
Abstract
Conventional chemotherapy- and radiotherapy-induced cancer senescence, which is characterized by poor proliferation, drug resistance, and senescence-associated secretory phenotype, has gained attention as contributing to cancer relapse and the development of an immunosuppressive tumor microenvironment. However, the association between cancer senescence and anti-tumor immunity is not fully understood. Here, we demonstrate that senescent cancer cells increase the level of PD-L1 by promoting its transcription and glycosylation. We identify ribophorin 1 as a key regulator of PD-L1 glycosylation during cancer senescence. Ribophorin 1 depletion reduces this elevated level of PD-L1 through the ER-lysosome-associated degradation pathway, thereby increasing the susceptibility of senescent cancer cells to T-cell-mediated killing. Consistently, ribophorin 1 depletion suppresses tumor growth by decreasing PD-L1 levels and boosting cytotoxic T lymphocyte activity in male mice. Moreover, ribophorin 1-targeted or anti-PD-1 therapy reduces the number of senescent cancer cells in irradiated tumors and suppresses cancer recurrence through the activation of cytotoxic T lymphocytes. These results provide crucial insights into how senescent cancer cells can escape T-cell immunity following cancer treatment and thereby contribute to cancer recurrence. Our findings also highlight the therapeutic promise of targeting senescent cancer cells for cancer treatment.
Similar content being viewed by others

Introduction
Cancer cell senescence can arise as a result of anti-cancer treatments, such as chemotherapy, radiotherapy, and targeted therapy, which can cause DNA damage and other cellular stress in cancer cells1. Senescent cancer cells are characterized by limited growth, drug resistance, and the senescence-associated secretory phenotype (SASP)2. Poor proliferation of senescent cancer cells can serve as a barrier to tumor growth, and SASP can elicit an immune response by attracting and activating innate immune cells in tumors; thus, senescence is expected to contribute to the therapeutic effectiveness3. Conversely, however, accumulating evidence indicates that senescent cancer cells can also play detrimental roles in cancer treatment. Recent studies showed that SASP can disrupt antitumor immunity by creating an immune-suppressive tumor microenvironment (TME) that interrupts the ability of immune system to recognize and attack cancer cells4,5. Furthermore, like chronic inflammation, SASP can directly enhance the survival and growth of tumor cells to contribute to treatment resistance and cancer recurrence6. Recently, Saleh et al. demonstrated in animal models that targeting senescent cancer cells for senolysis after chemotherapy can help overcome the harmful effects of senescent cells and enhance therapeutic efficacy7,8. Interestingly recent aging studies found that the accumulation of senescent cells with aging is related to T-immune evasion caused by increased programmed cell death ligand-1 (PD-L1) in existing senescent cells9,10. These studies showed that the proportion of PD-L1-expressing senescent cells increases with age in several organ tissues, suggesting that PD-L1 targeting could provide new therapeutic opportunities to treat certain aging-related diseases. These studies imply that PD-L1 could be involved with the ability of senescent cancer cells to interfere with antitumor immunity.
PD-L1 is an important immune checkpoint molecule that regulates T-cell activity by binding to the programmed cell death protein 1 (PD-1) receptor on activated T-cells; thus, proper PD-L1 expression is critical for maintaining the balance of T-cell immunity. Abnormally low expression of PD-L1 in tissues can cause T-cells to become overly activated, resulting in autoimmune responses11, whereas abnormally high PD-L1 expression makes it difficult for T-cells to effectively eliminate abnormal cells12. Interestingly, cancer cells escape T-surveillance by hijacking PD-L1. Several types of cancer highly express PD-L1 and even well-primed cytotoxic T lymphocytes (CTLs) are effectively neutralized in PD-L1-expressing tumors13,14. Therefore, researchers have developed various immune checkpoint blockers (ICBs) targeting PD-L1/PD-1; these include Atezolizumab, Durvalumab, Nivolumab, and Pembrolizumab, which are in active use for cancer immunotherapy15.
Glycosylation, a post-translational modification (PTM) involving the addition of glycans to a protein, is critical for PD-L1 activity16. The glycan structure of PD-L1 affects its stability, membrane trafficking, and interaction with PD-117,18. PD-L1 glycosylation requires the staurosporine and temperature sensitive 3 (STT3), the catalytic subunit of the oligosaccharyltransferase (OST) complex19, which is responsible for catalyzing the transfer of pre-assembled glycans onto asparagine residues within the consensus sequence Asn-X-Ser/Thr (where X can represent any amino acid except proline) in nascent polypeptides during the initial stages of N-linked glycosylation20. The OST complex is composed of STT3, dolichyl-diphosphooligosaccharide protein glycosyltransferase (DDOST), ribophorin 1 (RPN1), and RPN2. It is differentiated into OST-A and OST-B complex based on the inclusion of either subunit A or subunit B of STT3, respectively. Cherepanova et al. reported that STT3B binds more readily to certain unfolded or misfolded proteins than STT3A, particularly where N-glycosylation is lacking during the initial translation and folding phases21, implying OST-B complex may have a critical role during cellular stress conditions.
RPN1 is an integral membrane protein located in the rough endoplasmic reticulum (ER) and plays a crucial role in facilitating the recruitment and correct localization of substrate polypeptides to the catalytic ___domain of the OST complex, specifically the STT3 subunit20. Wilson et al. found that RPN1 suppression changes glycosylation patterns of specific proteins without entirely disrupting the glycan profile, suggesting that role of RPN1 is substrate-specific22. They explained that RPN1 is not essential for normal glycosylation but plays an important role in ER stress conditions where glycosylation is delayed. That is, RPN1 appear to enhance the efficiency of OST-mediated glycosylation by improving substrate accessibility to the STT3 subunit under ER stress conditions. Furthermore, RPN1 interacts with Malectin, an ER quality control key player that selectively binds to misfolded glycoproteins23, implying that RPN1 may involve in the ER quality control of newly synthesized glycoproteins. Collectively, these reports have highlighted that RPN1 can enhance substrate-specific glycosylation under cellular stress conditions22, thereby alleviating ER stress associated with abnormal excessive accumulation of proteins.
When PD-L1 has an aberrant glycan structure, it is occupied by the quality check complex, accumulates in the ER, and ultimately undergoes degradation via the ER-associated degradation (ERAD) pathway24. The glycosylation of PD-L1 also appears to be essential for its recycling at the plasma membrane: non-glycosylated PD-L1 has a short half-life because it undergoes endocytosis and is degraded via the lysosomal pathway25. Zhu et al. recently uncovered the role of O-linked N-acetylgalactosamine (O-GlcNAc) in the lysosomal degradation of PD-L1. They demonstrated that O-GlcNAcylation involves in T-cell immunity via regulating lysosomal degradation of PD-L126. Consistently, several recent studies suggested that alterations in PD-L1 glycosylation can affect the level of responsiveness to ICBs targeting PD-1 or PD-L127,28. Therefore, improving our understanding of the regulation of PD-L1 glycosylation could support the development of more effective cancer immunotherapies.
In this study, we propose a molecular mechanism which is employed by senescent cancer cells to suppress antitumor immunity, specifically T-cell immunity. Through a comprehensive investigation using in vitro, in silico, and in vivo approaches, we demonstrate that therapy-induced cancer cell senescence upregulates PD-L1 levels to generate a “PD-L1 umbrella” that suppresses CTL activity to promote the survival of residual cancer cells and contribute to recurrence. Furthermore, we identify RPN1, a critical regulator of the “PD-L1 umbrella,” as a therapeutic target. These findings provide important insights into how senescent and residual cancer cells can evade T-cell immunity after cancer treatment and suggest the possibility of targeting PD-L1-expressing senescent cancer cells for therapeutic approaches.
Results
Cancer cell senescence decreases T-cell susceptibility by increasing PD-L1
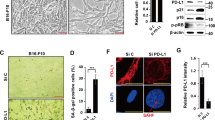
The immunosuppressive TME is likely to be alleviated when the tumor volume decreases following conventional cancer treatments. This raises the question: How can senescent cancer cells evade direct T-cell attack and persist long-term without the support of an immunosuppressive TME? Remarkably, our findings from syngeneic OT-cell killing assays show that therapy-induced senescent cancer cells are not susceptible to T-cell surveillance. We induced senescence in OVA-expressing B16F10 mouse melanoma spheroids and cells through ionizing radiation (IR) exposure (Supplementary Fig. 1a and b_IR-CS). These senescent cancer cells and spheroids were then co-cultured with OT-cells, which were expanded from the splenocytes of OT-1 mice (Supplementary Fig. 2a–e). We evaluated T-cell infiltration within these tumor spheroids (Fig. 1a and Supplementary Fig. 2f_upper) and measured caspase-3 activity in cancer cells (Fig. 1b and Supplementary Fig. 2f_bottom). Compared to control group (Cont), OVA-expressing B16F10 spheroids with IR-induced cancer senescence (IR-CS) exhibited reduced OT-cell infiltration (Fig. 1a_IR-CS) and OVA-expressing B16F10 cells showed diminished caspase-3 activity in the OT-cell killing assay (Fig. 1b_IR-CS), indicating that the senescent cancer cells had acquired the ability to evade T-cell surveillance independent of the TME.

