
Abstract
Hydrogen oxidation reaction in alkaline media is critical for alkaline fuel cells and electrochemical ammonia compressors. The slow hydrogen oxidation reaction in alkaline electrolytes requires large amounts of scarce and expensive platinum catalysts. While transition metal decoration can enhance Pt catalysts’ activity, it often reduces the electrochemical active surface area, limiting the improvement in Pt mass activity. Here, we enhance Pt catalysts’ activity without losing surface-active sites by using a Pd-Ru pair. Utilizing a mildly catalytic thermal pyrolysis approach, Pd-Ru pairs are decorated on Pt, confirmed by extended X-ray absorption fine structure and high-angle annular dark-field scanning transmission electron microscopy. Density functional theory and ab-initio molecular dynamics simulations indicate preferred Pd and Ru dopant adsorption. The Pd-Ru decorated Pt catalyst exhibits a mass-based exchange current density of 1557 ± 85 A g−1metal for hydrogen oxidation reaction, demonstrating superior performance in an ammonia compressor.
Similar content being viewed by others

Introduction
The sluggish kinetics of electrochemical hydrogen and oxygen redox reactions in alkaline electrolytes hinder the wide application of renewable energy conversion and storage devices1 that use alkaline fuel cells2,3 and electrochemical gas compressors4. State-of-the-art electrocatalysts for these electrochemical conversion systems are usually platinum group metal (PGM)-based nanoparticles. However, the high cost of the Pt-based electrocatalysts severely limits their widespread adoption. An effective method for reducing the cost is to substantially reduce the amount of Pt required by enhancing the catalytic activity per given mass of Pt, i.e., Pt mass activity (MA).
The MA depends on the electrochemically active surface area (ECSA) and the specific activity (SA, normalized by surface area). The ECSA can be enhanced by tailoring the geometrical structure, including ultra-small nanostructure5 and core-shell nanostructure with Pt skin6. The SA can be increased by modifying or decorating the Pt nanoparticle surface. Surface modification or decoration of Pt catalysts has been used for boosting the hydrogen evolution reaction (HER)7,8 and the oxygen reduction reaction (ORR)9. For instance, decorating the Pt nanostructure surface using Ni(OH)2 clusters8 greatly boosts the SA for the HER in alkaline electrolytes. However, such modification also reduces the ESCA by 30–50%. For surface modification, it is extremely challenging to increase the SA without substantially sacrificing the ECSA, which is necessary for achieving a high MA in both half-cells and full cells. The hydrogen oxidation reaction (HOR) enhancement by surface modification of Pt-based catalysts has rarely been reported before.
To boost the HOR (H2 + 2OH- → 2H2O + 2e-) on Pt, adsorption and activation of a large amount of OHads and Hads surrounding Pt atoms are critical, which can be realized by decorating with Ru and Pd on the Pt surface, respectively. On Ru- and Pd-decorated Pt nanoparticles, the Ru and Pd act as catalyst promoters to increase the availability of OHads and Hads surrounding Pt atoms, respectively. (Fig. 1 and Supplementary Fig. 1) It is critical to reduce the decorating species’ sizes to prevent surface Pt active sites from being blocked while producing as many activated Pt sites with adjacent Ru and Pd atoms as possible for improved SA for the HOR. The ultimate choice to achieve the highest MA is using surface atoms to decorate Pt, which allows the smallest number of Ru and Pd species to activate the most Pt atoms while minimizing blocking of surface Pt sites. However, the effect of Ru and Pd pair decoration on the Pt surface on hydrogen redox reaction has not been investigated insightfully due to the limitation of current synthetic approaches10,11,12,13,14.

The decoration of Pt nanoparticles with paired Pd-Ru species increases the availability of Hads and OHads on the Pt surface to boost the specific catalytic activity for the HOR without sacrificing the ECSA.
Here, we create paired Pd-Ru doped Pt nanoparticles (Pd-Ru@Pt) with a high exchange current density and long durability. The surface area and mass-based exchange current density (3.28 ± 0.23 mA cm−2Pt and 1557 ± 85 A g−1metal) of Pd-Ru@Pt are 7.8 and 7.7 times those of the Pt/C. Additionally, after degradation test, the surface area and mass-based exchange current density of the Pd-Ru@Pt/C were still 8.0 and 8.4 times those of the Pt/C. The uniform decoration structure of Pt nanoparticle surfaces by Pd-Ru pairs, without sacrificing electrochemical surface active area, is validated using extended X-ray absorption fine structure (EXAFS), aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), and electrochemical characterization. Density functional theory (DFT) calculations reveal that both transition metals, Pd and Ru, prefer to form bonds with Pt atoms at the Pt(110) surface rather than forming clusters. Also, ab-initio molecular dynamics (AIMD) simulations reveal that both atoms (Pd and Ru) are not very mobile at the (110) surface which reduces the probability of association.
Results and discussion
Synthesis and geometric structure
Inspired by the finding that nitrogen-rich carbon defect trapping Cu(NH3)x15 can be used to prepare a single Cu atom catalyst, we used a vacuum-assisted solid-phase synthetic approach to decorate Pd-Ru@Pt surfaces. As shown in Fig. 2a, we put carbon-supported Pt nanoparticles (Pt/C) on a silicon wafer, and loaded Ruthenium(III) acetylacetonate (Ru(acac)3) and Palladium(II) acetylacetonate (Pd(acac)2) in a combustion boat, and then transferred them into a vacuumed glass tube. The Ru(acac)3 and Pd(acac)2 were chosen as Pd and Ru precursors because of their low evaporation temperatures. The glass tube was heated to 300 °C for 3.0 h. The vaporized Pd and Ru precursors were trapped by the Pt nanoparticles’ surface defects and hollow sites at high temperatures, generating the isolated Pd and Ru atoms on the Pt surface via a catalytic pyrolysis effect of Pt forming Pd-Ru@Pt/C. As reference catalysts, atomic Pd doped Pt/C (Pd@Pt/C) and atomic Ru doped Pt/C (Ru@Pt/C) were also synthesized using the same method.

a Scheme for synthesizing Pd-Ru@Pt through a chemical vapor deposition (CVD) setup. Normalized Fourier Transform of the k³.χ(k) Extended X-Ray Absorption Fine Structure (EXAFS) spectra of (b) Pt, (c) Pd and (d) Ru. EXAFS of metallic Pt and PtO2 are shown in (b) as ref.37. EXAFS of metallic Pd and PdO are shown in (c) as ref. 38. EXAFS of metallic Ru and RuO2 are shown in (d) as ref. 39. Source data for (b)–(d) are provided as a Source Data file.
