many DNA repair factors, such as XRCC1, DDB1 and MRE11 to facilitate various DNA repair pathways [1]. We found that HU stimulation of cells transfected with wild-type USP10 led to an increase in the PARylation level of these factors, but HU stimulation of cells transfected with PARylation-defective USP10-3A mutant did not (Fig. 6I). In vitro PAR-binding assays confirmed that these proteins could directly bind to PAR polymers (Fig. 6J). In addition, cell survival assays revealed that expression of PARylation-defective USP10 in HEK293 cells led to enhanced cellular sensitivity to HU (Fig. 6K) and H2O2 (Fig. 6L) as compared with its wild-type counterpart. Together, these results suggest that PARP1-mediated USP10 PARylation promotes stabilization of PARP1 and contributes to DNA damage response.
USP10 inhibitor improves the efficacy of PARP1 inhibitor in breast cancer
Considering that USP10 deubiquitinates and stabilizes PARP1, particularly in the presence of DNA damage, we investigate whether combining a USP10 inhibitor (Spautin-1) with PARP1 inhibitor would increase the anti-tumor effectiveness. When Spautin-1 was combined with Olaparib or Rucaparib, a significant increase in apoptosis of MCF7 cells was observed (Fig. 7A, B). Effective treatment of Spautin-1 on USP10 was confirmed by examining the known USP10 substrates including p53, SIRT6 and Axin1 [27, 31, 40] (Supplementary Fig. 6A). The synergistic effect of Spautin-1 and Olaparib still exist in PARP2 knockdown cells, suggesting that the observed synergistic effect is not dependent on PARP2 (Fig. 7C). Likewise, the cell proliferation and colony formation assays also showed that Spautin-1 significantly reduced the survival rate and colony formation of MCF7 cells under PARP1 inhibitor treatment (Fig. 7D–F).

A MCF7 cells were treated with or without Spautin-1 (20 μM) or Olaparib (10 μM) for 24 h. Cells were stained with 7-AAD and APC-Annexin V and analyzed by FACS, ***P < 0.001. B MCF7 cells were treated with or without Spautin-1 (20 μM) or Rucaparib (10 μM) for 24 h. Cells were stained with 7-AAD and APC-Annexin V and analyzed by FACS, ***P < 0.001. C MCF7 cells transfected with or without PARP2 siRNA for 48 h were treated with or without Spautin-1 (10 μM) and Olaparib (10 μM) for 24 h. Cells were stained with 7-AAD and APC-Annexin V and analyzed by FACS; N.S., not significant. D, E Cell viability of MCF7 cells treated with or without Spautin-1 (20 μM) and Olaparib or Rucaparib at the indicated concentrations for 24 h. Cell viability was assessed by CCK8 assay. F MCF7 cells treated with Spautin-1 (20 μM), with or without different dose of PARP1 inhibitor Olaparib for 24 h. The representative images of cell colonies were shown. G The survival outcome analysis of patients with breast cancer in response to Olaparib treatment according to The Cancer Genome Atlas (TCGA) data. Correlation of USP10 and PARP1 expression in human breast cancer (H), ovarian cancer (I), pancreatic cancer (J) and prostate cancer (K) in the GEPIA database. Tumor growth assay in nude mice subcutaneously inoculated with MCF7 cells. The mice were treated with or without Spautin-1 (10 mg/kg) and Olaparib (40 mg/kg). The images of tumors were acquired (L), and their volume (M) and weight (N) were determined. Data are shown as mean ± SD (n = 5 for each group). ***P < 0.001.