a OT-cell infiltration assay in Control (Cont) or IR-induced senescent B16/F10-OVA melanoma spheroids (IR-CS). OT-cells and cancer spheroids are represented by green and red fluorescence, respectively (right). Infiltrated OT-cells in B16/F10-OVA melanoma spheroids were quantified (left). Scale bar: 200 µm. Mean ± SD of n = 3 independent samples. Two-sided Student’s t-test. b Syngeneic OT-cell killing assay of control or IR-induced Senescent B16/F10-OVA melanoma cells. Cancer cells were stained red, apoptotic cells were displayed by fluorescence of caspase-3 activity (green) (right). The percentage of apoptotic cells was quantified (left). Scale bar: 200 µm. Mean ± SD of n = 3 independent samples. Two-sided Student’s t-test. c Immunofluorescence staining of the B16/F10-OVA melanoma spheroid using an anti-PD-L1 antibody. Scale bar: 50 µm. Representative images of n = 3 independent replicates d–f Western blot showing the protein levels of PD-L1 in IR-induced H460 (d), Doxorubicin (50 ng/ml)-induced (Doxo) H460 (e), and PTEN loss-induced H460 (f). Representative Western blots of n = 3 independent replicates. g PD-L1 levels in various types of senescent cancer cells induced by IR. Representative Western blots of n = 3 independent replicates. h qRT-PCR analysis of PD-L1 mRNA levels in IR-induced senescent H460 cells. Mean ± SD of n = 3 independent samples. Two-sided Student’s t-test. i HEK293T cells co-transfected of pGIPz-hPD-L1sh and pCI-neo-PD-L1 Flag vectors were treated with IR. Created in BioRender. Cha, J. (2024) BioRender.com/i97z560. Representative Western blots of n = 3 independent replicates. Mean ± SD of n = 3 independent samples. Two-sided Student’s t-test. j CHX chase assay showing the degradation of exogenous PD-L1 in Cont or IR-induced senescent HEK293T cells. Western blot (upper) and the quantification (bottom) showing exogenous PD-L1 protein level. Representative Western blots of n = 3 independent replicates. Mean ± SD of n = 3 independent samples. Two-way ANOVA-Tukey multiple comparison test. Source data are provided as a Source Data file.
Cancer cells are known to evade T-cell surveillance through the overexpression of immune checkpoint proteins29, and recent reports suggest that PD-L1-mediated T-cell dysfunction could be an important mechanism in the aging process9,10. Indeed, we found that PD-L1 levels are upregulated in IR-induced senescent spheroids and H460 cells (Fig. 1c, d, and Supplementary Fig. 1c_ IR-CS). We further examined PD-L1 expression in cancer senescence induced by other types of stress and found that PD-L1 expression was significantly increased in cancer cell senescence induced by doxorubicin (DNA-damaging stress; Doxo-CS) (Fig. 1e and Supplementary Fig. 1c_Doxo-CS) or PTEN loss (non-DNA-damaging stress; PTEN-loss-CS) (Fig. 1f and Supplementary Fig. 1c_PTEN-loss-CS). Furthermore, increased PD-L1 expression following IR-exposure-based induction of cellular senescence was also observed in other types of cancer cells, including A549 lung carcinoma, U2OS osteosarcoma, H1299 lung cancer, and HCT116 colon carcinoma cells (Fig. 1g). IR increased PD-L1 expression in both cells with low (A549 and U2OS) and high (H1299 and HCT116) basal expression of PD-L1. These findings suggest that cancer cell senescence generally increases the PD-L1 level, enabling senescent cancer cells to evade T-cell surveillance.
Previous studies have shown that radiation therapy30,31 and chemotherapy32 can elevate the transcriptional levels of PD-L1 due to DNA damage and other cellular stresses early after treatment. Consistent with previous reports, PD-L1 mRNA levels increased gradually from three- to six-fold up to two days after IR exposure. Notably, on the fourth day post-IR, when cellular senescence became clearly evident, PD-L1 mRNA levels surged to eighty-fold (see Fig. 1h). This implies that the increase in PD-L1 mRNA may be more pronounced in the advanced stages of senescence33, rather than being merely a result of IR exposure. To investigate potential mechanisms that could amplify PD-L1 expression independently on IR-induced upregulation of its mRNA, we used the pCI-neo-PD-L1 wild type (WT)-Flag plasmid under endogenous PD-L1 knockdown condition. The promoter of this plasmid is unaffected by IR-induced stress34,35, allowing us to validate post-translational regulation of PD-L1 by cellular senescence (Fig. 1i). Interestingly, cancer senescence significantly increased exogenous PD-L1 level not regulated by transcriptional regulation of PD-L1. Furthermore, the half-life of the PD-L1 protein was notably extended in senescent cancer cells treated with cycloheximide (CHX) (Fig. 1j), indicating that cancer senescence may enhance the stability of PD-L1 protein. These results suggest that transcriptional and post-translational regulation may synergistically contribute to elevated PD-L1 levels in senescent cancer cells.
Cancer senescence could influence glycosylation, which is crucial for PD-L1 stabilization and membrane transport
PD-L1 is transported to the cell membrane through the intracellular transport system via the ER and Golgi apparatus. Functionally, since the binding with PD-1 occurs on the membrane, a substantial amount of membrane-bound PD-L1 is crucial for T-cell suppression. We closely examined the intracellular localization of PD-L1 in IR-CS, Doxo-CS, and PTEN-loss-CS cells. Compared to control cells, the PD-L1 signal was significantly increased in the ER, Golgi apparatus, and cell membrane of senescent cancer cells (Fig. 2a, Supplementary Fig. 3a and b; white arrow indicates position of the ER, yellow arrow indicates position of cell membrane, cyan arrow indicates position of Golgi), indicating that the upregulated PD-L1 in senescent cancer cells underwent active intracellular transport to the membrane. Flow cytometric analysis confirmed that the amount of membrane-bound PD-L1 was substantially increased in senescent cells (Fig. 2b_IR-CS, Supplementary Fig. 3c_Doxo-CS and d_PTEN-loss-CS). To measure the amount of functional PD-L1 capable of binding to its receptor PD-1 on the cell membrane, we performed a binding assay using recombinant PD-1-FC protein. The results showed that the binding of PD-L1 to PD-1-FC was significantly higher in senescent cancer cells compared to the control group (Fig. 2c_PD-1-FC green). These results suggest that cancer senescence can activate the transport of upregulated PD-L1 and thereby highly increase the level of functional cell-surface PD-L1.

a Immunofluorescence staining of PD-L1 and markers of ER (HSP90B1) and Golgi (cis, GM130) in Control (Cont) or IR-induced cancer senescence (IR-CS). Nuclei were counterstained with DAPI. Scale bar: 10 µm. Representative images of n = 3 independent replicates. b Cell surface levels of PD-L1 were analyzed by flow cytometry in IR-induced senescent H460 cells. The gating strategies are provided in Supplementary Fig. 13a. c PD-1 binding assay: representative images showing the binding of green fluorescence-labeled PD-1/Fc protein on IR-induced senescent H460 cells. Scale bar: 50 µm. Representative images of n = 3 independent replicates. d A Venn diagram was created to compare the N-glycosyltransferase list obtained from our microarray data in this study with the list of N-glycosyltransferases known to interact with PD-L143. e Identification of N-glycosylation-related enzymes that are differentially expressed in IR-induced senescent H460 cancer cells. f Validation of increased mRNA expressions of N-glycosylation-related enzymes in IR-induced senescent H460 cells. Mean ± SD of n = 3 independent samples. One-way ANOVA-Tukey multiple comparison test. g qRT-PCR analysis of PD-L1 mRNA levels in IR-induced senescent H460 cells, which were transfected with each indicated siRNAs. Mean ± SD of n = 3 independent samples. One-way ANOVA-Tukey multiple comparison test. h Immunoblot analysis of PD-L1 levels in IR-induced senescent cancer cells, which were transfected with each indicated siRNAs. Representative immunoblots of n = 3 independent replicates. Statistical significance is represented as means ± SD. Source data are provided as a Source Data file.
