
Abstract
Most C–H bond activations of natural gas alkanes rely on transition metal complexes. Activations by using main-group systems have been reported but required heating or photo-irradiation under high atmospheric pressure with rather low regioselectivity. Here we report that Lewis acid-carbene adducts facilely undergo oxidative additions to C–H bonds of ethane, propane and n-butane with high selectivity under room temperature and atmospheric pressure. The Lewis acids can be moved by the addition of a base and the carbene-derived products can be easily converted into aldehydes. This work offers a route for main-group element compounds to selectively functionalise C–H bonds of natural gas alkanes and other small molecules.
Similar content being viewed by others
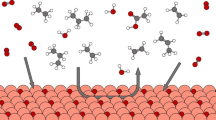


Introduction
Natural gas, particularly from shale, has substantial amounts of ethane, propane and n-butane, though it is primarily methane. These natural gas alkanes have been serving as low-cost hydrocarbon feedstocks. The efficient and selective activation of their C–H bonds can directly convert them into value-added chemicals and fuels for the needs of the industry1,2,3,4. Most of the C–H bond activations of natural gas alkanes rely on transition metal complexes (Fig. 1a)5,6,7,8,9. Alternative activations by main-group materials have been reported, such as superacids, main-group metal complexes, hexagonal boron nitrides, and frustrated radical pairs, etc. (Fig. 1b)10,11,12,13, but require high pressure upon heating or photo-irradiation. Moreover, the activation regioselectivity in many main-group cases is rather low, with little discrimination of primary and secondary C–H bonds of these light alkanes14,15,16,17. Notably, main-group compounds with unique structural and bonding behaviours have attracted extensive interest as potential alternatives to transition metals in recent years18,19,20,21,22. While activation of small molecules such as H2, N2, NH3, CO, C2H4, and P4 has emerged by main-group systems under mild conditions (Fig. 1c)23,24,25,26,27,28,29,30,31, natural gas alkanes are absent in the list for their strong undirected C(sp3)–H bonds and poor solubility in many solvents.

a Transition metal complexes activating C(sp3)–H bonds of natural gas alkanes. b Representative examples of activating C–H bonds of natural gas alkanes by main-group compounds. c Representative examples of small molecular activation by main-group compounds. d Reactions of Lewis acid-carbene adducts with C–H bonds of natural gas alkanes. Mes = mesityl; Dipp = 2, 6-diisopropylphenyl; Dur = 2, 3, 5, 6-tetramethylphenyl; ORF = OC(CF3)3.
Carbenes are species featuring a divalent carbon centre with a six-electron valence shell and normally considered reaction intermediates32,33,34. Since Bertrand et al. successfully synthesised stable carbenes and Arduengo et al. obtained isolable N-heterocyclic carbene (NHC)35,36, stable carbenes have attracted extensive attention in the fields of organic synthesis, organometallic chemistry, materials science and main-group chemistry37,38,39. Stable carbenes with strong electrophilicity such as cyclic (alkyl) (amino) carbenes (cAACs)40,41,42 and N, N’-diamidocarbenes (DACs)43,44,45, are capable of activating small molecules under mild conditions. Stable carbenes were also reported to insert into inert C–F, B–H, N–H bonds and C–H bonds in some acidic substrates, including PhC ≡ CH, C6F5H and Cl3CH at room temperature46. They may even cleave the C(sp3)–H bonds of much less acidic toluene47 (pKa = 41.2 in cyclohexylamine) upon heating up to 80–100 °C and liquid alkanes such as cyclohexane upon photo-irradiation48.
Recently we found that the electrophilicity of a N,N’-diamidocarbene (DAC1)47 could be considerably strengthened by the coordination with Lewis acid B(C6F5)3 (BCF)49. The BCF–DAC1 adduct has a significantly lower LUMO energy level compared to DAC1, accelerating its activation towards small molecules and inert bonds49. The work demonstrated the feasibility of regulating electron properties of stable carbenes with strong Lewis acids instead of designing a new carbene structure. Here we report that the BCF–DAC1 adduct facilely undergoes oxidative addition to C–H bonds of natural gas alkanes (ethane, propane and n-butane) under atmospheric pressure and at room temperature with high regioselectivity. The BCF–DAC1 adduct only inserts in secondary C–H bonds of propane and n-butane. Primary C–H bonds of ethane could be activated by DAC1 with more powerful Lewis acid Al(ORF)3 (ORF = OC(CF3)3) (Fig. 1d)50.