The morphology of Pd-Ru@Pt was characterized using transmission electron microscopy (TEM). Supplementary Fig. 2 shows the Pd-Ru@Pt catalysts with an average particle size of 2.9 ± 0.3 nm dispersed uniformly on the carbon support. Since the heat treatment only changed the average particle size of the Pt nanoparticle by 0.1 nm from 2.6 ± 0.3 nm (Supplementary Fig. 3) to 2.7 ± 0.4 nm (Supplementary Fig. 4), the thickness changes after surface decoration corresponds to a single particle coating of half atomic layer or less, considering the standard deviation. For the reference catalysts, the Pd@Pt/C (Supplementary Fig. 5) and Ru@Pt/C (Supplementary Fig. 6) showed good dispersion and crystal structure, with average particle sizes 2.9 ± 0.4.
Pd and Ru decoration on Pt for Pd-Ru@Pt/C catalysts was directly evidenced by extended X-ray absorption fine structure (EXAFS). The Pt EXAFS in Pd-Ru@Pt/C (Fig. 2b) displays a main peak at ∼2.638 Å, close to that of the Pt–Pt bond length in bulk Pt (2.641 Å in Pt foil). Pt-Pd and Pt-Ru bonds at 2.148 Å are also observed in the EXAFS. The Pd EXAFS in Pd-Ru@Pt/C (Fig. 2c) showed dominant features of a Pd-O peak at ∼1.473 Å and a Pd-Pt peak at ∼2.730 Å over the Pd-Pd bond at ∼2.466 Å (Fig. 2c), while the Ru EXAFS in Pd-Ru@Pt/C (Fig. 2d) showed dominant features of a Ru-Pt peak at ∼2.729 Å over a Ru-Ru bond at ∼2.382 Å. Therefore, Pd-Ru decoration dominated over Pd cluster or Ru cluster formation for Pd-Ru@Pt/C.
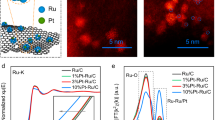
Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was used to probe the material’s atomic structure. As Pd and Ru have lower Z-contrast than that of Pt due to the large atomic number difference, the darker atomic columns on the edges (Fig. 3a) corresponds to the Pd-Ru decoration on the outmost surface of a Pt nanoparticle. Additionally, the image intensity line profile across the nanoparticle illustrates the significantly low HAADF contrast of the surface Pd/Ru atoms, as highlighted by black arrows in Fig. 3b. As shown in Fig. 3a, the Pd-Ru atoms on the outermost layer are not continuous. This implies that the Pt core may not be entirely covered by foreign doping atoms. After examining multiple particles from randomly picked sample locations (Supplementary Fig. 7a), this representative feature of low-contrast Pd-Ru single-layer shell surrounding high-contrast Pt core was generally observed, and no distinct multi-layer aggregation of Pd-Ru was found. Furthermore, STEM with simultaneously obtained energy-dispersive X-ray spectroscopy (EDS) provides the elemental mapping of Pd, Ru, and Pt (Fig. 3c, Supplementary Fig. 7b), further confirming the Pd-Ru@Pt core-shell structure. Pd and Ru’s relatively uniform spatial distribution implies that most Pd and Ru atoms are likely in the homogeneous and alternate atomic arrangement, confirming the Pd-Ru decoration derived from EXAFS (Fig. 2c, d).

a Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of Pd-Ru@Pt. The surface atoms in darker contrast are Pd-Ru sites. b Line profile of the image intensity along the arrow direction (indicated in the inset) across the individual nanoparticle shown in (a), showing Pd-Ru atoms located on the outmost surface (marked by black arrows). c HAADF-STEM image and the corresponding EDS mapping of individual Pd-Ru@Pt particles with the same type of elemental distribution. Individual elemental distribution of Pd, Ru, and Pt, and the overlaid mixture of Pd+Pt, Ru+Pt, and Pd+Ru+Pt illustrate the spatial distributions of Pd and Ru on the surface and Pt in the core.
Structure and thermodynamic stability
To gain an atomic-level understanding of the dopant decoration, the preferred adsorption sites of the Pd and Ru atoms at the top (T), short bridge (SB), and long bridge (LB) of the pristine and defective Pt(110) surface were analyzed from a thermodynamic standpoint using DFT calculations (Supplementary Fig. 8a–f). In Supplementary Fig. 8b, the outermost Pt atoms are labeled as the crest, and the bottom accessible Pt atoms are labeled as the trough region. The defective Pt(110) (herein, rPt(110)) was constructed by removing four Pt atoms from a crest row. Details regarding the construction of the surfaces and all sites tested are provided as SI. It is noteworthy that the models were constructed based on the [110] facet since it represents the highly active and stable surface for HOR in alkaline solutions16,17,18. The rPt(110) surface slab is akin to the reconstruction of well-ordered surfaces that become prominent upon annealing. The analysis was restricted to the adsorption of dopants to the detriment of the absorption process. The ground state energy geometry found under vacuum conditions predicted that the optimized site for both Pd and Ru adsorption on top of the pristine Pt(110) surface is the hollow site in the trough region (Supplementary Fig. 8c-f)), for both the pristine and the rPt(110) models. For the adsorption of Pd on the pristine Pt(110), the optimized configuration at the hollow site is 1.82 eV more thermodynamically stable than the adsorption at the top site (the least preferred configuration). Regarding the Ru adsorption, the optimized configuration is 1.03 eV more stable than Ru adsorbing at a top site. On the other hand, for the rPt(110) model, the Pd and Ru adsorptions at the hollow sites are 1.25 eV and 1.72 eV more stable than the least preferred sites, respectively.
To explore whether the Pd or Ru atom prefers to deposit on the Pt(110) surface as a dopant or as a clustered dopant, we calculated the binding energy of a Pd (or Ru) dopant on the Pt (110) surface and the energy required to add a dopant once a Pd (or Ru) dopant was already adsorbed at the hollow site in the trough region. The binding energy (Ebinding) per atom (n) was calculated as the difference between the total energy of the optimized substrate-adsorbate and the sum of the relaxed, clean surface (Eslab) and isolated dopants (ETM).
Upon optimization of the Pd (or Ru) adsorption on the pristine and rPt (110) surfaces (herein, Pd-Pt or Ru-Pt), the average binding energies of Pd-Pt (−3.82 eV) are larger than either of the Pd-Pd (−0.54 eV) and Pd-Ru (−1.07 eV) binding energy. Also, the average binding energy of Ru-Pt (−6.75 eV) is larger than either of the Ru-Ru (−2.80 eV) and Ru-Pd (−1.07 eV) binding energies. Therefore, both transition metals, Pd and Ru, prefer to form bonds with Pt atoms at the Pt(110) surface rather than forming clusters. To further investigate the long-term Pd-Ru@Pt(110) stability, the energetic cost of sequential dopant addition (assuming a Ru or Pd is already adsorbed at the (110) surface) was simulated. For these calculations, the energy of the added dopant was set to its calculated cohesive energy value. The average binding energy to add a Ru atom and form a Ru-Ru on the Pt surface is −0.06 eV. The average binding energy to add a Pd atom and form a Pd-Pd on the Pt surface is −0.19 eV. Meanwhile, the average binding energy to add a Pd atom and form Ru and Pd dopants adsorbed at the Pt (110) surface is −0.77 eV. Inspection of the individual energies of both surfaces shows more negative energy on the pristine Pt(110) surface (−1.42 eV) when compared to the rPt(110) surface (−0.12 eV). In other words, using a Pd-Ru@Pt(110) as a baseline, further stabilizes the decoration system because the atoms might sit in deeper potential wells of the hollow site that inhibits the atoms from clustering.