Since recent reports indicate that the protein glycosylation profile is significantly altered during cell senescence36,37, we focused on the relationship between cancer cell senescence and PD-L1 glycosylation. To identify key regulators of PD-L1 glycosylation in senescent cancer cells, we examined the expression profiles of glycosyltransferases and their cofactors in a microarray dataset generated from IR-induced senescent cancer cells. This dataset was cross-referenced with factors related to N-glycosylation previously identified as potential interactors with PD-L138. Out of 35 N-glycosylation-related factors that exhibited elevated expression in IR-induced senescent H460 cells, nine coincided with candidates previously suggested to interact with PD-L1 (Fig. 2d). The expression changes of these nine selected candidates were visualized using a heatmap (Fig. 2e) and validated by qRT-PCR (Fig. 2f). Notably, four of nine candidates STT3B, DDOST, RPN1, and RPN2 were components of the OST-B complex which has a higher affinity for certain types of unfolded or misfolded proteins compared to OST-A complex21, implying that increased expression of OST-B complex facilitates glycosylation processing in response to stress conditions during cancer cell senescence.
To assess the impact of each candidate on PD-L1 transcription and protein stability in senescent cells, H460 cells were transfected with siRNAs targeting nine candidates and then exposed to IR to induce therapy-induced senescence. Although depletion of all candidates did not affect to PD-L1 mRNA levels elevated by IR-CS (Fig. 2g), silencing OST-B complex components clearly reduced the IR-induced elevation of PD-L1 protein levels (Fig. 2h). Particularly, silencing STT3B and RPN1 led to a notable reduction in PD-L1 levels (Fig. 2h, RPN1 and STT3B). Silencing DDOST, RPN1, and STT3B reduced protein stability in the CHX assay (Supplementary Fig. 4a) and functional membrane level of PD-L1 (Supplementary Fig. 4b). Silencing RPN1 and DDOST showed a clear band shift, implying abnormal glycosylation (Fig. 2h, red arrows for DDOST and RPN1). In the case of RPN2, although its silencing reduced PD-L1 levels, it did not alter band shifts, stability, and membrane levels of PD-L1 (Fig. 2h, Supplementary Fig. 4a and b, RPN2). Given the broad role of STT3B as a glycosyltransferase across many glycoproteins19 and the essential role of DDOST for the assembly of the OST complex39, therapeutically targeting them could disrupt general N-glycosylation, potentially causing severe side effects. In contrast, inhibiting RPN1 expression alters the N-glycosylation patterns of specific proteins without affecting the overall glycan profile22, presenting a more targeted approach with fewer side effects. Thus, RPN1 could be a promising target to mitigate PD-L1 elevation in cancer senescence, balancing efficacy with safety. Elevated RPN1 levels were consistently observed in cancer senescence induced by both doxorubicin treatment (Supplementary Fig. 5a) and PTEN-loss (Supplementary Fig. 5b).
In silico TCGA analysis indicates that RPN1 may play an important role in tumorigenesis and anti-tumor immunity
To investigate the role of RPN1 as a potential key regulator of PD-L1 glycosylation and stabilization in senescent cancer cells, we conducted an in silico analysis using datasets from The Cancer Genome Atlas (TCGA). The analysis focused on tumor and antitumor immunity-related factors and was performed using the R package-based program, TIMER2.040,41,42. We first compared RPN1 mRNA expression levels between tumor and matched normal tissues in all TCGA tumors using the Gene_DE module (Fig. 3a). Our results showed that RPN1 mRNA levels were significantly increased in various cancer types, including breast, colorectal, glioblastoma, gastric, and lung cancers, when compared to normal tissues.

a RPN1 expression levels between tumor and tumor-matched normal tissues in all TCGA tumors. The box plots display the distribution of expression levels in tumor (red) and adjacent normal (blue) tissues. The center line in each box plot represents the median, the bounds of the box represent the first (25th percentile) and third quartiles (75th percentile), and the whiskers extend to 1.5 times the interquartile range (IQR). Outliers are displayed as individual points. Statistical significance, calculated by the Wilcoxon test, is indicated by stars (*p-value < 0.05; **p-value < 0.01; ***p-value < 0.001). b Spearman rank correlation analysis to assess the correlation between RPN1 and PD-L1 mRNA levels, as well as between RPN1 and cancer senescence markers across all TCGA tumor samples. c Heatmap table showing multiple associations between IFN-β, HIF-1α, or RPN1 and CD8+ tumor-infiltrating T-cell levels, estimated by six algorithms across cancer types in the TCGA database. Each cell in the heatmap indicates the statistical significance of the correlation coefficient based on Spearman’s correlation analysis. Red: statistically significant positive correlation; blue: statistically significant negative correlation.
Subsequently, we investigated the correlation between PD-L1 and RPN1 levels, as well as cancer senescence markers that exhibited increased transcription levels upon senescence including matrix metalloproteinase (MMP)-2/3, interleukin (IL)-6, insulin-like growth factor-binding protein (IGFBP) 3/5/7, CDKN1A (p21) and chemokine (C-C motif) ligand (CCL5)43 (Fig. 3b). Interestingly, Spearman’s rank correlation values displayed on the heatmap indicated a positive correlation for most cancers, especially the major cancers such as breast invasive carcinoma (BRCA), colorectal adenocarcinoma (COAD), skin melanoma (SKCM), pancreatic adenocarcinoma (PAAD), and prostate adenocarcinoma (PRAD). These results imply a potential close relationship between RPN1, PD-L1 regulation, and the senescence of cancer cells.
Finally, we used the cellular composition estimation module to generate a heatmap table of the Spearman correlations between the expression levels of RPN1 and the proportion of CD8 + T-cells in all TCGA tumors. To increase the accuracy of this analysis, we also analyzed interferon (IFN)-β144,45 and hypoxia inducible factor (HIF)-1α46,47, which are known to be positively and negatively correlated with CD8 + T-cell immunity in tumors, respectively. Our results showed that, similar to the well-known antitumor immunity-inhibiting factor, HIF-1α, the expression of RPN1 showed a significant negative correlation with tumor-infiltrating CD8 + T cell populations in various tumor types (Fig. 3c). Considering the results presented in Figs. 1 and 2, these findings from bioinformatic analysis of the TCGA database suggest that RPN1 could be a key regulator contributing to PD-L1 stabilization during cancer senescence, with potentially deleterious effects on the tumorigenesis process and antitumor immunity.
RPN1 is essential for N-glycosylation of PD-L1 increased by cancer senescence
PD-L1 possesses four N-glycosylation sites: N35, N192, N200, and N21917,24, as illustrated in Fig. 4a (top panel). These sites are highly glycosylated, as demonstrated by comparing WT PD-L1 with its non-glycosylated form, 4NQ (N35Q, N192Q, N200Q, and N219Q) PD-L1 (Fig. 4a). Following exposure to IR-CS, we observed a significant increase in the levels of fully glycosylated WT PD-L1 (50-55 kDa). Transfection with RPN1 siRNA resulted in decreased levels of WT PD-L1 and a distinct band shift indicative of abnormal glycosylation. Treatment with tunicamycin, a broad-spectrum N-glycosylation inhibitor, led to a shift in WT PD-L1 bands to sizes corresponding to the 4NQ mutants (34 kDa), which lack N-glycosylation, confirming the complete inhibition of N-glycosylation in PD-L1 by tunicamycin. Notably, under all experimental conditions tested, no bands appeared below the size of the 4NQ mutants. Further transfection with RPN1 siRNA had no effect on 4NQ PD-L1. These results suggest that the band shift observed in PD-L1 due to RPN1 depletion results from aberrant N-glycosylation, further implicating RPN1 in the N-glycosylation process of PD-L1.