Results
DAC1 is stable in n-hexane. As a matter of fact, many reactions of DAC1 were performed in n-hexane. To our surprise, the red colour of the solution of the BCF–DAC1 adduct in n-hexane immediately faded at room temperature, affording a large amount of light-yellow precipitate. Recrystallisation of the precipitate in toluene and n-pentane led to a BCF–DAC1-derived product as a racemate with the carbon centre of the carbene inserted into the secondary C–H bond of n-hexane, identified by single-crystal X-ray diffraction (Fig. 2a and Fig. 3a). This reaction could also be carried out in benzene with 2.5 equivalent of n-hexane, indicating this reaction does not require excess amount of alkane as feedstock. One-pot reaction of BCF–DAC1 with n-hexane directly followed by the addition of CH3CN gave DAC1-derived alkanes in 98% yield with a 4:1 ratio of β to γ site, tracked by 1H-NMR analysis and GC-MS (Fig. 2a, Supplementary Fig. 34 and Supplementary Fig. 64). The terminal sites have C–H bonds with smallest steric hindrance, but those primary C–H bonds activated product was not observed.

a Reaction of BCF–DAC1 and hexane in benzene. b Reaction of Lewis acid-DAC1 adducts with natural gas alkanes and selected alkane substrates. c The HOMO and LUMO energy of DAC1, BCF–DAC1 and Al(ORF)3–DAC1, computed with B3LYP/6-31 G(d). The yield was determined from crude 1H-NMR spectra and isolated yield in the parenthesis. The structures were all confirmed by single-crystal XRD. ‡ 1H NMR yield (see Supplementary Fig. 48). ORF = OC(CF3)3.

a The main product from the reaction of BCF-DAC1 with n-hexane; b The product from the reaction of BCF–DAC1 with propane; c Al(ORF)3–DAC1; d The product from the reaction of Al(ORF)3–DAC1 with ethane followed by quenching by addition of CH3CN. ORF = OC(CF3)3. Thermal ellipsoid and stick drawings are set at 30% probability and hydrogen atoms are omitted for clarity. Colour codes: carbon (grey); boron (brown); nitrogen (navy blue); oxygen (red); aluminium (light blue); fluorine (green sticks).
Encouraged by this initial finding, we systematically investigated reactions of the BCF–DAC1 adduct with natural gas alkanes (CnH2n+2, n = 1–4). These gaseous alkanes (0.1 MPa) were introduced into the benzene solution of the BCF–DAC1 adduct upon drying through a dehydration column filled with colour-changing silica gel and P2O5. The red reaction solution of BCF–DAC1 with propane gradually became pale yellow after 10 h. Crystallisation led to a DAC1-derived product with BCF, showing secondary C–H insertion (Fig. 2b and Fig. 3b). BCF can be removed by adding CH3CN, affording a DAC1–derived propane. One-pot reaction of BCF–DAC1 with propane directly followed by quenching with CH3CN gave same product in a 1H-NMR yield of 80%, and could be purified by column chromatography on silica gel using acetone/petroleum as eluent. A similar insertion occurred in the reaction of BCF-DAC1 with n-butane (Fig. 2b).
Resembling n-hexane, the C–H bond insertion at terminal position of propane and butane was not observed for higher energy barrier of primary C–H bonds compared with secondary C–H bonds51. For the same reason, the BCF–DAC1 adduct failed to react with methane and ethane. The design of a new adduct with a stronger Lewis acid is desired. Our calculation shows that DAC1 coupled with Al(ORF)350 possesses lower LUMO energy than that with BCF (Fig. 2c). Consequently, a colour change from red to purple was observed upon the addition of one equivalent of Al(ORF)3 into the fluorobenzene solution of DAC1. Adduct Al(ORF)3–DAC1 was isolated as dark purple crystals in a separation yield of 67% upon concentration and crystallisation at –20 °C in 1, 2-dichlorobenzene (Fig. 3c). Only one Al(ORF)3 is coordinated to one oxygen atom of DAC1 due to steric crowding. The reaction of the Al(ORF)3–DAC1 adduct with ethane was treated by a similar manner to the above. The C–H bond of ethane was activated within 6 h, affording DAC1–derived ethane in a yield of 30% (based on 1H-NMR spectroscopy) upon the addition of CH3CN (Fig. 2b), which is identified by single crystal X-ray diffraction (Fig. 2b and Fig. 3d). Additionally, many other alkanes with multiple reacting sites are also compatible in our method, giving the desired products with excellent regioselectivity (Fig. 2b). It demonstrates that the secondary C–H bond in alkane was privileged in our conditions (2–5), while the tertiary C–H bond was activated in the absence of secondary C–H bond (1) or it has larger steric hindrance (6).
