
Abstract
Saturated three-dimensional carbocycles have gained increasing prominence in synthetic and medicinal chemistry. In particular, bicyclo[2.1.1]hexanes (BCHs) have been identified as the molecular replacement for benzenes. Here, we present facile access to a variety of BCHs via a stepwise two-electron formal (3 + 2) cycloaddition between silyl enol ethers and bicyclo[1.1.0]butanes (BCBs) under Lewis acid catalysis. The reaction features wide functional group tolerance for silyl enol ethers, allowing the efficient construction of two vicinal quaternary carbon centers and a silyl-protected tertiary alcohol unit in a streamlined fashion. Interestingly, the reaction with conjugated silyl dienol ethers can provide access to bicyclo[4.1.1]octanes (BCOs) equipped with silyl enol ethers that facilitate further transformation. The utilities of this methodology are demonstrated by the late-stage modification of natural products, transformations of tertiary alcohol units on bicyclo[2.1.1]hexane frameworks, and derivatization of silyl enol ethers on bicyclo[4.1.1]octanes, delivering functionalized bicycles that are traditionally inaccessible.
Similar content being viewed by others
Introduction
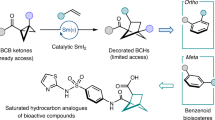
The strategic replacement of benzene with conformationally rigid and stable C(sp3)-enriched polycyclic scaffolds in small molecules represents an emerging trend in medicinal chemistry. Attributed to their constrained geometries and precisely oriented pendant substituents, these saturated polycycles effectively emulate the topological characteristics of substituted benzenes, which allows for the preservation of desired interactions with biomacromolecules while enhancing the pharmacokinetics, solubility, and metabolic stability of drug candidates1,2,3,4,5. Recent studies have identified 1,2-disubstituted bicyclo[2.1.1]hexanes as potential bioisosteres for ortho-disubstituted benzenes with retained biological activity validated by in vitro experiments (Fig. 1a)6,7. Hence, there is an increasing demand for development of efficient strategies for streamlined access to these bicycles6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32. One of most common methods to construct BCH skeleton is by an intramolecular [2 + 2] cycloaddition of 1,5-diene under the irradiation of light6,7,8,9,10,11,12,13,14. Alternatively, an intermolecular cycloaddition between bicyclo[1.1.0]butanes (BCBs) and alkenes is highly desirable since it allows the efficient construction of bicyclic ring through the fusion of two readily available starting materials. Pioneering studies were disclosed by Blanchard16 in 1966 and De Meijere17 in 1986. Subsequently, Wipf group reported an intramolecular variant of this cycloaddition under thermal condition in 200618.

a BCHs behave as bioisosteres of ortho- and meta-substituted benzenes. b State of the art in the cycloaddition of BCBs and alkenes to access BCHs. c Cycloaddition of BCBs and silyl enol ethers to access BCHs.
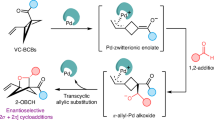
More recently, by taking advantage of the ready availability and inherent ring strain of BCBs20,33, the exploration of new strategies to the cycloaddition between BCBs and alkenes in the generation of various BCHs has attracted intensive attentions19,20,21,22,23,24,25,26,27,28,29,30,31,32,34,35,36,37,38,39. According to the reported reaction processes, most methods could be categorized into two modes: 1) radical pathway; and 2) two-electron pathway (Fig. 1b). By utilizing the photoinduced energy transfer strategy, Glorius19 and Brown20 groups respectively described elegant cycloaddition of BCBs and alkenes toward bicyclo[2.1.1]hexanes. The reaction was initiated by the excitation of either alkene or BCB to generate a diradical intermediate. Subsequently, Bach and coworkers achieved an enantioselective cycloaddition of 2(1H)‑quinolones and BCBs with a chiral mediator22. Recently, Jiang and coworkers developed a highly enantioselective cycloaddition of vinylazaarenes and BCBs under photosensitized chiral phosphoric acid catalysis23. Li24 and Wang26 groups developed a boryl-pyridine catalytic system to activate BCB as a cyclobutyl radical intermediate. Meanwhile, Procter group applied SmI2 as a single electron reductant to achieve the insertion of electron-deficient alkene into BCB25. Very recently, Zheng group described a Ti-catalyzed formal cycloaddition of BCB and 2-azadienes to synthesize aminobicyclo[2.1.1]hexanes27. Lately, Glorius accomplished the coupling of phenol and BCB by leveraging a photoredox process, which has been applied in the formal cycloaddition of non-activated alkenes (Fig. 1b, 1)28,29. All the reactions above entailed the generation of radical species, which limited the substrate scope.
