
Abstract
Transcription coupled-nucleotide excision repair (TC-NER) removes DNA lesions that block RNA polymerase II (Pol II) transcription. A key step in TC-NER is the recruitment of the TFIIH complex, which initiates DNA unwinding and damage verification; however, the mechanism by which TFIIH is recruited during TC-NER, particularly in yeast, remains unclear. Here, we show that the C-terminal ___domain (CTD) of elongation factor-1 (Elf1) plays a critical role in TC-NER in yeast by binding TFIIH. Analysis of genome-wide repair of UV-induced cyclobutane pyrimidine dimers (CPDs) using CPD-seq indicates that the Elf1 CTD in yeast is required for efficient TC-NER. We show that the Elf1 CTD binds to the pleckstrin homology (PH) ___domain of the p62 subunit of TFIIH in vitro, and identify a putative TFIIH-interaction region (TIR) in the Elf1 CTD that is important for PH binding and TC-NER. The Elf1 TIR shows functional, structural, and sequence similarities to a conserved TIR in the mammalian UV sensitivity syndrome A (UVSSA) protein, which recruits TFIIH during TC-NER in mammalian cells. These findings suggest that the Elf1 CTD acts as a functional counterpart to mammalian UVSSA in TC-NER by recruiting TFIIH in response to Pol II stalling at DNA lesions.
Similar content being viewed by others


Introduction
Transcription by RNA polymerase II (Pol II) is essential for cellular viability but can be blocked by the presence of DNA lesions on the transcribed strand (TS) of genes. Transcription-blocking lesions (TBLs) include ultraviolet (UV) light-induced cyclobutane pyrimidine dimers (CPDs) and bulky, helix-distorting DNA adducts1,2. TBLs cause elongating Pol II to stall, triggering a cellular repair pathway known as transcription coupled-nucleotide excision repair (TC-NER)3,4,5,6. Efficient removal of TBLs by the TC-NER pathway is critical to genome stability and cellular homeostasis, as genetic defects in TC-NER result in elevated sensitivity to DNA damaging agents such as UV light and can cause development defects, neurodegeneration, and rapid aging syndrome in humans4,5,7. However, despite its importance and nearly four decades of intense study, key gaps remain in our understanding of the basic mechanism of TC-NER in eukaryotic cells2,8.
TC-NER is initiated when Pol II stalling at a DNA lesion is detected by the Cockayne Syndrome B (CSB) protein in human cells or its homolog Rad26 in yeast. CSB/Rad26 binding eventually leads to the critical step of recruiting TFIIH, a helicase and ATPase complex that is required for DNA unwinding and damage verification, and which promotes subsequent excision of the damaged DNA strand1,2,9. While CSB/Rad26 binds to stalled Pol II, neither directly binds to TFIIH. In human cells, CSB, in conjunction with Cockayne Syndrome A protein and its associated Cullin Ring Ubiquitin Ligase complex, promotes Pol II ubiquitination, as well as recruitment and ubiquitination of a third TC-NER factor known as UVSSA10,11,12. UVSSA then recruits TFIIH via its TFIIH interacting region (TIR), which directly binds to the pleckstrin homology (PH) ___domain of the p62 subunit of TFIIH11,13. Notably, the UVSSA TIR resembles TFIIH-interacting regions in XPC, which is required for global genomic-nucleotide excision repair (GG-NER) in human cells9,14, and in transcription factors (e.g., TFIIEα, p53, and DP1) that recruit TFIIH during transcription initiation by also binding the PH ___domain of p6213,15,16,17,18. However, yeast and many other species lack a UVSSA homolog or other TIR-containing TC-NER factor(s). Hence, the mechanism by which TFIIH is recruited during TC-NER in these species is unknown. Moreover, previous studies have revealed that the TC-NER pathway in yeast continues to repair UV damage, albeit less efficiently, in the absence of the key TC-NER factor Rad26. However, the molecular mechanism by which Rad26-independent TC-NER is initiated in yeast remains unclear.
Recently, a new TC-NER factor known as ELOF1 in human cells and Elf1 in yeast was identified19,20,21. ELOF1/Elf1 are transcription elongation factors that are associated with Pol II during transcription elongation and stimulate elongation rates19,20,22,23,24,25,26. In yeast, Elf1 binds to the coding regions of genes in a transcription-dependent manner24, and its genome-wide binding profile resembles that of other transcription elongation factors25. Consistently, structural studies indicate that Elf1 functions as a key component of the DNA entry channel in the yeast Pol II elongation complex22,23. In human cells, ELOF1 is required for TFIIH recruitment during TC-NER because it stimulates ubiquitination of stalled Pol II and subsequent recruitment and ubiquitination of UVSSA19,20. While Elf1 also plays a critical role in TC-NER in the yeast Saccharomyces cerevisiae, the molecular mechanism(s) by which yeast Elf1 promotes TC-NER is unclear.
Here, we use both genome-wide repair studies in yeast (S. cerevisiae) and in vitro biochemistry to show that the C-terminal ___domain (CTD) of yeast Elf1 contains a TIR that binds the PH ___domain of the p62 subunit of TFIIH and promotes TC-NER in genes throughout the yeast genome.
Results
Elf1 is required for Rad26-independent TC-NER
Our previous study indicated that Elf1 not only plays a genome-wide role in TC-NER in WT cells but may also specifically contribute to Rad26-independent TC-NER, at least at the RPB2 locus19. To characterize the role of Elf1 in Rad26-independent TC-NER across the yeast genome, we used the CPD-seq method27,28 (Fig. 1a) to map the genome-wide distribution of CPD lesions both immediately after UV irradiation (0 h) and following 2 h of repair (2 h) in a rad26∆ elf1∆ double mutant. Analysis of putative lesion-forming dinucleotide sequences associated with CPD-seq reads revealed enrichment at CPD-forming dipyrimidines (i.e., TT followed by TC, CT, and CC) in the UV-exposed samples but not in the No UV control (Fig. 1b), consistent with our previous results28,29. To characterize the impact of elf1∆ mutant on Rad26-independent TC-NER, we analyzed the fraction of CPDs remaining following 2 h of repair (2 h) relative to 0 h control in both the rad26∆ elf1∆ double mutant and a previously published rad26∆ single mutant30 (Fig. 1c, d). Analysis of the fraction of unrepaired CPDs adjacent to the transcription start site (TSS) of ~5200 yeast genes in a rad26∆ mutant30 revealed a defect in repair of the TS, with peaks of unrepaired CPDs along both the TS and non-transcribed strand (NTS) associated with the centers of yeast nucleosomes (Fig. 1c), likely due to nucleosome inhibition of CPD repair by the GG-NER pathway28,29,31,32,33. However, there were still fewer unrepaired CPDs along the TS than the NTS in the rad26Δ strain immediately downstream of the TSS (~120 base pairs (bp)) and particularly on the TSS-proximal side of the +1 nucleosome (Fig. 1c, d and Supplementary Fig. 1a). Analysis of published CPD-seq data for catalytically inactive ATPase mutant in rad26 (i.e., rad26-K328R), which is also defective in TC-NER30, showed similar results (Supplementary Fig. 1b). These findings are consistent with previous analysis30, and are indicative of Rad26-independent TC-NER near the TSS of yeast genes. In contrast, CPD-seq data for rad26Δ elf1Δ double mutant revealed a higher fraction of CPDs remaining on the TS immediately downstream of the TSS, so that the fraction of unrepaired CPDs along the TS was similar to the NTS (Fig. 1e, f). Taken together, these findings indicate that Elf1 is required for Rad26-independent TC-NER across the yeast genome.

a Schematic of the CPD-seq protocol for mapping UV-induced CPD lesions at single nucleotide resolution. b CPD-seq reads in elf1∆ rad26∆ mutant cells are enriched at dipyrimidine sequences in UV-irradiated samples (0 h and 2 h) and not in non-irradiated (No UV) control. c Analysis of CPD-seq data from rad26∆ cells30 near the transcription start site (TSS) for ~5200 genes at single nucleotide resolution on both the transcribed strand (TS) and non-transcribed strand (NTS). The data depicts regions 200 bp upstream of and 640 bp downstream from the TSS of each yeast gene. The number of unrepaired CPDs after 2 h of repair is normalized to the initial damage counts (0 h) to calculate the fraction of CPDs remaining. The gray peaks (right y axis) correspond to average nucleosome coverage from MNase nucleosome map64. NCP, nucleosome core particle. d CPD-seq data from rad26∆ (data from ref. 30) were analyzed immediately adjacent to the transcription start site (TSS) of ~5200 yeast genes on both the transcribed strand (TS) and non-transcribed strand (NTS). Fraction of unrepaired CPDs after 2 h repair relative to 0 h control is depicted. Gray background (right y-axis) depicts dyad positions of +1 nucleosome based on MNase nucleosome map64. e Same as (c), except analysis of CPD-seq data from rad26∆ elf1∆ cells. f Same as (d), except analysis of CPD-seq data from elf1∆ rad26∆. Source data are provided as a Source Data file.
