
Abstract
Up to 30% of acute myeloid leukemia (AML) patients face unfavorable outcomes due to the FMS-like receptor tyrosine kinase-3 (FLT3) internal tandem duplication (ITD) mutation. Although FLT3 inhibitors show encouraging outcomes in treatment, they fail to eliminate leukemia stem cells, the origin of persistent and resistant lesions. Exploration of the mechanism in FLT3-ITD+ AML maintenance and chemoresistance is crucial for the development of novel therapeutic approaches. The manifestation of pluripotency transcription factors (TFs) and their link to clinical outcomes have been documented in various tumors. This study investigates the correlation between core pluripotency TF and treatment in AML. We discovered that FLT3 inhibition induced upregulation of OCT4 and NANOG in FLT3-ITD+ AML cells. Subsequently, we demonstrated that downregulation of OCT4 or NANOG inhibited cell growth, promoted apoptosis, and induced G0/G1 cell cycle phase arrest in FLT3-ITD+ AML cells. Knockdown of OCT and NANOG inhibited tumor growth in a mouse tumor model. OCT4 promotes the malignant biological behavior of FLT3-ITD+ AML by enhancing the abnormal FLT3 signaling pathway through transcriptional activation of NANOG. Importantly, downregulation of OCT4 or NANOG increased responsiveness to FLT3-tyrosine kinase inhibitor (TKI) (Gilteritinib), implying that OCT4 and NANOG may contribute to TKI resistance in FLT3-ITD+ AML. Our study verifies the involvement of OCT4/NANOG in regulating TKI sensitivity and targeting them may improve the cytotoxicity of FLT3-TKIs in FLT3-ITD+ AML.

Similar content being viewed by others

Introduction
Acute myeloid leukemia (AML) is a hematologic malignancy affecting the whole body [1]. Despite growing research on the pathogenesis of AML and the emergence of targeted medicines such as FMS-like receptor tyrosine kinase 3 (FLT3) tyrosine kinase inhibitors (TKIs) [2,3,4], isocitrate dehydrogenase (IDH) inhibitors [5,6,7], and B-cell leukemia/lymphoma 2 (BCL2) inhibitors [8], most patients still rely on conventional chemotherapy and hematopoietic stem cell transplantation [9]. As the most frequent mutant subtype, the FLT3-internal tandem replication (ITD) mutation in AML leads to ongoing activation of subsequent signaling pathways and increases the risk of recurrence [10]. New-generation FLT3 inhibitors, such as gilteritinib, only partially inhibit AML cell growth and temporary clinical responses when employed as a single agent [11]. Consequently, it is urgent to explore the potential mechanisms of chemotherapy resistance in FLT3-ITD+ AML to find targets that restore cancer sensitivity.
Transcription factors (TFs) are key regulators of cancers [12, 13]. Octamer-binding transcription factor 4 (OCT4), NANOG Homeobox (NANOG), SRY-box transcription factor 2 (SOX-2), and Krüppel-like factor 4 (KLF4) are among the most important stemness-associated TFs that regulate the stemness properties of cancer cells, including processes of chemotherapy-induced dormancy and reactivation [14, 15]. OCT4 and NANOG are pluripotency markers that not only regulate gene expression during embryonic growth but also preserve the self-renewal and pluripotency of stem cells [16, 17]. Deregulation of OCT4 and NANOG has been detected in numerous malignant tumors, leading to malignant progression [18, 19].
The overexpression of OCT4 and NANOG was positively associated with higher grades, metastasis formation, and poor survival of tumors, underscoring their clinical importance [16, 20]. OCT4 expression was increased in AML patients, which was linked to the FLT3-ITD mutation, complex karyotypes, and poor risk categorization [21]. Elevated levels of OCT4 expression independently forecasted poor overall survival (OS) and event-free survival (EFS). Whereas OCT4 expression was reduced in patients who achieved complete response (CR) compared with non-CR patients [21]. However, the direct regulating mechanisms of OCT4 and NANOG in AML are unclear.
Increased OCT4/NANOG expression shows a positive association with chemoresistance in cancer patients [16, 22]. OCT4 and NANOG expression is thought to be correlated with cisplatin resistance in gastric cancer cells [23]. Knockdown of OCT4 and NANOG improved the cisplatin sensitivity of head and neck squamous cell carcinoma and breast cancer [22, 24]. There are no studies on whether OCT4 and NANOG have an influential role in the TKI resistance of AML cells.
The aim of our study is to investigate the underlying mechanisms of TKI response and the role of pluripotency TFs in FLT3-ITD+ AML. We examined the alteration in the expression levels of pluripotency factors at transcript and protein levels after FLT3 inhibition in FLT3-ITD+ AML cells. In addition, the role of OCT4 and NANOG on cancer survival and TKI treatment was investigated by knocking down OCT4 or NANOG expression in FLT3-ITD+ AML cells. We propose that simultaneous targeting of FLT3 and pluripotency TFs may be an effective strategy to improve outcomes in AML patients.
Results
OCT4 and NANOG expression upregulated after TKI treatment
MV4-11 and MOLM-13 cells carry the FLT3-ITD mutation. FLT3 inhibitors are used for the treatment of FLT3-ITD+ AML. We evaluated the expression of four core pluripotency TFs, including OCT4, NANOG, KLF4, and SOX-2, in both cell lines after FLT3 inhibitor treatment. Remarkably, OCT4 and NANOG transcript levels were significantly increased after gilteritinib (4 nM) exposure, while KLF4 and SOX-2 changed little (Fig. 1A, B). Similarly, OCT4 and NANOG transcript levels were upregulated to varying degrees in FLT3-ITD+ AML cells upon exposure to another FLT3-TKI (quizartinib at 2 nM) (Fig. 1C, D). We next examined the alterations in OCT4 and NANOG expression after different concentrations of gilteritinib at 0, 2, 4, and 8 nM. Transcript Levels of both OCT4 (Fig. 1E, F) and NANOG (Fig. 1G, H) increased in response to upregulation of the administered concentration. Protein expression levels in OCT4 (Fig. 1J) and NANOG (Fig. 1K) were dose-dependent when MV4-11 and MOLM-13 (Fig. 1I) cells were treated with gilteritinib at different concentrations.