Bicyclo[1.1.0]butanes could be activated as an enolate nucleophile upon central σ-bond cleavage mediated by Lewis acid to attack electrophilic reagents such as aryl aldimine by Leitch group30, aldehyde by Glorius group32, or ketene by Studer group31, followed by the intramolecular cyclization to complete the formal cycloadditions. We demonstrated that Lewis acid could activate BCBs as electrophiles to react with indoles as the nucleophiles to construct complex indoline polycycles (Figs. 1b, 2)34. Notably, if a wide variety of nucleophiles could be utilized, this approach could be developed into a versatile strategy that complements existing methods involving radical or ionic intermediates to access BCHs.
More recently, Feng reported the use of silver triflate to promote reactions of BCBs and indoles but with opposite regioselectivity35,36. Very recently, Leitch group described a formal cycloaddition of BCBs via an enolate intermediate Int 4 by treatment of glycine imine or arylacetate derivatives with stoichiometric amount of LHMDS37. Despite these successful examples, there is a huge and unexplored chemical space for these bicycles, and a more general strategy to expediently construct such moieties with simple alkenes is highly demanded.
Here, we envisaged that the silyl enol ether, as the nucleophile, would be a suitable candidate in this scenario due to its importance in different cycloaddition reactions40,41,42,43,44,45,46,47,48. It is noteworthy that silyl enol ethers could be easily prepared by a one-step silylation of simple ketone motifs, which are presented in numerous compounds and widely used in synthetic chemistry. Although silyl enol ethers exhibit robust nucleophilicity in various synthetic contexts, such as Mukaiyama-aldol reaction and Michael addition, their nucleophilicity towards the BCB remains unexplored37,39. Furthermore, the subsequent intramolecular aldol-type cyclization presents a challenge due to the formation of sterically hindered vicinal quaternary carbon centers. Despite these concerns, we aim to explore the formal (3 + 2) cycloaddition between silyl enol ether and BCB to realize one-step access to a variety of BCH frameworks (Fig. 1c).
Results and discussion
Reaction optimization
With these considerations in mind, we began our investigations with screening studies of Lewis acids on their abilities to promote the reaction of triisopropylsilyl phenyl enol ether (1a) from acetophenone and naphthyl BCB (2a). In the presence of late transition metal-derived triflate salts, only trace amount of desired cycloadduct 3a was observed by NMR analysis of the crude reaction mixture (Table 1, entries 1–4). The major product was identified to be cyclobutyl silyl enol ether 4, presumably derived from nucleophilic addition followed by silyl migration.
Additionally, B(C6F5)3 would only give byproduct 4 (Table 1, entry 6). On the other hand, we found that with lanthanide triflates such as Eu(OTf)3, Gd(OTf)3, Tm(OTf)3, Lu(OTf)3 and Yb(OTf)3 the reaction afforded the desired product 3a in high yield (Table 1, entries 7–11). Among them, Yb(OTf)3 was the optimal catalyst, allowing the reaction to proceed in 97% yield (Table 1, entry 11). Interestingly, switching the counterion from triflate to chloride or acetate is detrimental to this transformation, illustrating the acidity of Lewis acid is the key to success. (Table 1, entries 12–13). Further optimization studies identified toluene as another suitable solvent (Table 1, entries 14–16). Pleasantly, the catalyst loading could be decreased to 5 mol% without negative impact (Table 1, entries 11 and 14). Control experiment illustrated that no reaction occurred in the absence of Lewis acid (Table 1, entry 17).
Substrate scope
With the optimal conditions in hand, the generality of substrates in this cycloaddition was investigated as depicted in Fig. 2. The reactions of BCB 2a and various silyl enol ethers 1a–d derived from acetophenones were examined, which proceeded to completion, affording the desired products 3a–d in 45–93% yields. Within these substrates, those bearing more stable silyl groups resulted in higher yields. The reactions tolerated a wide variety of aromatics with substituents on various positions and with different electronic properties (3e–p). 1-Naphthyl and 2-naphthyl substituted silyl enol ethers worked well to form the corresponding products (3q and 3r). The structure of cycloadduct 3r was unambiguously confirmed by single crystal X-ray diffraction analysis.