We then used The Cancer Genome Atlas (TCGA) data to analyze the potential survival outcome of patients with breast cancer who received PARP1 inhibitor treatment. OncoPredic is a method to evaluate the IC50 index of a sample for a drug based on RNA-seq sequencing data [41]. Using the RNA-seq data for TCGA-BRCA, we calculated the IC50 index of each breast cancer patient sample for Olaparib. According to the USP10 expression level and the IC50 score, patients were divided into four groups based on high or low USP10 expression and the response to Olaparib. Finally, the combined effects of the two variables on tumor prognosis were analyzed using the Kaplan–Meier algorithm. A significant difference was found among these four groups of breast cancer patients with regard to prognosis (P = 0.018). For the “Response + low USP10” group, survival was significantly more favorable compared to that of “NonResponse + high USP10” (HR = 0.36, P = 0.004) (Fig. 7G, Supplementary Fig. 6B). Importantly, data from The Cancer Genome Atlas (TCGA) indicate a significant positive correlation between USP10 and PARP1 expression in multiple cancer types that the FDA has determined suitable for PARP1 inhibitor treatment, including breast cancer, ovarian cancer, pancreatic cancer, and prostate cancer (Fig. 7H–K).
Given the potential benefit indicated by the clinical data analysis, we used a nude mouse model to test if the block in USP10 activity by its specific inhibitor Spautin-1 sensitizes breast cancer cells to PARP1 inhibition. A total of 20 female nude mice (BALB/cA-nu Mice) at 4 weeks of age were subcutaneously inoculated with 5 × 106 MCF cells. After one week, the mice were randomly divided into Control, Spautin-1, Olaparib, Spautin-1+Olaparib treatment groups. Spautin-1 and Olaparib were administered twice a week at a concentration of 10 mg/kg and 40 mg/kg, respectively. The mice were sacrificed after 25 days, and the tumors were removed for subsequent analyses.
The results of our analyses showed that the tumor volume and weight were significantly decreased following Olaparib treatment, whereas no significant change was observed after Spautin-1 treatment alone. Importantly, for the “Spautin-1+Olaparib” group, the tumor volume and weight decrease were even more significant (Fig. 7L–N). We also extracted proteins from above inhibitor-treated tumors for co-IP and western blot assays and observed the ubiquitination of PARP1 and PARylation of USP10 (Supplementary Fig. 6C). These results further indicate that USP10 inhibition sensitizes breast cancer cells to PARP1 inhibitor. Overall, these findings suggest that the deubiquitination-PARylation positive feedback loop of the USP10-PARP1 axis promotes DNA damage repair (Fig. 8).

The schematic model shows how the deubiquitination-PARylation positive feedback loop of the USP10-PARP1 axis promotes DNA damage repair.
Discussion
Breast cancer is one of the most lethal malignancies in women due to its asymptomatic nature at early stages and chemotherapy resistance. Significant progress has been made in recent years in unraveling the molecular biology of breast cancer, and targeted therapy for certain proteins has been developed. Given that BRCA1/2 breast cancers are defective in DNA DSB repair mechanisms, treatment has included targeting PARP1, a protein that binds to DNA single-strand breaks (SSBs) and mediates their repair by recruiting multiple scaffolding proteins such as XRCC1 and TDP1 [42, 43]. This treatment strategy relies on the resulting synthetic lethality of compromising both DNA single-stranded and double-stranded break repair pathways. Several PARP1 inhibitors (Olaparib, Rucaparib, Niraparib, and Talazoparib) have been approved for this use by the FDA and the EMA. For instance, in 2014, Olaparib was approved as maintenance therapy for platinum-sensitive advanced ovarian cancer with germline mutations in the DNA repair genes BRCA1/2. In 2018, Olaparib and Talazoparib were approved for human epidermal growth factor receptor type 2 (HER2)-negative locally advanced or metastatic breast cancer with germline BRCA1/2 mutations. Clinical trials have shown the effectiveness of PARP1 inhibitors in treating breast cancer and ovarian cancer with BRCA mutations, as well as prostate, pancreatic cancer, and certain HR-deficient cancers [44,45,46,47,48]. However, the therapeutic efficacy of PARP1 inhibitors varies among individuals. Therefore, elucidating the molecular mechanisms of PARP1 stability upon DNA damage might lead to the development of novel therapeutic strategies for PARP1 inhibitors.