a Western blot showing the effect of RPN1 on N-glycosylation of PD-L1. A stable H460 cell line expressing both WT and 4NQ PD-L1-Flag was transfected with either control siRNA (Cont si) or RPN1 siRNA (RPN1 si) and then subjected to IR-induced cancer senescence conditions. Three days after IR treatment, the stable cell lines expressing WT and 4NQ PD-L1-Flag were treated with or without tunicamycin (TM, 5 µg/ml) and chloroquine (CQ, 40 µg/ml) for 12 h. Representative Western blots of n = 3 independent replicates. b The extracted compound chromatogram of glycans detected in PD-L1 is presented. Glycan class: Oligomannose (green); undecorated complex/hybrid (blue); fucosylated complex/hybrid (purple); sialylated complex/hybrid (orange); and fucosylated/sialylated complex/hybrid (red). Representative chromatogram of n = 3 independent replicates. c–g Detailed analysis of extracted compound chromatogram based on glycan structure types (c) Oligomannose, (d) undecorated Complex/Hybrid type, (e) Fucosylated Complex/Hybrid type, (f) Sialylated Complex/Hybrid type, (g) Fucosylated and Sialylated Complex/Hybrid type. The representative structure for each glycan type is illustrated on the right side of each graph. Mean ± SD of n = 3 independent biological samples. One-sided Student’s t-test. Source data are provided as a Source Data file.
To validate this, we performed glycan structure analysis of PD-L1. Purified N-glycans were chromatographically separated and analyzed using a nanoLC/Q-TOF MS system. The result demonstrates a decrease in the overall glycan levels of PD-L1 following RPN1 knockdown, further illustrating that RPN1 depletion affects total glycan levels (Fig. 4b) rather than altering specific terminal glycan structures of PD-L1 (Fig. 4c–g). Although RPN1 does not exhibit glycosyltransferase activity20, its significant role lies in facilitating the alignment and positioning of substrate polypeptides into the catalytic ___domain of the OST complex48, thereby promoting the efficiency and precision of protein glycosylation. Together with these previous reports, our results support the conclusion that RPN1 primarily influences the early stages of N-glycosylation, facilitating the incorporation of pre-assembled glycans into PD-L1 polypeptides immediately post-translation.
RPN1 plays a crucial role in enabling senescent cancer cells to evade immune surveillance by elevating PD-L1 levels
To investigate the effect of RPN1 depletion on cancer senescence, we examined the senescence phenotypes of cells treated with three different siRNAs targeting RPN1 through a senescence-associated β-galactosidase (SA-β-Gal) activity assay (Fig. 5a). These RPN1 siRNAs effectively reduced PD-L1 levels (Fig. 5b) but did not impact IR-induced senescence per se (Fig. 5a). Additionally, RPN1 depletion effectively neutralized the increased PD-L1 expression observed in Doxo-CS and PTEN-loss-CS cells (Fig. 5c_Doxo-CS and 5d_PTEN-loss-CS). Furthermore, we examined PD-L1 protein levels in human cancer cell lines (Fig. 5e) and analyzed the effects of knocking down or overexpressing RPN1 in cancer cell lines with high (H1975 and BT549) or low (H460, A549, and HCC827) basal PD-L1 levels (Fig. 5f, g). Even in the absence of senescence, knocking down RPN1 reduced PD-L1 levels (Fig. 5f), and overexpressing it increased PD-L1 levels (Fig. 5g), indicating that RPN1 is critical for the stabilization of PD-L1 under both stress and normal conditions. RPN1 depletion in IR-CS resulted in decreased cell surface PD-L1 levels (Fig. 5h_Cont Si + IR-CS vs. RPN1 Si + IR-CS, yellow arrow indicates position of cell membrane; and Fig. 5j) and co-localization of PD-L1 with markers of the ER (Fig. 5h_Cont Si + IR-CS vs. RPN1 Si + IR-CS, white arrow indicates position of ER) and Golgi apparatus (Fig. 5i_Cont Si + IR-CS vs. RPN1 Si + IR-CS, cyan arrow indicates position of Golgi).

a H460 cells transfected with three different RPN1 siRNAs prior to IR exposure were subjected to SA-β-Gal staining. Scale bar: 50 µm. Mean ± SD of n = 3 independent samples. One-way ANOVA-Tukey multiple comparison test. Representative images of n = 3 independent replicates. b Immunoblot analysis of Control (Cont) or IR-induced senescent H460 cells (IR-CS), which were transfected with Cont si or three different RPN1 siRNAs (#1-3). Representative immunoblots of n = 3 independent replicates. c Cells were transfected with either Cont siRNA (Cont si) or RPN1 siRNA (RPN1 si), followed by treatment with Doxorubicin (50 ng/ml for 4 days). d H460 cells transfected with PTEN siRNA (PTEN si) underwent further transfection with either Cont si or RPN1 si. e Immunoblot analysis of PD-L1 levels in lung, breast and colon cancer cells. f Western blot showing PD-L1 levels in H1975 and BT549 cells transfected with either Cont si or RPN1 si. g Immunoblot analysis of PD-L1 in H460, A549, and HCC827 cells transfected with either mock vectors or RPN1-HA vectors. c–g Representative immunoblots of n = 3 independent replicates. h,i Immunofluorescence of PD-L1 and markers of ER (HSP90B1) (h) and Golgi (cis, GM130) (i) in IR-induced senescent H460 cells transfected with either Cont si or RPN1 si. Nuclei were counterstained with DAPI. Scale bar: 10 µm. Representative images of n = 3 independent replicates. j Median fluorescence intensity (MFI) of membrane-bound PD-L1 in IR-induced senescent H460 cells transfected with either Cont si or RPN1 si were analyzed by flow cytometry. Mean ± SD of n = 3 independent samples. One-way ANOVA-Tukey multiple comparison test. The gating strategies are provided in Supplementary Fig. 13b. k H460 cells transfected with either Cont si or RPN1 si were exposed to IR. On day 4 post-IR, the binding of green fluorescence-labeled PD-1/Fc protein was measured in each si-treated/irradiated cell group. Mean ± SD of n = 3 independent samples. One-way ANOVA-Tukey multiple comparison test. l B16/F10-OVA melanoma cells transfected with either Cont si or RPN1 si were allowed to form spheroids for 3 days and then exposed to IR. On day 4 post-IR, activated OT-cells were co-cultured with B16/F10-OVA melanoma spheroids. The next day, the number of infiltrated OT-cells in the spheroids was quantified. Mean ± SD of n = 3 independent samples. One-way ANOVA-Tukey multiple comparison test. m B16/F10-OVA melanoma cells transfected with either Cont si or RPN1 si prior to IR were co-cultured with OT-cells. After 48 hours, the quantitative ratios of dead cells to total cells were measured by the fluorescence of caspase-3 activity. Mean ± SD of n = 3 independent samples. One-way ANOVA-Tukey multiple comparison test. Source data are provided as a Source Data file.
To determine the role of RPN1 depletion in the immunosuppressive activity of senescent cancer cells, we examined PD-1 binding, T-cell infiltration, and T-cell-mediated killing activity. Depletion of RPN1 reduced the IR-CS-enhanced levels of membrane-bound PD-L1 (Fig. 5j) and consistently decreased the binding of PD-1 protein to the cell surface (Fig. 5k). RPN1 depletion significantly restored the infiltration rate of OT-1 cells into B16F10-OVA spheroids (Fig. 5l and Supplementary Fig. 2f_upper, 6a) and the B16F10-OVA cancer cell killing activity of OT-1 cells (Fig. 5m and Supplementary Fig. 2f_bottom, 6b), which had been suppressed by IR-CS. Taken together, these findings suggest that RPN1 plays a significant role in enabling senescent cancer cells to evade T-cell surveillance by increasing PD-L1 levels on the cell membrane.
RPN1 modulates PD-L1 degradation through the ERLAD pathways in senescent cancer cells
PD-L1 with an intact glycan structure is transported to the cell surface through the intracellular ER-to-Golgi transport system. Conversely, aberrant or non-glycosylated PD-L1 is degraded through proteasomal degradation related to ERAD and/or the lysosomal degradation pathway24,49. To determine the role of RPN1 in the homeostatic regulation of PD-L1 protein, we conducted a CHX chase assay. We found that RPN1 depletion markedly diminished PD-L1 protein stability in IR-induced senescent cancer cells treated with CHX (Fig. 6a). While the lysosomal inhibitor, chloroquine (CQ), reversed the PD-L1 downregulation caused by RPN1 depletion in senescent cancer cells, the proteasome inhibitor, MG132, had no discernible effect on PD-L1 protein levels (Fig. 6b). This indicates that the effect of RPN1 depletion on the level of PD-L1 may involve the lysosomal degradation pathway rather than ERAD.