aReaction conditions: unless indicated otherwise, the reaction of silyl enol ether 1 (0.2 mmol), and BCB 2 (0.26 mmol) was carried out in DCM (2 mL) or toluene (2 mL) in the presence of Yb(OTf)3 (0.01 mmol) at room temperature for 2–4 h. The yield was of isolated and purified products. b6 h. c12 h. β-Me, methyl group syn to ethyl group; α-Me, methyl group anti to ethyl group. Naph 2-naphthyl, TIPS triisopropylsilyl, TES triethylsilyl, TBS tert-butyldimethylsilyl, TBDPS tert-butyldiphenylsilyl, o ortho, m meta, p para.
Heterocycles commonly used in medicinal chemistry such as furan and thiophene were compatible, giving their corresponding bicycles (3s and 3t) in good yields. Additionally, silyl enol ethers bearing alkenyl and alkynyl groups were readily converted into the corresponding cycloadducts (3u and 3v) in high yields. Importantly, the reactions with aliphatic substituted silyl enol ethers with pendant functional groups such as chloride (3y), silyl ether (3z), olefin (3aa), electron-rich arene (3ab), and cyclopropane (3ac) performed well to furnish the corresponding cycloadducts in 47–90% yields.
Next, a series of cyclic silyl enol ethers derived from 5-, 6-, 7-membered cyclic ketones including those fused with aromatic and heteroaromatic rings were assessed, producing the corresponding assortment of bridged polycycles (3ad–an) in synthetically useful yields. Interestingly, these reactions could convert cyclic silyl dienol ethers to tricyclic alkenes 3al and 3am, which are suitable for further elaborations to synthesize more complex molecular frameworks.
Notably, acyclic tri- and tetra-substituted silyl enol ethers are viable substrates, allowing the rapid construction of highly substituted and compact bicycles 3ao–aq. It is worth noting that the reaction with different trisubstituted silyl enol ethers resulted in distinct diastereoselectivities. In particular, 3ao were obtained in high diastereoselectivity (13:1 dr) from the corresponding silyl enol ether 1ao (Z/E = 10:1) derived from 1-phenyl-1-butanone, while 3ap was formed as a mixture of diastereomers (1.3:1 dr) with silyl enol ether 1ap (Z/E = 7.4:1) derived from 3-pentanone (Supplementary Figs. 4–8).
Finally, we turned our attention to the evaluation of BCB scope. BCBs with aromatic, heteroaromatic and aliphatic substituents such as benzene (3ba–bb), 1,2-methylenedioxybenzene (3bc), furan (3bd), thiophene (3be), and n-butyl (3bf–bg) are well tolerated in current conditions. To our delight, the reaction with 3-substituted BCB 2g and 2h also proceeded smoothly to give the cycloadducts bearing three quaternary carbon stereocenters (3bh and 3bi). The ability to tolerate variations of both silyl enol ethers and BCBs indicated the potential of this method to generate a wide range of BCHs that are not easily accessible by previously reported methods.
Interestingly, structurally intriguing bicyclo[4.1.1]octane (BCO) architecture 5a was formed in the reaction of cyclohexenyl silyl dienol ether and BCB 2a under standard conditions (Fig. 3)49. Grygorenko et al. analyzed the structural characteristics of BCOs and suggested that they could serve as novel isosteres for multi-substituted benzenes50. However, regioselectivity issues between formal (4 + 3) and (3 + 2) cycloaddition might occur in this reaction. Further optimization studies identified Sc(OTf)3 as an alternative efficient Lewis acid that promotes formal (4 + 3) cycloaddition. With both Sc(OTf)3 and Yb(OTf)3 catalysts, we examined the generality of formal (4 + 3) cycloaddition with both reaction components. The reaction conditions were compatible with silyl dienol ether containing 5 and 7-membered rings, affording tricycles 5b and 5c bearing bicyclo[4.1.1]octane units. Silyl dienol ethers derived from aliphatic and aromatic vinyl ketones gave good yields (5d–g). Notably, silyl trienol ether also underwent the formal (4 + 3) cycloaddition selectively in synthetically useful yield (5i). Different substituents on BCBs such as benzene (5j), 1,2-methylenedioxybenzene (5k), thiophene (5 l), and n-butyl (5 m) groups are well tolerated, delivering desired (4 + 3) cycloadducts in medium to good yields. The divergent synthesis of both bicyclo[2.1.1]hexanes and bicyclo[4.1.1]octanes demonstrated the utility of silyl enol ethers and their derivatives in the construction of different bicycles.