The stability of the adsorbed dopants and the evolution of the Pd-Ru distance on the Pd-Ru@Pt catalyst were investigated using AIMD simulations. The effect of an aqueous solution was considered by constructing a neutral electrode/alkaline electrolyte interface containing thirteen H2O molecules and two K+/OH- ions. The K+ ions were fixed a few angstroms above the surface, while the OH- ions were allowed to evolve with the system. As shown in Fig. 4a–c (top view) and d-f (side view), the Pd and Ru atoms remained adsorbed at a hollow site in the trough region on the outermost surface of the Pd-Ru@Pt (110) over time.

Snapshots of the time evolution of the Pd-Ru@Pt (110) slab shown at (a–c) for the top view configurations, and side view configurations for (d) 0 fs, (e) 10,000 fs and (f) 20,000 fs. Color code: Gray, blue, yellow, white, red, and purple spheres represent Pt, Pd, Ru, H, O, and K, respectively. The model is represented in a ball and stick style for clarity.
Electronic structure and hydrogen adsorption energy as descriptors for the HOR
An XPS experiment was used to study the dopant’s electronic effect on Pt (Supplementary Fig. 9 and Supplementary Table 1). The Pt0: Pt2+ ratio of Pd-Ru@Pt/C (83.15: 16.85) from deconvoluted high-resolution Pt XPS spectra was larger than either of Pd@Pt/C (80.34: 19.66) or Ru@Pt/C (80.87: 19.13) Pt0: Pt2+ ratio, which favors Hads activation and subsequent oxidation reaction on Pd-Ru@Pt/C2,19,20. The Pt0 electron binding energy, which is higher in Pd-Ru@Pt (72.05 eV) than in Pt (71.84 eV), enhances the HOR kinetics by weakening hydrogen adsorption on the surface5. In contrast, the Pt0 electron binding energy in Pd@Pt/C negatively shifts to 71.62 eV from 71.84 eV in Pt/C. The Ru-doping on Pt/C does not significantly change the Pt0 electron binding energy, ruling out Ru’s electronic effect on the HOR performance for Ru@Pt/C. Furthermore, after cycling, the high ratio of Pt0: Pt2+ and a Pt0 electron binding energy that is higher for Pd-Ru@Pt/C than for Pt/C are maintained, confirming the stability of as-prepared catalysts. Additionally, Pt, Pd, and Ru’s oxidation states were probed by the white line intensity in the XANES spectra. The Pt L3-edge white line intensity of Pd-Ru@Pt/C is close to that of Pt foil (Fig. 5a), indicating that the average oxidation state of Pt is primarily zero. However, the white line intensity of the Pd K-edge (Fig. 5b) from Pd-Ru@Pt/C features a combination of metallic Pd and PdO. For the Ru K edge (Fig. 5c), the energy absorption edge fell between metallic Ru foil and RuO2, suggesting the co-existence of metallic and oxidized states.

a Pt L3-edge, (b) Pd K-edge, and (c) Ru K edge X-ray Absorption Near-Edge Structure (XANES) spectra. XANES of metallic Pt and PtO2 are shown in (a) as ref.37. XANES of metallic Pd and PdO are shown in (b) as ref.38. XANES of metallic Ru and RuO2 are shown in (c) as Ref.39. Source data for (a)–(c) are provided as a Source Data file.
In addition to XPS and XANES data, the effects of the dopants on local environment of Pt (110) were studied with a theoretical approach. Specifically, the effect of dopants on the catalyst surface was quantified in terms of hydrogen adsorption energy. The hydrogen adsorption energy is defined as the total energy difference between the pristine and doped surface slab and the sum of the slab with hydrogen atoms adsorbed. Different arrangements of H adsorption at the pristine and doped (110) surface were investigated and described in Supplementary Table 2 and Supplementary Table 3. In this respect, our calculations show that the preferred hydrogen adsorption site at the pristine surface is the top site (T) with an −0.56 eV hydrogen binding energy. The next preferred site is the SB site, with a slightly lower value of −0.54 eV. Finally, LB is the least preferred site with a −0.22 eV. In a reconstructed surface, new sites become available for hydrogen adsorption, as shown in Supplementary Fig. 8b. The trough region becomes the preferred adsorption site with a value of −0.60 eV, and the crest sites (top and short-bridge) have an equal binding energy of −0.43 eV. The T site at the edge atoms is the second preferred site with a −0.50 eV. Upon the Pd-Ru dopant addition, our calculations show that the hydrogen binding energies on the Pd-Ru@Pt(110) catalyst are similar to those obtained for the pristine catalyst. The SB sites show slightly reduced hydrogen binding energy values of −0.50 eV and −0.48 eV in the scenarios where the H atoms are adsorbed at SB sites near the Pd and Ru atoms, respectively. Interestingly, in the defective catalyst (Pd-Ru@rPt(110)), the hydrogen binding energies at the SB sites for Pd and Ru atoms are lowered to −0.24 eV and −0.25 eV, respectively. The energy for hydrogen binding at the top site of the edge row is also reduced to −0.41 eV and −0.35 eV for the Pd and Ru atoms, respectively. Finally, the hydrogen binding energy at the SB and T sites of the crest row was slightly reduced to −0.38 eV and −0.40 eV, respectively.
Electrochemical performance
The effects of surface decoration on electrocatalytic behavior in electrolytes with different pH were explored using a rotating disk electrode (RDE) test in both 0.1 M KOH and 0.1 M HClO4. Supplementary Fig. 10a confirms that the decreased HOR activities of Pt in alkaline electrolytes compared to that in acidic electrolytes is due to the increased HBE2,17,21,22 or destabilized OH adsorption16,17, as evidenced by the 145 mV positive shift of the OH and H associated peak in the cyclic voltammogram (CV)2,23. After the Pd-Ru co-doping on the Pt surface, the OH and H associated peak of the Pd-Ru@Pt/C in the alkaline electrolyte shifted negatively to 122 mV vs. RHE from 274 mV vs. RHE for the Pt/C (Fig. 6a), which is even comparable to that of the Pd-Ru@Pt/C in acidic electrolytes (Supplementary Fig. 10b). In contrast, only Pd or Ru doping on a Pt surface does not significantly shift the peak-current potential of Pt in alkaline electrolytes (Supplementary Fig. 10a, c, d). In 0.1 M KOH, the Pd-Ru co-doping on Pt negatively shifted the desorption peak potential indicating the weakened dissociated Hads or stabilized OH adsorption compared with the reference Pt/C (Fig. 6a), which contributed to HOR performance improvement. From a computational standpoint, we conduct a mechanistic study to shed light on the bond breaking and reforming processes.

