
Abstract
Inhibiting glycogen synthase kinase 3 (GSK3) improves muscle function, metabolism, and bone health in many diseases and conditions; however, whether GSK3 should be targeted for Duchenne muscular dystrophy (DMD), a severe muscle wasting disorder with no cure, remains unknown. Here, we show the effects of GSK3 inhibition in male DBA/2J (D2) and C57BL/10 (C57) mdx mice. Treating D2 mdx mice with GSK3 inhibitors alone or in combination with aerobic exercise improves muscle strength, endurance, and morphology, attenuates the hypermetabolic phenotype, and enhances insulin sensitivity. GSK3 inhibition in C57 mdx mice also improves muscle fatigue resistance and increases cage ambulation. Moreover, muscle-specific GSK3 knockdown in mdx mice augments muscle force production and endurance. In both mdx strains, GSK3 inhibition increases bone mineral content and density. Overall, these improvements to muscle, metabolic, and bone health with GSK3 inhibition in mdx mice may have clinical implications for patients with DMD, where the current standard of care, glucocorticoids, delay the loss of ambulation but increase the risk for insulin resistance and osteoporosis. Along with our observation of lowered β-catenin content in DMD myoblasts, a known cellular target for GSK3, this study provides ample evidence in support of inhibiting GSK3 for this disease.
Similar content being viewed by others


Introduction
Duchenne muscular dystrophy (DMD) is a severe muscle-wasting disorder caused by an X-linked recessive mutation to the dystrophin gene1 that affects 1 in 5000 males worldwide2. The absence of functional dystrophin compromises muscle integrity, leading to progressive muscle wasting and weakness that causes mobility impairments and a shortened lifespan of 3 to 4 decades in patients with DMD3. While there is no cure for DMD, the FDA recently approved restorative dystrophin gene therapy, however, it has only been approved for a subset of patients and, at best, can only partly increase dystrophin content. As a result, this therapy may only improve muscle health to a certain extent and essentially transform DMD into a milder disease known as Becker muscular dystrophy (BMD)4. Thus, pharmacological interventions will remain a clinical need for patients with DMD and BMD. In this context, glucocorticoids are the gold standard treatment and most widely prescribed drugs for DMD and BMD as they improve muscle function in the short term, lower muscle inflammation, and delay the loss of ambulation by an average of 3 years5. However, glucocorticoids are also associated with adverse metabolic side effects such as insulin resistance and a heightened risk for type 2 diabetes and osteoporosis6,7,8, which can hasten disease outcomes and lead to early drug withdrawal. Therefore, alternative therapeutic options for patients with DMD should be explored.
Glycogen synthase kinase 3 (GSK3) is a serine/threonine kinase first identified for its role in regulating glycogen synthase in muscle9. There are two GSK3 isoforms, GSK3α and GSK3β, with the latter being the most expressed and active isoform found within muscle10. It is a constitutively active enzyme, and its activity is regulated by phosphorylation, where Ser phosphorylation (Ser 21, GSK3α; and Ser9, GSK3β) inhibits GSK3 activity11. We and others have shown that inhibiting GSK3 can increase muscle mass, strength, regeneration, and the proportion of slow oxidative fibers in skeletal muscle12,13,14,15,16, which altogether can help attenuate dystrophic pathology given that DMD is a condition where fast-twitch fibers are most affected17,18. In addition, GSK3 acts on several pathways to limit insulin-mediated glucose disposal, such as phosphorylation and inactivation of the insulin receptor, insulin receptor substrate, and trafficking regulator of the glucose transporter type 4 (GLUT-4)19,20. With respect to bone, GSK3 has been largely implicated in osteoporosis due to its inhibition of the anabolic Wnt signaling pathway21,22,23. In turn, its inhibition has been shown to increase bone formation, bone healing, and bone strength21,22,24. Thus, inhibition of GSK3 could benefit patients with DMD, while also circumventing some of the negative side effects associated with glucocorticoid therapy25,26,27. To our knowledge, there has been no study investigating whether inhibiting GSK3 can alleviate DMD pathology; however, previous studies have shown that GSK3 inhibition can alleviate other muscle wasting conditions such as myotonic dystrophy28,29,30,31, limb-girdle muscular dystrophy32, and muscle unloading12,14. Furthermore, GSK3 has been shown to be overactive in muscles obtained from the preclinical mdx mouse model for DMD—suggesting GSK3 as a plausible therapeutic target33.
The purpose of this study was to investigate the effects of pharmacological and genetic inhibition of GSK3 on skeletal muscle function and morphology in the severe DBA/2J (D2) and milder C57BL/10J (C57) mdx mouse models of DMD. In addition, we explored the effects of GSK3 inhibition on insulin sensitivity, whole-body metabolism, and bone mineral density and content as those are measures adversely affected by glucocorticoid treatment. Overall, our work shows that GSK3 inhibition improves several metrics of muscle performance in mdx mice without negatively affecting whole-body metabolism, insulin sensitivity, and bone mineral density and content, thereby revealing a potential alternative therapy for DMD.
Results
Tideglusib improves D2 mdx muscle health and performance
We first examined the effects of tideglusib treatment on skeletal muscle function and morphology in 10–12-week-old and 28–30-week-old D2 mdx mice. Tideglusib is a clinically advanced GSK3 inhibitor34,35 currently being tested in clinical trials for conditions such as supranuclear palsy, autism spectrum disorders, and myotonic dystrophy. In this study, we used a dose of 10 mg/kg body mass/day, which is in line with a 600 mg dose for a 60 kg patient. This dose has been used for patients with myotonic dystrophy and is proven to be highly tolerable with little adverse effects36. For the 10–12 week-old cohort, tideglusib treatment started at 6–8 weeks of age and lasted a total of 4 weeks, and as such, treatment occurred in the midst of the initial bout of myonecrosis found in D2 mdx mice37. For the 28–30 week old cohort, tideglusib treatment also lasted 4 weeks and was initiated at 26–28 weeks, an age characterized by a phase of heightened muscle regeneration37. Tideglusib’s primary mode of inhibition is to irreversibly bind to GSK3 and lock it in an inactive state38. However, tideglusib and other thiadiazolidindione compounds can also reduce total GSK3 content39. In support of this, we found that tideglusib-treated mdx mice had lower total GSK3β content in the extensor digitorum longus (EDL) muscle compared with vehicle controls and regardless of age (Supplementary Fig. 1a, c). We did not find any main effects of tideglusib or age on the phosphorylation status of GSK3β (Ser9) relative to total GSK3β content (Supplementary Fig. 1b, d).
The EDL is a fast-twitch muscle that has been shown to be weakened in D2 mdx mice40. Here, we found that twitch and tetanic specific force production of the EDL was significantly greater in 10–12 week old and 28–30 week old tideglusib-treated mdx mice compared with age-matched vehicle-treated mdx mice (Fig. 1a-b). We also assessed the fatiguability of the EDL muscle and found a main effect of tideglusib in the 10–12 week old mdx mice, indicating that tideglusib treatment improved fatigue resistance (Fig. 1c). However, there was no effect of tideglusib on EDL fatiguability in 28–30 week old mdx mice (Fig. 1d). Muscle performance and endurance, measured as hangwire time normalized to body mass (e.g., impulse), was significantly lower in 28–30 week old mdx mice compared with 10–12 week old mdx mice, irrespective of treatment (Fig. 1e). However, there was a main effect of tideglusib, indicating that treatment with the GSK3 inhibitor improved hangwire performance relative to vehicle-treated mice, albeit this effect was minor in the 28–30 week old cohort (Fig. 1e). It is worth noting that while the hangwire test can provide a non-invasive and readily accessible measure of muscle function, it is difficult to control for whether a mouse uses two or four limbs to hang, the direction and speed of movement, or whether the mouse falls off the wire when it reaches the end of a traditional linear hanging apparatus41. These factors can lead to inconsistent results and emphasizes the need to provide added measures of muscle function and health such as ex vivo muscle contractile analyses and measures of muscle quality. In this respect, we also found a significant main effect of tideglusib when assessing serum creatine kinase (CK) levels, indicating that tideglusib treatment lowered muscle degeneration and damage (Fig. 1f). A significant main effect of age for serum CK (Fig. 1f) was also found, which is in agreement with previous findings showing that older D2 mdx mice have lower levels of circulating CK when compared with younger mdx mice42.

a, b Tideglusib treatment increases twitch and tetanic (160 Hz) specific force production (mN/mm2) in the EDL muscle (10–12 weeks, vehicle n = 11, tideglusib n = 10; 28–30 weeks, vehicle n = 9, tideglusib n = 9). c, d Tideglusib treatment improves EDL muscle fatigue with a rightward shift in the fatigue curve in 10–12 week old mdx mice, but not in 28–30 week old mdx mice (10–12 weeks, vehicle n = 9, tideglusib n = 10; 28–30 weeks, vehicle n = 7, tideglusib n = 9). The fatigue protocol consisted of 70 Hz stimulation every 2 s for 160 s and force is presented as percent of initial force over time. e Hangwire impulse is lower in 28–30 week-old mdx mice compared to 10–12 week-old mdx mice, and greater with tideglusib treatment regardless of age (10–12 weeks, vehicle n = 10, tideglusib n = 10; 28–30 weeks, vehicle n = 9, tideglusib n = 8). f Serum creatine kinase (CK) levels are lower with tideglusib treatment and with age (10–12 weeks, vehicle n = 12, tideglusib n = 10; 28–30 weeks, vehicle n = 6, tideglusib n = 5). g, h Muscle histological analysis with H&E staining shows a significant reduction in % necrosis in EDL muscles obtained from 10 to 12 week old tideglusib treated mdx mice (n = 4 per group). Scale bar set to 200 μm. i–k Tideglusib treatment increases myofiber cross-sectional area (CSA) and the proportion of oxidative fibers (type I and IIA) in 10–12 week-old mdx mice (n = 4 per group). Blue, type I fibers; green, type IIA fibers; red, type IIB fibers; unstained (black), type IIX fibers. Scale bar set to 1000 μm. l Tideglusib treatment increases utrophin expression in mdx plantaris muscles from 10 to 12-week-old mdx mice (vehicle, n = 7 and tideglusib n = 10). m, n Tideglusib increases Pax7 and myogenin content in EDL muscles obtained from 28 to 30-week-old mdx mice (n = 6 per group for Pax7 and n = 5 per group for myogenin). For (a–f), a two-way ANOVA was used to assess the main effects of age or time and tideglusib treatment. Significant main effects are denoted in the text above. For (h, j–l and n), a two-tailed Student’s t-test was used. All values are mean ± SEM. *p < 0.05.