MV4-11 cells (A) and MOLM-13 cells (B) underwent a 24-h treatment with vehicle (dimethyl sulfoxide) or gilteritinib (4 nM). MV4-11 cells (C) and MOLM-13 cells (D) underwent a 24-h treatment with vehicle (dimethyl sulfoxide) or quizartinib (2 nM). QRT-PCR was employed to examine the transcription of the four primary transcription factors. MV4-11 (E, G) and MOLM-13 cells (F, H) underwent treatment with gilteritinib in concentrations of 0, 2, 4, and 8 nM with gilteritinib for 24 h. QRT-PCR was employed to examine the transcription of OCT4 (E, F) and NANOG (G, H). I Varying doses (0, 2, 4, 8 nM) of gilteritinib were administered to MV4-11 and MOLM-13 cells over a period of 48 h, followed by western blotting analysis to detect the indicated proteins. The protein expression of OCT4 (J) and NANOG (K) in MV4-11 and MOLM-13 cells underwent treatment with varying concentrations of gilteritinib (0, 2, 4, and 8 nM) for 48 h using western blotting analysis. The statistical result is the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Downregulation of OCT4 suppresses growth and induces apoptosis in FLT3-ITD+AML cells
To further investigate the role of OCT4 in AML progression, we evaluated the effect of OCT4 downregulation on AML cell proliferation and apoptosis. We first generated stable OCT4 knockdown and control vector cell lines in MV4-11 and MOLM-13 cells by lentiviral (shOCT4-1, shOCT4-2, and shCtrl) transduction. It is showing that OCT4 and NANOG protein expression decreased when lenti-OCT4 was infected with MV4-11 and MOLM-13 cells (Fig. 2A–C). Notably, knockdown of OCT4 suppressed MV4-11 and MOLM-13 cell growth (Fig. 2D, E) and induced apoptosis (Fig. 2F, G) compared with the control. To explore the regulatory role of OCT4 in other AML cells, infection of two AML cell lines with lenti-OCT4 resulted in reduced expression of OCT4 protein (Fig. 2H, I). Knockdown of OCT4 also inhibited the proliferation of THP-1 and OCI-AML3 cells compared with the control group (Fig. 2J, K). These results suggest that OCT4 confers cell viability and that OCT4 downregulation inhibits the malignant proliferation of AML cells.

A OCT4’s suppression effectiveness and the protein expression of NANOG were assessed using western blotting analysis after transfecting MV4-11 (B) and MOLM-13 cells (C) with lentiviral vectors of OCT4 shRNA (shOCT4-1 and shOCT4-2) or control shRNA (shCtrl) for 48 h. The growth of MV4-11 (D) and MOLM-13 (E) cells was evaluated by the CCK-8 assay. F Examination of apoptosis via flow cytometry following 48 h of MV4-11 and MOLM-13 cell transduction. G Apoptosis was examined through the application of annexin V/propidium iodide (PI) markers. H Lentiviral vectors of OCT4 shRNA or shCtrl were transfected to THP-1 and OCI-AML3 cells for 48 h, followed by western blotting assay to detect the indicated proteins. I The protein expression of OCT4 after transfecting THP-1 and OCI-AML3 cells with lentiviral vectors of OCT4 shRNA or control shCtrl for 48 h using western blotting analysis. The growth of THP-1 (J) and OCI-AML3 (K) cells was evaluated by the CCK-8 assay. The statistical result is the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Downregulation of NANOG suppresses growth and induces apoptosis in FLT3-ITD+AML cells
To further investigate the role of NANOG in AML progression, we evaluated the effect of NANOG downregulation on AML cell growth and apoptosis. We first stably transfected MV4-11 and MOLM13 cells using shRNA (shNANOG-1 and shNANOG-2) targeting NANOG or shCtrl as controls. NANOG protein levels were reduced in both cell lines compared to controls (Fig. 3A–C). However, there was no change in the expression of OCT4. NANOG knockdown significantly suppressed cell growth (Fig. 3D, E) and induced apoptosis in both cell lines (Fig. 3F, G) compared to controls. To explore the regulatory role of NANOG in other AML cells, infection of two AML cell lines with lenti-NANOG resulted in reduced expression of NANOG protein (Fig. 3H, I). Knockdown of NANOG also inhibited the proliferation of THP-1 and OCI-AML3 cells compared with the control group (Fig. 3J, K). The findings verify that NANOG downregulation inhibits the malignant proliferation of AML cells.