Deletion of the Elf1 C-terminal ___domain imparts UV sensitivity in GG-NER deficient yeast
Inspection of the yeast Elf1 protein sequence revealed not only N-terminal and C4 zinc finger core domains, which are also present in human ELOF1, but also an extended CTD that is absent from the human ELOF1 sequence (Fig. 2a). To characterize the role of these different domains in TC-NER, we mutated each ___domain in a GG-NER deficient rad16∆ mutant background since it has been previously shown that deletion of TC-NER factors typically imparts significant UV sensitivity only in cells lacking GG-NER34,35. UV spotting assays revealed that while deletion of the Elf1 N-terminus (elf1-NΔ, corresponding to deletion of residues 2–16) did not affect UV sensitivity in the rad16∆ background, mutating critical cysteine residues in the Elf1 zinc finger ___domain (elf1-ZnΔ, corresponding to elf1-C49A, C52A) or deletion of the Elf1 CTD (elf1-CTDΔ, corresponding to deletion of residues 85–145) significantly enhanced UV sensitivity (Supplementary Fig. 2a and Fig. 2b). To determine if these Elf1 domains also affected Rad26-independent TC-NER, we analyzed their UV sensitivity in a rad16Δ rad26Δ background. Our results indicated that both the elf1-ZnΔ and elf1-CTDΔ cells exhibited increased UV sensitivity in rad16Δ rad26Δ strains (Supplementary Fig. 2b and Fig. 2b), indicating these domains of Elf1 are important for Rad26-independent TC-NER.

a Alignment of yeast Elf1 and human ELOF1 protein sequences. NTD, N-terminal ___domain; arrows, key cysteine residues in C4 zinc finger ___domain; and CTD, C-terminal ___domain. b Indicated mutant yeast strains were 10-fold serially diluted, spotted on the YPD plates, and exposed to the indicated doses of UVC light. Plates were photographed after 3 days of incubation in the dark. c Representative alkaline gel of bulk repair analysis of CPD lesions in WT cells at different time points following UVC irradiation. The genomic DNA was isolated at the indicated time points and treated with or without (+/−) T4 endonuclease V digestion and resolved on denaturing alkaline gels. First lane is the lambda DNA-HindIII digest molecular weight markers (Invitrogen). d Same as (c), except analysis of data from elf1-CTD∆ cells. e Quantification of the repair of CPDs in WT and elf1-CTD∆ yeast strains based on alkaline gel analysis. The plot represents mean ± SEM of three independent experiments. *P ≤ 0.05 analyzed by unpaired two-sided t-test with Holm–Sidak correction. f Analysis of CPD-seq data from WT cells19 for ~5200 genes at single nucleotide resolution on both the transcribed strand (TS) and non-transcribed strand (NTS). The plot depicts regions, 200 bp upstream of and +640 bp downstream from the transcription start site (TSS). The number of unrepaired CPDs after 2 h of repair is divided by the initial damage counts (0 h) to depict the fraction of CPDs remaining. The fraction of CPDs remaining was normalized to the bulk fraction of CPDs remaining determined by alkaline gel analysis. g Same as (f), except analysis of data from elf1-CTD∆ cells. h, i Gene cluster plot analysis of CPD-seq repair data from h WT and i elf1-CTD∆ cells for ~4500 genes, ordered based on transcription frequency69. Plots depict the fraction of unrepaired CPDs after 2 h of repair relative to the initial damage counts (0 h) for both TS and NTS. Data was normalized using alkaline gel analysis data, as described above. Source data are provided as a Source Data file.
While the zinc finger ___domain of Elf1 (and ELOF1) is required for binding to Pol II19,20,22,23,36, the function of the Elf1 CTD is unknown. To test whether the Elf1-CTD is required for Elf1 protein stability, we performed western blot analysis of yeast extracts derived from strains with tagged Elf1. These data indicated that the elf1-CTDΔ mutant did not affect Elf1 protein levels (Supplementary Fig. 3a), suggesting that the Elf1 CTD may instead play a direct functional role in TC-NER. Deletion of ELF1 is lethal in combination with mutations in genes encoding other transcription elongation factors, such as Spt424, consistent with a role for Elf1 in transcription elongation. While our data confirmed that the elf1Δ spt4Δ double mutant is lethal in yeast, as it is unable to grow on media containing 5-FOA, the elf1-CTDΔ spt4Δ double mutant grew normally in medium with 5-FOA (Supplementary Fig. 3b). This observation suggests that, unlike full-length Elf1, the Elf1 CTD is not required for transcription elongation, and that the UV sensitivity of the elf1-CTD∆ mutant does not arise from a defect in transcription elongation.
Elf1 C-terminal ___domain is required for efficient TC-NER
Since the UV sensitivity assays indicated the Elf1 CTD may play a role in TC-NER, we analyzed the impact of the Elf1 CTD on repair of UV-induced CPD lesions. Our initial assay used a T4 endonuclease V (T4 endoV) digestion and alkaline gel electrophoresis assay to measure repair of CPDs in bulk genomic DNA (Fig. 2c, d). The results indicated that the elf1-CTDΔ mutant did not cause a significant repair defect in bulk genomic DNA relative to WT (P > 0.05, Fig. 2e), suggesting that the Elf1-CTD is likely not required for global repair of UV lesions by the GG-NER pathway, which is primarily responsible for repair of CPD lesions in yeast.
To specifically study the role of Elf1-CTD in TC-NER, we analyzed the repair of CPDs across the genome in elf1-CTDΔ using CPD-seq. We examined repair of CPDs at single-nucleotide resolution around the TSS of ~5200 yeast genes for both the elf1-CTD∆ cells and a previously published WT control19, which were normalized using the T4 endoV alkaline gel data (Fig. 2e). In WT cells, there were fewer unrepaired CPDs remaining after 2 h repair on the TS of yeast genes than the NTS, and unrepaired CPDs on the NTS were elevated near the central dyad of nucleosomes (Fig. 2f). In the elf1-CTDΔ mutant, the fraction of unrepaired CPDs along the TS was elevated relative to WT, particularly near the TSS of yeast genes, while repair of the NTS was largely unaffected (Fig. 2g). These data reveal that, similar to our published elf1∆ data19, deletion of the Elf1-CTD causes a defect in the repair of the TS of yeast genes.
To further characterize the role of the Elf1-CTD on TC-NER, we analyzed the fraction of unrepaired CPDs on both the TS and NTS in six equally sized bins between the TSS and transcription end site (TES) of ~5000 yeast genes. In the elf1-CTDΔ strain, the fraction of CPDs remaining on the NTS after 2 h repair was roughly similar to WT (Supplementary Fig. 4a, b). However, there were more unrepaired CPDs along the TS in the elf1-CTDΔ strain throughout the transcribed region (i.e., TSS to TES). To test whether the Elf1 CTD is required for repair of the TS of all yeast genes or just a subset, we performed gene plot analysis of the fraction of the CPDs remaining after 2 h repair across each of the ~5000 yeast genes. The results revealed significantly more unrepaired CPDs along the TS in the elf1-CTDΔ mutant relative to WT for nearly all yeast genes (Fig. 2h, i). Taken together, these findings indicate that the Elf1 CTD is required for efficient repair of the TS of nearly all yeast genes.
CPDs located on the TS of yeast genes are repaired by both the TC-NER and GG-NER pathways. To specifically measure the impact of the Elf1 CTD on TC-NER, we analyzed asymmetry in repair of the transcribed and non-transcribed DNA strands, which quantifies TC-NER activity, as TC-NER only repairs the TS. Repair asymmetry can be quantified using the log2 ratio of unrepaired CPDs on the TS relative to the NTS (see “Methods”), as described previously29. In the elf1-CTDΔ mutant, the average log2 TS/NTS ratio for the six transcribed bins was −0.34, lower than the −0.67 value for WT (Supplementary Fig. 4c), indicating reduced repair asymmetry. This analysis is consistent with the hypothesis that the Elf1 CTD regulates TC-NER.