We next examined the effects of tideglusib treatment on muscle histology in 10–12 week-old mdx mice, given that this age is characterized by excessive muscle degeneration and necrosis37. Strikingly, we found that tideglusib treatment significantly lowered myonecrosis in the EDL (Fig. 1g, h). This result was accompanied by a significant increase in myofiber cross-sectional area (Fig. 1i-j), which is consistent with the known hypertrophic effect of GSK3 inhibition15,16. Moreover, and in agreement with the repressive action of GSK3 on the oxidative fiber type13,16, tideglusib treatment significantly lowered the proportion of glycolytic fibers (IIB and IIX) while increasing the proportion of oxidative (I and IIA fibers) (Fig. 1i, k).
Further, plantaris muscles from tideglusib-treated mdx mice had greater utrophin expression when compared with vehicle-treated mdx mice (Fig. 1l). Utrophin is a dystrophin homolog that can provide compensatory membrane stability in the absence of dystrophin. It is found predominantly in oxidative muscle fibers18 as its expression is largely controlled by the calcineurin/nuclear factor of activated T-cell (NFAT) signaling axis17,18. Calcineurin is a Ca2+-dependent phosphatase that dephosphorylates NFAT, permitting its localization to the nucleus where it can promote the expression of genes associated with oxidative phenotype, including utrophin18. In addition to regulating utrophin expression, calcineurin can also promote the expression of genes involved with muscle regeneration, and therefore, its activation provides multiple benefits that can attenuate dystrophic pathology in mdx mice17,43,44. Given that we and others have shown that GSK3 counteracts calcineurin signaling13,45,46, we attribute at least part of the benefits observed with GSK3 inhibition in mdx mice to the activation of calcineurin. With respect to muscle regeneration, we next assessed the protein levels of Pax7 and myogenin, which are markers of muscle regeneration14. We focused on 28–30-week-old mice for these measures since at this age, mdx mice are known to be undergoing a period of heightened muscle regeneration37. Our results show that tideglusib treatment significantly increased the protein levels of Pax7 and myogenin in EDL muscles when compared with vehicle controls (Fig. 1m, n). These findings are consistent with previous work showing that GSK3 inhibition accelerates muscle regeneration12,14,15. Collectively, these data show that GSK3 inhibition in the D2 mdx mouse model can impart several benefits to muscle strength and quality by lowering muscle necrosis, increasing muscle size, and promoting the oxidative fiber type and muscle regeneration.
Tideglusib improves whole-body metabolism in D2 mdx mice
It has been previously established that D2 mdx mice present with a hypermetabolic phenotype47,48, expending a significant amount of energy on a daily basis to support the costs of both muscle degeneration and regeneration. In 10–12-week-old and 28–30-week-old D2 mdx mice, tideglusib-treated mice had lower energy expenditure than vehicle control mice (Fig. 2a, b). This effect was not due to any changes in daily cage ambulation (Fig. 2c, d). Furthermore, the hypermetabolic phenotype in mdx mice has been previously shown to lower body fat content, resulting in less whole-body lipid supply, thereby raising the respiratory exchange ratio (RER)48. In our hands, D2 mdx mice also had a higher RER when compared with WT mice (Supplementary Fig. 2). However, and perhaps cohesive with the reduction in energy expenditure, tideglusib-treated mdx mice had higher body fat content when compared with vehicle-treated mice, though this effect was most prominent at 28–30 weeks of age (Fig. 2e, f). Moreover, at both 10–12 weeks and 28–30 weeks of age, tideglusib-treated mdx mice had lower RER values when compared with vehicle-treated mice (Fig. 2g, h).

a, b Tideglusib lowers daily energy expenditure in 10–12 week-old and 28–30-week-old mdx (10–12 weeks, vehicle n = 5, tideglusib n = 4; 28–30 weeks, vehicle n = 9, tideglusib n = 9). c, d Tideglusib treatment did not alter cage ambulation (10–12 weeks, vehicle n = 5, tideglusib n = 4; 28–30 weeks, vehicle n = 9, tideglusib n = 9). e, f Body fat content (g) and body fat composition (% of body mass) are elevated in tideglusib-treated mdx mice compared to vehicle controls, and this effect is most prominent in 28-30 week old mdx mice (10–12 weeks, vehicle n = 12, tideglusib n = 10; 28–30 weeks, vehicle n = 8, tideglusib n = 9). g, h Tideglusib lowers the respiratory exchange ratio (RER) in 10–12 week-old and 28–30 week-old mdx mice (10–12 weeks, vehicle n = 5, tideglusib n = 4; 28–30 weeks, vehicle n = 9, tideglusib n = 9). i–l Insulin tolerance tests and corresponding area-under-the-curve (AUC) analysis, baseline glucose levels and % decline in blood glucose in 28–30 week old mdx-vehicle and mdx-tidgelusib mice. Glucose traces are displayed relative to baseline glucose levels. AUC was obtained from the normalized traces. There are no differences in baseline glucose levels (mM) (vehicle n = 10 and tideglusib n = 8). For (a–d), a two-way ANOVA was used to assess the main effects of time period and tideglusib treatment. For (e–h), a two-way ANOVA was used to assess the main effects of age and tideglusib treatment with a Tukey’s post-hoc in the event of a significant interaction. For i, a two-way repeated ANOVA was used for the insulin tolerance test. Significant main effects and interaction terms are denoted in the text above. For (j–l), a two-tailed Student’s t-test was used. All values are mean ± SEM. *p < 0.05, **p < 0.01.
In addition to the hypermetabolic phenotype, mdx mice are well-known to be insulin resistant49. Here, we show that vehicle-treated mdx mice showed very little response to insulin during an insulin tolerance test, and many of these mice even displayed a slight increase in blood glucose levels after insulin injection (Fig. 2i–l). In contrast, tideglusib-treated mdx mice were more responsive to insulin, leading to a ~20% drop in blood glucose levels over time and a significant reduction in the area-under-the-curve (AUC, Fig. 2i–l). Altogether, our results show that tideglusib treatment improves muscle strength, lowers muscle necrosis, and enhances insulin sensitivity in D2 mdx mice. Indeed, these results are likely related as skeletal muscle accounts for more than 90% of whole-body insulin-stimulated glucose removal50, and thus, improving muscle health can positively impact glucose regulation.
Effects of tideglusib treatment in C57 mdx mice
While the D2 mdx mouse model exhibits a severe dystrophic phenotype that is relatively more representative of the human condition than the C57 mdx mouse model37, we also examined the effects of tideglusib on 10–12 week old C57 mdx mice, as this mouse model is most commonly used to study DMD. Twitch and tetanic specific force of the EDL was not different between tideglusib-treated and vehicle-treated mdx mice (Fig. 3a-b). However, EDL muscle endurance was improved with tideglusib treatment (Fig. 3c). Impulse (hangwire time normalized to body mass) was greater in tideglusib-treated mdx mice than in vehicle-treated mdx mice, though this did not reach statistical significance, which we believe could be due to the high degree of variability that can be observed with this test (p = 0.09; Fig. 3d). Serum CK was also not different between groups (Fig. 3e). We found a trending main effect of tideglusib on energy expenditure, where tideglusib-treated C57 mdx mice had higher energy expenditure than vehicle-treated C57 mdx mice (p = 0.09; Fig. 3f). This is in contrast with our tideglusib experiments in D2 mdx mice. However, a main effect of tideglusib and time were detected for RER, where tideglusib-treated C57 mdx mice had lower RER than vehicle-treated C57 mdx mice, which is consistent with our observations in D2 mdx mice (Fig. 3g). The differential response to tideglusib observed in C57 mdx mice with respect to energy expenditure (when compared to D2 mdx mice) could be explained by the higher levels of cage ambulation found in tideglusib-treated C57 mdx mice compared with vehicle-treated C57 mdx mice (Fig. 3h). Moreover, the increase in cage ambulation may be indicative of attenuated muscle pathology and enhanced muscle performance with tideglusib treatment in the C57 mdx mice. With respect to insulin sensitivity, and unlike that with D2 mdx mice, an insulin tolerance test found no differences between C57 vehicle and C57 tideglusib-treated mdx mice for blood glucose levels over time, the AUC, baseline blood glucose, and % decline in blood glucose (Fig. 3i–l). Nonetheless, it is important to consider that the improvements in muscle endurance and performance that we observed in C57 mdx mice treated with tideglusib was not met with any significant deleterious effects on insulin sensitivity or glucose regulation. This is in stark contrast with what can be expected with chronic daily exposure to glucocorticoids in these mice51,52.