A NANOG’s suppression effectiveness and the protein expression of OCT4 were assessed using western blotting analysis after transfecting MV4-11 (B) and MOLM-13 (C) cells with lentiviral vectors of NANOG shRNA (shNANOG-1 and shNANOG-2) or control shRNA (shCtrl) for 48 h. The growth of MV4-11 (D) and MOLM-13 (E) cells was evaluated by the CCK-8 assay. F Examination of apoptosis via flow cytometry following 48 h of MV4-11 and MOLM-13 cell transduction. G Apoptosis was examined through the application of annexin V/propidium iodide (PI) markers. H Lentiviral vectors of NANOG shRNA or control shCtrl were transfected to THP-1 and OCI-AML3 cells for 48 h, followed by western blotting assay to detect the indicated proteins. I The protein expression of NANOG after transfecting THP-1 and OCI-AML3 cells with lentiviral vectors of NANOG shRNA or shCtrl for 48 h using western blotting analysis. The growth of THP-1 (J) and OCI-AML3 (K) cells was evaluated by the CCK-8 assay. The statistical result is the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
The effect of reduced OCT4/NANOG on cell cycle distribution
Subsequently, we examined whether reducing OCT4/NANOG levels influences the cell cycle in FLT3-ITD+ AML cells. Flow cytometry was conducted to evaluate OCT4/NANOG function in the cell cycle distribution. The results demonstrate that reduced OCT4 causes MV4-11 (Fig. 4A) and MOLM-13 (Fig. 4C) cell cycle arrest at the G0/G1 phase and a shortened S phase. As compared with 52.75% in cells treated with shCtrl, the G0/G1 phase was 63.84% and 64.63% in MV4-11 cells treated with shOCT4-1 and shOCT4-2 (Fig. 4B). As compared with 48.59% in cells treated with shCtrl, the G0/G1 phase was 74.62% and 68.38% in MOLM-13 cells treated with shOCT4-1 and shOCT4-2 (Fig. 4D). Similarly, cell cycle analysis was conducted to evaluate the role of NANOG in cell cycle distribution. The results show that reduced NANOG causes MV4-11 (Fig. 4E) and MOLM-13 (Fig. 4G) cell cycle arrest at the G0/G1 phase and a shortened S phase. As compared with 49.21% in the control cells, the proportion of G0/G1 phase in MV4-11 cells undergoing treatment with shNANOG-1 and shNANOG-2 was 64.40% and 63.93% (Fig. 4F). As compared with 45.38% in the control cells, the proportion of G0/G1 phase was 71.21% and 66.72% in MOLM-13 cells undergoing treatment with shNANOG-1 and shNANOG-2 (Fig. 4H). Taken together, OCT4/NANOG knockdown inhibited AML cell growth by inducing G0/G1 phase arrest of the cell cycle. These results confirmed that OCT4 and NANOG contribute to enhancing the inherent carcinogenicity and malignant transformation of FLT3-ITD+ AML cells.

MV4-11 (A, B) and MOLM-13 (C, D) cells were transferred with OCT4 shRNA (shOCT4-1 and shOCT4-2) or control shRNA (shCtrl). MV4-11 (E, F) and MOLM-13 (G, H) cells were transduced with lentiviral vectors of NANOG shRNA (shNANOG-1 and shNANOG-2) or shCtrl. Examination of the cell cycle via flow cytometry following 48 h of cellular transduction. The statistical result is the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Downregulation of OCT4 enhances gilteritinib inhibition of FLT3-ITD+ AML
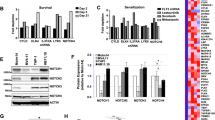
Given the involvement of deregulated OCT4 and NANOG in cancer chemoresistance, it was hypothesized that upregulating OCT4 and NANOG in FLT3-ITD+ AML cells subsequent to the inhibition of FLT3 could be significantly influential in counteracting the effect of TKIs on eradicating leukemia. The expression of OCT4 was reduced in cells by using lentivirus vectors of OCT4 shRNA (shOCT4-1 or shOCT4-2). We examined whether OCT4 downregulation affected downstream STAT5, AKT, and ERK pathways activated by mutant FLT3-ITD. Knockdown treatment with OCT4 failed to affect the total protein level of FLT3 but reduced the level of activation (phosphorylated FLT3) (Fig. 5A). Moreover, downregulation of OCT4 significantly inhibited the phosphorylation but had no effect on the total expression of STAT5, AKT, and ERK in both cell lines (Fig. 5A). In conclusion, downregulation of OCT4 increased apoptosis and showed an inhibitory effect on the abnormal phosphorylation pathway activated by the FLT3-ITD mutation but had little effect on the degradation of total FLT3 protein. The suppression of OCT4 induced apoptosis of two cell lines, and the anti-leukemia effect was more significant when in combination with gilteritinib in MV4-11 (Fig. 5B) and MOLM-13 cells (Fig. 5C). Knockdown of OCT4 (shOCT4-1 and shOCT4-2) increased the apoptosis rate of MV4-11 cells treated with gilteritinib by 35.79% and 33.23% (Fig. 5B). Knockdown of OCT4 (shOCT4-1 and shOCT4-2) increased the apoptosis rate of MOLM-13 cells after gilteritinib treatment by 34.39% and 32.35% (Fig. 5C). Knockdown of OCT4 increased gilteritinib sensitivity of both cell lines to gilteritinib (Fig. 5D, E). Downregulation of OCT4 activity significantly increased gilteritinib-induced apoptosis and growth suppression in FLT3-ITD+ AML cells. These data support that OCT4 expression level is associated with TKI susceptibility in FLT3-ITD+ AML cells.

A FLT3 protein and downstream targets of FLT3 activation were assessed using western blotting analysis after transfecting MV4-11 and MOLM-13 cells with lentiviral vectors of OCT4 shRNA (shOCT4-1 and shOCT4-2) or control shRNA (shCtrl) for 48 h. OCT4 shRNA or shCtrl was transduced in MV4-11 and MOLM-13 cells. Cells were then treated with gilteritinib (2 nM) or vehicle (dimethyl sulfoxide) for 48 h. Apoptosis was examined through the application of flow cytometry analysis. The ratio of apoptotic MV4-11 (B) and MOLM-13 (C) cells was measured. The viability of MV4-11 (D) and MOLM-13 (E) cells in treatment with varying doses of gilteritinib were examined by CCK-8 assay. The statistical result is the mean ± SEM. ****p < 0.0001.