aReaction conditions: unless indicated otherwise, the reaction of silyl dienol ether 1 (0.2 mmol), and BCB 2 (0.26 mmol) was carried out in DCM (2 mL) or toluene (2 mL) in the presence of Yb(OTf)3 (0.01 mmol) at room temperature for 3 h. The yield was of isolated and purified products. bThe reaction was run in the presence of Sc(OTf)3 (0.02 mmol) at room temperature for 30 min. rr: regioselectivity ratio of (4 + 3) to (3 + 2) cycloadduct. Naph 2-naphthyl, TIPS triisopropylsilyl.
Synthetic applications
To illustrate the utilities of this method, we conducted this reaction on a preparative scale with silyl enol ether 1w and BCB 2a, which afforded the cycloadduct 3w in 91% yield (770 mg) (Fig. 4a). Both the ketone and tertiary alcohol moieties in 3w provided handles for further synthetic elaborations to procure a wide array of new bicyclo[2.1.1]hexanes (Fig. 4b). Specifically, subjecting bicycle 3w to the Wittig reaction condition led to olefin 6. After deprotection of the silyl group, the free alcohol 7 was proved versatile for various transformations, including the dehydration to olefin 8 by treatment with Burgess reagent and fluorination to 9 in the presence of diethylaminosulfur trifluoride (DAST) reagent.

a Scale-up synthesis of 3w. b Derivatization of 3w and 5a. c Late-stage modification of natural products. Naph 2-naphthyl, TIPS triisopropylsilyl, TBAF tetrabutylammonium fluoride, DAST diethylaminosulfur trifluoride, Pyr pyridine, NMO 4-methyl morpholine N-oxide, HMDS bis(trimethylsilyl)amide.
Furthermore, isonitrile 10 would be readily obtained using Shenvi’s methodology51. All of derivatizations above further extended the attainability of new bicyclo[2.1.1]hexanes using this developed manifold. Additionally, the resulting silyl enol ether unit from the (4 + 3) cycloadducts could be further transformed to ketone 11 via desilylation and α-hydroxyketone 12 via dihydroxylation. Importantly, owing to its generality with respect to silyl enol ethers, this method could be applied to late-stage modifications of natural products, as bridged polycycles 13 and 14 were formed in synthetically useful yields from commercially available dihydro-β-ionone and cholestenone through ketone silylation and formal cycloaddition sequence (Fig. 4c).
To gain more insights into the reaction mechanism, we conducted 13C kinetic isotope effect experiments by employing Singleton’s method52,53,54 with natural abundance of 13C. We found only carbon c showed a pronounced kinetic isotope effect, which suggested the reaction possibly underwent a stepwise process instead of a concerted pathway (Fig. 5a). Therefore, a possible mechanism was proposed and illustrated in Fig. 5b. The reaction started from the nucleophilic addition of silyl enol ether with Lewis acid-activated BCB. The formed zwitterionic intermediate Int 5 then underwent an intramolecular aldol reaction to give the cycloadduct 3. Otherwise, silyl migration might occur after the in situ generation of Int 5, which leads to the cyclobutyl silyl enol ether 4. Additionally, when BCB reacts with silyl dienol ether to generate Int 5’, the reaction could proceed through 1,4-addition, delivering bicyclo[4.1.1]octane 5. The regioselectivity of formal (3 + 2) and (4 + 3) cycloadditions with silyl dienol ethers appears to be substrate-dependent. As demonstrated by the formation of (3 + 2) and (4 + 3) cycloadducts (3u, 3al, 3am, 5a-5h), the steric and electronic properties of the substituents on silyl dienol ethers have a significant influence on which pathway is favored. Considering the proposed reaction process, the origin of distinction in diastereoselectivity possibly arose from the steric bulk of R-substituent geminal to silyloxyl group on silyl enol ether 1 in the aldol-type cyclization step, which was elucidated by the proposed Felkin-Anh type model (Supplementary Fig. 9).