a, b Twitch and tetanic (160 Hz) specific force production (mN/mm2) is unchanged in the EDL muscle with tideglusib treatment (n = 5 per group). c Tideglusib treatment improves EDL muscle fatigue with a rightward shift in the fatigue curve in 10-12 week old C57 mdx mice (n = 5 per group). d Hangwire impulse is greater in tideglusib-treated mdx mice, which approached statistical significance (p = 0.09) (vehicle, n = 6 and tideglusib, n = 7). e Serum creatine kinase (CK) (U/L) is not different between vehicle and tideglusib-treated mdx mice (n = 8 per group). f Energy expenditure is greater in tideglusib-treated mdx mice with this effect approaching significance (p = 0.09), across light, dark, and daily cycles (vehicle, n = 8 and tideglusib, n = 7). g Tideglusib lowers the respiratory exchange ratio (RER) in C57 mdx mice (vehicle, n = 8 and tideglusib, n = 7). h Tideglusib elevates cage ambulation (meters traveled) in C57 mdx mice (vehicle, n = 8 and tideglusib, n = 7). i–l Insulin tolerance tests and corresponding area-under-the-curve (AUC) analysis, baseline glucose levels and % decline in blood glucose in mdx-vehicle and mdx-tideglusib mice (n = 8 per group). Glucose traces are displayed relative to baseline glucose levels. AUC was obtained from the normalized traces. There is no difference in the AUC, basline blood glucose (mM), and % decline in blood glucose. For (a, b, d, e, j, k, and l), a two-tailed Student’s t-test was used. For (c, f, g, and h), a two-way ANOVA was used to assess the main effects of time and tideglusib treatment. For (i), a two-way repeated ANOVA was used for the insulin tolerance test. Significant main effects and interaction terms are denoted in the text above. All values are mean ± SEM.
Effects of muscle-specific GSK3 knockdown in mdx mice
To further demonstrate the benefits of GSK3 inhibition in the mdx mouse, we then bred male homozygous muscle-specific GSK3 knockdown (KD) mice (C57BL/6J background14,53) with female homozygous D2 mdx mice. This cross produced male offspring that were 100% mdx mice on a 50:50 D2:C57BL/6J hybrid strain with either GSK3 protein left intact (mdx/GSK3flox, herein referred to as mdx) or GSK3 protein reduced by 30–40% specifically in their skeletal muscles (mdx/GSK3KD). We characterized muscle function, performance, and quality at 4-6 weeks and 10–14 weeks of age. We selected 4–6 weeks alongside 10–14 weeks of age since mdx mice typically experience an early phase of degeneration and myonecrosis; and thus, testing the potential benefits of GSK3 inhibition at this particular age may add clinical relevance. A main effect of genotype was detected on GSK3α and GSK3β content where mdx/GSK3KD had significantly lower GSK3α and GSK3β protein levels in their EDL muscles compared to mdx mice, regardless of age (Fig. 4a–c). Twitch and tetanic specific force production in the EDL were significantly greater in 4-6 week old and 10-14 week old mdx/GSK3KD mice compared to age-matched mdx controls (Fig. 4d, e). There was also a main effect of genotype on EDL fatiguability for both 4–6 week-old and 10–14 week-old mdx mice, where mdx/GSK3KD mice had enhanced fatigue resistance (Fig. 4f, g). Further, serum CK analysis revealed a main effect of genotype and significant interaction between genotype and age, where mdx/GSK3KD mice had significantly lower serum CK levels than mdx control mice, being statistically significant at 4–6 weeks but not at 10–14 weeks (Fig. 4h). Altogether, we demonstrate that muscle-specific KD of GSK3 improves skeletal muscle function, enhances fatigue resistance, and may reduce muscle damage in a D2:C57BL/6J hybrid mdx mouse model.

a–c GSK3α and GSK3β content are lower in 4–6 week old and 10–14 week-old mdx/GSK3KD mice compared with their respective mdx counterparts (mdx, n = 3 and mdx/GSK3KD n = 4). d–e Twitch and tetanic force production of the EDL is improved in 4–6 week-old and 10–14 week old mdx/GSK3KD mice compared with mdx mice (4–6 weeks mdx, n = 7 and mdx/GSK3KD n = 6; 10–14 weeks mdx, n = 4 and mdx/GSK3KD n = 6). f, g EDL muscle fatigue is improved in mdx/GSK3KD mice at 4–6 weeks and 10–14 weeks of age with a rightward shift in the fatigue curve (4–6 weeks mdx, n = 7 and mdx/GSK3KD n = 8; 10-14 weeks mdx, n = 4 and mdx/GSK3KD n = 6). h mdx/GSK3KD mice have lower serum creatine kinase (CK) (U/L) than mdx mice at 4–6 weeks, but not at 10–14 weeks (4–6 weeks mdx, n = 7 and mdx/GSK3KD n = 6; 10–14 weeks mdx, n = 4 and mdx/GSK3KD n = 6). For (b–e and h), a two-way ANOVA was used to assess the main effects of genotype and age. For (f, g), a two-way ANOVA was used to assess the main effects of genotype and time. All values are mean ± SEM. Significant main effects and interaction terms are denoted in the text above. *p < 0.05, **p < 0.01.
Combined effects of GSK3 inhibition and aerobic exercise
The improvements in muscle health and performance observed with GSK3 inhibition (pharmacological or genetic) could have downstream benefits that ultimately enhance mobility. Indeed, this was observed in C57 mdx mice treated with tideglusib, which we believe could have implications for those living with DMD. Given that DMD primarily affects muscle, the role of exercise in disease management has remained controversial54. That is, though the intentions are good, certain exercises may cause more harm and thus, for the most part, the recommended exercises have been limited to mild or moderate intensity regimes55,56. This highlights a potential opportunity to develop adjuvant therapies that can be combined with low-intensity forms of exercise. Thus, we next investigated the combined effects of exercise and GSK3 inhibition on muscle form and function in 10–12 week old D2 mdx mice. Voluntary wheel running (VWR) was chosen as the mode of exercise given its low intensity and minimal stress added to the mouse57. To further minimize stress, we treated the mice with the GSK3 inhibitor, lithium (Li), as it could be provided easily through drinking water rather than performing oral daily gavage as with tideglusib. Li is a natural inhibitor of GSK3 and is most commonly used for the treatment of bipolar disorder58; however, its clinical use must occur within a narrow therapeutic range of 0.5–1.0 mM serum Li concentration to avoid potential adverse effects on various tissues58. Here, we began treatment at 6 weeks of age and provided a low dose of lithium chloride (50 mg/kg/day via drinking water) for 6 weeks, which resulted in a serum Li concentration of 0.15 ± 0.05 mM (n = 9). Treatment did not result in any differences between groups on total or phosphorylated GSK3β content (Supplementary Fig. 3). However, this does not discount GSK3 inhibition since Li can regulate GSK3 in two ways: a secondary and indirect path that relies on the activation of kinases (e.g., Akt, PKA, PKC) which phosphorylate and inhibit GSK3; and a primary and direct path whereby Li competes for a magnesium (a co-factor for GSK3 activity) binding site on GSK359. Corresponding well with this, we found an increase in β-catenin content with Li supplementation (Supplementary Fig. 3). β-catenin is part of the canonical Wnt signaling pathway, and its phosphorylation by GSK3 signals for its cellular degradation, and thus an increase in β-catenin content can be used as a downstream marker of GSK3 inhibition9,15,24,60,61.
In further support of effective GSK3 inhibition, our results with Li supplementation are consistent with those found with tideglusib treatment and genetic reduction of GSK3. Specifically, we examined the effects of VWR with and without Li supplementation on EDL and soleus muscle contractility, given that the soleus muscle is responsive to VWR62. In the soleus, twitch and tetanic force were significantly depressed in mdx-SED mice when compared with WT mice (Fig. 5a, b). Though VWR appeared to improve soleus force production in mdx mice, it did not lead to any significant differences when compared with mdx-SED mice (p = 0.07 and 0.09 for twitch and tetanic force comparisons, respectively). Furthermore, the mdx-VWR soleus force outputs were still significantly lower when compared with WT mice (Fig. 5a, b). In contrast with this, the combination of VWR and Li provided additional benefits to soleus force production, where twitch and tetanic forces were higher than mdx-SED mice, and though still lower than WT levels, the difference between the two groups was not significant (Fig. 5a, b). In the EDL, twitch and tetanic force were significantly lower in all mdx groups compared with WT mice; however, force output was improved with Li supplementation such that twitch and tetanic force production were greater in the mdx-VWRLi group when compared with mdx-VWR (Fig. 5c, d). A fatigue protocol was applied to the soleus and EDL where significant main effects of time and condition were detected in both muscles (Fig. 5e, f). In the soleus, fatigue resistance was significantly worse in mdx-SED mice and while VWR benefited soleus muscle endurance, this effect was amplified with Li treatment (Fig. 5e). In the EDL, mdx-VWR mice had the lowest level of endurance, highlighting a possible negative effect of aerobic exercise on this muscle type; however, this was prevented in the mdx-VWRLi mice (Fig. 5f).