a CV curves of different catalysts. Test condition: 0.05 to 1.00 V vs. RHE, 50 mV s−1, N2-saturated 0.1 M KOH. b The linear fits (lines) in the micro-polarization region for different catalysts at 10 mV s−1 and 1600 rpm in H2-saturated 0.1 M KOH. All measurements were carried out at 298 K, corrected for the mass-transport and ohmic drop. The resistance of the electrolyte solution in the respective setup was determined to be 51.7 ± 0.5 Ω. c Comparison of ECSA (black arrow, left axis), surface area-based (SA, normalized by ECSA, orange arrow, right axis) and mass-based exchange current density (MA, normalized by metal mass, purple arrow, right axis) for HOR in H2-saturated 0.1 M KOH, obtained by linear fitting in the micro polarization region for all tested catalysts. Error bars represent the standard deviation of three independent tests for each catalyst. d Comparison of limit current density of Pt/C and Pd-Ru@Pt/C under NH3 pump mode in electrochemical cell. Source data for Fig. 6a-d are provided as a Source Data file.
The exchange current densities are obtained by linearly fitting the kinetic current versus the mass-transport and ohmic resistance-corrected24,25 potential between -5 and 5 mV vs. RHE (Method part, Fig. 6b). In addition, the ECSAs of catalysts were obtained by measuring the under-potential copper deposition (Cuupd)20 (Supplementary Fig. 11). The similar ECSAs after Pd and Ru decoration on Pt/C (Fig. 6c) prove that the distribution of Pd and Ru dopants does not block the active surface. Furthermore, from Fig. 6c, Supplementary Table 4 and Supplementary Table 5, the surface area and mass-based exchange current density of Pt/C were determined to be 0.42 ± 0.03 mA cm−2Pt and 201 ± 17 A g−1metal, respectively, in good agreement with those reported by other groups17,25,26,27,28. The surface area-specific exchange current density (3.28 ± 0.23 mA cm−2Pt) of Pd-Ru@Pt/C is 7.8 times that of the Pt/C (0.42 ± 0.03 mA cm−2Pt), 3.2 times that of Pd@Pt/C (1.03 ± 0.06 mA cm−2Pt) and 1.9 times that of Ru@Pt/C (1.72 ± 0.13 mA cm−2Pt). Normalized to metal mass, the mass-based exchange current density (1557 ± 85 A g−1metal) of Pd-Ru@Pt/C is 7.7 times that of the Pt/C (201 ± 17 A g−1metal), 3.2 times that of Pd@Pt/C (493 ± 21 A g−1metal) and 1.9 times that of Ru@Pt/C (829 ± 78 A g−1metal). Therefore, compared with either Pd or Ru modification, the Pd-Ru pair more efficiently improves the specific and mass exchange current density of Pt. Moreover, the electrochemical durability of the Pd-Ru@Pt/C in an alkaline electrolyte was also assessed by accelerated stability tests between 0.05 and 0.4 V (vs. RHE) at 100 mV s−1 in N2-saturated 0.1 M KOH. The surface area and mass-based exchange current density of the Pd-Ru@Pt/C after 10,000 cycles were still much higher than those for the Pt/C (8.0 and 8.4 times, respectively) (Supplementary Fig. 12, Supplementary Fig. 13).
We also used AIMD simulations combined with the slow-growth sampling approach to shed light on the structure and dynamics of the pristine and Pd-Ru@Pt(110) systems, focusing on assessing the free energy profiles of key reactions. Specifically, we investigated the H2 decomposition process, comparing the behavior at pristine and PdRu-doped Pt(110) surfaces. For these simulations, we used the equilibrated models of the pristine Pt(110) and Pd-Ru@Pt(110) systems (this latter model is shown in Fig. 4). Regarding the H2 decomposition process, we note that the H2 decomposition in a standard AIMD simulation is only a few femtoseconds long, in accordance with the ab-initio studies by Ishikawa and co-workers29. In the following, two slow-growth simulations that compare the H2 decomposition process at the pristine and PdRu-doped Pt(110) surface are discussed in detail. Here, Supplementary Fig. 14a–f summarizes the slow-growth trajectory of a simulation using the pristine Pt(110) surface, where an H2 molecule is adsorbed at the top position of the (110) surface. At the pristine Pt(110) surface, our simulations revealed the steps involved in H2 decomposition, showing an initial elongation of the H–H bond followed by movement towards adjacent Pt atoms. This process exhibited an energy barrier of approximately 0.27 eV. The free energy profile of these dynamics is shown in Supplementary Fig. 14f. The initial H–H bond elongation is facile, but then it overcomes a 0.27 eV barrier. The free energy of the final state shows a value of approximately −0.11 eV: thus, resulting in a strengthened Pt-H interaction.
Meanwhile, at the Pd-Ru@Pt(110) surface (Fig. 7), H2 decomposition process overcomes an energy barrier of approximately 0.13 eV. This is a 0.14 eV reduction in the energy barrier when compared to the pristine model. In this decomposition process, the moving H atom is quickly located at a bridge-like position between a top Pt atom and the Pd atom. Thus, the presence of the Pd atom modifies the bonding environment of H atoms. This finding suggests that Pd dopants aid in the H2 decomposition.

a–e The H2 dissociation and mobility reaction mechanism across the Pd-Ru@Pt(110) catalyst. f The free energy associated with this reaction mechanism. Color code: Gray, white, red, purple, yellow, and blue spheres represent Pt, H, O, K, Ru, and Pd atoms, respectively. The model is represented in a ball and stick style for clarity. Source data for (f) are provided as a Source Data file.
Supplementary Fig. 15a–e show the entire H2 decomposition and Supplementary Fig. 15f shows the free energy profile of this reaction. Considering the weakened adsorption of hydrogen atoms on the Pd-Ru@rPt(110) catalyst, we extended our simulations to investigate H2 dissociation at this surface, revealing similarities to the process observed in the Pd-Ru@Pt(110) catalyst. Despite a slightly larger energy barrier of approximately 0.16 eV, the final state energy value was comparable.