a 13C kinetic isotope effect experiment. b Catalytic cycle.
In summary, we developed a Lewis acid-catalyzed formal (3 + 2) cycloaddition of silyl enol ethers and BCBs55. This reaction exhibits high efficiency along with mild condition, operative simplicity, and broad substrate scope with respect to silyl enol ethers, which are readily available from ketone precursors. Notably, ketone is one of the most widely presented functional groups in organic molecules including natural products. Moreover, this formal (3 + 2) cycloaddition can be further extended to a (4 + 3) variant with silyl dienol ethers. This discovery enables the divergent syntheses of different structurally intriguing polycyclic frameworks and highlights the great potential of silyl enol ethers in the synthesis of polycycles. Importantly, this method should provide a broadly useful entry into bicyclo[2.1.1]hexanes from easily accessible starting materials covering a wide range of structural complexity. Further studies for the development of new reactions involving other widely available starting materials and BCBs are ongoing in our laboratory.
Methods
General procedure for the formal cycloaddition of silyl (di)enol ethers and bicyclo[1.1.0]butanes
A 1-dram screw-cap vial with a stir bar was charged with silyl (di)enol ether 1 (0.2 mmol), BCB 2 (0.26 mmol) and Yb(OTf)3 (0.01 mmol). Then the vial was sealed with a septum, evacuated, and backfilled with N2 (×3). Under N2 atmosphere, dry DCM or toluene (2 mL) was added rapidly. The septum was quickly replaced with a screw cap. The reaction was stirred for 2‒4 h until silyl enol ether was completely consumed. The reaction mixture was filtered through a pad of silica-gel and washed with DCM. The filtrate was concentrated on rotary evaporator and the residue was directly purified by silica-gel flash column chromatography to afford the desired product 3 or 5.
General procedure for the formal cycloaddition of silyl dienol ethers and bicyclo[1.1.0]butanes
A 1-dram screw-cap vial with a stir bar was charged with silyl dienol ether 1 (0.2 mmol), BCB 2 (0.26 mmol). Then the vial was sealed with a septum, evacuated, and backfilled with N2 (×3). Under N2 atmosphere, dry DCM or toluene (2 mL) was added rapidly. When fully dissolved, Sc(OTf)3 (0.02 mmol) was added in one portion, and the septum was quickly replaced with a screw cap. The reaction was stirred for 30 min until silyl dienol ether was completely consumed. After quenching with triethylamine (0.2 mL), the reaction mixture was filtered through a pad of deactivated silica-gel and washed with DCM. The filtrate was concentrated on rotary evaporator and the residue was directly purified by silica-gel flash column chromatography to afford the product 5.
Data availability
The details of experimental procedures and the data about the findings of this study are available within the article and its supplementary information. Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2329559 (3r) and 2358618 (5a). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. All data are available from the corresponding author upon request.
References
Lovering, F., Bikker, J. & Humblet, C. Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 52, 6752–6756 (2009).
Lovering, F. Escape from Flatland 2: complexity and promiscuity. MedChemComm 4, 515–519 (2013).
Bauer, M. R. et al. Put a ring on it: application of small aliphatic rings in medicinal chemistry. RSC Med. Chem. 12, 448–471 (2021).
Mykhailiuk, P. K. Saturated bioisosteres of benzene: where to go next? Org. Biomol. Chem. 17, 2839–2849 (2019).
Subbaiah, M. A. M. & Meanwell, N. A. Bioisosteres of the phenyl ring: recent strategic applications in lead optimization and drug design. J. Med. Chem. 64, 14046–14128 (2021).
Denisenko, A., Garbuz, P., Shishkina, S. V., Voloshchuk, N. M. & Mykhailiuk, P. K. Saturated bioisosteres of ortho-substituted benzenes. Angew. Chem. Int. Ed. 59, 20515–20521 (2020).
Denisenko, A., Garbuz, P., Makovetska, Y., Shablykin, O. & Mykhailiuk, P. K. 1,2-Disubstituted bicyclo[2.1.1]hexanes as saturated bioisosteres of ortho-substituted benzene. Chem. Sci. 14, 14092–14099 (2023).
Kleinnijenhuis, R. A. et al. Formal synthesis of solanoeclepin A: enantioselective allene diboration and intramolecular [2 + 2] photocycloaddition for the construction of the tricyclic core. Chem. Eur. J. 22, 1266–1269 (2016).