a–d Twitch and tetanic specific force production in isolated soleus and EDL muscles is improved in the mdx-VWRLi group (soleus, WT n = 8, mdx-sedentary [SED] n = 9, mdx-VWR n = 10, mdx-VWRLi n = 11; EDL WT n = 8, mdx-SED n = 8, mdx-VWR n = 9, mdx-VWRLi n = 8). e, f Soleus fatigue resistance is enhanced in the mdx-VWR and mdx-VWRLi groups, while in the EDL, fatigue resistance is worse in the mdx-VWR group and restored with lithium supplementation (soleus, mdx-SED n = 9, mdx-VWR n = 10, mdx-VWRLi n = 10; EDL mdx-SED n = 9, mdx-VWR n = 10, mdx-VWRLi n = 10). g, h Muscle histological analysis with H&E staining shows a significant reduction in % necrosis in soleus and EDL muscles obtained from 12-week old mdx-VWRLi mice vs. mdx-SED mice. A significant main effect of muscle type also indicates that EDL muscles have greater % necrosis vs. soleus muscles (soleus, mdx-SED n = 6, mdx-VWR n = 6, mdx-VWRLi n = 5; EDL mdx-SED n = 5, mdx-VWR n = 5, mdx-VWRLi n = 6). i Utrophin expression is higher in in quadricep muscles from mdx-VWR and mdx-VWRLi groups when compared with mdx-SED mice (mdx-SED n = 9, mdx-VWR n = 11, mdx-VWRLi n = 11). j Respiratory exchange ratio (RER) is lowest in the mdx-VWRLi mice when compared to all other mdx groups (n = 10 per group). k–n Insulin tolerance tests and corresponding area-under-the-curve (AUC) analysis, baseline glucose levels and % decline in blood glucose in 12-week old WT, mdx-SED, mdx-VWR, and mdx-VWRLi mice (n = 11 per group; mdx-SED vs. WT = *, mdx-SED vs. mdx-VWR = †; mdx-VWR vs. WT = $; mdx-SED vs. mdx-VWRLi = #). Glucose traces are displayed relative to baseline glucose levels. AUC were obtained from the normalized traces. There were no differences in baseline glucose levels. o, p Average daily and total distance traveled with a cagewheel was lower in mdx-VWRLi mice vs. mdx-VWR mice (n = 11 per group). For (a–d, i, l and n), a Welch’s one-way ANOVA with a Dunnett’s T3 post-hoc test was used. For (e, f), a two-way ANOVA was used to assess main effects of force and time. For (h, j), a two-way ANOVA with a Tukey’s post-hoc test was used; and significant main effects and interactions denoted in the text above. For (k), a two-way repeated ANOVA was used with a Tukey’s post-hoc test. For (m), a one-way ANOVA was used. Significant main effects are denoted in the text above. For (o, p), a a two-tailed Student’s t test was used. All values are mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001. † p< 0.05, †† p < 0.01. # p < 0.05, ## p < 0.01, ### p < 0.0001. $ p < 0.05.
These improvements in soleus and EDL contractile function observed in the mdx-VWRLi mice was accompanied by reduced muscle necrosis compared with mdx-SED mice (Fig. 5g, h), which is consistent with our tideglusib experiments. Furthermore, a significant main effect of muscle type for muscle necrosis was found, indicating that across treatment groups, the EDL had greater % necrosis when compared with the soleus (Fig. 5h). This was expected as glycolytic muscles are more susceptible to dystrophic pathology17,18. We also found that VWR increased utrophin expression in the quadriceps, which is consistent with previous work63, however, Li supplementation provided no added benefit to this muscle (Fig. 5i).
With respect to the metabolic impairments observed in D2 mdx mice, and similar to that observed with tideglusib treatment (Figs. 2 and 3), the mdx-VWRLi mice had lower RER when compared to the mdx-SED and mdx-VWR mice, which is indicative of enhanced lipid oxidation (Fig. 5j). The mdx-VWRLi mice were also more insulin sensitive when compared with the other mdx groups (Fig. 5k, l). Specifically, insulin tolerance tests showed that mdx-SED mice were insulin resistant compared with WT, mdx-VWR, and mdx-VWRLi mice (Fig. 5k). While VWR certainly improved insulin tolerance in mdx mice, Li supplementation provided an additive effect, whereby mdx-VWRLi mice had the lowest AUC and largest percent decline (~67%) in blood glucose levels compared with all mdx groups and without any differences in baseline glucose levels (Fig. 5l–n). Furthermore, the metabolic and contractile benefits of Li were found with half the exercise volume compared with mdx-VWR mice (Fig. 5o, p). Though we cannot provide an explanation for the lowered exercise volume with Li supplementation, these results show that despite running less, Li supplementation combined with VWR can significantly improve specific force production, lower muscle necrosis, and enhance oxidative lipid metabolism and insulin sensitivity. As some patients with DMD may not be able to engage in adequate amounts of physical activity, our results may be clinically impactful, as they suggest that exercise and treatment with a GSK3 inhibitor may provide synergistic benefits to muscle and metabolic health.
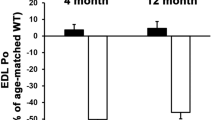
GSK3 inhibition positively affects bone health in mdx mice
Finally and in addition to insulin sensitivity, which we found was either improved (D2 mdx mice treated with tideglusib or Li) or unchanged (C57 mdx mice treated with tideglusib) with pharmacological GSK3 inhibition, we also examined the potential effects on bone mineral content and density. This is important given that chronic and daily glucocorticoid use can accelerate bone turnover and increase the risk of osteoporosis25,26,27. In contrast, GSK3 inhibition is known to have osteoprotective effects that can for the most part be attributed to the downstream activation of the canonical Wnt signaling pathway28,29,36,64. Our results in mdx mice are consistent with this. We used weight-adjusted bone mineral content and bone mineral density as we (Supplementary Fig. 4) and others65,66,67 have shown that body mass is positively associated with bone mineral content and density. In D2 mdx mice, we found that weight-adjusted bone mineral content and density were significantly higher in the tideglusib-treated group, particularly at 28–30 weeks of age (Fig. 6a, b). In 10–12 week old C57 mdx mice, those treated with tideglusib for 4 weeks had significantly higher weight-adjusted bone mineral content and density when compared with vehicle controls (Fig. 6c, d). Further, in our VWR study, we found that mdx-VWR mice had significantly lower bone mineral content and density than mdx-SED mice (Fig. 6e, f), which indicates that 6 weeks of VWR had a negative effect on bone health in D2 mdx mice. We speculate that this may be due to the stress of added muscle activation, and presumably damage, which is consistent with the negative effects of VWR that we observed on EDL muscle function. In contrast, these negative effects on muscle (Fig. 5c–e) and bone (Fig. 6e, f) were not apparent when VWR was combined with Li supplementation. This result is not entirely surprising given the known osteoprotective effects of Li68. Nevertheless, these findings are important given that prescribing exercise as a way to manage DMD remains controversial. Here, we show that while VWR improved insulin sensitivity in D2 mdx mice, it did not improve and even appeared to worsen EDL muscle and bone outcomes. In contrast, VWR combined with Li supplementation provided an additive benefit to insulin sensitivity in D2 mdx mice, all while restoring or improving EDL muscle performance and bone mineral content/density.

a, b Tideglusib-treated D2 mdx mice have greater weight-adjusted bone mineral content (mg/g body weight, wBMC) and bone mineral density (mg/cm2, wBMD) at 10–12 weeks and 28–30 weeks of age, though this effect was more prominent in 28–30 week old mice (D2 10–12 weeks, vehicle n = 12, tideglusib n = 10; D2 28–30 weeks, vehicle n = 8, tideglusib n = 10). c, d wBMC and wBMD are greater in tideglusib-treated 10–12 week old C57 mdx mice than vehicle-treated mdx mice (n = 8 per group). e, f D2 mdx-VWR have lower wBMC and wBMD than mdx-SED mice, and lithium supplementation attenuated this effect (mdx-SED n = 4, mdx-VWR n = 4, mdx-VWRLi n = 5). For (a, b), a two-way ANOVA was used to assess main effects of age and tideglusib treatment or genotype. For (c, d), a a two-tailed Student’s t- est was used. For (e, f), a one-way ANOVA was used. All values are mean ± SEM. Significant main effects are denoted in the text above. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
GSK3 signaling in DMD myoblasts
Collectively, our findings from the various mdx mouse models suggest that inhibiting GSK3 could be a potential therapeutic approach for the management of DMD. To provide additional support for the role of GSK3 in DMD pathology, we measured the content and activation status of GSK3 in myoblasts previously derived from two patients living with DMD. Interestingly, when compared with myoblasts obtained from a healthy control, we found that DMD myoblasts have lowered GSK3β content (Fig. 7a, b). Additionally, the levels of Ser9 phosphorylated GSK3β were also lower in DMD myoblasts compared with controls (Fig. 7a), however, the reduction in Ser9 phosphorylated GSK3β was primarily due to the reduction in total GSK3β as the calculated ratio of phosphorylated GSK3β relative to total GSK3β revealed no differences between groups (Fig. 7c).

a Representative western blot images for GSK3β, Ser9 phosphorylated GSK3β, and β-catenin in myoblasts derived from a healthy control (HC), 18 year-old DMD patient (DMD18) and a 9-year-old DMD patient (DMD9) (n = 3 technical replicates per group). b Total GSK3β content is elevated in myoblasts from HC compared to myoblasts from DMD18 and DMD9 (n = 3 technical replicates per group). c Phosphorylated GSK3β content (relative to total) is not different between groups (n = 3 technical replicates per group). Data are presented relative to HC. d β-catenin content is significantly greater in myoblasts from HC compared to myoblasts from DMD18 and DMD9 (n = 3 technical replicates per group). The ponceau stain for phosphorylated GSK3β and β-catenin are the same as both proteins were probed on the same membrane. e β-catenin content is significantly greater in myoblasts from the isogenic control (IC) of DMD9 compared with myoblasts derived from DMD9 (n = 3 technical replicates per group). IC is the DMD9 myoblast with the dystrophin gene re-introduced. Data are presented relative to IC. For (b–d), a one-way ANOVA was used. For (e), a a two-tailed Student’s t-test was used. All values are mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001.