a CHX chase assay showing the degradation of PD-L1 protein in Cont si or RPN1 si-transfected H460 cells after exposure to IR. Western blot (top) and the quantification (bottom) showing PD-L1 protein level. Representative Western blots of n = 3 independent replicates. Means ± SD of n = 3 independent samples. Two-way ANOVA-Tukey multiple comparison test. b Western blot showing the effect of RPN1 on PD-L1 protein levels in Cont si or RPN1 si-transfected cells after treatment with MG132 (20 μM), a proteasomal degradation inhibitor, or CQ (40 μg/ml), a lysosomal inhibitor. Representative Western blots of n = 3 independent replicates. c Western blot showing the depletion effects of FAM134B, Rab5, and Rab11 on PD-L1 protein levels in Cont si or RPN1 si-treated cells after exposure to IR. Representative Western blots of n = 3 independent replicates. d–f Immunofluorescence showing the colocalization of PD-L1 with FAM134B (d), LC3B (e), or LAMP1 (f) in cells treated with CQ after transfection with Cont si or RPN1 si. Cell nuclei were counterstained with DAPI. Scale bar: 10 µm. Representative images of n = 3 independent replicates. g A model illustrating the regulatory mechanism of PD-L1 through ERLAD pathway during cancer senescence. Senescent cancer cells have elevated expression of the glycosyltransferase RPN1, facilitating the complete glycosylation of PD-L1. This increases the levels of membrane-bound PD-L1 by enhancing the stability of PD-L1. RPN1 depletion induces incomplete glycosylation of PD-L1, leading to increased degradation by ERLAD pathway, resulting in lower overall PD-L1 levels. Created in BioRender. Kang, D. (2023) BioRender.com/e62x895. Source data are provided as a Source Data file.
PD-L1 is recycled on the cell membrane through endocytosis; during this process, dysfunctional PD-L1 is degraded via the lysosomal pathway within the formed endosome29,50. Knockdown of Rab5 and Rab11, which are integral to endocytosis and endosome formation51, failed to restore PD-L1 levels in RPN1-depleted senescent cells (Fig. 6c_line 5 and 6), implying that the RPN-1-regulated pathway might be associated with an alternative lysosomal degradation mechanism that is distinct from the recycling process. Recent studies indicate that certain aberrant proteins are routed to lysosomes and eliminated via the ER-lysosome-associated degradation (ERLAD) pathway52,53. Notably, IR-CS increased the level of family with sequence similarity 134 member B (FAM134B) (Fig. 6c_line 2), which is an LC3-binding ER-phagy receptor that acts as a key component of ERLAD. When we depleted FAM134B, the reduced PD-L1 level was restored in RPN1-depleted senescent cells (Fig. 6c_line 3 vs. line 4). Consistent with this, immunofluorescent (IF) staining also revealed that the colocalization between PD-L1 and FAM134B (Fig. 6d_inset A vs. inset B) or LC3B (Fig. 6e_inset C vs. inset D) was enhanced by CQ treatment of RPN1-depleted senescent cells. Furthermore, in the presence of CQ, RPN1 depletion significantly increased the colocalization of PD-L1 with the lysosomal marker, lysosomal associated membrane protein 1 (LAMP1), indicating accumulation of PD-L1 in the lysosome (Fig. 6f_inset E vs. inset F, white arrow). To ensure the reliability of these intracellular localization changes of PD-L1 induced by IR-CS, the specificity of the two different PD-L1 antibodies used for IF staining was further validated in PD-L1 knockdown cells (Supplementary Fig. 7). These findings suggest that RPN1 depletion mediates the degradation of PD-L1 via the ERLAD pathway in senescent cancer cells, providing further evidence for the crucial role of RPN1 in regulating of PD-L1 protein homeostasis (Fig. 6g).
RPN1 might facilitate cancer recurrence by inhibiting T-cell immunity through PD-L1 in an irradiated tumor model
To verify the impact of RPN1 on PD-L1 expression and T-cell activation in tumor tissue following radiation therapy, we performed an in vivo study using an immunocompetent syngeneic mouse tumor model. Initially, we confirmed that RPN1 depletion does not affect the cell proliferation or viability of the CT26 cells used for in vivo validation (Supplementary Fig. 8) and that IR exposure does indeed induce cell senescence in CT26 tumors (Supplementary Fig. 9). BALB/c mice were engrafted with CT26 cells transfected with either Cont siRNA (Cont si) or RPN1 siRNA (RPN1 si). On day 10, they were exposed to a 12 Gy dose of IR. Then, Cont si or RPN1 si were administered through intratumoral transfection, as depicted in the schematic (Fig. 7a_bottom of graph). Following IR exposure, we observed a significant decline in tumor size, with a regression of up to 50% within the first week. By the end of the second week post-irradiation, however, the tumors had resumed growing and almost returned to their original sizes. This trend was consistent across all samples (Fig. 7a_right upper graph, red lines). Remarkably, however, tumors in the RPN1 si-treated group displayed a continuous size reduction, showing no signs of regrowth throughout the observation period (Fig. 7a_right bottom graph, blue lines). At the end of the experiment, while there was no notable size difference between tumors in the non-irradiated Cont si and RPN1 si groups, tumors of the IR-treated Cont si group were considerably larger than those of the IR-treated RPN1 si group (Fig. 7b and c).

a CT26 cancer cells transfected with either Cont si or RPN1 si were injected into BALB/c mice on day 0 (TI: treatment initiation). On day 10, the mice received a 12 Gy IR. Intratumoral siRNA transfection was carried out as described. The tumor size (mm³) was measured at the indicated times. Data is presented as means ± SD, 6 mice per group. Two-way ANOVA with Tukey’s multiple comparison test. b,c Pictures of excised (b) and the average weights (c) of tumors harvested at the endpoint. Mean ± SD of n = 6 mice per group. Two-sided Student’s t-test. d Immunofluorescence staining of tumor sections for PD-L1 and RPN1, with DAPI used as a nuclear counterstain. Representative images from tumor sections in each group. Scale bar: 20 µm. e Relative protein levels of RPN1 and PD-L1 in tumor tissue were quantified from the results in Supplementary Fig. 8 using ImageJ. Mean ± SD of n = 6 mice per group. Two-sided Student’s t-test. f Correlation analysis between PD-L1 and RPN1 expression in Supplementary Fig. 8. Mean ± SD of n = 6 mice per group. Spearman’s correlation test. g Representative images of immunostaining of CD8, Granzyme B, and caspase-3 in tumor sections with DAPI as nuclear counterstain. Scale bar: 20 µm. h–j Quantitative analysis of CD8 (h), Granzyme B (GB) (i), and cleaved caspase-3 (CCA3) (j) using ImageJ. Data is shown as means ± SD, n = 6 mice per group. Two-sided Student’s t-test. Analysis unit = 12,945 μm2. k, l On day 0 (Treatment Initiation, TI), BALB/c mice were inoculated with CT26 cancer cells transfected with either control siRNA (Cont si) or RPN1 siRNA (RPN1 si), followed by a 12 Gy dose of IR on day 10. Intratumoral siRNA transfection and injection of antibodies against CD8 + T cells (using clone 2.43) were conducted as described in the Methods section. Data are presented as means ± SD, with 6 mice per group. Tumor size (in mm³) was measured at specified intervals. Two-way ANOVA with Tukey’s multiple comparison test (k). Survival curves for each treatment group are provided. Log-rank test (l). Source data are provided as a Source Data file.
Consistent with our proposed mechanism, IR-CS significantly increased the levels of PD-L1 and RPN1 in tumor samples (Fig. 7d, e and Supplementary Fig. 10_Cont si vs. Cont si + IR-CS). Furthermore, RPN1 si treatment inhibited the IR-CS-induced upregulation of PD-L1 expression (Fig. 7d, e and Supplementary Fig. 10_ Cont si + IR-CS vs. RPN1 si + IR-CS). In alignment with our in vitro results (Figs. 5 and 6), immunoblotting of tumor lysates showed a clear positive correlation between PD-L1 and RPN1 in tumors exposed to IR (Fig. 7f and Supplementary Fig. 10). Consistent with these results, RPN1 depletion notably increased the infiltration and activity (as indicated by the expression of granzyme B; GB) of CD8 + T-cells in tumor tissues under IR-CS (Fig. 7g–i), which in turn promoted apoptosis in senescent tumor tissues (Fig. 7j).