Due to these similarities, next, we calculated the H2O formation at the Pd-Ru@Pt(110) catalyst only. Moving to the formation of water (H2O), we observed differences in the behavior of OH molecules at the two surfaces. At the pristine Pt(110) catalyst surface, OH was adsorbed in Supplementary Fig. 16a, with subsequent water formation exhibiting a high energy barrier compared to the Pd-Ru@Pt(110) surface (0.91 eV versus 0.48 eV shown in Supplementary Fig. 16). We note that the OH is adsorbed at the catalyst’s surface (Supplementary Fig. 16a). However, Supplementary Fig. 16b–d shows the OH molecule slightly away from the surface. Thus, this Pt-O bond is dynamic, and the formed water molecule can stay adsorbed at the surface, as shown in Supplementary Fig. 16e, f. However, due to the absence of a potential in the calculation, we will not discuss the rate-limiting steps in the HOR. Instead, based on the relative reduction in the energy barrier observed at the Pd-Ru@Pt(110) surface we speculate that the dopants may form catalytic centers able to enhance the adsorption of OH molecules thus facilitating the HOR. It is noteworthy that our simulations at the Pt(110) surface reveal at some instances the interactions between an adsorbed OH and a water molecule in the interface region. This dynamic interaction leads to a proton from water bonding to the adsorbed OH forming adsorbed water and an OH molecule in the interface region. Thus, we speculate that the adsorbed H atom may compete with this exchange for an available OH molecule, making the water formation more difficult at this catalyst surface.
Meanwhile, at the Pd-Ru@Pt(110) catalyst, one feature that differentiates this catalytic surface from the pristine one is that the Ru atom attracts OH molecules. As shown in Supplementary Fig. 17, the adsorbed OH molecule does not need to interact with Pt atoms to form a water molecule. However, it is important to point out that although our simulations did not reveal proton exchange with adsorbed OH molecules on the Pd-Ru@Pt(110) catalyst, the possibility of water molecule adsorption remains. Furthermore, regardless of the absence of an applied potential in our calculation, the observed differences in energy barriers for key reactions between the pristine and doped surfaces suggest potential enhancements in catalytic activity due to the presence of dopants.
Regarding the water adsorption strength, its increase on the Pd-Ru@Pt surface in alkaline electrolytes was validated by Fourier-transform infrared spectroscopy (FTIR). The relative absorbance of the OH stretching bands can be used to estimate the overall water organization changes. Water adsorption changes OH stretching modes in the region 3000 − 3500 cm−1, highly sensitive to water’s hydrogen bond (HB) network. The broad band consists of two main components peaking at 3250 (ν1) and 3400 cm−1 (ν2), which relate to water molecules in tetrahedral-bonded ice-like organization and an HB-distorted liquid environment, respectively30. The increase of the bands absorbance at 3250 cm−1 demonstrates a higher coordination of water molecules. As shown in Supplementary Fig. 18, the ratio R of ν1: ν2 increases in the order: R(0.1 M KOH) < R(Pt/C 0.1 M KOH) < R(Pd-Ru@Pt/C 0.1 M KOH), indicating the increased coordination and water-substance interaction strength.
It is important to note, in light of the fundamental insights provided by Koper et al.14, which underscore the enthalpic barriers on polycrystalline Pt, our work extends these findings by employing AIMD simulations to elucidate the synergistic effects of Pd-Ru pair modifications on Pt within an aqueous environment. This approach not only complements the microsolvation method but also provides a comparative baseline analysis of the HOR process, thereby enhancing our understanding of the dynamic interactions at play and contributing to a broader perspective on catalytic performance in alkaline media. Our results, which highlight the relative improvements in catalytic activity in the presence of Pd and Ru, serve to reinforce the potential of such modified catalysts for practical applications in fuel cell technologies.
In a proof-of-principle demonstration of practical application, we compared the bifunctional hydrogen electrocatalytic activity of Pt/C and Pd-Ru@Pt/C using a 5 cm2 NH3 pump cell (Supplementary Fig. 19). The Pd-Ru@Pt/C showed a 1.4 times higher steady current value than the Pt/C in the NH3 pump mode in alkaline environments (Supplementary Figs. 20 and 6d), which is consistent with the advantage of optimized surface micro-environments via Pd-Ru doping in the presence of OH-.
In this work, Pd-Ru decoration on Pt nanoparticles (Pd-Ru@Pt) was synthesized by trapping the high-temperature-evaporated Pd and Ru atoms from precursors onto the Pt nanoparticle surface. The stable Pd-Ru@Pt structure is verified by both EXAFS and DFT simulations. The surface area and mass specific exchange current density of Pd-Ru pair were 7.8 and 7.7 times those of the Pt/C. DFT and AIMD simulations indicate potential dopant stability and preferred adsorption mode in the presence of a liquid environment. The AIMD simulations, coupled with slow-growth sampling, offered insights into the structural and dynamic differences between pristine and Pd-Ru@Pt(110) systems, especially concerning key reactions like H2 decomposition and subsequent H2O formation. The decomposition of H2 on pristine and Pd-Ru doped Pt(110) surfaces shows different energy barriers, with the pristine system encountering a barrier of approximately 0.27 eV and the doped system a reduced barrier of about 0.13 eV. Similarly, the energy barrier for water formation varies markedly between the two surfaces. On the pristine Pt(110) catalyst, the process exhibits a high barrier of 0.91 eV, which is substantially lowered to 0.48 eV on the Pd-Ru@Pt(110) catalyst. The simulations revealed variations in energy barriers between the two surfaces, which may indicate potential catalytic benefits of Pd and Ru dopants in facilitating these reactions. While we acknowledge that the presence of an applied potential in the simulation would provide additional input into the electron transfer steps, our findings highlight the importance of dopants in modifying the bonding environment and potentially enhancing catalytic activity. Further investigations may shed more light on these mechanisms and their implications for practical applications in catalysis. Practically, the enhanced HOR and HER performances of the Pd-Ru@Pt/C is validated by NH3 electrochemical pump performance. The demonstrated electrochemical device can easily be scaled up on demand. With renewable energy as the power input, our method represents an efficient, distributed, and sustainable NH3 separation and/or compression with broad scientific and technical impacts.
Methods
Synthesis of electrocatalysts
All chemicals were purchased from Sigma-Aldrich unless otherwise specified. Pt nanoparticles supported on carbon black were prepared via a solvothermal technique following previously reported procedures31. Typically, 2.5 mL of H2PtCl6 (99.99%) solution (40 mM in ethylene glycol (EG, 99.8%)) was added into 60 mL EG. The pH of the solution was adjusted to 12 by adding 1 M NaOH (99.99%) in EG. Then, 80 mg of carbon black was added to the above solution and stirred for 1 h. The solution was heated at 120 °C for 4 h. After cooling down, the pH of the reaction solution was adjusted to <3 using 2 M HNO3 aqueous solution, then 40 mL water was added and stirred for 10 h. The resulting catalyst was washed with DI water until Cl− was not detected by silver nitrate titration method and then vacuum dried overnight at 80 °C, denoted as Pt/C.
Atomic Pd-Ru doped Pt/C was synthesized by the catalytic pyrolysis method. In detail, 0.6 mg of Ru(acac)3 (97%) and 0.4 mg of Pd(acac)2 (99.9%) were mixed and transferred into a combustion boat, which was transferred to a glass tube. A silicon wafer with 40 mg of Pt/C was put into the tube. Then the tube was sealed under vacuum and underwent thermal treatment before being cooled to room temperature in the tube furnace. Thermal treatment detail: Increase temperature from 30 to 300 °C over 2 h and hold at 300 °C for 1 h before cooling to room temperature. Pd or Ru doped Pt/C was obtained using similar procedure, except that 1.2 mg Ru(acac)2 or 0.9 mg Pd(acac)2 was used instead of bi-salt mixture.