In light of the findings presented in this study, we view the reduction in GSK3 content to be an adaptive response aimed at improving muscle health and function in DMD patients. Yet, when we measured the protein levels of β-catenin as a downstream marker of GSK3 activation, we found that β-catenin content was also lower in DMD myoblasts compared with healthy control (Fig. 7a, d). In fact, similar observations of lowered β-catenin content were made when comparing DMD myoblasts with their isogenic control, which are DMD myoblasts with the dystrophin gene re-introduced69 (Fig. 7e). This demonstrates that the absence of dystrophin leads to reductions in β-catenin content and could suggest one of two things. First, despite the reduction in GSK3 content found in DMD myoblasts, its activation may still be enhanced when compared with controls. Secondly, or perhaps congruently, the lowered levels of β-catenin found in DMD myoblasts may be due to upstream impairments in Wnt activation. Once Wnt ligands bind to their receptors, the activation of the canonical Wnt signaling pathway prevents GSK3 phosphorylation of β-catenin, thereby allowing for its cellular accumulation9. In either case, our findings highlight the importance of targeting GSK3 for DMD, as pharmacological inactivation of GSK3 may help restore β-catenin levels and the canonical Wnt signaling pathway in DMD muscle cells. Indeed, this pathway is clinically relevant in the context of DMD since Wnt signaling, through β-catenin, acts as a critical molecular switch regulating the transition from cell proliferation to myogenic differentiation – a vital muscle development and repair process70. It was previously shown that these myoblasts derived from DMD patients fail to differentiate and fuse into myotubes69, and our results indicate that this may be due to a reduction in β-catenin content. Moreover, we and others have shown that inhibiting GSK3 in murine and porcine muscle cells augments myogenic differentiation and myoblast fusion, an effect largely attributed to an accumulation of β-catenin15,61,71,72,73. Thus, future studies from our laboratory will determine if inhibiting GSK3 in DMD myoblasts restores β-catenin content and rescues myoblast differentiation and fusion.
Discussion
In the present study, we demonstrate that inhibiting GSK3 in severe D2 and milder C57 mdx mouse models, either through pharmacological means or genetic intervention, can enhance skeletal muscle health and function. The improvements in muscle health and performance most notably include an increase in specific force production and a consistent improvement in fatigue resistance. We attribute these benefits to a shift towards the oxidative fiber type as indicated by immunofluorescent fiber type experiments as well as lowered RER, which is indicative of enhanced oxidative metabolism with GSK3 inhibition. Additionally, GSK3 inhibition in mdx mice reduced myonecrosis and increased myogenesis. However, other cellular mechanisms, such as enhanced mitochondrial biogenesis and respiration, as well as increased glycogen synthesis, may also play a role in these benefits and should be investigated in future studies.
Additionally, the positive effects on muscle health and performance resulting from GSK3 inhibition did not negatively impact insulin sensitivity or bone mineral content and density. In the severe D2 mdx mouse model, we observed improvements in insulin sensitivity and markers of bone health in mice treated with tideglusib or Li (in combination with VWR). These findings are in contrast with results typically found with glucocorticoid treatment, the current standard of care for DMD, which is known to negatively affect insulin sensitivity and bone health, increasing risk of developing type 2 diabetes and osteoporosis25,26,27. Therefore, based on our current findings, future research should investigate whether combining glucocorticoid treatment with GSK3 inhibition can synergistically improve muscle health and performance without negatively impacting glucose regulation and skeletal health. Indeed, since tideglusib is currently being tested for other diseases and Li is already FDA-approved, there is a strong potential to repurpose these drugs for DMD.
To further support the role of GSK3 in DMD pathology, we measured GSK3 content and Ser9 phosphorylation in myoblasts previously derived from two patients living with DMD. While GSK3 content was lowered, there were no differences in Ser9 phosphorylation in these myoblasts when compared with the healthy control. However, we did observe evidence of impaired Wnt/ß-catenin signaling, with DMD myoblasts having significantly lower ß-catenin content. This reduction in ß-catenin content was also evident in our comparisons made between DMD myoblasts and their isogenic control, where the dystrophin gene was reintroduced. We believe that this may be contributing to the previously characterized impaired myogenic differentiation and fusion in these cells69. While our manuscript was under review, another study was published demonstrating impaired Wnt/ß-catenin signaling in mdx mice, which could be resolved by GSK3 inhibition via isolecanoric acid (ILA)74. Specifically, treatment with ILA was found to improve muscle regeneration and performance and is therefore in strong agreement with our findings presented here. Furthermore, it was found that GSK3 inhibition via ILA treatment promotes myogenic differentiation and ß-catenin signaling in murine and human stem cells74, which is again consistent with previous findings from our lab and others15,61,71,72,73. Altogether, even though GSK3 levels are reduced in DMD myoblasts, its activation within the canonical Wnt signaling pathway could still be elevated. This increase might stem from disrupted upstream Wnt signaling events; and consequently, inhibiting GSK3 could potentially mimic Wnt signaling to help restore ß-catenin levels and muscle regeneration in DMD muscle.
We acknowledge that our study has several limitations. First, we only assessed the acute effects of tideglusib treatment. Thus, future studies should investigate long-term tideglusib treatment in mdx mice to fully assess its efficacy and potential side effects. We also only assessed the effects of pharmacological GSK3 inhibition (tideglusib or Li) in mdx mice at an age past the initial bout of necrosis (>4 weeks of age). However, we did find significant benefits of muscle-specific GSK3 deletion in relatively younger (4–6 week old) mdx mice. The mdx/GSK3KD mice are a 50:50 hybrid of DBA/2J and C57BL/6J, thus future studies should investigate whether deleting GSK3 specifically in the muscles can provide benefits to a mdx mouse that is 100% DBA/2J. This will help determine if GSK3 knockdown can be beneficial for the severe mdx mouse model. Additionally, our experiments involving DMD myoblasts are limited because we could only acquire healthy control myoblasts sourced from human embryonic stem cells, as opposed to the DMD myoblasts that were derived from human pluripotent stem cells (converted from fibroblasts). It is possible that this difference could confound our results; however, we note that we also found lowered levels of ß-catenin in DMD myoblasts compared to their own isogenic control.
In conclusion, our study showcases the therapeutic potential of inhibiting GSK3 for DMD. Here, we have shown benefits to mdx muscle health and performance with GSK3 inhibition alone or combined with exercise. Given that these benefits with GSK3 inhibition were not associated with negative adverse effects to glucose regulation and bone health, future studies should also explore other combinatorial therapies, especially the potential combination of GSK3 inhibition and glucocorticoid treatment. We expect that such treatments have the potential to significantly improve the quality of life for those with DMD.
Methods
Study approval
All animal procedures were approved by the Brock University Animal Care and Utilization Committee (file #17-06-03, 20-07-01, 22-08-03, 22-04-01, and 23-03-02) and were carried out in accordance with the Canadian Council on Animal Care guidelines. Work with human-derived myoblasts was also approved by the Brock University Research Ethics Board (File # 21-355).
Mice and design
D2-mdx (stock #013141), D2 WT (stock #000671), C57-mdx (stock #001801), and C57 WT (stock #000664) were ordered from Jackson Laboratories at 5–7 weeks of age. For the aged tideglusib experiments, mdx mice were ordered from Jackson Laboratories at 7–9 weeks of age with treatment starting at 24–26 weeks of age. To generate the mdx/GSK3KD and mdx/GSK3flox control mice, homozygous male GSK3KD mice (C57BL/6J) were bred with homozygous D2 mdx female mice (stock #013141, Jackson Laboratories). Muscle-specific GSK3 deletion was achieved using the Cre-Lox system with heterozygous ACTA-1 Cre driving muscle-specific GSK3 knockdown14. When homozygous GSK3 floxed (+heterozygous ACTA-1 Cre) male mice (sourced from a breeding colony at Brock University) were bred with homozygous D2 mdx mice, it produced male offspring that were 100% mdx (X chromosome derived from homozygous mdx female mouse) and heterozygous for GSK3 flox on a DBA/2J:C57BL/6J (50:50) hybrid strain. Half of the male offspring harbored the ACTA-1Cre gene thereby leading to partial muscle-specific GSK3KD on an mdx background (mdx/GSK3KD), whereas the other half of male offspring did not have the ACTA-1Cre gene giving rise to our mdx/GSK3flox controls. All mice were housed in Brock University’s Animal Facility, in an environmentally controlled room with a standard 12:12 h light-dark cycle, at room temperature (22–24 °C) and allowed access to food (2014 Teklad, Inotiv, IN, USA) and water ad libitum.
Tideglusib treatment
6–8 week-old D2 and C57 and 24–26 week old D2 mdx mice were treated with tideglusib at a dose of 10 mg/kg/day via oral gavage, whereas a separate vehicle group of mdx mice were given 20% Kolliphor (v/v) for 5 days per week for 4 weeks. In vivo assessments began 2 weeks after treatment started.