We also used CPD-seq to measure the impact of the elf1-CTDΔ mutant on repair in a rad16Δ background, which is deficient in GG-NER. In the rad16Δ elf1-CTDΔ double mutant, there were more unrepaired CPDs along the NTS of ~5200 yeast genes after 2 h repair than the elf1-CTD∆ mutant alone, and the absence of the periodic pattern of unrepaired CPDs associated with nucleosomes (Fig. 3a), consistent with a loss of GG-NER activity33. The rad16Δ elf1-CTDΔ strain showed a marginal defect in the repair of the TS (Supplementary Fig. 5a), as quantified by an average log2 TS/NTS ratio of the transcribed bins of −0.68 for the rad16Δ elf1-CTDΔ mutant, relative to average log2 TS/NTS ratios of −0.86 and −1.04 for rad16Δ controls32,33 (Fig. 3b and Supplementary Fig. 5b), indicating diminished repair asymmetry.

a Analysis of CPD-seq data from rad16∆ elf1-CTD∆ cells adjacent to the TSS for ~5200 genes. The fraction of unrepaired CPDs after 2 h of repair relative to the 0 h control is plotted. Nucleosome positioning data from ref. 64. b Log2 ratio of unrepaired CPDs (after 2 h repair relative to 0 h control) on the TS relative to the NTS in the rad16∆ elf1-CTD∆ and rad16∆ cells plotted between TSS and TES of ~5000 genes. Data for rad16∆ cells from ref. 33. Each gene was divided in six equally sized bins, and three 167 bp bins in flanking DNA upstream of the TSS and downstream of the transcription end site (TES) are also depicted. c Same as (a), except CPD-seq data from rad26∆ elf1-CTD∆ cells are depicted. d Close up of CPD-seq data from rad26∆ elf1-CTD∆ cells immediately flanking the TSS and +1 nucleosome of ~5200 yeast genes. e Same as (b), except analysis of data from rad26∆ elf1-CTD∆ and rad26∆ cells. Data for rad26∆ cells from ref. 30. Source data are provided as a Source Data file.
Our UV sensitivity data indicate that the Elf1 CTD may also be required for Rad26-independent TC-NER (Fig. 2b). To test this hypothesis, we analyzed repair in rad26Δ elf1-CTDΔ using CPD-seq and compared the repair to our published rad26∆ CPD-seq data30. Analysis of the CPD-seq data near the TSS of ~5200 yeast genes revealed a higher fraction of CPDs remaining along the TS in rad26Δ elf1-CTDΔ cells (Fig. 3c) compared to rad26Δ cells30, particularly immediately downstream of the TSS (compare Fig. 3d with Fig. 1d). The role of the Elf1 CTD in Rad26-independent TC-NER was further confirmed by diminished asymmetry of repair in transcribed bins in the rad26Δ elf1-CTDΔ compared to rad26Δ alone (Supplementary Fig. 5c and Fig. 3e), as reflected in the average log2 TS/NTS ratio of 0.069 for the rad26Δ elf1-CTDΔ mutant, but −0.061 for the rad26Δ control. A similar loss of repair asymmetry was observed relative to the catalytically inactive rad26-K328R mutant (Supplementary Fig. 5d, average log2 TS/NTS ratio of −0.096). However, the elf1-CTD∆ (or elf1∆) in a rad16∆ rad26∆ background was less UV sensitive than an rad16∆ rad26∆ rpb9∆ mutant (Supplementary Fig. 6), a mutant that should render cells completely defective in TC-NER37. These findings indicate that the elf1-CTD∆ mutant significantly reduces but likely does not completely eliminate Rad26-independent TC-NER, consistent with a previous analysis of elf1∆ mutant cells19. In summary, these results imply that the Elf1 CTD plays an important role in TC-NER in WT and rad26∆ mutant cells across the yeast genome.
Elf1 CTD binds to the PH ___domain of the p62 subunit of TFIIH
Previous studies indicate that human ELOF1 facilitates TFIIH recruitment indirectly by promoting Pol II ubiquitination and UVSSA recruitment19,20. Since yeast lacks a UVSSA homolog, we wondered if Elf1 might directly interact with TFIIH, perhaps through its CTD. To test this hypothesis, we expressed and purified either full-length Elf1, Elf1 without CTD, or Elf1 CTD alone fused to glutathione S-transferase (GST) and measured their binding to purified PH ___domain of the p62 subunit of TFIIH in vitro. We tested binding to the PH ___domain of the p62 subunit since previous studies have shown that both transcription factors (e.g., TFIIE and p53) and NER factors, including Rad4/XPC and UVSSA, specifically bind to the PH ___domain to recruit TFIIH13,15,16,17,18. SDS-PAGE analysis of GST pull-down assays revealed that while full-length or the N-terminal half of Elf1 (i.e., residues 1–85) did not bind the PH ___domain of p62 in vitro, the Elf1 CTD (residues 85–145) did bind the PH ___domain (Fig. 4a). That full-length Elf1 was unable to bind the PH ___domain suggests that the Elf1 CTD may be inhibited by other regions of Elf1, which may have important ramifications for Elf1 activity during TC-NER (see “Discussion”). Quantitative binding assays of GST-Elf1-CTD and purified PH ___domain using Biolayer Interferometry (BLI) yielded a dissociation constant (Kd) of 3.82 ± 1.09 μM (mean ± standard deviation; see Supplementary Fig. 7 and Supplementary Table 1).

a, b SDS polyacrylamide gel electrophoresis (PAGE) analysis of pull-down assays, in which purified Glutathione S-transferase (GST) fusion proteins containing the indicated Elf1 regions were incubated with purified pleckstrin homology (PH) ___domain of the p62 subunit of TFIIH (see Input). Output indicates proteins present after GST pull-down. Band corresponding to PH ___domain is indicated with arrow. Only the Elf1-CTD interacts with the PH ___domain. Lanes 1 and 7 in (a, b) are Precision Plus Protein Dual Color Standards molecular weight (MW) markers (Bio-Rad). The experiment was repeated three times with similar results. c Quantification of pull-down assay based on SDS-PAGE analysis in (b). Data depicted in (c) are mean and standard deviation (n = 3). d UV spotting assay of elf1 mutant strains exposed to the indicated doses of UV light. Source data are provided as a Source Data file.
To determine which region(s) of the Elf1 CTD are important for TFIIH binding, we expressed and purified different truncated versions of the Elf1 CTD fused to GST, and tested their binding to the PH ___domain of p62 in vitro. The GST pull-down data (Fig. 4b,c) indicated that Elf1 residues 85–122 and 85–113 bound to the PH ___domain similarly as the Elf1-CTD (i.e., residues 85–145), indicating that Elf1 residues 85–113 likely contain the TFIIH-interacting region. Deletion of Elf1 residues 85–113 in yeast imparted elevated UV sensitivity in a rad16∆ rad26∆ mutant background (Fig. 4d), suggesting that this region is important for TC-NER in yeast.
Elf1 CTD has a TFIIH-interacting region important for Rad26-independent TC-NER
Multiple sequence alignment of the Elf1 sequences from related yeast species indicated that residues 95–106 in the CTD are highly conserved (Supplementary Fig. 8). This region contains a conserved aromatic residue (Y99) flanked by acidic residues. This region, as well as the rest of the Elf1 CTD, is predicted by the PONDR software38 to be intrinsically disordered (Supplementary Fig. 9). Notably, these sequence features are characteristic of TFIIH-interacting regions (TIRs) found in XPC, UVSSA, and other proteins that bind the PH ___domain of p62 subunit of TFIIH13,15,16,17,18. Sequence alignment of the putative Elf1 TIR (residues 89–106) with TIRs from XPC, UVSSA, and other TFIIH-interacting proteins showed considerable sequence similarity (Fig. 5a), with each TIR containing a central aromatic residue (Y99 in Elf1) flanked by acidic residues.

a Alignment of putative TFIIH-interacting region (TIR) in yeast Elf1-CTD (residues 89–106) with TIRs from other PH ___domain interacting proteins, namely UVSSA (residues 398–415), XPC (residues 123–140), TFIIEα (residues 377–394). Red arrow indicates ___location of conserved aromatic residue critical for PH binding. b UV spotting assay of elf1-CTD mutant strains exposed to the indicated doses of UV light. c Quantitative UV survival assay of the indicated elf1 mutants (or ELF1 WT) in a rad16∆ rad26∆ background. The graph represents the quantification of UV survival from three (n = 3) independent experiments. Mean ± SEM is depicted, *P ≤ 0.05, based on two-sided t-test with Holm–Sidak correction for different UV doses for each pair of strains compared. d SDS-PAGE analysis of GST pull-down assays in which the purified GST-Elf1-CTD (residues 85–145) fusion proteins containing the indicated WT or mutant sequences were incubated with purified PH ___domain of p62 subunit of TFIIH (indicated by the arrow). Lanes 1 and 7 are Precision Plus Protein Dual Color Standards molecular weight (MW) markers (Bio-Rad). e Quantification of Elf1-CTD interaction with PH ___domain from replicate GST pull-down experiments. Mean and standard deviation (n = 3) are depcited. f Biolayer Interferometry (BLI) assay was used to determine the dissociation constant (Kd) values for the interaction between PH ___domain of p62 and WT and Y99A GST-Elf1-CTD (residues 85–145). Kd measurements are for three independent assays conducted at four concentrations of PH-p62, ranging from 30 μM to 3.75 μM, for a total of twelve data points per variant. P-value is 2.49 × 10−7 from two-sided t-test. Error bars represent standard deviation. g Analysis of CPD-seq data after 2 h repair relative 0 h control in rad26∆ elf1-Y99A cells adjacent to the TSS of ~5200 genes. Average nucleosome coverage from ref. 64. h Close up of CPD-seq data from rad26∆ elf1-Y99A cells immediately flanking the TSS of ~5200 yeast genes. i Plot of transcriptional asymmetry in repair of TS and NTS of ~5000 genes using CPD-seq data from rad26∆30, rad26∆ elf1-Y99A, and rad26∆ elf1-CTD∆ cells.