Downregulation of NANOG improves gilteritinib-mediated inhibition of FLT3-ITD+ AML
The expression of NANOG was reduced in MV4-11 and MOLM-13 cells by using lentiviruses (shNANOG-1 or shNANOG-2). We examined whether NANOG downregulation affected downstream STAT5, AKT, and ERK pathways activated by mutant FLT3-ITD. Knockdown treatment with NANOG failed to affect the total protein level of FLT3 but reduced the level of activation (phosphorylated FLT3) (Fig. 6A). Moreover, downregulation of NANOG significantly inhibited the phosphorylation but had no effect on the total expression of STAT5, AKT, and ERK in both cell lines (Fig. 6A). In conclusion, downregulation of NANOG increased apoptosis and showed an inhibitory effect on the abnormal phosphorylation pathway activated by the FLT3-ITD mutation but had little effect on the degradation of total FLT3 protein. The suppression of NANOG led to notable apoptosis in both cell lines, with its anti-leukemia impact being more intense when combined with gilteritinib (Fig. 6B, C). Knockdown of NANOG (shNANOG-1 and shNANOG-2) increased the apoptosis rate of MV4-11 cells treated with gilteritinib by 41.2% and 30.94% (Fig. 6B). Knockdown of NANOG (shNANOG-1 and shNANOG-2) increased the apoptosis percentage of MOLM-13 cells treated with gilteritinib by 27.81% and 18.2% (Fig. 6C). Knockdown of NANOG increased gilteritinib sensitivity of both cell lines to gilteritinib (Fig. 6D, E). Downregulation of NANOG significantly improved gilteritinib-induced apoptosis and growth inhibition of FLT3-ITD+ AML cells. These results indicate that NANOG is associated with TKI susceptibility in FLT3-ITD+ AML cells.

A FLT3 protein and downstream targets of FLT3 activation were assessed using western blotting analysis after transfecting MV4-11 and MOLM-13 cells with lentiviral vectors of NANOG shRNA (shNANOG-1 and shNANOG-2) or control shRNA (shCtrl) for 48 h. NANOG shRNA or shCtrl was transduced in MV4-11 and MOLM-13 cells. Cells were then treated with gilteritinib (2 nM) or vehicle (dimethyl sulfoxide) for 48 h. Apoptosis was examined through the application of flow cytometry analysis. The ratio of apoptotic MV4-11 (B) and MOLM-13 (C) cells was measured. The viability of MV4-11 (D) and MOLM-13 (E) cells in treatment with varying doses of gilteritinib were examined by CCK-8 assay. The statistical result is the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
OCT4 transcriptionally regulates NANOG expression
Studies have shown that OCT4 can interact with NANOG to regulate the occurrence and development of different cancers through specific mechanisms. We have defined the effects of OCT4 and NANOG on FLT3-ITD+ AML cells, whereas previous studies have demonstrated the effects of OCT4 on tumor cells and the interaction between OCT4 and NANOG. Therefore, we hypothesized that the interaction between OCT4 and NANOG could also affect FLT3-ITD+ AML. Firstly, we analyzed the correlation between OCT4 and NANOG through the GEPIA (Gene Expression Profiling Interactive Analysis) database, and the results showed that the expression of the two was positively correlated (R = 0.85, p < 0.001) (Fig. 7A). The JASPAR database was used to predict the binding sites of transcription factor OCT4 to the upstream promoter of NANOG (Fig. 7B). The MV4-11 cell was used to perform a dual-luciferase assay to clarify the effect of transcription factor OCT4 on NANOG gene expression (Fig. 7C). The results showed that the luciferase activity of the wild-type NANOG promoter was significantly higher than that of the control and mutant NANOG promoters, and the luciferase activity of the mutant NANOG promoter was not significantly different from that of the control. This indicates that OCT4 binds to the NANOG promoter region and positively regulates NANOG. In conclusion, OCT4 promotes NANOG expression through transcriptional regulation during FLT3-ITD+ AML development.

A The GEPIA database analysis showed a positive correlation between OCT4 and NANOG expression. B Schematic representation of the wild-type (WT) and mutant (Mut) sequences of OCT4 at the three putative binding sites on the NANOG promoter. C Relative luciferase activity was detected in MV4-11 cells co-transfected with luciferase reporter plasmids containing wild-type or mutant NANOG promoter sequences and OCT4 overexpression plasmids. The statistical result is the mean ± SEM. ***p < 0.001, ****p < 0.0001.
Down-regulation of OCT4 or NANOG has an antitumor effect in vivo
Finally, we determined the role of OCT4 and NANOG in regulating FLT3-ITD+ AML tumor proliferation in vivo. To confirm the role of OCT4 in vivo, we established a mouse transplantation model using the MV4-11 cell line (Fig. 8A). First, the model results showed that OCT knockdown significantly inhibited tumor growth (Fig. 8B, C). OCT4 knockdown reduced the volume and weight of leukemia cell-derived tumors (Fig. 8E, F). Paraffin-embedded tumor sections were examined by immunohistochemistry and terminal deoxynucleotidyl transferase-mediated dUTP Nick end labeling (TUNEL). We found downregulation of OCT4/NANOG expression and an increased percentage of apoptotic cells in the knockdown OCT4 groups (shOCT4-1 and shOCT4-2) (Fig. 8G).

A In vivo subcutaneous implantation of NCG mouse models induced by MV4-11 cells transfected with OCT4 shRNA (shOCT4-1 and shOCT4-2) or control shRNA (shCtrl), tumor volume was measured daily after injection of tumor cells. B Representative image of tumor-bearing mice in different groups. C Representative images of xenograft tumors in each group. D OCT4 expressions were detected by western blotting analysis. E, F Quantification of tumor size and weight in different groups. G OCT4 and NANOG expression in paraffin-embedded continuous sections were detected by immunohistochemistry, and apoptotic cells were counted by TUNEL (bar, 50 μm). The A was created with BioRender.com. The statistical result is the mean ± SEM. **p < 0.01, ****p < 0.0001.