Voluntary wheel running and Li supplementation
6 week-old D2 mdx mice were divided into three groups: 1) mdx-SED, 2) mdx-VWR, and 3) mdx-VWRLi based on lean mass (g). The mdx-SED group was used as a diseased control group and a WT group was used as a healthy control. The VWR group was provided with a cage wheel for 6 weeks and all mice were housed in the Techniplast DVC80 with GYM500 software to track cage wheel distance traveled 24/7. The mdx-VWR mice were provided lithium chloride (50 mg/kg/day) in their drinking water with 2 water changes per week for the entire duration of the study. To attain the desired dose of 50 mg/kg/day, the concentration of lithium chloride in the drinking water was calculated based on a presumed daily water intake of 5 ml/day. By the end of the treatment schedule, the average daily water consumption for each group was as follows: mdx-SED, 4.585 ± 0.063 ml/day; mdx-VWR, 4.834 ± 0.134 ml/day; and mdx-VWRLi, 4.838 ± 0.133 ml/day.
Cell culture
DMD patient and control-derived myoblasts were obtained from Dr. Gabsang Lee using a previously published protocol69. DMD myoblasts include those from a 9-year-old patient with an EX4-43DEL mutation (05169) as well as an 18-year-old patient with a c.5533G>T, p.E1845X mutation (05127). The DMD myoblasts were originally obtained from fibroblasts (Coriell Institute for Medical Research Catalog #GM05169 and #GM05127, respectively) that were reprogrammed into human pluripotent stem cells (hPSC), whereas the control myoblasts were originally obtained from human embryonic stem (hESC). The H9 hESC line was purchased from WiCell (WA09) and is listed on the Human Pluripotent Stem Cell Registry (hPSCReg ID: WAe009-A), where details of informed consent are provided. Details surrounding patient informed consent for the fibroblasts purchased from Coriell #GM05169 and #GM05127 could not be provided due to confidentiality. Both DMD hPSCs and control hESCs were directed towards a skeletal muscle cell lineage and converted into myoblasts using a chemical compound-based approach69. In addition to the healthy control, an isogenic control was also included. The isogenic control myoblasts are genetically corrected DMD myoblasts whereby the entire dystrophin gene was inserted into the GM05169 DMD fibroblast before its myogenic conversion. Once the myoblasts were shipped to Brock University, they were cultured in growth medium [Dulbecco’s modified Eagle’s medium (DMEM), D6429 Sigma, Oakville, ON, Canada] supplemented with 10% fetal bovine serum (Sigma F1051), 1% penicillin/streptomycin (Sigma P4333), and 2% non-essential amino acids (Sigma M7145) at 37 °C in a humidified atmosphere with 5% CO2 and 5% O2 levels15. After reaching confluence, the cells were trypsinized and pelleted. The pellets were then lysed in RIPA buffer and stored at −80 °C for western blot analysis.
RER analysis
Mice were housed in the Promethion Metabolic Screening System for a period of 36–48 h to track fuel utilization. The cages are connected to a flow regulator and O2 and CO2 gas analyzers which measure the volume of carbon dioxide produced (VCO2) and the volume of oxygen consumed (VO2). Respiratory exchange ratio (RER) was calculated as VCO2 divided by VO2.
Insulin tolerance test
Insulin tolerance testing was performed as previously described53. Briefly, concentrations of blood glucose were determined by sampling blood from the tail vein at 0, 15, 30, 60, and 120 min following the IP insulin injection (0.5 U/kg) with the use of a hand-held glucometer (Freestyle Lite; Abbott). Plots of the average changes in plasma glucose over time were made for each group, and the average total area under the curve was calculated. Area under the curve is presented in mmol/l × time, and baseline values are set to X = 0. The % decline in blood glucose was calculated by taking the largest drop in blood glucose throughout the 120 min divided by the baseline glucose levels (timepoint 0).
Hangwire impulse
Whole-body muscle performance was measured using a hangwire test according to the Neuromuscular Disease Network Standard Operating Procedure: DMD_M.2.1.004. All mice were gently placed on the wire situated 12 in. high and were left suspended on the wire until they reached exhaustion and dropped from the wire to the base of the cage. The time they remained suspended was recorded for three trials, separated by a 60 s recovery period, and impulse (s*g) was calculated by multiplying the average time suspended by body mass.
Body fat content and bone mineral content and density analysis
A small animal dual-energy X-ray absorptiometry (DXA) scanner (OsteoSys InSIGHT; Scintica) was used to measure body composition, including body fat content and bone mineral content and density at the baseline and endpoint of each study.
Sample and tissue collection
All mice were euthanized via exsanguination under general anesthetic (vaporized isoflurane) and their tissues and blood were collected. Blood was spun at 5000 × g for 8 min (4 °C) and serum was collected and stored at −80 °C. EDL, soleus, plantaris, and quadriceps were isolated and stored at −80 °C for further analysis.
Isolated muscle contractility
Soleus and EDL muscles were carefully dissected and mounted onto an Aurora Scientific contractile apparatus (model 305B&701B) to assess muscle force production and fatigue as previously described13. Muscles were subjected to a twitch and tetanic (160 Hz) isometric contraction. For data analyses, peak isometric force amplitude (mN) was determined across the range of stimulation frequencies. Peak isometric force was then normalized to muscle cross‐sectional area (CSA), which was calculated using the following formula: CSA = m/l*d*(Lf/Lo), where m, muscle mass (mg); l, muscle length (mm); d, mammalian skeletal muscle density (1.06 mg/mm3)75. Lf/Lo is the fiber length‐to‐muscle length ratio (0.44 for the EDL and 0.71 for the soleus)76. The fatigue protocol consists of a 70 Hz stimulation every 2 s for 160 s. Data were presented as a percent of initial force throughout the fatigue protocol.
Serum CK and Li analysis
Serum creatine kinase (CK) activity was measured with a M2 Molecular Device plate reader and a commercially available assay (Cat. #C7522, Pointe Scientific Inc., Canton, MI, USA) fitted onto a 96-well plate and calibrated with a standard curve of purified creatine kinase (Sigma, Oakville, ON, Canada, Cat. 10127566001). Serum Li analysis in the mdx-VWRLi mice were measured using the commercially available kit (ab235613, Abcam).
Immunofluorescent fiber typing
Fiber typing was accomplished through immunofluorescent staining of myosin heavy chain (MHC) isoforms in 10 μm sections of the EDL and soleus muscles as previously described77 with primary antibodies MHC I (1:50), IIa (1:600), and IIb (1:100) obtained from Developmental Studies Hydrobioma Bank. A cocktail of immunofluorescent secondary antibodies which included: Alexa Fluor 350 (IgG2b for MHCI, 1:100, A-21140 ThermoFisher Scientific), Alexa Fluor 488 (IgG1 for MHCIIa, 1:100, A-21121 ThermoFisher Scientific), and Alexa Fluor 555 (IgM for MHCIIb, 1:100, A-21426 ThermoFisher Scientific, MI, USA) were used to visualize the various fiber types. Slides were imaged using a BioTek Cytation 5 Multimode Plate Reader at 10× magnification using auto-exposure settings for three filters: DAPI, GFP, and Texas Red. Images were then stitched, processed and saved using the Gen5 image processing functions, and then analyzed using ImageJ (NIH) software to estimate fiber type composition and CSA.
Histological assessment
For EDL and soleus muscles, necrosis was measured using Hematoxylin and Eosin (H&E) staining. All slides were imaged using a BioTek Cytation 5 Multimode Plate Reader. All images were acquired using 10x magnification using auto-exposure settings for the Color Brightfield setting. Images were then saved and then analyzed using ImageJ (NIH) software to estimate necrotic area. This was done by calculating the total surface area of the muscle, then calculating the sum of the area of all necrosis (areas occupied by collagen and immune cell infiltration) within the sample. This total necrosis was then divided by the total area of the sample.
mRNA analysis
mRNA analyses for utrophin (forward, GGGGAAGATGTGAGAGATTT; reverse, GTGTGGTGAGGAGATACGAT; primers obtained from Integrated DNA Technologies (IDT), IA, USA) was accomplished by homogenizing plantaris (mdx-vehicle and mdx-tideglusib) and quadriceps (mdx-SED, mdx-VWR, mdx-LiVWR) samples with 1 mL of TRIzol. A commercially available Qiagen RNeasy kit (Quiagen, Hilden, Germany, ID #74104) and a DNase Max kit (Quiagen, Hilden, Germany, ID #15200-50) were then used to isolate RNA. This was then quantified using a NanoVue Plus spectrophotometer (Biochrom Ltd., Cambridge, UK), followed by the use of EcoDry RNA to cDNA (Takara Bio Inc., Kusatsu, Shiga, Japan, ID #639547) reaction tubes and a SimpliAmp Thermal Cylinder (ThermoFisher, MA, USA, ID #A24811) to facilitate the generation of cDNA. Diluted cDNA was then analyzed using a qPCR reaction 96-well plate and the StepOnePlus Real-Time PCR System (ThermoFisher, MA, USA, ID #4376600). Threshold cycle (Ct) values were recorded, and data were analyzed using the ΔΔCt method and reported as a fold-change (2ΔCt), using expression of the Gapdh housekeeping gene as a reference (forward, CGGTGCTGAGTATGTCGTGGAGTC; reverse, GGGGCTAAGCAGTTGGTGGTG; primers obtained from IDT).