So far, many E3 ligases that target PARP1 have been identified. However, there are few reports on the factors that mediate deubiquitination and stabilization of PARP1. One recent report showed that AEG-1 recruits USP10 to deubiquitinate PARP1 at Lys425, preventing its degradation and promoting homologous recombination-mediated DNA repair, ultimately leading to radioresistance in esophageal squamous cell carcinoma (ESCC) [24]. Our study investigates the USP10-PARP1 axis in breast cancer, emphasizing a ROS-triggered, ATM-dependent mechanism by which USP10 deubiquitinates PARP1 at Lys418. We further identify a novel positive feedback loop involving PARP1-mediated PARylation of USP10, which enhances the deubiquitination activity of USP10 and promotes DNA damage repair. In our study, we identified USP10 as a possible DUB for PARP1 through mass spectrometry analysis of co-purifying proteins. We discovered that USP10 could interact with and stabilize PARP1 by deubiquitinating PARP1 at K418. Meanwhile, PARP1 can PARylate USP10 at D634, D645, and E648 sites, which further promotes the deubiquitination activity of USP10 and DNA damage response, forming a positive feedback loop. We showed that these activities operate under genotoxic stress, which leads to increased interaction between USP10 and PARP1, and increased stabilization of PARP1. This positive feedback loop is dependent on the ROS signal generated by DNA damage as well as ATM-mediated USP10 phosphorylation at Thr42 and Ser337. Moreover, we found that both USP10 and PARP1 are highly expressed in breast cancer tissues compared to paired peri-tumor tissues, and there is a significant positive correlation between USP10 and PARP1 expression in breast cancer cases. Thus, the USP10-PARP1 positive feedback loop that is triggered upon DNA damage might turn out to be a key disadvantage for the success of PARP1 inhibitor therapy.
The discovery of the USP10-PARP1 axis prompted us to explore whether inhibiting USP10 could improve the therapeutic efficacy of PARP1 inhibitors. Several studies have indicated that administering Spautin-1, a specific inhibitor of USP10, either alone or in conjunction with other DNA damage agents, leads to a decrease in cell survival and colony formation in cancers [49,50,51]. Moreover, another study showed that Spautin-1 can inhibit EGFR signaling and induce cell death in cases of prostate cancer [52]. We observed that, compared to the control groups, combining Olaparib therapy with Spautin-1 significantly reduced the survival rate and colony formation of breast cancer cells. Moreover, the combination treatment with Spautin-1 significantly increased the apoptosis of breast cancer cells. Similar results were observed using a nude mouse model. These findings indicate that a blockade of USP10 by its specific inhibitor Spautin-1 sensitizes breast cancer cells to PARP1 inhibitor.
It has been reported that PARP1 deletion is less effective at cell killing than PARP1 inhibition, and the ability of the inhibitors to “trap” PARP1 on the DNA contribute to their cytotoxicity [53,54,55]. On the one hand, PARP1 deletion is rare in tumor cells suitable for PARP1 inhibitor treatment. On the other hand, we found that PARP1 is highly expressed in breast cancer, and PARP1 expression is significantly increased in response to both ROS and DNA damage, which frequently occur in tumorigenesis. Therefore, PARP1 inhibitor might not efficiently “trap” excessive PARP1 on the DNA to exert their cytotoxicity, which would probably leave “untrapped” PARP1 to promote DNA damage repair and cell survival of tumor cells. In this study, we observed that combination of Spautin-1 and PARP1 inhibitor led to enhanced cell death, probably because that inhibition of USP10 decreases PARP1 expression to the amount that could not successfully accomplish DNA damage repair but PARP1 inhibitor could still “trap” for cytotoxicity.
Overall, our study suggested that the deubiquitination-PARylation positive feedback loop of the USP10-PARP1 axis promotes DNA damage repair. DNA damage induces a ROS signal that promotes the deubiquitinaiton-PARylation positive feedback loop, mediated by the USP10-PARP1 axis in an ATM-dependent manner. PARP1 is highly expressed in breast cancer tissues and positively correlates with USP10 levels. USP10 inhibitor sensitizes breast cancer cells to PARP1 inhibitor both in vivo and in vitro.