To further validate the T-cell-based therapeutic efficacy of RPN1-targeted therapy, we measured tumor growth and survival rates long-term after combining IR with Cont si or RPN1 si intratumoral transfection, with or without CD8 T-cell depletion. As a result, intratumoral RPN1 depletion suppressed cancer recurrence after IR treatment (Fig. 7k_Cont si + IR-CS + IgG vs RPN1 si + IR-CS + IgG) and greatly improved animal survival (Fig. 7l_Cont si + IR-CS + IgG vs RPN1 si + IR-CS + IgG). Consistent with our mechanism study, CD8 + T-cell depletion with anti-CD8 mAb treatment effectively neutralized the benefits of RPN1-targeted therapy (Fig. 7k_RPN1 si + IR-CS + IgG vs RPN1 si + IR-CS + α-CD8 mAb and Fig. 7l_Cont si + IR-CS + IgG vs RPN1 si + IR-CS + α-CD8 mAb). These findings collectively suggest that inhibition of RPN1 expression could suppress the radiotherapy-induced increase of PD-L1 in senescent tumors, potentially preventing cancer recurrence by enhancing anti-tumor immunity.
The elimination of therapy-induced senescent cancer cells through RPN1 depletion or PD-L1/PD-1-targeted therapy can enhance therapeutic efficacy by increasing CTL activity
The results presented in Fig. 7 demonstrate that RPN1-targeted therapy applied via intratumoral siRNA delivery leads to activation of T-cell immunity with PD-L1 downregulation within the tumor tissue. To assess the impact of RPN1-targeted therapy on senescent cancer cells, we analyzed the levels of SA-β-Gal activity and p21 expression, established markers for cancer senescence. Tumor samples exposed to IR exhibited pronounced increases in the levels of SA-β-Gal activity and p21 expression, whereas those treated with RPN1 si showed significant decreases in the IR-induced enhancements of these parameters (Fig. 8a–c). Immunoblot analyzes of tumor lysates revealed a distinct positive correlation between p21 and RPN1 in IR-exposed tumors (Fig. 8d and Supplementary Fig. 10). Consistently, when we examined expression level of various senescence markers such as PCNA, LMNB1, MMP2, MMP3, IL-6, CCL5, IGFBP3 IGFBP5, and IGFBP7 in the tumors using qPCR, the changes in the senescence markers of induced by IR-CS were effectively neutralized in the RPN1 si-treated group (Supplementary Fig. 11a). Interestingly, while intratumoral RPN1 depletion inhibited tumor growth via activated T-cell immunity, it also restored the expression of cell cycle regulators such as PCNA and LMNB1, which were reduced by IR-CS (Supplementary Fig. 11a_PCNA and LMNB1). As PCR quantifies mRNA levels per unit volume of the tumor, the ratio of proliferative cancer cells to non-proliferative senescent cancer cells per unit volume would be higher in the RPN1 siRNA-treated group compared to the control group, due to the elimination of senescent cancer cells. Therefore, the increased mRNA levels of PCNA and LMNB1 observed in RPN1 si-treated group can be interpreted a reduction in the proportion of senescent cancer cells within the tumor. Furthermore, in contrast to the in vivo results (Supplementary Fig. 11b), RPN1 si treatment did not affect the changes in senescence markers induced by IR-CS under in vitro cell culture conditions (Supplementary Fig. 11b), implying that the effect of intertumoral RPN1 depletion is mediated by effectors in the TME. Collectively, these results suggest that senescent cancer cells were likely eliminated by T-cell immunity due to the RPN1 depletion-induced loss of PD-L1.

a–d Tumor tissues from Fig. 6 were used for analysis. (a) Representative images of SA-β-Gal staining and p21 immunostaining in tumor tissues. DAPI was used for nuclear counterstaining. Scale bar: 20 µm (top), 50 µm (bottom). b,c (b) Quantification of SA-β-Gal staining intensity and (c) p21 protein levels in tumor sections using ImageJ. Data are presented as means ± SD; n = 6 mice per group. Analysis unit: (b) 317,850 μm2, (c) 77,428 μm2. Two-sided Student’s t-test. d Correlation analysis between p21 and RPN1 shown in Supplementary Fig. 10. Mean ± SD of n = 6 mice per group. Spearman’s correlation test. e–j CT26 cancer cells were inoculated into BALB/c mice on day 1 (TI). Mice were irradiated with 12 Gy IR on day 13 and treated with 200 µg anti-PD-1 antibody (ICB) per mouse through intraperitoneal (IP) injection for 9 mice per group. The results of tumor growth are presented in Supplementary Fig. 12. e Representative images of SA-β-Gal staining and p21 immunostaining in tumor sections. DAPI was used for nuclear counterstaining. Scale bar: 20 µm (top), 50 µm (bottom). f, g Quantification of SA-β-Gal staining intensity (f) and p21 protein levels (g) in tumor sections from Supplementary Fig. 12 using ImageJ. Data are presented as means ± SD; (f) n = 7 mice per group, (g) n = 6 mice per group. Analysis unit: (f) 317,850 μm2, (g) 77,428 μm2. Two-sided Student’s t-test. h Representative images of immunostaining of CD8, Granzyme B, and caspase-3 in tumor sections with DAPI as nuclear counterstain. Scale bar: 20 µm. i, j Quantitative analysis of CD8 (i), and Granzyme B (GB) (j) using ImageJ. Data is shown as means ± SD, n = 6 mice per group. Analysis unit: 12,945 μm2. Two-sided Student’s t-test. k, l CT26 cancer cells were inoculated into BALB/c mice on day 1 (TI). The mice were treated with 250 µg of anti-CD8 antibody on day 10, administered once every three days for a total of four doses. On day 11, the mice were irradiated with 12 Gy IR and subsequently treated with 200 µg of anti-PD-1 antibody (ICB) per mouse through IP injection (n = 9 mice per group). k Tumor size (in mm³) was measured at the indicated times. Two-way ANOVA with Tukey’s multiple comparison test. l Survival rates for each group until 123 days. Log-rank test. m A model showing how senescent cancer cells suppress anti-tumor immunity by modulating PD-L1 levels, potentially leading to cancer relapse. In the absence of T-cell suppressive TME with tumor volume reduction post conventional treatments, senescent cancer cells may contribute to potential cancer recurrence by acting as primary protectors of residual cancer cells by providing a PD-L1 umbrella against attack by activated T cells. Created in BioRender. Cha, J. (2023) BioRender.com/n74o053. Source data are provided as a Source Data file.
To further explore this hypothesis, we analyzed senescent cancer cell populations after the administration of an anti-PD-1 therapeutic antibody in an IR-CS animal model. BALB/c mice were engrafted with CT26 cells and irradiated with 12 Gy IR on day 13 post-inoculation. The anti-PD-1 therapeutic antibody was administered via intraperitoneal injection, as presented in the schematic (Supplementary Fig. 12_bottom of graph). Considering the proper tumor size for analysis and high CTL activity, we conducted sampling one day after the second administration of the anti-PD-1 antibody. Interestingly, similar to the results obtained with RPN1-targeted therapy, the anti-PD-1 therapeutic antibody-treated group showed decreased levels of SA-β-Gal activity (Fig. 8f) and p21 expression (Fig. 8g), accompanied by increased CTL activity in the tumors (Fig. 8h–j). These results support that IR-induced senescent cancer cells can be effectively targeted and eliminated via T-cell immunity with PD-L1/PD-1-specific therapeutics.
Finally, the correlation of T-cell immunity with the elimination of senescent cancer cells by ICB treatment and its therapeutic value were assessed through long-term observation over four months. BALB/c mice were engrafted with CT26 cells and irradiated with 12 Gy IR on day 11 post-inoculation. The anti-PD-1 therapeutic antibody and anti-CD8 depletion antibody were administered via intraperitoneal injection, as presented in the schematic (Fig. 8k_bottom of graph). The IR combined with PD-1 blockade group showed more effective tumor suppression and longer survival without recurrence compared to the IR single-treated group (Fig. 8k, l_α-PD-1 mAb + IR-CS + IgG), and these benefits of combination therapy were neutralized by CD8 + T-cell depletion (Fig. 8k, l_α-PD-1 mAb + IR-CS + α-CD8 mAb). These results clearly show that the elimination of PD-L1-positive senescent cancer cells can enhance therapeutic efficacy by inhibiting cancer recurrence via T-cell immunity.
Taken together, our results suggest that conventional therapies should restore intratumoral CTL activity by relieving the T-cell-suppressive TME. However, therapy-induced senescent cancer cells have increased PD-L1 expression; this creates an “umbrella” that can potentially shield residual cancer cells from CTL surveillance. The few surviving residual cancer cells can then proliferate and re-establish the T-cell-suppressive TME, leading to relapse. Targeting these PD-L1-expressing senescent cancer cells with RPN1-targeted and/or PD-L1/PD-1-targeted therapies may therefore emerge as a promising approach to enhance therapeutic efficacy and prevent cancer recurrence (Fig. 8m).