Takao, K.-i et al. Total syntheses of (+)-aquatolide and related humulanolides. Angew. Chem., Int. Ed. 58, 9851–9855 (2019).
Rigotti, T. & Bach, T. Bicyclo[2.1.1]hexanes by visible light-driven intramolecular crossed [2 + 2] photocycloadditions. Org. Lett. 24, 8821–8825 (2022).
Herter, L., Koutsopetras, I., Turelli, L., Fessard, T. & Salomé, C. Preparation of new bicyclo[2.1.1]hexane compact modules: an opening towards novel sp3-rich chemical space. Org. Biomol. Chem. 20, 9108–9111 (2022).
Paul, S., Adelfinsky, D., Salome, C., Fessard, T. & Brown, M. K. 2,5-Disubstituted bicyclo[2.1.1]hexanes as rigidified cyclopentane variants. Chem. Sci. 14, 8070–8075 (2023).
Reinhold, M., Steinebach, J., Golz, C. & Walker, J. C. L. Synthesis of polysubstituted bicyclo[2.1.1]hexanes enabling access to new chemical space. Chem. Sci. 14, 9885–9891 (2023).
Posz, J. M. et al. Synthesis of borylated carbocycles by [2 + 2]-cycloadditions and photo-ene reactions. J. Am. Chem. Soc. 146, 10142–10149 (2024).
Yang, Y. et al. An intramolecular coupling approach to alkyl bioisosteres for the synthesis of multisubstituted bicycloalkyl boronates. Nat. Chem. 13, 950–955 (2021).
Cairncross, A. & Blanchard, E. P. Jr. Bicyclo[1.1.0]butane chemistry. II. cycloaddition reactions of 3-methylbicyclo[1.1.0]butanecarbonitriles. The formation of bicyclo[2.1.1]hexanes. J. Am. Chem. Soc. 88, 496–504 (1966).
De Meijere, A. et al. Cycloadditions of methylenecyclopropanes and strained bicyclo[n.1.0]alkanes to radicophilic olefins. Tetrahedron 42, 1291–1297 (1986).
Wipf, P. & Walczak, M. A. A. Pericyclic cascade reactions of (bicyclo[1.1.0]-butylmethyl)amines. Angew. Chem., Int. Ed. 45, 4172–4175 (2006).
Kleinmans, R. et al. Intermolecular [2π+2σ]-photocycloaddition enabled by triplet energy transfer. Nature. 605, 477–482 (2022).
Guo, R. et al. Strain-release [2π+2σ] cycloadditions for the synthesis of bicyclo[2.1.1]hexanes initiated by energy transfer. J. Am. Chem. Soc. 144, 7988–7994 (2022).
Kleinmans, R. et al. ortho-Selective dearomative [2π + 2σ] photocycloadditions of bicyclic aza-arenes. J. Am. Chem. Soc. 145, 12324–12332 (2023).
de Robichon, M. et al. Enantioselective, intermolecular [π2+σ2] photocycloaddition reactions of 2(1H)-quinolones and bicyclo[1.1.0]butanes. J. Am. Chem. Soc. 145, 24466–24470 (2023).
Fu, Q. et al. Enantioselective [2π + 2σ] cycloadditions of bicyclo[1.1.0]butanes with vinylazaarenes through asymmetric photoredox catalysis. J. Am. Chem. Soc. 146, 8372–8380 (2024).
Xu, M. et al. Diboron(4)‐catalyzed remote [3+2] cycloaddition of cyclopropanes via dearomative/rearomative radical transmission through pyridine. Angew. Chem., Int. Ed. 61, e202214507 (2022).
Agasti, S. et al. Catalytic alkene insertion approach to bicyclo[2.1.1]hexane bioisosteres. Nat. Chem 15, 535–541 (2023).
Liu, Y. et al. Pyridine-boryl radical-catalyzed [2π + 2σ] cycloaddition of bicyclo[1.1.0]butanes with alkenes. ACS Catal. 13, 5096–5103 (2023).
Ren, H. et al. Ti-catalyzed formal [2π + 2σ] cycloadditions of bicyclo[1.1.0]butanes with 2-azadienes to access aminobicyclo[2.1.1]hexanes. Org. Lett. 26, 1745–1750 (2024).