To better understand the mechanism of this C–H activation, we performed DFT calculations on the reaction using propane and DAC1 in the presence or absence of BCF (Fig. 4a). The calculations indicate that the activation barriers for concerted carbene insertion into the internal and terminal C–H bonds of propane by DAC1 were prohibitively high at 38.3 kcal/mol (TS-a) and 40.1 kcal/mol (TS-b), respectively. These barriers suggest that such processes cannot occur under typical reaction conditions, consistent with experimental findings. However, in the presence of Lewis acid, the coordination of carbene DAC1 and BCF is found to be an exergonic process, resulting in the formation of stable adduct INT-1, which has been previously characterised crystallographically49. The corresponding activation barriers can be dramatically lowered to 24.8 kcal/mol (TS-B-a) and 28.6 kcal/mol (TS-B-b), respectively, in the presence of BCF. Analysis of the carbene’s electrophilicity revealed that the charge on the carbon atom increased from 0.248 for free DAC1 to 0.288 in the BCF–DAC1 adduct (Fig. 4b). This increased electrophilicity enhances the carbene’s ability to abstract a hydride from the alkane, thereby reducing the energy barrier for the reaction (TS-B-a vs. TS-a). Given that the barriers depend heavily on the electrophilicity of the carbene adduct, we propose that insertion proceeds initially via hydride transfer from the alkane to the carbene, generating a partial positive charge on the alkane carbon. This results in the selectivity relating to the stability of the corresponding alkyl carbocation, and therefore a preference for internal C–H bonds and predominant formation of branched isomers.

a Reaction of BCF–DAC1 and propane. b The NPA charge of carbon and oxygen atoms in free carbene, adducts and products. c Reaction of Al(ORF)3–DAC1 and ethane. All calculations were carried out at SMD(benzene)-M06-2X/6-311 + G(d,p)//B3LYP-D3/6-31 G(d) level of theory. Most hydrogen atoms are omitted for clarity. All distances are in Å. Colour codes: hydrogen (white); boron (pink); carbon (grey); nitrogen (blue); oxygen (red); fluorine (green); aluminium (tawny).
Furthermore, the carbon atom in INT-4 possesses a charge of 0.292, indicating higher reactivity compared to BCF-DAC1 under the reaction conditions. Ethane, being a more challenging substrate for C–H activation, was examined. In the presence of Al(ORF)3, the activation of ethane required a free energy of 24.4 kcal/mol via TS-Al-Et, whereas with BCF, the energy barrier increased to 26.6 kcal/mol via TS-B-Et (Fig. 4c). Notably, we successfully achieved the desired product using ethane only when INT-4 was present. These findings underscore the critical role of adduct electrophilicity in determining the reactivity of simple alkanes.
Derivatizations were conducted to further evaluate the synthetic utilisation and practicality of this methodology. The DAC1-derived alkanes were treated with LiAlH4/HCl (aq) at room temperature. After 10 h, corresponding aldehydes elongated by one carbon atom were obtained in 34-50% yield, achieving transformation from natural gas alkanes to aldehydes under mild condition (Fig. 5a–c). As shown in Fig. 5d, the DAC1-propane can be reduced by LiAlH4, giving the corresponding compound 752. In the acidic conditions, compound 7 was transformed into isobutyraldehyde via intermediates 8 and 953,54. (Fig. 5d). This provides an opportunity to complete hydroformylation reactions directly from alkanes in the industry instead of using alkene and syngas (CO/H2) catalysed by transition metals under high temperature and pressure55. Furthermore, butylene and propylene will give linear regioisomers valeraldehyde and butyraldehyde as major products in the transition metal catalysed carbonylation, respectively. However, hydroformylation of alkenes to form branched aldehydes is highly desired56,57. Significantly, branched 2-methylbutyraldehyde and isobutyraldehyde can be obtained from butane and propane in our conditions, which should be an importantly complementary chemistry compared with oxo-synthesis.