Discussion
Recently, the relationship between senescent cancer cells and tumor malignancy has emerged as a critical topic in cancer research. While the role of senescent cancer cells in tumors has been controversial, several recent pieces of evidence strongly support that cellular senescence contributes to tumor malignancy6,54,55. Therapy-induced senescent cells no longer proliferate but continue to metabolize and secrete pro-inflammatory substances2. This can lead to TME that suppresses anti-tumor immunity, making cancer treatment less effective4,5. Consequently, efforts are being made to reduce cancer malignancy by targeting genes involved in cellular senescence56. Additionally, the potential of using senolytic drugs to eliminate senescent cancer cells is gaining attention as an innovative strategy for cancer treatment7,8. In line with these researches, our current study suggests the important concept that senescent cancer cells may effectively defend the remaining cancer cells against CTL attacks, thereby contributing to cancer recurrence (Fig. 8m). Specifically, we observed that radiotherapy or chemotherapy-induced senescent cancer cells express high levels of PD-L1, enabling them to evade T-cell surveillance (Fig. 1). Senescent cancer cells promote not only the transcription of PD-L1, but also the glycosylation of PD-L1 through the induction of RPN1, which stabilizes PD-L1 and supports its translocation to the cell membrane (Figs. 1–4). The increased PD-L1 on senescent cancer cells directly induces T-cell exhaustion (Figs. 1a,b and 5l,m). Knocking down RPN1 expression decreases PD-L1 levels via ERLAD in senescent cancer cells (Fig. 6), rendering them more susceptible to T-cell killing. Indeed, depletion of intratumoral RPN1 with specific siRNA treatment effectively inhibits cancer recurrence in vivo with long-term effects (Fig. 7), including reductions of senescent cancer cells (Fig. 8a–d) and enhancement of antitumor immunity (Fig. 7g–j). These results suggest that “PD-L1 umbrella” provided by senescent cancer cells may play an important role in allowing cancer cells to evade T-cell surveillance and support recurrence after chemotherapy and radiation therapy.
This study also highlights the role of RPN1 in maintaining PD-L1 stability, revealing its potential as a key therapeutic target in cancer immunotherapy. Our data demonstrate that IR-induced cancer senescence increases not only PD-L1 transcription (Fig. 1h) but also the expression of RPN1 along with other OST-B components such as STT3B, DDOST, and RPN2 (Fig. 2d–f). The enhanced expression of RPN1 likely ensures the proper glycosylation of the markedly increased PD-L1 polypeptides (Fig. 4), facilitating its transport to the cell membrane (Fig. 5h–j). This process aids senescent cancer cells in evading T-cell surveillance (Fig. 5k–m). As a therapeutic approach, inhibiting RPN1 expression may disrupt the access of PD-L1 to the STT3 catalytic site, resulting in the accumulation of nascent PD-L1 polypeptides within the ER (Fig. 6d–f) and consequently inducing ER stress. To alleviate this stress, cancer cells may activate the ERLAD pathway to degrade excess PD-L1 (Fig. 6), leading to reduced levels of PD-L1 on the cell membrane and heightened sensitivity of cancer cells to T-cell detection (Fig. 7). Based on our experimental evidence and previous reports on RPN1, we interpreted that RPN1 promotes the glycosylation process of PD-L1 by acting as an adapter in a substrate-specific manner. The detail mechanisms by which RPN1 achieves this substrate specificity and facilitates glycosylation need to be elucidated through further studies. While the role of RPN1 is prominent under cellular stress conditions during cancer senescence, our preliminary data show that RPN1 also contributes to PD-L1 stabilization in various cancer types under normal conditions (Fig. 5e–g). If the mechanisms by which RPN1 contributes to PD-L1 stability in non-stress conditions are elucidated, the value of RPN1 as a therapeutic target in cancer immunotherapy is expected to increase significantly.
Our current study has shown that PD-L1 levels in tumors can be upregulated through a process of cellular senescence triggered by conventional treatments. Consistently, Shahbandi et al. demonstrated that mRNA levels of various immune regulatory proteins, including PD-L1 and CD80, were increased in both doxorubicin-induced senescent cancer cells and senescent-like cells from treated tumors33. Chemotherapy combined with PD-1 and/or CD80 blockade enhanced therapeutic efficacy through T cell accumulation. Ruscetti et al. also showed that PD-L1 levels are increased in endothelial cells, immune cells, and cancer cells in tumors treated with a combination of trametinib and palbociclib (T/P)57. Similarly, induction of senescence with doxorubicin improves the efficacy of PD1 blockade in breast cancer brain metastasis in a CD8 + T-cell-dependent manner58. Together with these previous reports, our findings suggest that it may be necessary to reconsider the criteria of administering PD-1/PD-L1-targeted ICBs based solely on the PD-L1 immunohistochemistry (IHC) score of the tumor. In subgroup analyzes of certain clinical trials that combined radiotherapy or chemotherapy with PD-1/PD-L1-targeted ICBs, even patients with low PD-L1 expression demonstrated superior efficacy with the combination therapy compared to chemo- or radio-therapy alone59,60,61. For instance, in the KEYNOTE-021 and -024 trials, the combination treatments had response rates of 57% and 11%, respectively, in the subgroup with less than 1% PD-L1 expression. It is worth considering the possibility that conventional therapies can increase PD-L1 levels through cancer senescence, and combining ICBs may counteract this side effect. To prevent the immune evasion associated with cancer senescence, patients with low PD-L1 expression may be considered for PD-1/PD-L1-targeted ICBs as a potential second-line therapy following conventional treatment.
Our findings support the therapeutic potential of eliminating senescent cancer cells following cancer treatment. Intertumoral RPN1 depletion effectively suppressed senescent parameters induced by IR-CS in the tumor (Supplementary Fig. 11: PCNA, LMNB1, MMP2, MMP3, IL-6, CCL5, IGFBP3, IGFBP5, and IGFBP7). Indeed, when RPN1 siRNA or anti-PD-1 therapeutic antibody was administered, a substantial reduction in senescent cells was observed (Fig. 8a–g), along with improved T-cell immunity (Fig. 7g–I and Fig. 8h–j) and tumor regression (Fig. 7k, a and Fig. 8k) in the IR-treated CT26 tumor syngeneic mouse model. Consistent with our current study, it has been experimentally demonstrated that targeted senolysis combined with chemotherapy can increase therapeutic efficiency and prevent recurrence. Demaria et al. elucidated the role of senescent cancer cells in cancer therapy using a p16-3MR BAC transgenic mouse model capable of detecting and eradicating senescent cells56, showing that the removal of senescent cells can enhance treatment efficacy with reduced metastasis. Similarly, Saleh et al. revealed that administration of the senolytic agent, ABT-263 (Navitoclax), to tumor-bearing mice after etoposide or doxorubicin treatment significantly inhibited tumor growth7,8. These findings strongly support the idea that senolysis may mitigate some limitations and side effects of conventional cancer treatments, such as chemotherapy and radiotherapy.
Although our result suggest that senescent cancer cells can suppress T-cell function, they may also play a positive role in maintaining anti-tumor immunity. Recent studies have obtained promising results in efforts to activate anti-tumor immunity by inducing innate immunity in low-immunogenicity cold tumors by using pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) to trigger inflammation62. Within this context, we cannot completely disregard the possibility that pro-inflammatory cytokines released from senescent cancer cells could enhance tumor immunogenicity by inducing inflammation within the tumor. Indeed, Marin et al. reported that immunization with senescent cancer cells can elicit a stronger systemic anti-tumor response compared to immunization with dying cancer cells63. Furthermore, Ruscetti et al. demonstrated that SAPS secreted from therapy-induced senescent cancer cells can induce tumor vessel remodeling through angiogenic factors. This enhances drug delivery and the efficacy of gemcitabine, and promotes CD8 + T-cell accumulation, transitioning a cold tumor to become more sensitive to PD-1 blockade in a preclinical mouse model of pancreatic ductal adenocarcinoma57. Importantly, Chen et al. recently showed that senescent cells upregulate IFNγ receptors, are hypersensitive to microenvironmental IFNγ, and induce antigen presentation mechanisms more strongly. This implies that senescent cancer cells may differ in their function depending on the composition of SASP in the TME of senescence-induced tumors64.
Despite the great potential of senescent cancer cells as a therapeutic target, studies on their function have shown conflicting results, leaving their role unclear. Therefore, it is necessary to conduct follow-up research to elucidate their specific functions in various TMEs and to assess the dual nature of senescent cancer cells as a target for cancer treatment. This will help fully understand the potential of therapies targeting senescent cancer cells and optimize their safety and efficacy.