Data availability
All data generated or analyzed during this study are included in this article and its Supplementary Information Files.
References
Ray Chaudhuri A, Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol. 2017;18:610–21.
Alemasova EE, Lavrik OI. Poly(ADP-ribosyl)ation by PARP1: reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019;47:3811–27.
Kraus WL, Lis JT. PARP goes transcription. Cell. 2003;113:677–83.
Liu C, Vyas A, Kassab MA, Singh AK, Yu X. The role of poly ADP-ribosylation in the first wave of DNA damage response. Nucleic Acids Res. 2017;45:8129–41.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7.
Zimmer AS, Gillard M, Lipkowitz S, Lee JM. Update on PARP Inhibitors in Breast Cancer. Current Treat options Oncol. 2018;19:21.
Sun R, Luo H, Su J, Di S, Zhou M, Shi B, et al. Olaparib Suppresses MDSC Recruitment via SDF1α/CXCR4 Axis to Improve the Anti-tumor Efficacy of CAR-T Cells on Breast Cancer in Mice. Mol Ther J Am Soc Gene Ther. 2021;29:60–74.
Ratnam K, Low JA. Current development of clinical inhibitors of poly(ADP-ribose) polymerase in oncology. Clin Cancer Res. 2007;13:1383–8.
Ossovskaya V, Koo IC, Kaldjian EP, Alvares C, Sherman BM. Upregulation of Poly (ADP-Ribose) Polymerase-1 (PARP1) in Triple-Negative Breast Cancer and Other Primary Human Tumor Types. Genes Cancer. 2010;1:812–21.
Juvekar A, Burga LN, Hu H, Lunsford EP, Ibrahim YH, Balmañà J, et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012;2:1048–63.
Sun X, Tang H, Chen Y, Chen Z, Hu Z, Cui Z, et al. Loss of the receptors ER, PR and HER2 promotes USP15-dependent stabilization of PARP1 in triple-negative breast cancer. Nat Cancer. 2023;4:716–33.
Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nature Med. 2014;20:1242–53.
Scheffner M, Nuber U, Huibregtse JM. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature. 1995;373:81–83.
Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–83.
Sadowski M, Suryadinata R, Tan AR, Roesley SN, Sarcevic B. Protein monoubiquitination and polyubiquitination generate structural diversity to control distinct biological processes. IUBMB Life. 2012;64:136–42.
Qian H, Zhang N, Wu B, Wu S, You S, Zhang Y, et al. The E3 ubiquitin ligase Smurf2 regulates PARP1 stability to alleviate oxidative stress-induced injury in human umbilical vein endothelial cells. J Cell Mol Med. 2020;24:4600–11.
Zhang N, Zhang Y, Qian H, Wu S, Cao L, Sun Y. Selective targeting of ubiquitination and degradation of PARP1 by E3 ubiquitin ligase WWP2 regulates isoproterenol-induced cardiac remodeling. Cell Death Differ. 2020;27:2605–19.
Gatti M, Imhof R, Huang Q, Baudis M, Altmeyer M. The Ubiquitin Ligase TRIP12 Limits PARP1 Trapping and Constrains PARP Inhibitor Efficiency. Cell Rep. 2020;32:107985.
Yu JT, Hu XW, Yang Q, Shan RR, Zhang Y, Dong ZH, et al. Insulin-like growth factor binding protein 7 promotes acute kidney injury by alleviating poly ADP ribose polymerase 1 degradation. Kidney Int. 2022;102:828–44.
Zhang Y, Liao XH, Xie HY, Shao ZM, Li DQ. RBR-type E3 ubiquitin ligase RNF144A targets PARP1 for ubiquitin-dependent degradation and regulates PARP inhibitor sensitivity in breast cancer cells. Oncotarget. 2017;8:94505–18.