Since previous studies have suggested that the central aromatic residue in the TIR is important for binding TFIIH13,16,17,18, we used CRISPR genome editing39,40 to construct an elf1-Y99A mutant and tested its UV sensitivity in rad16∆ and rad16∆ rad26∆ mutant backgrounds. Spotting assays indicated that the elf1-Y99A caused a mild-to-moderate increase in UV sensitivity in these mutant backgrounds (Fig. 5b and Supplementary Fig. 10). Quantitative analysis of the UV sensitivity of the elf1-Y99A in a rad16∆ rad26∆ mutant background revealed that the elf1-Y99A mutant imparted significant UV sensitivity, although to a lesser extent than the elf1∆ or elf1-CTD∆ (85-145∆) mutants (Fig. 5c), suggesting other residues in the Elf1 CTD also play a role in UV resistance. In summary, these UV sensitivity assays indicate that Elf1-Y99 residue in the putative TIR plays a role in TC-NER.
To determine whether the putative Elf1 TIR mediates binding to TFIIH, we expressed and purified a GST-fused Elf1-CTD containing the Y99A mutant and tested its binding to the PH ___domain of p62. As a control, we also expressed and purified a GST-Elf1-CTD containing a mutation in a hydrophobic residue that lies outside the putative TIR (i.e., Elf1-I115, see Supplementary Fig. 8). GST pull-down assays indicated that the Y99A mutant caused a significant decrease in binding to the p62 PH ___domain relative to the WT CTD (Fig. 5d, e). In contrast, the I115A control did not significantly affect binding (Fig. 5d, e). Quantitative BLI assays indicated that the Kd of the GST-Elf1-CTD Y99A mutant increased to 7.32 ± 1.24 μM (Fig. 5f and Supplementary Table 2), confirming that the Y99A mutant CTD has a significantly weaker interaction with the p62 PH ___domain than the WT CTD (P < 0.0001, Fig. 5f). Taken together, these findings indicate that the Y99 residue in the putative Elf1 TIR is important for binding to TFIIH.
To test whether the elf1-Y99A mutant affected TC-NER, we used CPD-seq to measure repair of UV damage. We analyzed repair in a rad26∆ mutant background since the rad16∆ rad26∆ elf1-Y99A mutant showed elevated UV sensitivity (Fig. 5b, c). Analysis of the CPD-seq data revealed a partial defect in repair of the TS relative to the NTS, particularly immediately downstream of the TSS (Fig. 5g, h). Moreover, the transcriptional asymmetry along the transcribed regions of ~5000 yeast genes also showed a partial defect in the rad26∆ elf1-Y99A double mutant relative to the rad26∆ or rad26-K328R mutants alone (Fig. 5i and Supplementary Fig. 11), as reflected in an average log2 TS/NTS of −0.0049 (compared to −0.061 and −0.096 for rad26∆ and rad26-K328R). However, the defect in transcriptional asymmetry was not as significant as observed in rad26∆ elf1-CTD∆ double mutant (average log2 TS/NTS of 0.069, see also Fig. 5i), consistent with our UV sensitivity results (Fig. 5c) and in vitro binding assays (Fig. 5d, e).
To determine whether Elf1 Y99 promotes UV resistance through its interaction with the PH ___domain of the p62 subunit of TFIIH (encoded by the yeast TFB1 gene), CRISPR was used to specifically delete the PH ___domain (residues 2–114) in TFB1. UV spotting assays indicated that the tfb1-PH∆ mutant showed elevated UV sensitivity in a rad16∆ mutant background (Supplementary Fig. 12a), suggesting that the PH ___domain plays a role in TC-NER. The tfb1-PH∆ mutant also showed elevated UV sensitivity in a rad16∆ rad26∆ mutant background (Supplementary Fig. 12b), indicating that the PH ___domain also functions in Rad26-independent TC-NER. These findings are consistent with a recent report indicating that the Tfb1-PH ___domain regulates TC-NER in yeast41. Mutating elf1-Y99A did not increase the sensitivity of the tfb1-PH∆ mutant in either a rad16∆ or rad16∆ rad26∆ mutant background. (Supplementary Fig. 12), suggesting that the elf1-Y99A and tfb1-PH∆ mutants in these genetic backgrounds are epistatic. To confirm this finding, we performed quantitative UV sensitivity assays on each mutant strain in a rad16∆ rad26∆ mutant background. The elf1-Y99A mutant caused a significant increase in UV sensitivity at the 10 J/m2 dose (P < 0.05) and a marginal increase in UV sensitivity at the 15 J/m2 dose. In contrast, the elf1-Y99A mutant did not significantly alter the UV sensitivity of the tfb1-PH∆ mutant in a rad16∆ rad26∆ background (Supplementary Fig. 12c), confirming these two mutants are epistatic. The elf1-CTD∆ mutant also appeared to be largely epistatic with the tfb1-PH∆ mutant (Supplementary Fig. 12a, b). Quantitative UV sensitivity assays indicated that the elf1-CTD∆ caused a significant increase in the UV sensitivity of the tfb1-PH∆ mutant in a rad16∆ rad26∆ background at the 7.5 and 10 J/m2 UV doses (P < 0.05, Supplementary Fig. 12d). However, the magnitude of the increase in sensitivity across all UV doses (~2.6-fold) was much less than observed in cells in which the TFB1-PH ___domain was intact (~23-fold; see Supplementary Fig. 12d), suggesting that the Elf1-CTD is largely epistatic with the Tfb1-PH ___domain in a rad16∆ rad26∆ background. In summary, these findings support a model in which the Y99 residue in the Elf1 CTD promotes UV resistance by interacting with the PH ___domain in TFIIH to facilitate TC-NER.
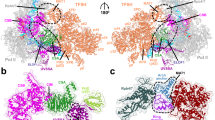
Finally, AlphaFold-multimer42 was used to predict the structure of the binding interaction between the Elf1-CTD and the PH ___domain of p62 (Fig. 6a). The resulting structure suggests that the Elf1-CTD binds to the PH ___domain primarily through Tyr99, which inserts in a pocket on the PH ___domain’s surface composed of β-sheets. This pocket, characterized by a hydrophobic environment, appears to stabilize the interaction. Notably, Tyr99 is observed to stack with Arg61 of the PH ___domain at an approximate distance of 3.3 Å. This structural prediction is consistent with our biochemical and genetic data. Importantly, alignment of the predicted Elf1-CTD/p62 interface with NMR structures of known TFIIH-interacting proteins bound to the PH ___domain of p62 (i.e., human UVSSA-TIR/p62 (PDB: 5XV8), yeast Rad4-TIR/p62 (PDB: 2M14), and human TFIIEα-TIR/p62 (PDB: 2RNR)) reveals a striking common binding mode of how the TIR binds to p62. Specifically, conserved aromatic residues (Tyr99 in Elf1, Phe408 in UVSSA, Phe95 in Rad4, and Phe387 in TFIIEα) bind to the same pocket of p62.

a Superposition of Elf1-CTD (yellow), Rad4 (magenta), TFIIEα (blue), and UVSSA (green) in binding the PH ___domain of p62 (grey). The model of Saccharomyces cerevisiae Elf1-CTD/p62 was generated by Alphafold-multimer42. The NMR models of Saccharomyces cerevisiae Rad4/p62 (PDB: 2M14), Homo sapiens TFIIEα/p62 (PDB: 2RNR), and Homo sapiens UVSSA (PDB: 5XV8) were used in the structural alignment. In the left panel with zoom-in views, the key residues, Y99 in Elf1-CTD, F95 in Rad4, F387 in TFIIEα, and F408 in UVSSA, are depicted as sticks. The p62 residues involved in the interaction with Elf1-CTD are also visualized as sticks. b Comparison of TFIIH recruitment between human and yeast during TC-NER. We propose the Elf1-CTD plays an equivalent role as human UVSSA to recruit TFIIH.