Western blotting
Western blotting was done to assess protein levels of total GSK3α and GSK3β, phosphorylated GSK3β, Pax7, myogenin, and β-catenin as previously described14. Standard SDS-PAGE was performed with BioRad 7-12% TGX gels (4561086; BioRad, Hercules, CA, USA) and polyvinylidine difluoride (PVDF) membranes were used for all proteins (BioRad). All membranes were blocked with Everyblot buffer (#12010020, BioRad) for 10 min at room temperature and incubated with their respective primary antibodies. Total GSK3α (4818, 1:2000) and GSK3β (9315, 1:2000), phosphorylated GSK3β (Ser9) (9336 m, 1:2000), β-catenin (8480, 1:2000) were obtained from Cell Signalling Technology. Pax7, (APAX7, 1:250), and myogenin, (Myog, 1:250), were obtained from Developmental Studies Hydrobioma Bank. Following primary incubation, membranes were washed three times for 5 min with TBST and then incubated for 60 min with the corresponding anti-mouse (7076, 1:2000) or anti-rabbit (7074, 1:2000) horseradish peroxidase secondary antibody (Cell Signaling). Membranes were then washed another three times with TBTS for 5 min each prior to imaging with a BioRad ChemiDoc Imager with Immobilon ECL Ultra Western HRP Substrate (WBKLS0500, Sigma-Aldrich). Images were analyzed using BioRad ImageLab software (BioRad) and were normalized to total protein analyzed on a Ponceau stain.
Statistical analysis
Statistical comparisons were made using a Student’s t-test, a one-way or two-way ANOVA with a Tukey’s post-hoc or a Welch’s ANOVA with a Dunnett’s T3 post-hoc test where appropriate. All statistical analyses were done using GraphPad Prism 10 software. Statistical outliers were identified using the ROUT method integrated into GraphPad Prism with Q = 5% across all experiments. Statistical significance was set to a p ≤ 0.05. All values are presented as means ± SEM.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data generated in this study are provided in the Supplementary Information/Source Data file. Source data are provided with this paper.
References
Falzarano, M. S., Scotton, C., Passarelli, C. & Ferlini, A. Duchenne muscular dystrophy: from diagnosis to therapy. Molecules 20, 18168–18184 (2015).
Gruber, D. et al. Newborn screening for Duchenne muscular dystrophy-early detection and diagnostic algorithm for female carriers of Duchenne muscular dystrophy. Am. J. Med. Genet. C Semin. Med. Genet. 190, 197–205 (2022).
Duan, D., Goemans, N., Takeda, S. I., Mercuri, E. & Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Prim. 7, 13 (2021).
Hammers, D. W. et al. Glucocorticoids counteract hypertrophic effects of myostatin inhibition in dystrophic muscle. JCI Insight 5, e133276 (2020).
McDonald, C. M. et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet 391, 451–461 (2018).
Zhang, T. & Kong, X. Recent advances of glucocorticoids in the treatment of Duchenne muscular dystrophy (Review). Exp. Ther. Med. 21, 447 (2021).
Birnkrant, D. J. et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 17, 347–361 (2018).
Phung, K. et al. Risk factors associated with prevalent vertebral fractures in Duchenne muscular dystrophy. Osteoporos. Int. 34, 147–160 (2023).
Beurel, E., Grieco, S. F. & Jope, R. S. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharm. Ther. 148, 114–131 (2015).
Wang, H., Brown, J. & Martin, M. Glycogen synthase kinase 3: a point of convergence for the host inflammatory response. Cytokine 53, 130–140 (2011).
Gillespie, J. et al. GSK-3β function in bone regulates skeletal development, whole-body metabolism, and male life span. Endocrinology 154, 3702–3718 (2013).
Pansters, N. A. et al. Muscle-specific GSK-3beta ablation accelerates regeneration of disuse-atrophied skeletal muscle. Biochim. Biophys. Acta 1852, 490–506 (2015).
Whitley, K. C. et al. GSK3 inhibition with low dose lithium supplementation augments murine muscle fatigue resistance and specific force production. Physiol. Rep. 8, e14517 (2020).
Baranowski, R. W. et al. Toward countering muscle and bone loss with spaceflight: GSK3 as a potential target. iScience 26, 107047 (2023).
Kurgan, N. et al. A low-therapeutic dose of lithium inhibits GSK3 and enhances myoblast fusion in C2C12 Cells. Cells 8, 1340 (2019).
Mirzoev, T. M., Sharlo, K. A. & Shenkman, B. S. The role of GSK-3β in the regulation of protein turnover, myosin phenotype, and oxidative capacity in skeletal muscle under disuse conditions. Int. J. Mol. Sci. 22, 5081 (2021).
Chakkalakal, J. V. et al. Stimulation of calcineurin signaling attenuates the dystrophic pathology in mdx mice. Hum. Mol. Genet. 13, 379–388 (2004).
Chakkalakal, J. V. et al. Expression of utrophin A mRNA correlates with the oxidative capacity of skeletal muscle fiber types and is regulated by calcineurin/NFAT signaling. Proc. Natl. Acad. Sci. USA 100, 7791–7796 (2003).
Duan, X. et al. Trafficking regulator of GLUT4-1 (TRARG1) is a GSK3 substrate. Biochem. J. 479, 1237–1256 (2022).
Lee, J. & Kim, M. S. The role of GSK3 in glucose homeostasis and the development of insulin resistance. Diabetes Res. Clin. Pract. 77, S49–S57 (2007).
Bai, J., Xu, Y., Dieo, Y. & Sun, G. Combined low-dose LiCl and LY294002 for the treatment of osteoporosis in ovariectomized rats. J. Orthop. Surg. Res. 14, 177 (2019).
Vachhani, K., Whyne, C., Wang, Y., Burns, D. M. & Nam, D. Low-dose lithium regimen enhances endochondral fracture healing in osteoporotic rodent bone. J. Orthop. Res. 36, 1783–1789 (2018).
Marsell, R. et al. GSK-3 inhibition by an orally active small molecule increases bone mass in rats. Bone 50, 619–627 (2012).
Kurgan, N. et al. Low dose lithium supplementation activates Wnt/beta-catenin signalling and increases bone OPG/RANKL ratio in mice. Biochem. Biophys. Res. Commun. 511, 394–397 (2019).
Matthews, E., Brassington, R., Kuntzer, T., Jichi, F. & Manzur, A. Y. Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst. Rev. 2016, Cd003725 (2016).
Birnkrant, D. J. et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 17, 251–267 (2018).
Weber, D. R., Hadjiyannakis, S., McMillan, H. J., Noritz, G. & Ward, L. M. Obesity and endocrine management of the patient with duchenne muscular dystrophy. Pediatrics 142, S43–s52 (2018).
Jones, K. et al. GSK3beta mediates muscle pathology in myotonic dystrophy. J. Clin. Investig. 122, 4461–4472 (2012).
Lutz, M. et al. Therapeutic targeting of the GSK3β-CUGBP1 pathway in myotonic dystrophy. Int. J. Mol. Sci. 24, 10650 (2023).
Wei, C., Jones, K., Timchenko, N. A. & Timchenko, L. GSK3β is a new therapeutic target for myotonic dystrophy type 1. Rare Dis. 1, e26555 (2013).
Wei, C. et al. Correction of GSK3beta at young age prevents muscle pathology in mice with myotonic dystrophy type 1. FASEB J. 32, 2073–2085 (2018).
Findlay, A. R. et al. Lithium chloride corrects weakness and myopathology in a preclinical model of LGMD1D. Neurol. Genet. 5, e318 (2019).
Villa-Moruzzi, E., Puntoni, F. & Marin, O. Activation of protein phosphatase-1 isoforms and glycogen synthase kinase-3 beta in muscle from mdx mice. Int. J. Biochem. Cell Biol. 28, 13–22 (1996).
Khan, I., Tantray, M. A., Alam, M. S. & Hamid, H. Natural and synthetic bioactive inhibitors of glycogen synthase kinase. Eur. J. Med. Chem. 125, 464–477 (2017).
Noori, M. S. et al. Identification of a novel selective and potent inhibitor of glycogen synthase kinase-3. Am. J. Physiol. Cell Physiol. 317, C1289–c1303 (2019).
Horrigan, J. et al. A phase 2 study of AMO-02 (Tideglusib) in congenital and childhood-onset myotonic dystrophy type 1 (DM1). Pediatr. Neurol. 112, 84–93 (2020).
Hammers, D. W. et al. The D2.mdx mouse as a preclinical model of the skeletal muscle pathology associated with Duchenne muscular dystrophy. Sci. Rep. 10, 14070 (2020).
Balasubramaniam, M. et al. Structural modeling of GSK3β implicates the inactive (DFG-out) conformation as the target bound by TDZD analogs. Sci. Rep. 10, 18326 (2020).
Jones, K. et al. GSK3β mediates muscle pathology in myotonic dystrophy. J. Clin. Investig. 122, 4461–4472 (2012).
Coley, W. D. et al. Effect of genetic background on the dystrophic phenotype in mdx mice. Hum. Mol. Genet. 25, 130–145 (2016).
Hoffman, E. & Winder, S. J. A Modified wire hanging apparatus for small animal muscle function testing. PLoS Curr. 8 ecurrents.md.1e2bec4e78697b7b0ff80ea25a1d38be (2016).
van Putten, M. et al. Natural disease history of the D2-mdx mouse model for Duchenne muscular dystrophy. FASEB J. 33, 8110–8124 (2019).
Fajardo, V. A. et al. Sarcolipin deletion in mdx mice worsens dystrophic pathology by impairing calcineurin signaling and reducing utrophin expression. FASEB J. 30, 1224.1226 (2016).
Stupka, N., Schertzer, J. D., Bassel-Duby, R., Olson, E. N. & Lynch, G. S. Stimulation of calcineurin Aalpha activity attenuates muscle pathophysiology in mdx dystrophic mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R983–R992 (2008).
Rommel, C. et al. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat. Cell Biol. 3, 1009–1013 (2001).