De Vos M, El Ramy R, Quénet D, Wolf P, Spada F, Magroun N, et al. Poly(ADP-ribose) polymerase 1 (PARP1) associates with E3 ubiquitin-protein ligase UHRF1 and modulates UHRF1 biological functions. J Biol Chem. 2014;289:16223–38.
Zhang G, Tan R, Wan S, Yang R, Hu X, Zhao E, et al. HECTD3 regulates the tumourigenesis of glioblastoma by polyubiquitinating PARP1 and activating EGFR signalling pathway. Br J Cancer. 2022;127:1925–38.
Zhao X, Ma Y, Li J, Sun X, Sun Y, Qu F, et al. The AEG-1-USP10-PARP1 axis confers radioresistance in esophageal squamous cell carcinoma via facilitating homologous recombination-dependent DNA damage repair. Cancer Lett. 2023;577:216440.
Zeng Z, Wu HX, Zhan N, Huang YB, Wang ZS, Yang GF, et al. Prognostic significance of USP10 as a tumor-associated marker in gastric carcinoma. Tumour Biol. 2014;35:3845–53.
Bhattacharya U, Neizer-Ashun F, Mukherjee P, Bhattacharya R. When the chains do not break: the role of USP10 in physiology and pathology. Cell Death Dis. 2020;11:1033.
Yuan J, Luo K, Zhang L, Cheville JC, Lou Z. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell. 2010;140:384–96.
He Y, Jiang S, Mao C, Zheng H, Cao B, Zhang Z, et al. The deubiquitinase USP10 restores PTEN activity and inhibits non-small cell lung cancer cell proliferation. J Biol Chem. 2021;297:101088.
Bomberger JM, Barnaby RL, Stanton BA. The deubiquitinating enzyme USP10 regulates the post-endocytic sorting of cystic fibrosis transmembrane conductance regulator in airway epithelial cells. J Biol Chem. 2009;284:18778–89.
Deng M, Yang X, Qin B, Liu T, Zhang H, Guo W, et al. Deubiquitination and Activation of AMPK by USP10. Mol Cell. 2016;61:614–24.
Lin Z, Yang H, Tan C, Li J, Liu Z, Quan Q, et al. USP10 antagonizes c-Myc transcriptional activation through SIRT6 stabilization to suppress tumor formation. Cell Rep. 2013;5:1639–49.
Weisberg EL, Schauer NJ, Yang J, Lamberto I, Doherty L, Bhatt S, et al. Inhibition of USP10 induces degradation of oncogenic FLT3. Nature Chem Biol. 2017;13:1207–15.
Niu J, Shi Y, Xue J, Miao R, Huang S, Wang T, et al. USP10 inhibits genotoxic NF-κB activation by MCPIP1-facilitated deubiquitination of NEMO. EMBO J. 2013;32:3206–19.
Hassa PO, Hottiger MO. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front Biosci J Virtual Libr. 2008;13:3046–82.
Davidovic L, Vodenicharov M, Affar EB, Poirier GG. Importance of poly(ADP-ribose) glycohydrolase in the control of poly(ADP-ribose) metabolism. Exp Cell Res. 2001;268:7–13.
Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol. 2012;13:411–24.
Yan Y, Zhang D, Zhou P, Li B, Huang SY. HDOCK: a web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017;45:W365–w373.
Kapor S, Čokić V, Santibanez JF. Mechanisms of Hydroxyurea-Induced Cellular Senescence: An Oxidative Stress Connection?. Oxid Med Cell Longev. 2021;2021:7753857.
DeSesso JM. Cell death and free radicals: a mechanism for hydroxyurea teratogenesis. Med Hypotheses. 1979;5:937–51.
Wang Y, Mao A, Liu J, Li P, Zheng S, Tong T, et al. USP10 strikes down β-catenin by dual-wielding deubiquitinase activity and phase separation potential. Cell Chem Biol. 2023;30:1436–.e1410.
Maeser D, Gruener RF, Huang RS. oncoPredict: an R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief Bioinformatics. 2021;22:bbab260.
El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003;31:5526–33.