Taken together, these findings suggest that the TIR of Elf1-CTD functions in a similar manner as other known TFIIH-interacting proteins (such as UVSSA, Rad4/XPC, and TFIIEα) in the recruitment of TFIIH. This is consistent with our biochemical data indicating that the Elf1 CTD binds to the PH ___domain of the TFIIH p62 subunit, and our CPD-seq data indicating that the Elf1-CTD is required for efficient repair of the TSs of yeast genes, both in RAD26 (WT) and rad26∆ cells.
Discussion
TC-NER plays a critical role in maintaining genome stability by removing DNA lesions that would otherwise impede Pol II transcription. We and others have previously identified the elongation factor Elf1 as playing an important role in TC-NER in yeast19,43, but the molecular mechanism by which Elf1 promotes TC-NER was previously unclear. Here, we have shown that the intrinsically disordered CTD of Elf1 promotes TC-NER across the yeast genome. Our data indicate that the Elf1 CTD binds the PH ___domain of the p62 subunit of TFIIH in vitro, suggesting it may play a direct role in recruiting TFIIH to initiate repair. We further demonstrate that the Elf1 CTD sequence contains a TFIIH-interacting region (TIR) that resembles similar regions in the human NER factors XPC and UVSSA and that the Elf1 TIR is important for TFIIH binding and TC-NER in yeast. Taken together, these findings suggest a model in which Elf1 recruits the TFIIH complex during TC-NER by directly binding the p62 PH ___domain (Fig. 6b).
Recruitment of the TFIIH complex is a critical step during NER since TFIIH both unwinds the DNA in preparation for incision of the damaged strand and verifies that damage is present2,6,9. During GG-NER, TFIIH is recruited by the key damage sensor proteins XPC in human cells and Rad4 in yeast9,14. However, the protein responsible for recruiting TFIIH during TC-NER in yeast was previously unclear. Our data indicate that the Elf1 CTD likely promotes TFIIH recruitment during repair by directly binding the PH ___domain of the p62 subunit of TFIIH. This same PH ___domain is bound by XPC/Rad4 to promote TFIIH recruitment during GG-NER17,44, suggesting that a common biochemical mechanism is utilized to recruit TFIIH in each NER subpathway. Since Elf1 proteins in many other species, including yeasts (e.g., S. pombe) and plants (e.g., Arabidopsis), have an extended CTD24, we hypothesize that despite significant sequence divergence these domains may have similar functions in TC-NER.
Our data indicate that the Elf1 interacts with the PH ___domain of the p62 subunit of TFIIH via a consensus TIR in its CTD. Mutation of a key aromatic residue in the TIR (elf1-Y99A), caused elevated UV sensitivity, a partial TC-NER defect in a rad26∆ mutant background, and a defect in PH ___domain binding in vitro, supporting this model. Our data also indicate that the UV sensitivity of the elf1-Y99A mutant is epistatic with a deletion in the p62 PH ___domain (tfb1-PH∆), consistent with the model that Elf1 Y99 promotes UV resistance through its interaction with the PH ___domain. However, the elf1-Y99A phenotypes were not as severe as a complete CTD deletion, suggesting the hypothesis that other regions of the Elf1-CTD may also contribute to the TFIIH recruitment and TC-NER. This hypothesis may explain our preliminary CPD-seq data for the elf1-Y99A mutant in a RAD26 WT background, which unlike the elf1-CTD∆ mutant did not have a significant TC-NER defect (Supplementary Fig. 13). This hypothesis is consistent with previous studies of the yeast GG-NER sensor Rad4, which revealed that disruption of the Rad4 TIR region alone caused only mild UV sensitivity unless the TIR mutant was combined with mutations in the Rad4 CTD, which was also important for NER and TFIIH binding44. It will be important in future studies to explore which residues in the Elf1-CTD, in addition to Y99, contribute to TFIIH binding and TC-NER.
The AlphaFold-multimer structure of the Elf1-CTD bound to the p62 PH ___domain (Fig. 6a) reveals a similar binding mode to that previously observed for other TIR regions13,15,17,18. The Elf1 Y99 residue inserts into the same pocket bound by UVSSA F408, Rad4 F95, and TFIIEα F387, aromatic residues that have also been shown to be important for PH binding. In the UVSSA, Rad4, and TFIIEα structures, a neighboring valine residue (i.e., UVSSA V411, Rad4 V98, or TFIIEα V390) inserts into a second pocket in the PH ___domain, but Elf1 lacks this valine residue. This may explain why the binding affinity of the Elf1 CTD to the p62 PH ___domain is significantly weaker than the TIRs of UVSSA (Kd = 71 nM), Rad4 (Kd = 50 nM), and TFIIEα (Kd = 45 nM)13,44,45. However, the Elf1 CTD has a roughly similar binding affinity to p62 PH ___domain (Kd = 3.82 μM) as the unmodified TIRs in the transcription factors p53 (Kd = 3.18 μM to 24.21 μM)15,46 and DP1 (Kd = 35.3 μM)16), which are known to recruit TFIIH during transcription activation by binding the p62 PH ___domain. Like Elf1, the p53 and DP1 TIRs lack a neighboring valine residue to bind the second PH ___domain pocket, which can potentially explain the similarity in their binding affinities.
Our genetic data in yeast indicate that the PH ___domain mutant (tfb1-PH∆) is more UV sensitive in a GG-NER deficient background than the elf1-CTD∆ or elf1-Y99A mutants (Supplementary Fig. 12). One interpretation of these findings is that other TIR-containing proteins may also function to bind the PH ___domain and recruit TFIIH during TC-NER. A likely candidate is Rad4, since previous studies have indicated that Rad4 is also required for TC-NER47,48, and a recent study indicates that the Rad4 TIR regulates TC-NER in yeast through its interaction with the p62 PH ___domain41. This hypothesis may explain why the elf1-CTD∆ mutant does not completely abolish TC-NER in yeast, since Rad4 may serve as a functional replacement in binding and recruiting TFIIH. Consistent with this hypothesis, our preliminary data indicate that the Elf1-CTD shows enhanced UV sensitivity when mutated in combination with a rad4-TIR∆ mutant (deletion of Rad4 residues 85–102; see Supplementary Fig. 14), suggesting that they function in parallel and partially redundant pathways to recruit TFIIH during TC-NER. This mechanism may play a particularly important role in rad16∆ strains, since Rad4 presumably no longer functions to recruit TFIIH during GG-NER, and therefore can prioritize recruiting TFIIH during TC-NER. This model can explain previous observations indicating that TC-NER activity is elevated (and TC-NER defects partially mitigated) in rad16∆ cells49,50, consistent with own rad16∆ elf1-CTD∆ CPD-seq data. Alternatively, the higher UV sensitivity of the PH ___domain mutant may reflect its role in subsequent steps of the NER reaction. For example, the Rad2 endonuclease, which makes the 3’ incision during NER51, has been reported to interact with the PH ___domain of p62 and this interaction is important for NER in yeast41,44,52. The elf1-CTD∆ mutant is also less UV sensitive than a complete ELF1 deletion. A recent report, which indicated that the core ___domain of Elf1 also facilitates TC-NER by facilitating the binding of Rad26 to lesion-stalled Pol II53, can potentially explain this observation. These data, in conjunction with our findings reported herein, indicate that Elf1 promotes TC-NER by two distinct mechanisms, namely promoting initial recognition of stalled Pol II by Rad26 and facilitating subsequent TFIIH recruitment.
ELOF1, the human homolog of yeast Elf1, is also required to recruit TFIIH to stalled Pol II during TC-NER. However, unlike yeast Elf1, human ELOF1 lacks a CTD and is not thought to directly bind TFIIH (Fig. 6b). Instead, ELOF1 promotes recruitment of a second TC-NER factor known as UVSSA by facilitating ubiquitination of a single lysine residue (Rpb1 K1268) in the largest subunit of Pol II19,20. Once UVSSA is stably recruited to stalled Pol II by ELOF1, UVSSA subsequently binds the PH ___domain of p62 to recruit TFIIH11,13. This mechanism likely does not operate in budding yeast, since it (and many other species) lacks a UVSSA homolog and our preliminary data (Supplementary Fig. 15) indicate that mutations in the homologous lysine residue in yeast Pol II (i.e., rpb1-K1246R) do not impart UV sensitivity, consistent with a previous report54. Instead, we propose that the Elf1 CTD is the functional homolog of UVSSA since both contain a TIR that directly binds the same subunit of TFIIH.