Dutta, S. et al. Photoredox-enabled dearomative [2π + 2σ] cycloaddition of phenols. J. Am. Chem. Soc. 2024, 2789–2797 (2024).
Tyler, J. L. et al. Bicyclo[1.1.0]butyl radical cations: synthesis and application to [2π + 2σ] cycloaddition reactions. J. Am. Chem. Soc 146, 16237–16247 (2024).
Dhake, K. et al. Beyond bioisosteres: divergent synthesis of azabicyclohexanes and cyclobutenyl amines from bicyclobutanes. Angew. Chem. Int. Ed. 61, e202204719 (2022).
Radhoff, N., Daniliuc, C. G. & Studer, A. Lewis acid catalyzed formal (3+2)-cycloaddition of bicyclo[1.1.0]butanes with ketenes. Angew. Chem. Int. Ed. 62, e202304771 (2023).
Liang, Y., Paulus, F., Daniliuc, C. G. & Glorius, F. Catalytic formal [2π+2σ] cycloaddition of aldehydes with bicyclobutanes: expedient access to polysubstituted 2-oxabicyclo[2.1.1]hexanes. Angew. Chem. Int. Ed. 62, e202305043 (2023).
Schwartz, B. D., Zhang, M. Y., Attard, R. H., Gardiner, M. G. & Malins, L. R. Structurally diverse acyl bicyclobutanes: valuable strained electrophiles. Chem. Eur. J. 26, 2808–2812 (2020).
Ni, D. et al. Intermolecular formal cycloaddition of indoles with bicyclo[1.1.0]butanes by Lewis acid catalysis. Angew. Chem. Int. Ed. 62, e202308606 (2023).
Tang, L. et al. Silver-catalyzed dearomative [2π+2σ] cycloadditions of indoles with bicyclobutanes: access to indoline fused bicyclo[2.1.1]hexanes. Angew. Chem. Int. Ed. 62, e202310066 (2023).
Wang, J.-J. et al. Switching between the [2π+2σ] and hetero-[4π+2σ] cycloaddition reactivity of bicyclobutanes with Lewis acid catalysts enables the synthesis of spirocycles and bridged heterocycles. Angew. Chem. Int. Ed. 63, e202405222 (2024).
Woelk, K. J., Dhake, K., Schley, N. D. & Leitch, D. C. Enolate addition to bicyclobutanes enables expedient access to 2-oxo-bicyclohexane scaffolds. Chem. Commun. 59, 13847–13850 (2023).
Zhang, J., Su, J.-Y., Zheng, H., Li, H. & Deng, W.-P. Eu(OTf)3‐catalyzed formal dipolar [4π + 2σ] cycloaddition of bicyclo[1.1.0]butanes with nitrones: access to polysubstituted 2‐oxa‐3‐azabicyclo[3.1.1]heptanes. Angew. Chem. Int. Ed. 63, e202318476 (2024).
Liang, Y., Nematswerani, R., Daniliuc, C. G. & Glorius, F. Silver-enabled cycloaddition of bicyclobutanes with isocyanides for the synthesis of polysubstituted 3-azabicyclo[3.1.1]heptanes. Angew. Chem. Int. Ed. 63, e202402730 (2024).
Yamaoka, Y. & Takasu, K. Catalytic [2+2] cycloaddition of silyl enol ethers. In Methods and Applications of Cycloaddition Reactions in Organic Syntheses (eds Nishiwaki, N.) 115–134 (Wiley, 2014).
Takasu, K. Synthesis of multisubstituted silyloxy-based donor-acceptor cyclobutanes by an acid-catalyzed [2+2] cycloaddition. Isr. J. Chem. 56, 488–498 (2016).
Bach, T., Jödicke, K., Kather, K. & Fröhlich, R. 1,3-Allylic strain as a control element in the Paternò−Büchi reaction of chiral silyl enol ethers: synthesis of diastereomerically pure oxetanes containing four contiguous stereogenic centers. J. Am. Chem. Soc. 119, 2437–2445 (1997).
Kang, T. et al. A chiral N,N’-dioxide-Zn(II) complex catalyzes the enantioselective [2+2] cycloaddition of alkynones with cyclic enol silyl ethers. Angew. Chem. Int. Ed. 55, 5541–5544 (2016).