a Transformation from n-butane to 2-methylbutyraldehyde. b Transformation from propane to isobutyraldehyde. c Transformation from ethane to propionic aldehyde. d Reaction mechanism of transformation from DAC1-propane to isobutyraldehyde. Yields were determined by 1H-NMR spectra using 1, 3, 5-trimethoxybenzene as internal standard. ORF = OC(CF3)3.
In conclusion, the reactivity of a stable carbene has been greatly improved to directly insert into the C–H bonds of natural gas alkanes (ethane, propane and n-butane) and other simple alkanes through the coordination with strong Lewis acids, offering a route for main-group element systems to C–H functionalization of natural gas alkanes. This kind of reaction only requires mild conditions at room temperature and one atmospheric pressure, without the need for excessive liquid or solid alkanes as feedstock. The work reported here indicates stable carbenes are able to mimic the behaviour of transition metals towards the activation of natural gas alkanes. It features that the electronic properties of the carbon centre of a carbene could be regulated by the strength of Lewis acids, as demonstrated by the different reactivity of Lewis acid-diamidocarbene adducts with ethane and propane. The carbene-derived products can be converted into aldehydes after reduction, achieving carbon chain growth of natural gas alkanes. This finding shows that the Lewis acid-carbene adduct is not only complementary to transition metal activation but also an exciting paradigm in transformation from natural gas alkanes to value-added chemicals.
Methods
General methods for the synthesis of DAC1–hexane, DAC1–propane, DAC1–butane, DAC1–ethane, other DAC1–alkane products and Al(ORF)3–DAC1 are as follows. More details can be found in the Supplementary Information file.
General synthesis of DAC1–hexane
DAC1 (1 eq), B(C6F5)3 (1 eq) and benzene were charged in a Schlenk flask. Then benzene and n-hexane was added into the flask. The solution was stirred for 10 h at 25°C and the solution turned from red to yellow with some yellow residue. Acetonitrile (1 mL) with a drop of water was added to quench the reaction. The product was isolated as a white solid after purification by column chromatography on silica gel.
General synthesis of DAC1–propane, DAC1–butane and DAC1–ethane
DAC1 (1 eq), Lewis Acid (B(C6F5)3 or PhF→Al(ORF)3 (1 eq) and benzene were charged in a Schlenk flask. Ethane, propane or butane (0.1 MPa) was transferred into the flask via vacuum. The solution was stirred for about 10 h at 25 °C and the dark red solution turned yellow with some yellow residue. Acetonitrile (1 mL) with a drop of water was added to quench the reaction. The product was isolated as a white solid after purification by column chromatography on silica gel.
General synthesis of DAC1–alkane products (1–6)
DAC1 (1 eq), B(C6F5)3 (1 eq) and benzene were charged in a Schlenk flask. Then benzene and alkanes (2.5 eq) was added into the flask. The solution was stirred for 10 h at 25 °C and the solution turned from red to yellow with some yellow residue. Acetonitrile (1 mL) with a drop of water was added to quench the reaction. The product was isolated as a white solid after purification by column chromatography on silica gel.
General synthesis of Al(ORF)3–DAC1
DAC1 (1 eq) and PhF→Al(ORF)3 (1 eq) were charged in a Schlenk flask, then 1 mL 1, 2-dichlorobenzene was added and stirred for 15 min. The flask was placed at −20 °C and dark purple crystals were obtained. The crystals would be fairly unstable under room temperature and are required to store in −20 °C in glove box.
Transformation from DAC1–alkane products into aldehydes
DAC1-alkane products (1 eq) and LiAlH4 (2.5 eq) was charged in a tube with a magnetic stirring bar, and dried THF was added. After reacting for 10 h at room temperature, HCl aqueous solution was added to quench the reaction. The product was then extracted by CDCl3, and the ratios and yields were determined by 1H-NMR using 1, 3, 5-trimethoxybenzene as internal standard.