Unlike UVSSA, the Elf1 CTD does not need to be recruited by Pol II ubiquitination, since Elf1 is a transcription elongation factor that is bound to Pol II22,23,24,25,55. Instead, we hypothesize that the activity of the Elf1 CTD must be regulated so that it only binds and recruits TFIIH when Pol II stalls at a DNA lesion. Notably, our in vitro binding data indicate that the Elf1 CTD is unable to bind TFIIH in the context of full-length Elf1, suggesting that other domains in Elf1 may regulate its TFIIH binding activity, potentially as a mechanism to prevent spurious binding to TFIIH. It is also possible that other factors or post-translational modifications of Elf1 (e.g., phosphorylation56) serve as additional layers in regulating the selective binding of Elf1 CTD to TFIIH when a DNA lesion is present, and prevent its binding to TFIIH in the absence of DNA lesions during normal productive transcription elongation. Future studies will be required to understand the mechanism of this regulation.
In summary, our data reveal that a common structural mechanism, which operates to recruit TFIIH during transcription (Supplementary Fig. 16a) and GG-NER (Supplementary Fig. 16b), also mediates TFIIH recruitment by the Elf1-CTD during TC-NER in yeast (Fig. 6). That the human TC-NER factor UVSSA uses a similar mechanism to recruit TFIIH in mammalian cells13 suggests that the Elf1 CTD and UVSSA likely have equivalent and conserved functions in TC-NER.
Methods
Strains and plasmids
All yeast strains were constructed in the BY4741 strain background. Complete gene deletions and 3xFLAG gene tagging were done using homologous recombination-based methods57 and confirmed by PCR. Specific ___domain deletions (e.g., elf1-CTD∆, elf1-N∆, tfb1-PH∆, rad4-TIR∆) and/or point mutations in ELF1 or TFB1 were done using our published yeast CRISPR system39,40 and confirmed by Sanger sequencing. For the plasmid-based expression of Spt4, we used a plasmid in which the promoter, coding sequence, and 3’ terminator sequences of SPT4 were inserted into the multiple cloning sites of the URA3 plasmid, pRS41658 (gift from Shisheng Li, Louisiana State University, USA). Yeast strains and other materials are available upon request to the corresponding authors. Oligonucleotide sequences are given in Supplementary Data 1.
Protein expression and purification
Expression and purification of yeast Elf1 and Elf1 truncations were performed essentially as previously described23,55. Briefly, GST-tagged Elf1 protein was expressed in Escherichia coli Rosetta 2(DE3) (Novagen, #71397) and purified by Glutathione Sepharose 4 Fast Flow resin (GE Healthcare, #17513202), and Superdex 200 10/300 GL column (GE Healthcare, #17517501). Expression and purification of the PH ___domain (1–115) of TFIIH p62 subunit was performed as previously described59 with certain modifications. Briefly, GST-PH ___domain was expressed in host strain Rosetta 2(DE3), purified by Glutathione Sepharose 4 Fast Flow resin (GE Healthcare). After extensive wash, the PH ___domain was released from the resin by overnight PreScission protease cleavage.
Yeast UV sensitivity test
The yeast cells were grown in Yeast extract Peptone Dextrose (YPD) medium at 30 °C overnight. For spotting assays, these cells were 10-fold serially diluted in fresh YPD medium with the first dilution containing ~1 × 108 cells/ml and spotted on YPD plates. The plates were incubated at 30 °C in the dark after exposing to different doses of UVC light (254 nm), based on our previous calibration, and images were taken after 3–5 days of incubation. For quantitative UV survival assay, diluted yeast cells were plated on YPD plates and exposed to the indicated UV dose, based on our previous calibration. The number of colonies was counted after incubating the plates for 3 days at 30 °C in the dark. The survival graph depicts the mean and SEM of three independent experiments.
CPD-seq library preparation and sequencing
The WT or the mutant yeast cultures were grown to an OD600 of ~0.8–1.0, pelleted, and re-suspended in dH2O. Cells were collected for a “No UV” sample, and the remaining cells were irradiated with 125 J/m2 UVC light (254 nm), based on previous calibration. Cells were collected immediately after UV exposure (0 h) and the remaining cells after UV treatment were incubated in the dark in pre-warmed fresh YPD medium for repair. The cells were collected after 2 h (2 h) repair incubation at 30 °C. The cells were pelleted and stored in the −80 °C freezer until genomic DNA isolation. Genomic DNA extraction was done using phenol:chloroform:isoamylalcohol (PCI) extraction and ethanol precipitation. The CPD-seq library preparation and quality control, sequencing with an Ion Proton sequencer, and data processing were performed essentially as previously described27,28. Briefly, genomic DNA was sonicated (30 s ON/OFF, 25 cycles; Diagenode Biorupter 300) to an average size of ∼400 bp, end-repaired (NEB, E6050L), and dA-tailed (NEB, E6053L) using the indicated NEB kits. DNA was then ligated to the double-stranded adapter DNA trP1 using Quick ligase (NEB, E6056L), and treated with terminal transferase (NEB, M0315L). DNA was then digested with T4 endonuclease V (T4 PDG, NEB, M0308S) and AP endonuclease (NEB, M0282S) to create 3′-OH groups immediately upstream of the CPD lesions, followed by treatment with Shrimp alkaline phosphatase (Thermo Fisher Scientific, 78390500UN). The resulting DNA fragments were ligated to a biotin-labeled second adapter (adapter A). Up to 6 different barcoded A adapters (barcode sequences are A1: AAGAGGAT; A2: TTCGTGAT; A3: CCTGAGAT A4: ATCGCGAT; A5: TACTGGAT; and A6: GAACTGAT) were used to generate multiplexed libraries for different experiments. Fragments ligated to the A adapter were selected for with streptavidin beads (Thermo Fisher Scientific, 11205D) with a high affinity for biotin on the second adapter. These fragments were PCR amplified and sent out for Ion Torrent sequencing. Ampure XP beads (Beckman Coulter, A63881) were used for size selection and cleanup between enzymatic steps.
The resulting sequencing reads were trimmed of the barcode (and typically one nucleotide at the 3′ end of the read), aligned to the yeast genome (SacCer3) using bowtie260, and processed using SAMtools61 and BEDTools62 to generate a BED file. CPD-seq reads associated with lesions at dipyrimidine sequences (i.e., TT, TC, CT, CC) were retained for further analysis. BED files were split into individual DNA strands and converted to wig files for subsequent analysis. Dipyrimidine sequences associated with no CPD-seq reads were assigned a read count of 0. CPD-seq data for WT, rad26Δ, rad16Δ controls are from19,30,32,33. Data processing for these data sets was generally performed as described previously29. For transcriptional asymmetry analysis, the fraction of CPDs remaining in each bin for each DNA strand was averaged for the two rad16∆33 or rad26∆30 replicates prior to calculating log2 TS/NTS ratio. The other rad16∆ replicate (i.e., Supplementary Fig. 5b) is derived from a control anchor-away strain in a W303 background treated with rapamycin32.
CPD-seq data analysis
Analysis of CPD repair around the transcription start site (TSS) of ~5200 yeast genes was performed as previously described19,29,30,32,33. Briefly, custom Perl scripts analyzed the number of CPD lesions at each position on the TS and NTS adjacent to the TSS positions for ~5200 yeast genes obtained from ref. 63, both for the 2 h repair and 0 h control. The fraction of CPDs remaining in the 2 h CPD-seq data relative to the 0 h control at each position relative to the TSS was plotted. For the elf1-CTD∆ and WT control data sets, the fraction of CPDs remaining in all analyses was normalized using the fraction of unrepaired CPDs after 2 h repair determined by alkaline gel electrophoresis. Positioning of nucleosome dyad positions relative to the TSS was analyzed using published MNase-seq data64, as previously described29,30,32,33. Bin plot analysis of repair between the TSS and TES was performed similarly using custom Perl scripts, as previously described19,29,30,32,33, except that a modified Perl script that fixed a small error related to bin boundaries was used to perform bin plots of individual yeast genes. Briefly, each of ~5000 yeast genes that have coordinates for the TSS and TES (derived from position of poly-adenylation site (PAS) coordinates63) were divided into six equally sized bins, dependent upon gene length, and the number of CPDs in each bin was counted. Three additional bins uniformly 167 bp in length were also analyzed upstream of the TSS and downstream of the TES of each yeast gene. The fraction of CPDs remaining after 2 h repair relative to the 0 h control was plotted for all yeast genes, and for individual yeast genes, which were visualized using the Java TreeView software65. The log2 ratio of the fraction of CPDs remaining in the TS and NTS was also plotted. TES coordinates were derived from the published ___location of the PAS for each yeast gene63. Finally, the average log2 ratio of CPDs remaining in the TS and NTS was calculated using the average of log2 TS/NTS ratio of the six transcribed bins between the TSS and TES of yeast genes.