To further confirm the role of NANOG in vivo, we established a mouse transplantation model using the MV4-11 cell line (Fig. 9A). Similarly, NANOG knockdown inhibited tumor growth (Fig. 9B, C). NANOG knockdown reduced tumor volume and weight (Fig. 9E, F). The NANOG expression level in the knockdown NANOG groups (shNANOG-1 and shNANOG-2) was decreased, corresponding to an increase in TUNEL-stained positive tumor cells, while OCT4 expression was unchanged (Fig. 9G). All these results indicated that knockdown of OCT/NANOG significantly reduced tumor growth capacity.

A In vivo subcutaneous implantation of NCG mouse models induced by MV4-11 cells transfected with NANOG shRNA (shNANOG-1 and shNANOG-2) or control shRNA (shCtrl), tumor volume was measured daily after injection of tumor cells. B Representative image of tumor-bearing mice in different groups. C Representative images of xenograft tumors in each group. D NANOG expressions were detected by western blotting analysis. E, F Quantification of tumor size and weight in different groups. G NANOG and OCT4 expression in paraffin-embedded continuous sections were detected by immunohistochemistry, and apoptotic cells were counted by TUNEL (bar, 50 μm). The A was created with BioRender.com. The statistical result is the mean ± SEM. **p < 0.01, ****p < 0.0001.
Discussion
Despite advances in AML therapy with the development of FLT3 TKIs for the treatment of FLT3-ITD+ AML, TKIs have reported only limited effects as monotherapy in clinics [25]. Most FLT3-ITD+ AML patients relapsed quickly, and 5-year survival remains poor [23]. Therefore, it is critically important to figure out the process of TKI resistance and develop novel approaches to eradicate FLT3-ITD+ AML cells. Several preclinical investigations indicate that TKI resistance may be driven by cellular adaptation mechanisms [26, 27]. Our study found that the pluripotency TFs OCT4 and NANOG were upregulated upon FLT3 inhibition.
Therapeutic agents associated with elevated TFs in pluripotency mainly fall into two categories, conventional chemoradiotherapy and TKIs [28,29,30,31]. Chemoradiotherapy results in an enrichment of cancer stem cells and an increase in cancer stem cell markers [32]. Studies of chemotherapy resistance in relation to OCT4 expression are confined to a few medicines, such as cisplatin, paclitaxel, doxorubicin, and tamoxifen [33,34,35]. Cisplatin, etoposide, doxorubicin, and paclitaxel γ-ray irradiation increase the expression level of OCT4 in lung cancer [36]. TKIs are another class of drugs that are prone to resistance. IL-6-DNMT3b-mediated OCT4 expression confers sorafenib resistance in hepatocellular carcinoma (HCC) patients [37]. We observed that OCT4 and NANOG were highly expressed in FLT3-ITD+ AML cells after TKI therapy in a dose-dependent manner, with a higher dose of gilteritinib (a common FLT3 TKI) inducing higher levels of OCT4 and NANOG expression. It suggests that OCT4 and NANOG may serve as molecular predictors of FLT3 inhibitor responses.
The malfunctioning of OCT4 and NANOG is pivotal in the development of tumors. Exogenous OCT4 expression increases tumorigenicity, while OCT4 knockdown reduces tumor malignancy. High OCT4 expression was positively linked with glioma grade [38]. OCT4 accelerates self-renewal, aggressiveness, and chemoresistance in HCC stem cells [39, 40]. High levels of OCT4 are linked to poor outcomes in hormone receptor-positive breast cancer patients [35]. Downregulation of OCT4 lowers stemness characteristics in germ cell cancers [41]. A multitude of studies have shown abnormal expression of NANOG in human cancers [22]. Colorectal cancer patients with high NANOG expression were linked to Dukes’ grade [42]. Gastric cancer patients with high NANOG levels have a poorer OS [43]. The Kaplan-Meier analysis demonstrated that the high expression of OCT4 and NANOG predicted a poor prognosis in pancreatic cancer [44]. It is critical to understand the function of the pluripotency TFs OCT4 and NANOG in FLT3-ITD+ AML.
Studies on the molecular processes underlying the association between pluripotency TFs and AML are limited. The role of TFs such as OCT4 and NANOG in AML remains unknown. Our findings provide compelling evidence for the potential therapy of targeting OCT4 and NANOG in FLT3-ITD+ AML. We generated two FLT3-ITD+ AML cell lines with OCT4 or NANOG knockdown using stable lentivirus strains. By using cell viability, apoptosis, and cell cycle analysis, we found that knockdown of OCT4 or NANOG inhibited cell proliferation and promoted apoptosis. We then investigated the effect of downregulating OCT4 or NANOG on the sensitivity of FLT3-ITD+ AML cells to gilteritinib. A significant rise in the efficacy of TKI treatment was observed. This suggests that the presence of OCT4 or NANOG contributes to AML resistance to FLT3 inhibition. By targeting these genes, we can increase the sensitivity of FLT3-TKIs in FLT3-ITD+ AML. This synergistic effect suggests that the combination of stemness gene silencing and FLT3i works together in a complementary mechanism to target FLT3-ITD+ AML more effectively.
It is well known that abnormal expression of TFs contributes to tumorigenesis, and oncogenic TFs alter the core self-regulatory circuits of cells. Several key TF chaperones form an interconnected autoregulatory circuit that promotes the sustained activation of the oncogenic program. OCT4, the transcription factor of NANOG, was predicted by online databases, and a positive correlation was found by immunohistochemical analysis in our study. At the same time, after OCT4 knockdown, NANOG, and its downstream FLT3 pathway proteins were correspondingly reduced, further indicating that OCT4 has a positive regulatory effect on NANOG. Finally, a dual-luciferase assay confirmed that OCT4 regulated NANOG at the transcriptional level, thereby promoting the malignant biological behavior of FLT3-ITD+ AML.