Data availability
The crystallographic data for compounds BCF–DAC1–hexane, BCF–DAC1–propane, Al(ORF)–DAC1, DAC1–butane, DAC1–ethane and other DAC1–alkane products 1–6 have been deposited in the Cambridge Crystallographic Data Centre (CCDC) database under deposition numbers 2327624-2327628, 2332622-2332627, respectively. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. Additional spectroscopic, crystallographic, and computational data generated in this study are provided in the Supplementary Information file. Source Data are included in this manuscript. All data are available from the corresponding author upon request. Source data are provided in this paper.
References
Pombeiro, A. J. L. in Alkane Functionalization 1–15 (John Wiley & Sons Ltd., 2019).
Labinger, J. A. & Bercaw, J. E. Understanding and exploiting C–H bond activation. Nature 417, 507–514 (2002).
Administration, U. S. E. I. International Energy Outlook 2023. (https://www.eia.gov/outlooks/ieo/pdf/IEO2023_Release_Presentation.pdf, 2023).
Bergman, R. G. C–H activation. Nature 446, 391–393 (2007).
Shilov, A. E. & Shul’pin, G. B. Activation of C–H bonds by metal complexes. Chem. Rev. 97, 2879–2932 (1997).
Jia, C., Kitamura, T. & Fujiwara, Y. Catalytic functionalization of arenes and alkanes via C−H bond activation. Acc. Chem. Res. 34, 633–639 (2001).
Periana, R. A., Mironov, O., Taube, D., Bhalla, G. & Jones, C. Catalytic, oxidative condensation of CH4 to CH3COOH in one step via CH activation. Science 301, 814–818 (2003).
Cybulskis, V. J. et al. Zinc promotion of platinum for catalytic light alkane dehydrogenation: insights into geometric and electronic effects. ACS Catal. 7, 4173–4181 (2017).
Chen, S. et al. Propane dehydrogenation: catalyst development, new chemistry, and emerging technologies. Chem. Soc. Rev. 50, 3315–3354 (2021).
Koppaka, A. et al. Selective C–H functionalization of methane and ethane by a molecular SbV complex. Angew. Chem. Int. Ed. 58, 2241–2245 (2019).
Shu, C., Noble, A. & Aggarwal, V. K. Metal-free photoinduced C(sp3)–H borylation of alkanes. Nature 586, 714–719 (2020).
Shao, B. et al. Arylation ofhydrocarbons enabled by organosilicon reagents and weakly coordinating anions. Science 355, 1403–1407 (2017).
Fokin, A. A. & Schreiner, P. R. Metal-free, selective alkane functionalizations. Adv. Synth. Catal. 345, 1035–1052 (2003).
Power, P. P. Main-group elements as transition metals. Nature 463, 171–177 (2010).
Martin, D., Soleilhavoup, M. & Bertrand, G. Stable singlet carbenes as mimics for transition metal centers. Chem. Sci. 2, 389–399 (2011).
Nesterov, V. et al. NHCs in main group chemistry. Chem. Rev. 118, 9678–9842 (2018).
Melen, R. L. Frontiers in molecular p-Block chemistry: from structure to reactivity. Science 363, 479–484 (2019).
Spikes, G. H., Fettinger, J. C. & Power, P. P. Facile activation of dihydrogen by an unsaturated heavier main group compound. J. Am. Chem. Soc. 127, 12232–12233 (2005).
Welch, G. C., Juan, R. R. S., Masuda, J. D. & Stephan, D. W. Reversible Metal-free hydrogen activation. Science 314, 1124–1126 (2006).
Frey, G. D., Lavallo, V., Donnadieu, B., Schoeller, W. W. & Bertrand, G. Facile Splitting of hydrogen and ammonia by nucleophilic activation at a single carbon center. Science 316, 439–441 (2007).
Masuda, J. D., Schoeller, W. W., Donnadieu, B. & Bertrand, G. NHC-mediated aggregation of P4: isolation of a P12 cluster. J. Am. Chem. Soc. 129, 14180–14181 (2007).
Liu, Y., Dong, W., Li, Z. H. & Wang, H. Methane activation by a borenium complex. Chem. 7, 1843–1851 (2021).