Preparation of electrolytes
A 0.1 M KOH electrolyte (pH=12.7) was prepared by dissolving 5.611 g of potassium hydroxide (KOH, Sigma-Aldrich, 99.99%) in deionized water to a final volume of 1 liter in a volumetric flask, while a 0.1 M HClO₄ electrolyte was prepared by diluting 14.351 g of 70% perchloric acid (HClO₄, Sigma-Aldrich) in deionized water to a final volume of 1 liter. Both solutions were mixed thoroughly to ensure complete dissolution. The prepared electrolytes were stored in tightly sealed polypropylene containers, labeled with the concentration and date of preparation, and kept in a cool, dry, and well-ventilated area to prevent contamination and degradation.
Material characterizations
The catalyst morphology was characterized by a JEOL 2100 F field emission TEM with an electron accelerating voltage of 200 keV. The atomic structure and elemental distribution were characterized by STEM-EDS using JEOL NEOARM aberration-corrected TEM operated at 80 kV. EDS was obtained on a Hitachi SU-70 FEG SEM coupled with a Bruker EDS system. X-ray photoelectron spectroscopy (XPS) was performed on a high sensitivity Kratos Axis 165 X-ray photoelectron spectrometer equipped with Mg Kα radiation. The binding-energy was calibrated using the C 1 s peak of carbon at 284.8 eV. Fourier transform infrared spectroscopy (FTIR) was recorded by a NEXUS 670 FT-IR Instrument. XAS measurements were performed at the 7-BM beamline of the National Synchrotron Light Source II (NSLS II) at Brookhaven National Laboratory in the transmission mode. The X-ray absorption near edge structure and Extended X-ray absorption fine structure spectra were processed using the Athena software package. The AUTOBK code was used to normalize the absorption coefficient, and separate the EXAFS signal, χ(k), from the isolate atom-absorption background. The extracted EXAFS signal, χ(k), was weighted by k3 to emphasize the high-energy oscillations and then Fourier-transformed in a k range from 3.0 to 10.5 Å−1 to analyze the data in R space.
Electrochemical measurements for the HOR and HER
To prepare the catalyst ink, 5 mg catalyst and 20 µL 5 wt% Nafion 117 solution were dispersed in 2.5 mL ethanol. Then, catalyst ink was drop-cast on a polished glassy carbon electrode (5 mm diameter and 0.19625 cm2, Pine Research Instrumentation, USA) for the RDE test. The electrochemical measurements were conducted using a Gamry Reference 1010E unless otherwise specified. For cyclic voltammetric measurements, higher loadings of ∼10.4 μgmetal cm−2geo were used. For details of catalyst loading calculation, 6 µL × (5 mg / 2.5 mL) / 0.19625 cm2 × 0.17 = 10.4 μgmetal cm−2geo. For HOR/HER kinetic measurements, lower loading of ∼3.5 μgmetal cm−2geo were used. For details of catalyst loading calculation, 2 µL × (5 mg / 2.5 mL) / 0.19625 cm2 × 0.17 = 3.5 μgmetal cm−2geo. A mercury/mercury oxide electrode was utilized as the reference electrode, with a graphite rod as the counter electrode. To obtain a clean and stable surface of the electrocatalyst, a voltage scan at the rate of 100 mV s−1 between 0.05 and 1.00 V (vs. RHE) was performed on the working electrode in N2‐saturated electrolyte for 20 cycles before electrochemical testing. Cyclic voltammetry (CV) was performed in a N2-saturated 100 mL 0.1 M KOH electrolyte with a potential scan rate of 50 mVs−1. The electrochemical active surface area (ECSA) of Pt/C was determined by integrating the hydrogen adsorption charge on the CV curve by assuming 210 µC/cm2 for the adsorption of a hydrogen monolayer. All of the other catalyst ECSAs were obtained by means of copper underpotential deposition (Cuupd). For the Cu underpotential deposition/stripping experiments, an N2-saturated solution containing 2 mM CuSO4 and 0.05 M H2SO4 was used as the electrolyte. For Pd-Ru@Pt/C and Ru@Pt/C, the electrode potential was first held at 0.95 V to oxidize all Ru sites. For other catalysts, there was no need to hold the electrode potential at 0.95 V before Cuupd. The potential was held at 0.3 V versus RHE for 300 s to form a Cuupd monolayer, and then followed by a linear scan to 0.95 V at 5 mV s−1. The ECSAs were calculated by subtracting the background CVs that were collected in the N2-saturated 0.05 M H2SO4 at 5 mV s−1, assuming a charge density of 470 μC cm−2. The HOR and HER tests were conducted with a working glassy carbon electrode rotation speed of 1,600 rpm in an H2-saturated 100 mL 0.1 M KOH electrolyte with a potential scan rate of 10 mVs−1. Accelerated durability tests were conducted by cycling between 0.05 V and 0.4 V versus RHE at 100 mV s−1 in N2-saturated 100 mL 0.1 M KOH. The resistance of the electrochemical cell was measured using Electrochemical Impedance Spectroscopy (EIS). The measurements were conducted using a Gamry Reference 1010E. The impedance spectra were recorded in the frequency range from 100 kHz to 1 Hz with an AC amplitude of 10 mV. The exchange current densities are obtained by linearly fitting the kinetic current versus the mass-transport and ohmic resistance-corrected potential between −5 and 5 mV vs. RHE. The calibration of Hg/HgO reference electrodes was conducted in a standard three-electrode system, with platinum wires as the working and counter electrodes, and the Hg/HgO electrode as the reference electrode. Electrolytes were saturated with high-purity H₂. Linear sweep voltammetry (LSV) was performed at a scan rate of 0.5 mV s⁻¹, and the potential at which the current crossed zero was taken as the thermodynamic potential for the hydrogen electrode reactions.
Electrochemical measurements for H2 pump and NH3 pump
The Nafion 211 membrane (25 μm) is boiled in a 3% hydrogen peroxide solution for about an hour. After boiling in hydrogen peroxide, the membrane is rinsed thoroughly with deionized water to remove any residual peroxide. The membrane is then boiled in a 0.5 M sulfuric acid solution for another hour. Finally, the membrane is rinsed again in deionized water to remove any remaining sulfuric acid. Both the cathode and anode catalysts were spray-coated onto the Nafion membrane, with a loading of 0.2 mgmetal cm−2 for Pt/C as the cathode and Pd-Ru@Pt/C as the anode. For the H2 pump, a Nafion 211 membrane (3.5 cm × 3.0 cm) was used with 5 cm2 active area (2.5 cm × 2.0 cm),76% relative humidity (RH), and 50 mL min–1 of H2. For the NH3 pump, a Nafion 211 membrane (3.5 cm × 3.0 cm) was used with 5 cm2 active area (2.5 cm × 2.0 cm) operated at 25 °C, 34% relative humidity (RH), 50 mL min–1 of H2 and 200 mL min–1 of NH3.