In summary, the study investigated the therapeutic relevance of OCT4/NANOG in FLT3-ITD+ AML, focusing on the molecular mechanism by which they mediate AML progression and TKI sensitivity. The pluripotency TFs OCT4 and NANOG expression were shown to correlate with treatment and TKI resistance in FLT3-ITD+ AML. Overcoming this resistance will improve the prognosis of AML patients. In addition, it is helpful for the establishment of molecular biomarkers that predict the FLT3-ITD+ AML response to FLT3i before treatment. Our findings suggest that OCT4 and NANOG may be molecular biomarkers of the AML response to TKIs and potential targets to overcome resistance.
Conclusion
Our findings identify novel OCT4/NANOG targets that promote leukemia maintenance and mediate TKI resistance. Functionally, OCT4 or NANOG silencing in FLT3-ITD+ AML cells inhibited growth, promoted apoptosis, and significantly improved TKI treatment sensitivity. Given that mutations in tyrosine kinase are frequent genetic alterations in numerous tumors, our study may have implications for the development of therapeutic approaches. This also updates the role of pluripotency TFs in promoting the resistance of tumor cells to TKIs. Our study suggests that combining stemness gene intervention with TKIs may be a viable approach for treating FLT3-ITD+ AML and other tyrosine kinase-mutated leukemias.
Materials and methods
FLT3 inhibitor
Gilteritinib (ASP2215) (Selleck Chemicals, USA) was mixed into dimethyl sulfoxide (Macklin, China) and subsequently diluted using RPMI-1640 medium to working concentrations.
Cells and cell culture
Human FLT3-ITD+ AML cell lines (MV4-11 and MOLM-13) were bought from American Type Culture Collection (ATCC, USA). Cells underwent cultivation in RPMI-1640 (Gibco, USA), which included 10% FBS (Gibco, USA) and 1% penicillin/streptomycin (Solaribio, China). Cells underwent cultivation in humidified environment at 37 °C and 5% CO2.
Lentiviral transduction
ShRNAs were used for stable knockdown of OCT4 and NANOG expression. MV4-11 and MOLM-13 cells were transfected using lentiviral shOCT4 (TranSheepBio, China) targeting OCT4 and shNANOG (TranSheepBio, China) targeting NANOG. An empty lentiviral vector control shRNA (shCtrl) was serving as a control. The shRNA sequences are in Table 1. Prior to lentiviral transduction, a single-cell suspension was prepared post-centrifugation and resuspension with fresh medium containing 4 μg/ml polybrene (TranSheepBio, China), and seeded 4 × 105 cells in 24-well plates with a volume of 500 μL per well. Lentivirus was mixed and added to the cell culture medium, which underwent an overnight incubation. After 15 h, the fresh medium was substituted. Successful transduction was verified through the detection of green fluorescent protein expression in fluorescence microscopy. For identifying shRNA-positive cells, they were grown in a full medium containing 1.5 μg/mL puromycin (Sparkjade, China) for a minimum of 3 days. The expression of target genes was assessed by qRT-PCR and western blotting analysis.
Cell Counting Kit (CCK) − 8 proliferation analysis
MV4-11 and MOLM-13 cells were counted at 1 × 104 in 96-well plates in triplicate. Each experimental group had at least three replicated wells. Cell viability was monitored at the times of 24, 48, 72, 96, and 120 h. Post-treatment for specified durations, each well received 10 μL of CCK-8 solution (Beyotime Institute of Biotechnology, China), was incubated for an additional 3 h, and the absorbance at 450 nm was gauged using the BioTek microplate reader (USA). For every group, the optical density (OD) of each well was calculated by subtracting the OD value of the blank control from that of each well.
RNA extraction and qRT-PCR
Total RNA from MV4-11 and MOLM-13 cells was obtained using a TRIzol reagent (Sparkjade, China). RNA was reverse transcribed to complementary DNA (cDNA) using the HiScript™ III RT SuperMix (Vazyme, China). For qRT-PCR analysis, ChamQ SYBR qPCR Master Mix (Vazyme, China) was used on DA7600 Real-time Nucleic Acid Amplification Fluorescence Detection System (Bio-Rad Laboratories Inc, USA). Gene expression was normalized by GAPDH. The determination of mRNA levels was expressed as the mean, and qRT-PCR experiments were repeated with at least three independent assays. PCR primer sequences are in Table 2. The primer sequences were synthesized by General Biol (Anhui, China). The final analysis was relatively quantified by 2−ΔΔCT method.
Western blotting analysis
Total proteins were extracted from MV4-11 and MOLM-13 cells using RIPA buffer (Beyotime, China), which contained protease inhibitors (Beyotime, China). The protein concentration in the samples was measured using the BCA kit (Epizyme, China). Proteins were isolated through 10% SDS-PAGE and electro-transferred onto PVDF membranes (Merck Millipore, USA). Subsequently, PVDF membranes underwent blocking using 5% skim milk for 2 h, followed by an overnight incubation with primary antibodies at 4 °C. PVDF membranes underwent a TBST wash followed by a 2-h incubation with horseradish peroxidase-tagged secondary antibodies. The detection of the protein bands was achieved through a highly sensitive ECL chemiluminescence substrate (Biosharp, China). Using GAPDH and β-actin as a control. All primary antibodies were bought from Proteintech (Wuhan, China), and secondary antibodies were from Epizyme (Shanghai, China). All western blotting analyses were performed at least three times.
Cell apoptosis analysis
The cells were collected through centrifugation, cleansed using chilled PBS (Phosphate Buffered Saline, Servicebio, China) at 4 °C, and then reconstituted in 500 μL of binding buffer to modify the concentration to 1 × 107/mL. The cells underwent incubation with 5 μL of Annexin V-FITC and 5 μL of propidium iodide (PI) (Biolegend, USA) for a quarter-hour at ambient temperature in darkness. Subsequently, leukemia cells underwent apoptosis analysis at an excitation wavelength (Ex) of 488 nm and an emission wavelength (Em) of 585 ± 21 nm, utilizing CytoFLEX (Beckman, USA). The FlowJo 7.6 software was utilized to assess the proportion of apoptosis cells.