Olah, G. A., Prakash, G. K. S. & Sommer J. Superacids. Sci. 206, 13–20 (1979).
Hashiguchi, B. G. et al. Main-group compounds selectively oxidize mixtures of methane, ethane, and propane to alcohol esters. Science 343, 1232–1237 (2014).
Grant, J. T. et al. Selective oxidative dehydrogenation of propane to propene using boron nitride catalysts. Science 354, 1570–1573 (2016).
Lu, Z. et al. Regioselective aliphatic C–H functionalization using frustrated radical pairs. Nature 619, 514–520 (2023).
Peng, Y., Ellis, B. D., Wang, X., Fettinger, J. C. & Power, P. P. Reversible reactions of ethylene with distannynes under ambient conditions. Science 325, 1668–1670 (2009).
Légaré, M.-A. et al. Nitrogen fixation and reduction at boron. Science 359, 896–900 (2018).
Légaré, M.-A. et al. The reductive coupling of dinitrogen. Science 363, 1329–1332 (2019).
Fujimori, S. & Inoue, S. Carbon monoxide in main-group chemistry. J. Am. Chem. Soc. 144, 2034–2050 (2022).
Chu, T. & Nikonov, G. I. Oxidative addition and reductive elimination at main-group element centers. Chem. Rev. 118, 3608–3680 (2018).
Davies, H. M. L. & Beckwith, R. E. J. Catalytic enantioselective C−H activation by means of metal−carbenoid-induced C−H insertion. Chem. Rev. 103, 2861–2904 (2003).
Díaz-Requejo, M. M. & Pérez, P. J. Coinage metal catalyzed C−H bond functionalization of hydrocarbons. Chem. Rev. 108, 3379–3394 (2008).
Doyle, M. P., Duffy, R., Ratnikov, M. & Zhou, L. Catalytic carbene insertion into C−H bonds. Chem. Rev. 110, 704–724 (2010).
Arduengo, A. J. III, Harlow, R. L. & Kline, M. A stable crystalline carbene. J. Am. Chem. Soc. 113, 361–363 (1991).
Igau, A., Grützmacher, H., Baceiredo, A. & Bertrand, G. Analogous α,α‘-bis-carbenoid, triply bonded species: synthesis of a stable λ3-Phosphinocarbene-λ5-Phosphaacetylene. J. Am. Chem. Soc. 110, 6463–6466 (1988).
Bourissou, D., Guerret, O., Gabbai, F. P. & Bertrand, G. Stable carbenes. Chem. Rev. 100, 39–91 (2000).
Hopkinson, M. N., Richter, C., Schedler, M. & Glorius, F. An overview of N-heterocyclic carbenes. Nature 510, 485–496 (2014).
Smith, C. A. et al. N-heterocyclic carbenes in materials chemistry. Chem. Rev. 119, 4986–5056 (2019).
Lavallo, V., Canac, Y., Prasang, C., Donnadieu, B. & Bertrand, G. Stable cyclic (alkyl)(amino)carbenes as rigid or flexible, bulky, electron-rich ligands for transition-metal catalysts: a quaternary carbon atom makes the difference. Angew. Chem. Int. Ed. 44, 5705–5709 (2005).
Melaimi, M., Jazzar, R., Soleilhavoup, M. & Bertrand, G. Cyclic (alkyl)(amino)carbenes (caacs): recent developments. Angew. Chem. Int. Ed. 56, 10046–10068 (2017).
Jazzar, R., Soleilhavoup, M. & Bertrand, G. Cyclic (Alkyl)- and (Aryl)-(amino)carbene Coinage Metal Complexes and Their Applications. Chem. Rev. 120, 4141–4168 (2020).
Hudnall, T. W., Bielawski, C. W. & An N,N’-Diamidocarbene: studies in C–H insertion, reversible carbonylation, and transition-metal coordination chemistry. J. Am. Chem. Soc. 131, 16039–16041 (2009).
Moerdyk, J. P. & Bielawski, C. W. Diamidocarbenes as versatile and reversible [2+1] cycloaddition reagents. Nat. Chem. 4, 275–280 (2012).
Moerdyk, J. P., Schilter, D. & Bielawski, C. W. N,N’-Diamidocarbenes: isolable divalent carbons with bona fide carbene reactivity. Acc. Chem. Res. 49, 1458–1468 (2016).