Computational details
Density functional theory (DFT)-based calculations and ab-initio molecular dynamics (AIMD) simulations were conducted with the Vienna ab-initio simulation package (VASP). For the exchange-correlation functional and pseudopotential treatment, the Perdew-Burke-Ernzerhof (PBE) functional within the generalized gradient approximation (GGA-PBE)32 and projector augmented wave (PAW) pseudopotentials were used, respectively22,33. The Grimme-D3 method was used to incorporate the van der Waals (vdW) interactions into the system34,35. The self-consistent field (SCF) and force convergence tolerance were set to 1×10−4 eV and 1×10−3 eV/Å, respectively. For geometry optimization calculations, a Γ-point-centered Monkhorst−Pack reciprocal grid of 5×5×1 k-points were used for Brillouin zone sampling. The geometry of bulk Pt was determined via first-principles calculations; then, the optimized–crystalline model was used to create (110) slab models. The optimized crystal structure at the GGA-PBE level has a calculated lattice parameter of 3.965 Å. This result leads to a small deviation between the experimental and the DFT-predicted unit cell volume. To model the pristine and the modified surfaces, surfaces with five–layer slabs were used consisting of 60 and 56 Pt atoms for the pristine and defective Pt(110) surfaces, respectively. The vacuum space normal to the surface was set to 15 Å to guarantee a sufficient separation between the periodic images. The size of the supercell in all models was kept to 11.89 Å × 11.21 Å × 20.60 Å. The crystallographic information files (CIF), containing the x, y, and z positions in Cartesian coordinates of the atoms in the system, represent the system’s time evolution, from the atomic positions at time 0 fs up to time 18000 fs in intervals of 3000 fs. The CIF files for the reconstructed PdRu Pt (110) slab show the evolution at the start of the simulation (files 0, 50, and 100) and include a final file containing the atomic position of the system at time 2000 fs. The files are provided as a supplementary data 1.
The AIMD simulations were carried out using the NVT (constant number of particles, volume, and temperature) ensemble at 300 K and a time step of 1 femtosecond. The Nose-Hoover thermostat was used to control the temperature oscillations during the simulations. Tritium masses were used for H atoms. AIMD simulations were performed at the Γ point only with a plane-wave energy cutoff of 400 eV. The slow-growth approach was used to calculate the free energy barriers of H2 decomposition and H2O formation at the pristine and Pd-Ru@Pt (110) surface. This approach has been successfully applied and detailed across several disciplines to observe the dynamic processes in a complex environment and explore the events in other catalytic and electrochemical reactions. The slow-growth approach is based on constraining selected reaction coordinates during the dynamics and slowly vary the reaction coordinate from an initial value tied to the initial state of the system to a characteristic value of a selected final state. The free energy difference between the final and initial states is calculated with Eq. 2 below:
Here, ξ is the velocity of transformation and F is the free energy which is evolving with time. The choice of reaction coordinate is the interatomic distance of the atoms involved in the H2 decomposition and H2O formation. For H2 decomposition, the reaction coordinate needs to be associated with the H-H bond’s elongation, the scission of one Pt-H or Pd-H bond, and a new Pt-H bond. For H2O formation, the reaction coordinates are associated with the scission of one Pt-H and formation of a O-H bond. Constraints during the molecular dynamics simulations were maintained by the SHAKE algorithm36. Total simulation times of 2 – 8 ps using a 1.0 fs time step within the NVT ensemble and assigning the mass of tritium to H atoms were carried out to complete the reactions. The average energy gradient was used for thermodynamic integration calculations using the trapezoidal rule to obtain the relative activation energy profile. The error margins on the forces are based on standard errors determined using the block averaging method. The slow-growth approach was conducted on previously equilibrated models with the NVT ensemble in standard AIMD simulations.
The effect of dopants on the Pt(110) surface was quantified in terms of hydrogen adsorption energy. The adsorption energy is defined as the total energy difference between the pristine and doped surface slab and the sum of the slab with adsorbates. Different arrangements of H adsorption sites at the pristine and doped (110) surface were investigated. Calculations of hydrogen binding energies (∆EHBE) were performed according to the following equation:
Here, ETOT (Pt slab + adsorbed species) is the total energy of the system. Then, ESlab(Pt slab) and ESpecies (H2) are the energies of the Pt(110) slab and the H2 molecule, respectively. Calculation of the H2 molecule was obtained by placing the atoms in a 1 nm cube. The Open Visualization Tool (OVITO)41 was used to visualize all the configurations and models.
Data availability
Source data are provided with this paper. The data generated in this study are provided in the Supplementary Information. Source data are provided with this paper.
Code availability
The code that supports the findings within this paper and other finding of this study are available from the corresponding authors upon reasonable request.
References
Oliveira, N. J., Malkani, A., Xu, B. & Yan, Y. Understanding electric and non-electric field effects on electrochemical double layer restructuring for the model platinum HOR. ECS Meet. Abstr. MA2020-02, 3161 (2020).
Sheng, W. et al. Correlating hydrogen oxidation and evolution activity on platinum at different pH with measured hydrogen binding energy. Nat. Commun. 6, 5848 (2015).
Strmcnik, D. et al. Improving the hydrogen oxidation reaction rate by promotion of hydroxyl adsorption. Nat. Chem. 5, 300–306 (2013).
Tao, Y., Gibbons, W., Hwang, Y., Radermacher, R. & Wang, C. Electrochemical ammonia compression. Chem. Commun. 53, 5637–5640 (2017).
Scofield, M. E. et al. Role of chemical composition in the enhanced catalytic activity of Pt-based alloyed ultrathin nanowires for the hydrogen oxidation reaction under alkaline conditions. ACS Catal. 6, 3895–3908 (2016).
Schwämmlein, J. N. et al. Origin of superior activity of Ru@Pt core-shell nanoparticles towards hydrogen oxidation in alkaline media. ECS Trans. 75, 971 (2016).
Li, M. et al. Single-atom tailoring of platinum nanocatalysts for high-performance multifunctional electrocatalysis. Nat. Catal. 2, 495–503 (2019).
Subbaraman, R. et al. Enhancing hydrogen evolution activity in water splitting by tailoring Li+-Ni(OH)2-Pt Interfaces. Science 334, 1256–1260 (2011).
Cao, L. et al. Enhancing the oxygen reduction reaction performance by modifying the surface of platinum nanoparticles. ChemElectroChem 3, 309–317 (2016).
Wang, Y. et al. Advanced electrocatalysts with single-metal-atom active sites. Chem. Rev. 120, 12217–12314 (2020).