Das BB, Huang SY, Murai J, Rehman I, Amé JC, Sengupta S, et al. PARP1-TDP1 coupling for the repair of topoisomerase I-induced DNA damage. Nucleic Acids Res. 2014;42:4435–49.
Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med. 2015;373:1697–708.
Mateo J, Lord CJ, Serra V, Tutt A, Balmaña J, Castroviejo-Bermejo M, et al. A decade of clinical development of PARP inhibitors in perspective. Ann Oncol Off J Eur Soc Med Oncol. 2019;30:1437–47.
Pilié PG, Gay CM, Byers LA, O’Connor MJ, Yap TA. PARP Inhibitors: Extending Benefit Beyond BRCA-Mutant Cancers. Clin Cancer Res. 2019;25:3759–71.
Sonnenblick A, de Azambuja E, Azim HA Jr, Piccart M. An update on PARP inhibitors-moving to the adjuvant setting. Nat Rev Clin Oncol. 2015;12:27–41.
Zhu H, Wei M, Xu J, Hua J, Liang C, Meng Q, et al. PARP inhibitors in pancreatic cancer: molecular mechanisms and clinical applications. Mol Cancer. 2020;19:49.
Shao S, Li S, Qin Y, Wang X, Yang Y, Bai H, et al. Spautin-1, a novel autophagy inhibitor, enhances imatinib-induced apoptosis in chronic myeloid leukemia. Int J Oncol. 2014;44:1661–8.
Correa RJ, Valdes YR, Peart TM, Fazio EN, Bertrand M, McGee J, et al. Combination of AKT inhibition with autophagy blockade effectively reduces ascites-derived ovarian cancer cell viability. Carcinogenesis. 2014;35:1951–61.
Guo J, Zhang J, Liang L, Liu N, Qi M, Zhao S, et al. Potent USP10/13 antagonist spautin-1 suppresses melanoma growth via ROS-mediated DNA damage and exhibits synergy with cisplatin. J Cell Mol Med. 2020;24:4324–40.
Liao Y, Guo Z, Xia X, Liu Y, Huang C, Jiang L, et al. Inhibition of EGFR signaling with Spautin-1 represents a novel therapeutics for prostate cancer. J Exp Clin Cancer Res. 2019;38:157.
D’Andrea AD. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair. 2018;71:172–6.
Michelena J, Lezaja A, Teloni F, Schmid T, Imhof R, Altmeyer M. Analysis of PARP inhibitor toxicity by multidimensional fluorescence microscopy reveals mechanisms of sensitivity and resistance. Nat Commun. 2018;9:2678.
Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012;72:5588–99.
Funding
This work was supported by the key project of the National Natural Science Foundation (82030091), the key project of LiaoNing Science Foundation (2022JH6/100100037, 2022JH2/20200034, 2022JH2/20200054, 2021JH2/10300023), the National Natural Science Foundation (82102740, 82073089), Shenyang Youth Science and Technology Innovation Talent Support Project (RC220139) and the Science Foundation for Outstanding Youth of Liaoning Province (2023JH3/10200017).
Author information
Authors and Affiliations
Contributions
LC, CX, ZW and XS conceived and designed the study. JL, SZ, LC, NZ, QG, YZ, RY, SD, LZ, YX, YW, SL, PJ, KZ, SC and DC conducted the experiments. HL, YZ and RS performed the statistical analysis. JL and SZ wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All methods were performed in accordance with the relevant guidelines and regulations. The approval of this study has been obtained from ethics committee in Outdo Biotech (SHYJS-CP-2210008). Informed consent was obtained from all participants. The animal studies were authorized by the Institutional Animal Care and Use Committee of China Medical University ([2022]146).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, J., Zhang, S., Cao, L. et al. The deubiquitination-PARylation positive feedback loop of the USP10-PARP1 axis promotes DNA damage repair and affects therapeutic efficacy of PARP1 inhibitor. Oncogene (2025). https://doi.org/10.1038/s41388-025-03428-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41388-025-03428-7