Global analysis of CPD repair in yeast by alkaline gel electrophoresis
The global repair of CPD lesions was analyzed by T4 endonuclease V digestion and alkaline gel electrophoresis, as described previously19,29,66. In brief, WT or elf1-CTDΔ mutant strains were grown in YPD to mid-log phase (OD600 ~ 0.65), pelleted, and re-suspended in dH2O. Cells were collected for a “No UV” control and the rest of the cells were exposed to 125 J/m2 UVC (254 nm). The UV-exposed cells were pelleted, re-suspended in YPD medium, and incubated at 30 °C with aliquots taken at different timepoints as indicated. Genomic DNA was isolated from these samples using PCI extraction and ethanol precipitation. Equal amounts of DNA (50 μg) were treated with T4 endonuclease V/PDG (NEB M0308S) and the digested samples were resolved using alkaline gel electrophoresis. The gel was subsequently stained with SYBR Gold (Invitrogen), imaged with Typhoon FLA 7000 (GE Healthcare), and quantified with ImageQuant 5.2. Graphs represent the mean and SEM of three independent experiments.
Western blot
Cells were cultured in YPD medium at 30 °C to O.D.600 ~ 1 and whole cell protein extracts were prepared from the cells using the procedure described previously67. The protein extracts were analyzed by western blot using antibodies against total histone H3 (anti-H3, Abcam, ab46765) and FLAG (anti-FLAG, Sigma-Aldrich, F1804) epitope. Both antibodies were used at a 1:10,000 dilution. Blots were scanned using Typhoon FLA 7000 (GE Healthcare).
In vitro pull-down assay
The quantities of the purified protein were assessed by Bradford assay. GST Beads containing estimated equivalent quantities of the tagged Elf1 protein truncations were mixed with the purified PH ___domain of TFIIH p62 subunit. The mixtures were incubated at 23 °C for 1 h with rotation. After centrifugation, the supernatants were removed, and the beads were washed twice in a buffer composed of 20 mM HEPES, pH 7.4, 5% glycerol. Interacting proteins were analyzed by SDS-PAGE.
Quantitative binding assays using Biolayer Interferometry
To elucidate the impact of the Elf1-Y99 residue on the binding affinity within the PH ___domain of p62, we quantified the dissociation constant (Kd) for both wild-type and Y99A mutant forms of Elf1-CTD (residues 85–145) using Biolayer Interferometry (BLI). The Octet® K2 system was utilized for these measurements, adhering strictly to the manufacturer’s guidelines. In brief, recombinant Elf1-CTD proteins (Supplementary Fig. 17; uncropped gel and blot images are in Source Data), both wild-type and Y99A mutant, tagged with glutathione S-transferase (GST), were immobilized onto GST-specific biosensors. Subsequently, the PH ___domain of p62 was serially diluted in BLI assay buffer (20 mM Tris-HCl, pH 7.5, 5% glycerol, and 1 mM DTT) to the following concentrations: 30 µM, 15 µM, 7.5 µM, 3.75 µM, and 0 µM. Each biosensor with bound Elf1-CTD was exposed to a different concentration of the PH-p62 protein for an association phase of 2 min, followed by a dissociation phase in BLI buffer for 3 min. This procedure was replicated in three independent experiments to ensure the reliability of the Kd values obtained. The response signals generated during the assay were automatically captured and processed by the Octet system’s software (Data Acquisition v12.0.2.11 and Data Analysis HT v12.0.2.59 from FORTEBIO), which was also used for the subsequent analysis and calculation of the Kd values.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The CPD-seq data generated in this manuscript have been deposited in NCBI’s Gene Expression Omnibus (GEO) database and are available under GEO series accession code GSE243603. The published CPD-seq data for WT, rad26Δ, and rad16Δ is available from GEO with the accession numbers: GSE161930GSE145911GSE149082GSE131101. Source data are provided with this paper.
Code availability
The custom Perl scripts used in the study have been deposited at GitHub: https://github.com/bmorledge-hampton19/ELF1_CTD68.
References
Lans, H., Hoeijmakers, J. H. J., Vermeulen, W. & Marteijn, J. A. The DNA damage response to transcription stress. Nat. Rev. Mol. Cell Biol. 20, 766–784 (2019).
Geijer, M. E. & Marteijn, J. A. What happens at the lesion does not stay at the lesion: transcription-coupled nucleotide excision repair and the effects of DNA damage on transcription in cis and trans. DNA Repair 71, 56–68 (2018).
Spivak, G. & Hanawalt, P. C. Photosensitive human syndromes. Mutat. Res. 776, 24–30 (2015).
Hanawalt, P. C. & Spivak, G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 9, 958–970 (2008).
van den Heuvel, D., van der Weegen, Y., Boer, D. E. C., Ogi, T. & Luijsterburg, M. S. Transcription-coupled DNA repair: from mechanism to human disorder. Trends Cell Biol. 31, 359–371 (2021).
Marteijn, J. A., Lans, H., Vermeulen, W. & Hoeijmakers, J. H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 15, 465–481 (2014).
Vermeulen, W. & Fousteri, M. Mammalian transcription-coupled excision repair. Cold Spring Harb. Perspect. Biol. 5, a012625 (2013).
Li, S. Transcription coupled nucleotide excision repair in the yeast Saccharomyces cerevisiae: the ambiguous role of Rad26. DNA repair 36, 43–48 (2015).
Scharer, O. D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 5, a012609 (2013).
Jia, N. et al. Dealing with transcription-blocking DNA damage: repair mechanisms, RNA polymerase II processing and human disorders. DNA Repair 106, 103192 (2021).
van der Weegen, Y. et al. The cooperative action of CSB, CSA, and UVSSA target TFIIH to DNA damage-stalled RNA polymerase II. Nat. Commun. 11, 2104 (2020).
Nakazawa, Y. et al. Ubiquitination of DNA damage-stalled RNAPII promotes transcription-coupled repair. Cell 180, 1228–1244.e1224 (2020).
Okuda, M., Nakazawa, Y., Guo, C., Ogi, T. & Nishimura, Y. Common TFIIH recruitment mechanism in global genome and transcription-coupled repair subpathways. Nucleic acids Res. 45, 13043–13055 (2017).
Friedberg, E. C. et al DNA repair and Mutagenesis 2nd edn (ASM Press, 2006).
Di Lello, P. et al. Structure of the Tfb1/p53 complex: insights into the interaction between the p62/Tfb1 subunit of TFIIH and the activation ___domain of p53. Mol. Cell 22, 731–740 (2006).
Okuda, M., Araki, K., Ohtani, K. & Nishimura, Y. The interaction mode of the acidic region of the cell cycle transcription factor DP1 with TFIIH. J. Mol. Biol. 428, 4993–5006 (2016).
Okuda, M., Kinoshita, M., Kakumu, E., Sugasawa, K. & Nishimura, Y. Structural insight into the mechanism of TFIIH recognition by the acidic string of the nucleotide excision repair factor XPC. Structure 23, 1827–1837 (2015).
Okuda, M. et al. Structural insight into the TFIIE-TFIIH interaction: TFIIE and p53 share the binding region on TFIIH. EMBO J. 27, 1161–1171 (2008).
Geijer, M. E. et al. Elongation factor ELOF1 drives transcription-coupled repair and prevents genome instability. Nat. Cell Biol. 23, 608–619 (2021).
van der Weegen, Y. et al. ELOF1 is a transcription-coupled DNA repair factor that directs RNA polymerase II ubiquitylation. Nat. Cell Biol. 23, 595–607 (2021).
Carnie, C. J. & Jackson, S. P. The ELOF(1)ant in the room of TCR. Nat. Cell Biol. 23, 584–586 (2021).
Ehara, H. et al. Structural insight into nucleosome transcription by RNA polymerase II with elongation factors. Science 363, 744–747 (2019).
Ehara, H. et al. Structure of the complete elongation complex of RNA polymerase II with basal factors. Science 357, 921–924 (2017).
Prather, D., Krogan, N. J., Emili, A., Greenblatt, J. F. & Winston, F. Identification and characterization of Elf1, a conserved transcription elongation factor in Saccharomyces cerevisiae. Mol. Cell. Biol. 25, 10122–10135 (2005).
Mayer, A. et al. Uniform transitions of the general RNA polymerase II transcription complex. Nat. Struct. Mol. Biol. 17, 1272–1278 (2010).
Joo, Y. J., Ficarro, S. B., Chun, Y., Marto, J. A. & Buratowski, S. In vitro analysis of RNA polymerase II elongation complex dynamics. Genes Dev. 33, 578–589 (2019).