Cheng, N., Zhang, L., Doyle-Davis, K. & Sun, X. Single-atom catalysts: from design to application. Electrochem. Energy Rev. 2, 539–573 (2019).
Wang, A., Li, J. & Zhang, T. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2, 65–81 (2018).
Mitchell, S. & Pérez-Ramírez, J. Single atom catalysis: a decade of stunning progress and the promise for a bright future. Nat. Commun. 11, 4302 (2020).
McCrum, I. T. & Koper, M. T. M. The role of adsorbed hydroxide in hydrogen evolution reaction kinetics on modified platinum. Nat. Energy 5, 891–899 (2020).
Qu, Y. et al. Direct transformation of bulk copper into copper single sites via emitting and trapping of atoms. Nat. Catal. 1, 781–786 (2018).
Feng, Z. et al. Role of hydroxyl species in hydrogen oxidation reaction: a DFT study. J. Phys. Chem. C. 123, 23931–23939 (2019).
Schmidt, T. J., Ross, P. N. & Markovic, N. M. Temperature dependent surface electrochemistry on Pt single crystals in alkaline electrolytes: Part 2. The hydrogen evolution/oxidation reaction. J. Electroanalytical Chem. 524-525, 252–260 (2002).
Gudmundsdóttir, S., Skúlason, E., Weststrate, K.-J., Juurlink, L. & Jónsson, H. Hydrogen adsorption and desorption at the Pt(110)-(1×2) surface: experimental and theoretical study. Phys. Chem. Chem. Phys. 15, 6323–6332 (2013).
Mercer, M. P. & Hoster, H. E. Electrochemical kinetics: a surface science-supported picture of hydrogen electrochemistry on Ru(0001) and Pt/Ru(0001). Electrocatalysis 8, 518–529 (2017).
Wang, Y. et al. Pt–Ru catalyzed hydrogen oxidation in alkaline media: oxophilic effect or electronic effect? Energy Environ. Sci. 8, 177–181 (2015).
Durst, J. et al. New insights into the electrochemical hydrogen oxidation and evolution reaction mechanism. Energy Environ. Sci. 7, 2255–2260 (2014).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
McCrum, I. T., Chen, X., Schwarz, K. A., Janik, M. J. & Koper, M. T. M. Effect of step density and orientation on the apparent ph dependence of hydrogen and hydroxide adsorption on stepped platinum surfaces. J. Phys. Chem. C. 122, 16756–16764 (2018).
Rheinländer, P. J., Herranz, J., Durst, J. & Gasteiger, H. A. Kinetics of the hydrogen oxidation/evolution reaction on polycrystalline platinum in alkaline electrolyte reaction order with respect to hydrogen pressure. J. Electrochem. Soc. 161, F1448 (2014).
Schwämmlein, J. N. et al. Origin of superior HOR/HER activity of bimetallic Pt-Ru catalysts in alkaline media identified via Ru@Pt Core-Shell Nanoparticles. J. Electrochem. Soc. 165, H229 (2018).
Davydova, E. S., Mukerjee, S., Jaouen, F. & Dekel, D. R. Electrocatalysts for hydrogen oxidation reaction in alkaline electrolytes. ACS Catal. 8, 6665–6690 (2018).
Wang, L., Mahoney, E. G., Zhao, S., Yang, B. & Chen, J. G. Low loadings of platinum on transition metal carbides for hydrogen oxidation and evolution reactions in alkaline electrolytes. Chem. Commun. 52, 3697–3700 (2016).
Sheng, W., Gasteiger, H. A. & Shao-Horn, Y. Hydrogen oxidation and evolution reaction kinetics on platinum: acid vs alkaline electrolytes. J. Electrochem. Soc. 157, B1529 (2010).
Feibelman, P. J. Surface-diffusion mechanism versus electric field: Pt/Pt(001). Phys. Rev. B 64, 125403 (2001).
Petit, T. et al. Unusual water hydrogen bond network around hydrogenated nanodiamonds. J. Phys. Chem. C. 121, 5185–5194 (2017).
Zhang, S., Shao, Y., Yin, G. & Lin, Y. Carbon nanotubes decorated with Pt nanoparticles via electrostatic self-assembly: a highly active oxygen reduction electrocatalyst. J. Mater. Chem. 20, 2826–2830 (2010).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Ryckaert, J.-P., Ciccotti, G. & Berendsen, H. J. C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 (1977).
Zhu, Y. et al. One-pot pyrolysis to N-doped graphene with high-density Pt single atomic sites as heterogeneous catalyst for alkene hydrosilylation. ACS Catal. 8, 10004–10011 (2018).
Li, X. et al. PdFe single-atom alloy metallene for N2 electroreduction. Angew. Chem. Int. Ed. 61, e202205923 (2022).
Zhang, J. et al. Competitive adsorption: reducing the poisoning effect of adsorbed hydroxyl on Ru single-atom site with SnO2 for efficient hydrogen. Evolution 61, e202209486 (2022).
Acknowledgements
L. C. and C. W. at the University of Maryland gratefully acknowledge funding support from the Advanced Research Projects Agency–Energy under Contract No. DEAR0000962. This research used beamlines 7-BM of the National Synchrotron Light Source II, a U.S. DOE Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. Computational resources from Texas A&M Supercomputing Center and from Texas Advanced Computing Center at UT Austin are gratefully acknowledged. X. L. and K. H. acknowledge the American Chemical Society Petroleum Research Fund (62493-DNI10) for partial support of this research. A portion of STEM-EDS research conducted as part of a user project at the Center for Nanophase Materials Sciences (CNMS), which is a US Department of Energy, Office of Science User Facility at Oak Ridge National Laboratory.
Author information
Authors and Affiliations
Contributions
L. Cao, F. Soto, and D. Li conceived the idea. L. Cao and D. Li performed the electrochemical experiments. L. Cao synthesized the catalysts. L. Cao, D. Li, and T. Deng carried out characterizations. E. Hu, and X.-Q. Yang performed and analyzed EXAFS and XANES analysis. X. Lu, K. He, and D. A. Cullen performed STEM-EDS analysis. F. Soto and P. Balbuena proposed the mechanism research and simulations. N.E. participated in the discussions. C.W. supervised all the studies. All authors contributed to writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Elena Davydova, Hany El-Sayed, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cao, L., Soto, F.A., Li, D. et al. Pd-Ru pair on Pt surface for promoting hydrogen oxidation and evolution in alkaline media. Nat Commun 15, 7245 (2024). https://doi.org/10.1038/s41467-024-51480-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51480-w
This article is cited by
-
Bifunctional Pt/TiO2-Ov catalysts for enhanced electron transfer and CO tolerance in acidic HOR and ORR
Frontiers in Energy (2025)
-
One-step synthesis of hollow Ce-doped ZnIn2S4/MOF(Ce) heterojunctions with hard-soft acid-base coordination for enhanced hydrogen evolution
Science China Chemistry (2025)