Fuchibe, K., Aono, T., Hu, J. & Ichikawa, J. Copper(I)-catalyzed [4 + 1] cycloaddition of silyl dienol ethers with sodium bromodifluoroacetate: access to β,β-difluorocyclopentanone derivatives. Org. Lett. 18, 4502–4505 (2016).
Takasu, K., Nagao, S. & Ihara, M. Construction of highly‐functionalized cyclopentanes from silyl enol ethers and activated cyclopropanes by [3+2] cycloaddition catalyzed by triflic imide. Adv. Synth. Catal. 348, 2376–2380 (2006).
de Nanteuil, F. & Waser, J. Catalytic [3+2] annulation of aminocyclopropanes for the enantiospecific synthesis of cyclopentylamines. Angew. Chem. Int. Ed. 50, 12075–12079 (2011).
Xu, H., Qu, J. P., Liao, S., Xiong, H. & Tang, Y. Highly enantioselective [3+2] annulation of cyclic enol silyl ethers with donor-acceptor cyclopropanes: accessing 3a-hydroxy [n.3.0]carbobicycles. Angew. Chem. Int. Ed. 52, 4004–4007 (2013).
Xu, H., Hu, J. L., Wang, L., Liao, S. & Tang, Y. Asymmetric annulation of donor-acceptor cyclopropanes with dienes. J. Am. Chem. Soc. 137, 8006–8009 (2015).
Nicolai, S. & Waser, J. Lewis Acid Catalyzed [4+2] Annulation of Bicyclobutanes with Dienol Ethers for the Synthesis of Bicyclo[4.1.1]octanes. Chem. Sci. https://doi.org/10.1039/D4SC02767A (2024).
Semeno, V. V. et al. Bicyclo[m.n.k]alkane building blocks as promising benzene and cycloalkane isosteres: multigram synthesis, physicochemical and structural characterization. Chem. Eur. J. 30, e202303859 (2024).
Pronin, S. V., Reiher, C. A. & Shenvi, R. A. Stereoinversion of tertiary alcohols to tertiary-alkyl isonitriles and amines. Nature 501, 195–199 (2013).
Singleton, D. A. & Thomas, A. A. High-precision simultaneous determination of multiple small kinetic isotope effects at natural abundance. J. Am. Chem. Soc. 117, 9357–9358 (1995).
Kwon, K.-H., Lee, D. W. & Yi, C. S. Chelate-assisted oxidative coupling reaction of arylamides and unactivated alkenes: mechanistic evidence for vinyl C−H bond activation promoted by an electrophilic ruthenium hydride catalyst. Organometallics 29, 5748–5750 (2010).
Bartelson, K. J., Singh, R. P., Foxman, B. M. & Deng, L. Catalytic asymmetric [4 + 2] additions with aliphatic nitroalkenes. Chem. Sci. 2, 1940–1944 (2011).
This manuscript was submitted on ChemRxiv. Hu, S., Pan, Y., Ni, D. & Deng, L. Facile access to bicyclo[2.1.1]hexanes by formal cycloaddition between silyl enol ethers and bicyclo[1.1.0]butanes with Lewis acids. Preprint at https://doi.org/10.26434/chemrxiv-2024-486kr (2024).
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (grant no. 22201233 to D.N., grant no. U22A20389 to L.D.), and the Foundation of Westlake University; the Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (2020R01004). We thank the Instrumentation and Service Center for Molecular Sciences and Physical Sciences at Westlake University for the assistance in measurement/data interpretation. We also thank Dr. Xiao-huo Shi at Westlake University for his assistance in the measurement of NMR and Dr. Yinjuan Chen at Westlake University for her assistance in the measurement of HRMS, Dr. Fucheng Leng at Westlake University for assistance with X-ray measurement.
Author information
Authors and Affiliations
Contributions
S.H., Y.P. and D.N. carried out the experiments and data analysis work. D.N. and L.D. designed the reaction and directed the project. The paper was written by D.N. and L.D. All authors contributed to discussions. S.H. and Y.P. contributed equally.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Honggen Wang and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hu, S., Pan, Y., Ni, D. et al. Facile access to bicyclo[2.1.1]hexanes by Lewis acid-catalyzed formal cycloaddition between silyl enol ethers and bicyclo[1.1.0]butanes. Nat Commun 15, 6128 (2024). https://doi.org/10.1038/s41467-024-50434-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50434-6