Mao, P. & Wyrick, J. J. Genome-wide mapping of UV-induced DNA damage with CPD-Seq. Methods Mol. Biol. 2175, 79–94 (2020).
Mao, P., Smerdon, M. J., Roberts, S. A. & Wyrick, J. J. Chromosomal landscape of UV damage formation and repair at single-nucleotide resolution. Proc. Natl Acad. Sci. USA 113, 9057–9062 (2016).
Selvam, K., Plummer, D. A., Mao, P. & Wyrick, J. J. Set2 histone methyltransferase regulates transcription coupled-nucleotide excision repair in yeast. PLoS Genet. 18, e1010085 (2022).
Duan, M., Selvam, K., Wyrick, J. J. & Mao, P. Genome-wide role of Rad26 in promoting transcription-coupled nucleotide excision repair in yeast chromatin. Proc. Natl Acad. Sci. USA 117, 18608–18616 (2020).
Duan, M. et al. High-resolution mapping demonstrates inhibition of DNA excision repair by transcription factors. Elife 11, e73943 (2022).
Bohm, K. A. et al. Distinct roles for RSC and SWI/SNF chromatin remodelers in genomic excision repair. Genome Res. 31, 1047–1059 (2021).
Mao, P., Smerdon, M. J., Roberts, S. A. & Wyrick, J. J. Asymmetric repair of UV damage in nucleosomes imposes a DNA strand polarity on somatic mutations in skin cancer. Genome Res. 30, 12–21 (2020).
Jansen, L. E. et al. Spt4 modulates Rad26 requirement in transcription-coupled nucleotide excision repair. EMBO J. 19, 6498–6507 (2000).
Gaillard, H. et al. Genome-wide analysis of factors affecting transcription elongation and DNA repair: a new role for PAF and Ccr4-not in transcription-coupled repair. PLoS Genet. 5, e1000364 (2009).
Kujirai, T. et al. Structural basis of the nucleosome transition during RNA polymerase II passage. Science 362, 595–598 (2018).
Li, S. & Smerdon, M. J. Rpb4 and Rpb9 mediate subpathways of transcription-coupled DNA repair in Saccharomyces cerevisiae. EMBO J. 21, 5921–5929 (2002).
Obradovic, Z., Peng, K., Vucetic, S., Radivojac, P. & Dunker, A. K. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins 61, 176–182 (2005).
Laughery, M. F. & Wyrick, J. J. Simple CRISPR-Cas9 genome editing in Saccharomyces cerevisiae. Curr. Protoc. Mol. Biol. 129, e110 (2019).
Laughery, M. F. et al. New vectors for simple and streamlined CRISPR-Cas9 genome editing in Saccharomyces cerevisiae. Yeast 32, 711–720 (2015).
Gong, W., Holmberg, H., Lu, C., Huang, M. & Li, S. Interplay of the Tfb1 pleckstrin homology ___domain with Rad2 and Rad4 in transcription coupled and global genomic nucleotide excision repair. Nucleic Acids Res. 52, 6333–6346 (2024).
Evans, R. et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv, 2021.10.04.463034 (2022).
Olivieri, M. et al. A genetic map of the response to DNA damage in human cells. Cell 182, 481–496.e421 (2020).
Lafrance-Vanasse, J., Arseneault, G., Cappadocia, L., Legault, P. & Omichinski, J. G. Structural and functional evidence that Rad4 competes with Rad2 for binding to the Tfb1 subunit of TFIIH in NER. Nucleic Acids Res. 41, 2736–2745 (2013).
Di Lello, P. et al. p53 and TFIIEalpha share a common binding site on the Tfb1/p62 subunit of TFIIH. Proc. Natl Acad. Sci. USA 105, 106–111 (2008).
Okuda, M. & Nishimura, Y. Extended string binding mode of the phosphorylated transactivation ___domain of tumor suppressor p53. J. Am. Chem. Soc. 136, 14143–14152 (2014).
Verhage, R. et al. The RAD7 and RAD16 genes, which are essential for pyrimidine dimer removal from the silent mating type loci, are also required for repair of the nontranscribed strand of an active gene in Saccharomyces cerevisiae. Mol. Cell. Biol. 14, 6135–6142 (1994).
Verhage, R. A., Van de Putte, P. & Brouwer, J. Repair of rDNA in Saccharomyces cerevisiae: RAD4-independent strand-specific nucleotide excision repair of RNA polymerase I transcribed genes. Nucleic Acids Res. 24, 1020–1025 (1996).
Ding, B., Ruggiero, C., Chen, X. & Li, S. Tfb5 is partially dispensable for Rad26 mediated transcription coupled nucleotide excision repair in yeast. DNA Repair 6, 1661–1669 (2007).
Li, S. et al. The roles of Rad16 and Rad26 in repairing repressed and actively transcribed genes in yeast. DNA Repair 6, 1596–1606 (2007).
Prakash, S. & Prakash, L. Nucleotide excision repair in yeast. Mutat. Res. 451, 13–24 (2000).
Lafrance-Vanasse, J. et al. Structural and functional characterization of interactions involving the Tfb1 subunit of TFIIH and the NER factor Rad2. Nucleic Acids Res. 40, 5739–5750 (2012).
Sarsam, R. D. et al. Elf1 promotes Rad26’s interaction with lesion-arrested Pol II for transcription-coupled repair. Proc. Natl Acad. Sci. USA 121, e2314245121 (2024).
Tufegdžić Vidaković, A. et al. Regulation of the RNAPII pool is integral to the DNA damage response. Cell 180, 1245–1261.e1221 (2020).
Xu, J., Chong, J. & Wang, D. Opposite roles of transcription elongation factors Spt4/5 and Elf1 in RNA polymerase II transcription through B-form versus non-B DNA structures. Nucleic Acids Res. 49, 4944–4953 (2021).
Kubinski, K., Zielinski, R., Hellman, U., Mazur, E. & Szyszka, R. Yeast elf1 factor is phosphorylated and interacts with protein kinase CK2. J. Biochem. Mol. Biol. 39, 311–318 (2006).
Brachmann, C. B. et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132 (1998).
Sikorski, R. S. & Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 (1989).
Di Lello, P. et al. NMR structure of the amino-terminal ___domain from the Tfb1 subunit of TFIIH and characterization of its phosphoinositide and VP16 binding sites. Biochemistry 44, 7678–7686 (2005).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Park, D., Morris, A. R., Battenhouse, A. & Iyer, V. R. Simultaneous mapping of transcript ends at single-nucleotide resolution and identification of widespread promoter-associated non-coding RNA governed by TATA elements. Nucleic Acids Res. 42, 3736–3749 (2014).
Weiner, A. et al. High-resolution chromatin dynamics during a yeast stress response. Mol. Cell 58, 371–386 (2015).
Saldanha, A. J. Java Treeview–extensible visualization of microarray data. Bioinformatics 20, 3246–3248 (2004).
Hodges, A. J., Plummer, D. A. & Wyrick, J. J. NuA4 acetyltransferase is required for efficient nucleotide excision repair in yeast. DNA Repair 73, 91–98 (2019).
Kushnirov, V. V. Rapid and reliable protein extraction from yeast. Yeast 16, 857–860 (2000).
Selvam, K. et al. Elf1 promotes transcription-coupled repair in yeast by using its C-terminal ___domain to bind TFIIH. Github https://doi.org/10.5281/zenodo.11403759 (2024).
Holstege, F. C. et al. Dissecting the regulatory circuitry of a eukaryotic genome. Cell 95, 717–728 (1998).
Acknowledgements
We are grateful to Dr. Shisheng Li for providing the pRS416-SPT4 plasmid. We thank Benjamin Morledge-Hampton for assistance in depositing the Perl scripts. This work was supported by grants from the National Institute of General Medical Sciences (R01 GM102362 to D.W.) and the National Institute of Environmental Health Sciences (R01ES028698 (J.J.W.), R01ES032814 (J.J.W.), and R21ES035139 (J.J.W.)).
Author information
Authors and Affiliations
Contributions
K.S., J.X., H.E.W., D.W., and J.J.W. designed the research and interpret results; K.S. performed the yeast UV sensitivity and CPD-seq experiments; J.X., J.O., and Q.L. purified proteins and perform in vitro pull-down and binding assays. K.S. and J.J.W. analyzed the CPD-seq data; H.E.W. performed the alkaline gel repair studies; K.S., J.X., H.E.W., J.O., Q.L., D.W., and J.J.W. wrote and edited the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Selvam, K., Xu, J., Wilson, H.E. et al. Elf1 promotes transcription-coupled repair in yeast by using its C-terminal ___domain to bind TFIIH. Nat Commun 15, 6223 (2024). https://doi.org/10.1038/s41467-024-50539-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50539-y