Cell cycle analysis
The cells underwent a cleansing process using PBS and were then reconstituted in a staining solution with 50 μg/mL PI (KeyGEN, China) and PBS, inclusive of 100 μg/mL RNase A. These suspensions were then left to incubate at 4 °C overnight. Data on the distribution of cell cycles were acquired through the use of CytoFLEX (Beckman, USA). The cell cycle distribution was examined using FlowJo 7.6 software.
Luciferase reporter assay
The binding site of the transcription factor OCT4 within 2000 bp before the NANOG transcription start sites was predicted from the online tool. PcDNA3.1 was used as the vector to construct OCT4-pcDNA3.1, and then pGL3-NANOG-WT and pGL3-NANOG-MUT with mutated binding sites were constructed using pGL3-basic as the vector (GenePharma, Shanghai, China). The vector pRL-TK renilla luciferase was set as the control plasmid. MV4-11 cells were co-transfected with different plasmids and lipofectamine TM3000 transfection reagent (Invitrogen, Carlsbad, CA, USA) according to experimental grouping. Firefly and renilla luciferase activities were measured after 48 h using the dual luciferase reporter assay kit (Promega, Madison, WI, USA).
In vivo functional measurements
The 5-week-old female NCG mice (NOD/ShiltJPGT-PrkDCEM26CD52il2rGEM26CD22/Gpt) were purchased from Chengdu Pharmacokang Biotechnology Co., Ltd., China, and raised under specific pathogen-free conditions. The experimental mice were randomly divided into different groups (n = 5) for subcutaneous injection of MV4-11 control, MV4-11-shOCT4, and MV4-11-shNANOG cells (5 × 106 cells per mouse). The transplanted mice were observed and palpated daily. After 5 weeks, the animals were euthanized by cervical dislocation. The tumor was separated, and the weight and volume of the tumor were recorded. Tumor tissues were fixed in 4% neutral buffered formalin and used for cryosection preparation.
Statistical analysis
Every experiment was conducted a minimum of three times. The GraphPad Prism software (Version 9.0, San Diego, CA, USA) was utilized for visualizing data and conducting statistical analyses. The variances among the experimental groups were examined through either a paired or unpaired student t-test, along with a two-way variance analysis incorporating multiple tests. A p value below 0.05 was deemed to hold statistical significance.
Data availability
Data supporting the findings from this study are available from the corresponding author on request.
References
Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373:1136–52.
Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377:454–64.
Perl AE, Altman JK, Cortes J, Smith C, Litzow M, Baer MR, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1-2 study. Lancet Oncol. 2017;18:1061–75.
Cortes J, Perl AE, Döhner H, Kantarjian H, Martinelli G, Kovacsovics T, et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: an open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol. 2018;19:889–903.
DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378:2386–98.
Pollyea DA, Tallman MS, de Botton S, Kantarjian HM, Collins R, Stein AS, et al. Enasidenib, an inhibitor of mutant IDH2 proteins, induces durable remissions in older patients with newly diagnosed acute myeloid leukemia. Leukemia. 2019;33:2575–84.
Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130:722–31.
Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016;6:1106–17.
Bazinet A, Kantarjian HM. Moving toward individualized target-based therapies in acute myeloid leukemia. Ann Oncol 2023;34:141–51.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–21.
Daver N, Kantarjian H. FLT3 inhibition in acute myeloid leukaemia. Lancet Oncol. 2017;18:988–9.
Kant R, Manne RK, Anas M, Penugurti V, Chen T, Pan BS, et al. Deregulated transcription factors in cancer cell metabolisms and reprogramming. Semin Cancer Biol. 2022;86:1158–74.
Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. The human transcription factors. Cell. 2018;172:650–65.
Sissung TM, Figg WD. Stem cell clinics: risk of proliferation. Lancet Oncol. 2020;21:205–6.
Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–7.
Wang Y-J, Herlyn M. The emerging roles of Oct4 in tumor-initiating cells. Am J Physiol Cell Physiol. 2015;309:C709–C718.
Iv Santaliz-Ruiz LE, Xie X, Old M, Teknos TN, Pan Q. Emerging role of nanog in tumorigenesis and cancer stem cells. Int J cancer. 2014;135:2741–8.
You L, Guo X, Huang Y. Correlation of cancer stem-cell markers OCT4, SOX2, and NANOG with clinicopathological features and prognosis in operative patients with rectal cancer. Yonsei Med J. 2018;59:35–42.
Jeter CR, Badeaux M, Choy G, Chandra D, Patrawala L, Liu C, et al. Functional evidence that the self-renewal gene NANOG regulates human tumor development. Stem Cells. 2009;27:993–1005.
Jeter CR, Yang T, Wang J, Chao H-P, Tang DG. Concise review: NANOG in cancer stem cells and tumor development: an update and outstanding questions. Stem Cells. 2015;33:2381–90.
Xiang Y, Zhou X. Octamer-binding transcription factor 4 correlates with complex karyotype, FLT3-ITD mutation and poorer risk stratification, and predicts unfavourable prognosis in patients with acute myeloid leukaemia. Hematology. 2018;23:721–8.
Nagata T, Shimada Y, Sekine S, Hori R, Matsui K, Okumura T, et al. Prognostic significance of NANOG and KLF4 for breast cancer. Breast Cancer. 2014;21:96–101.
Port M, Böttcher M, Thol F, Ganser A, Schlenk R, Wasem J, et al. Prognostic significance of FLT3 internal tandem duplication, nucleophosmin 1, and CEBPA gene mutations for acute myeloid leukemia patients with normal karyotype and younger than 60 years: a systematic review and meta-analysis. Ann Hematol. 2014;93:1279–86.