Turner, Z. R. Chemically non-innocent cyclic (alkyl)(amino)carbenes: ligand rearrangement, C−H and C−F bond activation. Chem. Eur. J. 22, 11461–11468 (2016).
Moerdyk, J. P. & Bielawski, C. W. Reductive generation of stable, five-membered N,N’-Diamidocarbenes. Chem. Commun. 50, 4551–4553 (2014).
Perera, T. A. et al. Photochemical reactions of a Diamidocarbene: cyclopropanation of bromonaphthalene, addition to pyridine, and activation of sp3 C–H bonds. Chem. Sci. 14, 7867–7874 (2023).
Pei, R., He, L., Zhao, Y. & Wang, X. The Dynamic lewis acid–carbene hybrid: pushing the electrophilicity of carbenes to the limit. J. Am. Chem. Soc. 145, 21733–21737 (2023).
Müller, L. O. et al. Simple access to the non-oxidizing lewis superacid PhF→Al(ORF)3 (RF=C(CF3)3). Angew. Chem. Int. Ed. 47, 7659–7663 (2008).
Schultz, J. C., Houle, F. A. & Beauchamp, J. L. Photoelectron spectroscopy of 1-Propyl, 1-Butyl, Isobutyl, Neopentyl, and 2-Butyl radicals: free radical precursors to high-energy carbonium ion isomers. J. Am. Chem. Soc. 106, 3917–3927 (1984).
Argese, M. et al. Synthesis and NMR characterization of cis and trans decahydro-2a,4a,6a,8a-tetraazacyclopent [fg] acenaphthylene. solid state structure of the trans stereoisomer. modelling studies. Tetrahedron 63, 6915–6923 (2007).
Hine, J., Narducy, K. W. & Imines imidazolidines, and imidazolidinium ions from the reactions of ethylenediamine derivatives with isobutyraldehyde and acetone. J. Am. Chem. Soc. 95, 3362–3368 (1973).
Kallen, R. G. & Jencks, W. P. The mechanism of the condensation of formaldehyde with tetrahydrofolic acid. J. Biol. Chem. 241, 5851–5863 (1966).
Franke, R., Selent, D. & Boerner, A. Applied hydroformylation. Chem. Rev. 112, 5675–5732 (2012).
Dingwall, P. et al. Understanding a hydroformylation catalyst that produces branched aldehydes from alkyl alkenes. J. Am. Chem. Soc. 139, 15921–15932 (2017).
Ning, Y., Ohwada, T. & Chen, F.-E. Transition metal-catalyzed branch-selective hydroformylation of olefins in organic synthesis. Green. Synth. Catal. 2, 247–266 (2021).
Acknowledgements
We thank Shichao Wang and Professor Shaolin Zhu at Nanjing University for GC-MS testing and analysing. We thank the National Natural Science Foundation of China (Grants 22231005 (X.W.)), the National Key R&D Programme of China (Grants 2018YFA0306004 (X.W.) and 2022YFA1503200 (Y.L.)), the Strategic Priority Research Programme of the Chinese Academy of Sciences (Grant XDB0610000 (X.W.)), the Natural Science Foundation of Jiangsu Province (BK20230018 (Y.L)), and the Fundamental Research Funds for the Central Universities (020514380253 (Y.L)) for financial support. The calculations were performed at the High-Performance Computing Center of Nanjing University.
Author information
Authors and Affiliations
Contributions
X.W. conceived the project. R.P. performed the chemical experiments of the BCF–DAC1 adduct. W.C. assisted in experiments and conducted theoretical calculations. L.H. performed the chemical experiments of the Al(ORF)3–DAC1 adduct. R.P. and T.W. collected the crystallographic data. Y.Z. refined crystal structures. X.W. and Y.L. supervised the experiment work. Y.L. supervised theoretical calculations. R.P., W.C., X.W. and Y.L. wrote the first draft of the manuscript. All authors discussed the results and manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Liu Leo Liu who co-reviewed with Yanbo Mei; and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pei, R., Chang, W., He, L. et al. Main-group compounds selectively activate natural gas alkanes under room temperature and atmospheric pressure. Nat Commun 15, 7943 (2024). https://doi.org/10.1038/s41467-024-52185-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52185-w