Shen, T., Cseresnyes, Z., Liu, Y., Randall, W. R. & Schneider, M. F. Regulation of the nuclear export of the transcription factor NFATc1 by protein kinases after slow fibre type electrical stimulation of adult mouse skeletal muscle fibres. J. Physiol. 579, 535–551 (2007).
Cleverdon, R. E. G. et al. Sarco(endo)plasmic reticulum Ca(2+)-ATPase function is impaired in skeletal and cardiac muscles from young DBA/2J mdx mice. iScience 25, 104972 (2022).
Strakova, J. et al. Integrative effects of dystrophin loss on metabolic function of the mdx mouse. Sci. Rep. 8, 13624 (2018).
Podkalicka, P. et al. miR-378 affects metabolic disturbances in the mdx model of Duchenne muscular dystrophy. Sci. Rep. 12, 3945 (2022).
DeFronzo, R. A. & Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 32, S157–S163 (2009).
Wintzinger, M. et al. Effects of glucocorticoids in murine models of Duchenne and limb-girdle muscular dystrophy. Methods Mol. Biol. 2587, 467–478 (2023).
Quattrocelli, M. et al. Pulsed glucocorticoids enhance dystrophic muscle performance through epigenetic-metabolic reprogramming. JCI Insight 4, e132402 (2019).
Geromella, M. S. et al. Low-dose lithium supplementation promotes adipose tissue browning and sarco(endo)plasmic reticulum Ca(2+) ATPase uncoupling in muscle. J. Biol. Chem. 298, 102568 (2022).
Hammer, S. et al. Exercise training in Duchenne muscular dystrophy: a systematic review and meta-analysis. J. Rehabil. Med. 54, jrm00250 (2022).
Lott, D. J. et al. Safety, feasibility, and efficacy of strengthening exercise in Duchenne muscular dystrophy. Muscle Nerve 63, 320–326 (2021).
Zelikovich, A. S., Quattrocelli, M., Salamone, I. M., Kuntz, N. L. & McNally, E. M. Moderate exercise improves function and increases adiponectin in the mdx mouse model of muscular dystrophy. Sci. Rep. 9, 5770 (2019).
Kim, Y. J., Kim, H. J., Lee, W. J. & Seong, J. K. A comparison of the metabolic effects of treadmill and wheel running exercise in mouse model. Lab. Anim. Res. 36, 3 (2020).
Hamstra, S. I., et al. Beyond its psychiatric use: the benefits of low dose lithium supplementation. Curr. Neuropharmacol. 21, 891–910 (2022).
Jope, R. S. Lithium and GSK-3: one inhibitor, two inhibitory actions, multiple outcomes. Trends Pharm. Sci. 24, 441–443 (2003).
Huang, J., Guo, X., Li, W. & Zhang, H. Activation of Wnt/β-catenin signalling via GSK3 inhibitors direct differentiation of human adipose stem cells into functional hepatocytes. Sci. Rep. 7, 40716 (2017).
Yang, Y. et al. Accumulation of β-catenin by lithium chloride in porcine myoblast cultures accelerates cell differentiation. Mol. Biol. Rep. 38, 2043–2049 (2011).
Tallis, J. et al. The effects of 8 weeks voluntary wheel running on the contractile performance of isolated locomotory (soleus) and respiratory (diaphragm) skeletal muscle during early ageing. J. Exp. Biol. 220, 3733–3741 (2017).
Delacroix, C. et al. Improvement of dystrophic muscle fragility by short-term voluntary exercise through activation of calcineurin pathway in mdx mice. Am. J. Pathol. 188, 2662–2673 (2018).
Wei, C. et al. Correction of GSK3β at young age prevents muscle pathology in mice with myotonic dystrophy type 1. FASEB J. 32, 2073–2085 (2018).
Rico, H., Revilla, M., Villa, L. F., Alvarez del Buergo, M. & Ruiz-Contreras, D. Determinants of total-body and regional bone mineral content and density in postpubertal normal women. Metabolism 43, 263–266 (1994).
Salamat, M. R., Salamat, A. H., Abedi, I. & Janghorbani, M. Relationship between weight, body mass index, and bone mineral density in men referred for dual-energy X-ray absorptiometry scan in Isfahan, Iran. J. Osteoporos. 2013, 205963 (2013).
Liu, T.-T., Li, X.-D., Wang, W.-Z., Zhang, J.-G. & Yang, D.-Z. Efficacy of weight adjusted bone mineral content in osteoporosis diagnosis in Chinese female population. Chin. Med. J. 132, 772–781 (2019).
Wong, S. K., Chin, K. Y. & Ima-Nirwana, S. The skeletal-protecting action and mechanisms of action for mood-stabilizing drug lithium chloride: current evidence and future potential research areas. Front. Pharm. 11, 430 (2020).
Choi, I. Y. et al. Concordant but varied phenotypes among duchenne muscular dystrophy patient-specific myoblasts derived using a human iPSC-based model. Cell Rep. 15, 2301–2312 (2016).
Tanaka, S., Terada, K. & Nohno, T. Canonical Wnt signaling is involved in switching from cell proliferation to myogenic differentiation of mouse myoblast cells. J. Mol. Signal 6, 12 (2011).
Pansters, N. A. et al. Segregation of myoblast fusion and muscle-specific gene expression by distinct ligand-dependent inactivation of GSK-3beta. Cell Mol. Life Sci. 68, 523–535 (2011).
van der Velden, J. L. et al. Inhibition of glycogen synthase kinase-3beta activity is sufficient to stimulate myogenic differentiation. Am. J. Physiol. Cell Physiol. 290, C453–C462 (2006).
van der Velden, J. L., Schols, A. M., Willems, J., Kelders, M. C. & Langen, R. C. Glycogen synthase kinase 3 suppresses myogenic differentiation through negative regulation of NFATc3. J. Biol. Chem. 283, 358–366 (2008).
Matias-Valiente, L. et al. Evaluation of pro-regenerative and anti-inflammatory effects of isolecanoric acid in the muscle: potential treatment of Duchenne Muscular Dystrophy. Biomed. Pharmacother. 170, 116056 (2024).
Mendez, J. & Keys, A. Density and composition of mammalian muscle. Metabolism 9, 184–188 (1960).
Brooks, S. V. & Faulkner, J. A. Contractile properties of skeletal muscles from young, adult and aged mice. J. Physiol. 404, 71–82 (1988).
Bloemberg, D. & Quadrilatero, J. Rapid determination of myosin heavy chain expression in rat, mouse, and human skeletal muscle using multicolor immunofluorescence analysis. PLoS One 7, e35273 (2012).
Acknowledgements
This work was supported by an unrestricted Brock University Grant, a Canada Research Chair (Tier 2) award to V.A.F., and by a Brock-Niagara Validation Prototyping and Manufacturing Applied Project Grant in partnership with AMO-Pharma. B.M.M. was supported by a CIHR CGS-M award and an NSERC USRA. S.I.H. was supported by a NSERC Doctoral Vanier Award, J.L.B. was supported by a CIHR CGS-D award, and B.L.H. was supported by an Ontario Graduate Scholarship. A.A.T.M. was supported by a NSERC USRA. We thank the animal care staff at Brock University for their assistance with animal care and handling.
Author information
Authors and Affiliations
Contributions
B.M.M. – Concept design, writing and drafting the manuscript, figure creation, data collection, data interpretation, particularly on skeletal muscle function. B.L.H. – Data collection, data interpretation, reviewing and revising the manuscript. J.L.B. – Data collection, data interpretation, reviewing and revising the manuscript. K.C.W. – Data collection, data interpretation, reviewing and revising the manuscript. M.S.G. – Data collection, data interpretation, reviewing and revising the manuscript. R.W.B. – Data collection, data interpretation, reviewing and revising the manuscript. C.J.F.W. – Data collection, data interpretation, reviewing and revising the manuscript. S.S. – Data collection, data interpretation, reviewing and revising the manuscript. S.I.H. – Data collection, data interpretation, reviewing and revising the manuscript. L.J.W. – Data collection, data interpretation, reviewing and revising the manuscript. R.W.E. – Data collection, data interpretation, reviewing and revising the manuscript. A.A.T.M. – Data collection, data interpretation, reviewing and revising the manuscript. K.H.K. - Data collection, reviewing and revising the manuscript. G.L. - Provision of reagents and resources, reviewing and revising the manuscript. R.V. – Provision of reagents and resources, reviewing and revising the manuscript. B.D.R. – Provision of reagents and resources, reviewing and revising the manuscript. A.J.M. – Provision of reagents and resources, reviewing and revising the manuscript. R.E.K.M. – Provision of reagents and resources, reviewing and revising the manuscript. V.A.F. – Concept design, supervision, provision of reagents and resources, acquiring funding, writing and drafting the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare the following competing interests. A portion of this project, specifically the aged (28–30 weeks) tideglusib experiments, was funded by AMO-Pharma in partnership with a Brock-Niagara Validation Prototyping and Manufacturing Applied Project Grant (1:2 monetary ratio). AMO-Pharma also provided the tideglusib for the aged tideglusib experiments. For all other experiments, tideglusib was purchased from Sigma Alrdich (SML0339).
Peer review
Peer review information
Nature Communications thanks Gordon Lynch and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Marcella, B.M., Hockey, B.L., Braun, J.L. et al. GSK3 inhibition improves skeletal muscle function and whole-body metabolism in male mouse models of Duchenne muscular dystrophy. Nat Commun 15, 10210 (2024). https://doi.org/10.1038/s41467-024-53886-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-53886-y