Yu CC, Chen YW, Chiou GY, Tsai LL, Huang PI, Chang CY, et al. MicroRNA let-7a represses chemoresistance and tumourigenicity in head and neck cancer via stem-like properties ablation. Oral Oncol. 2011;47:202–10.
Kindler T, Lipka DB, Fischer T. FLT3 as a therapeutic target in AML: still challenging after all these years. Blood. 2010;116:5089–102.
Park IK, Mundy-Bosse B, Whitman SP, Zhang X, Warner SL, Bearss DJ, et al. Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia. 2015;29:2382–9.
Piloto O, Wright M, Brown P, Kim KT, Levis M, Small D. Prolonged exposure to FLT3 inhibitors leads to resistance via activation of parallel signaling pathways. Blood. 2007;109:1643–52.
Yeh CT, Chen TT, Satriyo PB, Wang CH, Wu A, Chao TY, et al. Bruton’s tyrosine kinase (BTK) mediates resistance to EGFR inhibition in non-small-cell lung carcinoma. Oncogenesis. 2021;10:56.
Hu F, Li C, Zheng X, Zhang H, Shen Y, Zhou L, et al. Lung adenocarcinoma resistance to therapy with EGFR‑tyrosine kinase inhibitors is related to increased expression of cancer stem cell markers SOX2, OCT4 and NANOG. Oncol Rep. 2020;43:727–35.
Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–84.
Malik B, Nie D. Cancer stem cells and resistance to chemo and radiotherapy. Front Biosci. 2012;4:2142–9.
Lee S, Wottrich S, Bonavida B. Crosstalks between Raf-kinase inhibitor protein and cancer stem cell transcription factors (Oct4, KLF4, Sox2, Nanog). Tumour Biol. 2017;39:1010428317692253.
Kaufhold S, Garbán H, Bonavida B. Yin Yang 1 is associated with cancer stem cell transcription factors (SOX2, OCT4, BMI1) and clinical implication. J Exp Clin Cancer Res. 2016;35:84.
Blum W, Pecze L, Felley-Bosco E, Wu L, de Perrot M, Schwaller B. Stem cell factor-based identification and functional properties of in vitro-selected subpopulations of malignant mesothelioma cells. Stem Cell Rep. 2017;8:1005–17.
Gwak JM, Kim M, Kim HJ, Jang MH, Park SY. Expression of embryonal stem cell transcription factors in breast cancer: Oct4 as an indicator for poor clinical outcome and tamoxifen resistance. Oncotarget. 2017;8:36305–18.
Chen YC, Hsu HS, Chen YW, Tsai TH, How CK, Wang CY, et al. Oct-4 expression maintained cancer stem-like properties in lung cancer-derived CD133-positive cells. PloS ONE. 2008;3:e2637.
Lai SC, Su YT, Chi CC, Kuo YC, Lee KF, Wu YC, et al. DNMT3b/OCT4 expression confers sorafenib resistance and poor prognosis of hepatocellular carcinoma through IL-6/STAT3 regulation. J Exp Clin Cancer Res. 2019;38:474.
Du Z, Jia D, Liu S, Wang F, Li G, Zhang Y, et al. Oct4 is expressed in human gliomas and promotes colony formation in glioma cells. Glia. 2009;57:724–33.
Zhang MX, Gan W, Jing CY, Zheng SS, Yi Y, Zhang J, et al. High expression of Oct4 and Nanog predict poor prognosis in intrahepatic cholangiocarcinoma patients after curative resection. J Cancer. 2019;10:1313–24.
Wang XQ, Ongkeko WM, Chen L, Yang ZF, Lu P, Chen KK, et al. Octamer 4 (Oct4) mediates chemotherapeutic drug resistance in liver cancer cells through a potential Oct4-AKT-ATP-binding cassette G2 pathway. Hepatology. 2010;52:528–39.
Voutsadakis IA. The chemosensitivity of testicular germ cell tumors. Cell Oncol. 2014;37:79–94.
Meng HM, Zheng P, Wang XY, Liu C, Sui HM, Wu SJ, et al. Over-expression of Nanog predicts tumor progression and poor prognosis in colorectal cancer. Cancer Biol Ther. 2010;9:295–302.
Lin T, Ding Y-Q, Li J-M. Overexpression of Nanog protein is associated with poor prognosis in gastric adenocarcinoma. Med Oncol. 2012;29:878–85.
Lu Y, Zhu H, Shan H, Lu J, Chang X, Li X, et al. Knockdown of Oct4 and Nanog expression inhibits the stemness of pancreatic cancer cells. Cancer Lett. 2013;340:113–23.
Acknowledgements
The graphical abstract, Fig. 8A, and Fig. 9A were created with BioRender.com.
Author information
Authors and Affiliations
Contributions
Qi Zhou: Conceptualization, Methodology, Investigation, Writing—original draft, Visualization. Zijian Li: Conceptualization, Writing—review & editing. Pingping Zhao: Conceptualization, Visualization. Yongyu Guan: Formal analysis. Huiyuan Chu: Project administration. Yaming Xi: Project administration, Validation. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
Our study was approved by the Medical Ethics Committee of the First Hospital of Lanzhou University (Approval No.: LDYYLL-2024-422), and animal care and experimental protocols were performed in accordance with the guidelines established by the Medical Laboratory Animal Ethics Committee of Lanzhou University.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, Q., Li, Z., Zhao, P. et al. FLT3 inhibition upregulates OCT4/NANOG to promote maintenance and TKI resistance of FLT3-ITD+ acute myeloid leukemia. Oncogenesis 14, 7 (2025). https://doi.org/10.1038/s41389-025-00553-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41389-025-00553-6