Methods
Ethical statement
All animal care and experimental procedures were approved by the INHA University-Institutional Animal Care and Use Committee (INHAIACUC) (Approval Numbers INHA-221121-846-2 and INHA-220712-831-10). Subcutaneous tumor burden in animals ranging between 1500 and 2000 mm3 was euthanized according to the protocol of the Animal Care and Use Committee of Inha University.
Cell culture
H460 (HTB-177, ATCC, Manassas, VA, USA), A549 (CCL-185, ATCC), H1975 (CRL-5905, ATCC), HCC827 (CRL-2868, ATCC), and H1299 (CRL-5803, ATCC) cells were cultured in RPMI 1640 medium (WelGENE Inc., Daegu, Korea). OT-cells were also cultured in RPMI 1640 medium (WelGENE). U2OS (HTB-96, ATCC) and HCT116 (CCL-247, ATCC) cells were cultured in McCoy’s 5A medium (WelGENE). HEK293T (CRL-11268, ATCC), BT549 (HTB-122, ATCC), CT26 (CRL-2638, ATCC), and B16/F10-OVA melanoma cells (SCC420, Sigma, St. Louis, MO, USA) were cultured in DMEM medium (WelGENE). Fetal bovine serum (10%; GIBCO, Grand Island, NY, USA), 1% penicillin, and streptomycin were added to all media. All cell lines were maintained at 37 °C in a humidified atmosphere with 5% CO2.
Reagents and antibodies
Reagents were obtained from the following suppliers: Reagents were obtained from the following suppliers: Doxorubicin (Sigma Aldrich, D1515), MG132 (Sigma Aldrich, C2211), Cycloheximide (CHX) (Sigma Aldrich, C7698), Chloroquine (CQ) (Sigma Aldrich, C6628), Formaldehyde (Sigma Aldrich, F1636), Sodium chloride (NaCl) (Duchefa, Haarlem, The Netherlands, 7647-14-5), Magnesium chloride (MgCl2) (Sigma Aldrich, M2670), Citric acid (Sigma Aldrich, C1909), Sodium phosphate (Sigma Aldrich, S3264), Potassium ferrocyanide (Sigma Aldrich, P9387), Potassium ferricyanide (Sigma Aldrich, P8131), X-galactosidase (X-Gal) (Beamsbio, Sungnam, Korea, 7002), Tris (Duchefa, T1801), NP-40 (USB, CA, USA, 68987-90-6), Ethylenediaminetetraacetic acid (EDTA) (Sigma Aldrich, 03609), Sodium dodecyl sulfate (SDS) (Sigma Aldrich, 3771), Sodium deoxycholate (Sigma Aldrich, D6750), Triton X-100 (Sigma Aldrich, X-100), Protease inhibitors (Roche, Basel, Switzerland, 11873580001), Phosphatase inhibitor cocktail (Sigma Aldrich, P5726). Peptide N-glycosidase F (PNGase F) (New England Biolab, P0704L), Porous graphitic carbon (PGC) cartridge (Agilent Technologies, 12102201), Acetonitrile (ACN) (Sigma-Aldrich, 34851, HPLC grade), Formic acid(Honeywell, 94318)
Antibodies were obtained from the following suppliers: PD-L1 (E1L3N®) (1:1000, Cell Singing Technology; CST, Danvers, MA, USA, 13684), p21 (12D1) (1:1000, CST, 2947), LAMP1 (D4O1S) (1:1000, CST, 15665), LC3B (D11) (1:1000, CST, 3868), Rab5A (E6N8S) (1:1000, CST, 46449), Rab11 (D4F5) (1:1000, CST, 5589), β-Actin (8H10D10) (1:10000, CST, 3700), p53 (DO7) (1:1000, Leica, Wetzlar, Germany, NCL-L-p53-DO7), GM130 (Clone 35) (1:1000, BD Bioscience, Franklin lakes, NJ, USA, 610823), TGN46 (2F11) (1:100, Novus, St. Louis, MO, USA, H00010618-M02), gp96/HSP90B1/GRP94 (CL2647) (1:100, Novus, NBP2-42379), PD-L1 (PDL1/2746) (1:100, Novus, NBP2-80490), FAM134B (1:100, Proteintech, Rosemont, IL, USA, 21537-1-AP), Granzyme B (1:100, LS Bio, Shirley, MA, USA, LS-C332166), PD-L1 (clone OTI2C7) (1:100, LS Bio, LS-C338364), RPN1 (E-7) (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA, sc-48367), CD8 (LT8) (1:100, GeneTex, Alton Pkwy Irvine, CA, USA, GTX74773). Human Recombinant Fc Chimera PD-1 (1:40, R&D Systems, Minneapolis, MN, USA, 1086-PD), NucView 488 caspase-3 (BIOTIUM, San Francisco, CA, USA, 30029), PerCP-labeled CD8α (1:100, BioLegend, 100732), PE-labeled Vα2 TCR (1:100, BD Pharmingen, 553289), FITC-labeled Vβ5 TCR (1:100, BD Pharmingen, 553189), InVivo MAb rat IgG2b isotype control (BioXCell, West Lebanon, NH, USA, BE0090), InVivo MAb anti-mouse CD8α (BioXCell, BE0061), DAPI (Thermo scientific, Waltham, MA, USA, 62248), Alexa Fluor® 488 AffiniPure Donkey Anti-Rabbit IgG (H + L) (1:100, Jackson Immuno Research, West Grove, PA, USA, 711-545-152), Alexa Fluor® 488 AffiniPure Donkey Anti-Mouse IgG (H + L) (1:100, Jackson Immuno Research, 711-545-150), Alexa Fluor® 594 AffiniPure Donkey Anti-Rabbit IgG (H + L) (1:100, Jackson Immuno Research, 711-585-152), Alexa Fluor® 594 AffiniPure Donkey Anti-Mouse IgG (H + L) (1:100, Jackson Immuno Research, 711-585-150), Goat anti-Mouse IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 488 (1:200, Invitrogen, A11001).
Irradiation
The cells were irradiated with a 2-mm Al filter, 150 kV accelerating voltage, and a dosage rate of 0.72 Gy/min using an X-RAD iR160 X-ray irradiator (North Branford, CT., USA, Precision X-ray (PXi) Inc.).
Senescence-associated β-galactosidase staining
Cells were exposed to ionizing radiation to induce cellular senescence. Cells were fixed with 3.7% formaldehyde for 5 min, washed with PBS, and incubated with fresh staining solution consisting of 1 mg/ml 5-bromo-4-chloro-3-indolyl β-D-galactoside (X-Gal) in 40 mM citric acid/sodium phosphate (pH 6.0), 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 5 mM NaCl, and 2 mM MgCl2. Staining was evident after 12–16 h. Cell staining was analyzed using an Olympus CKX41 light microscope (Olympus, Tokyo, Japan). To evaluate senescence in tumor tissues, frozen tissue sections were stained with fluorogenic β-galactosidase detection kit-SPiDER-βGal (Dojindo Molecular Technologies, Inc., Kumamoto, Japan) according to the manufacturer’s instruction.
Immunoblot analysis
Protease inhibitors (Roche) and phosphatase inhibitors (Sigma Aldrich) were added to RIPA lysis buffer (Sigma-Aldrich, St. Louis, MO, USA). Lysates were combined with 2 x Laemmli sample buffer, boiled, and electrophoresed on a polyacrylamide gel with sodium dodecyl sulfate (SDS-PAGE). A nitrocellulose membrane was used to transfer all proteins, which were then blocked with 5% non-fat dried milk. After blocking, the membranes were sequentially incubated with various primary antibodies and secondary antibodies conjugated with horseradish peroxidase. ECL reagents were used for detection (Thermo scientific).
RNA interference
Cells were transfected with 100 nM siRNA duplexes by using RNAiMAX (Invitrogen, Karlsruhe, Germany). The sequences of the siRNA (Bioneer Inc., Daejeon, Korea) are listed in Supplementary Table 1.
qRT-PCR
Total RNA was extracted using TRIzol reagent (Molecular Research Center, Netherlands). RNA was reverse transcribed to cDNA using M-MLV reverse transcriptase (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instruction, and qPCR was performed using iQTM SYBR® Green Supermix (BioRad Laboratories, Hercules, CA, USA) in a CFX ConnectTM RT-PCR Detection System (BioRad Laboratories). Housekeeping gene Actin was used as internal controls to normalize target mRNAs. The primers used for qRT-PCR are listed in Supplementary Table 2.
Immunofluorescence staining
The cells were fixed with 3.7% formaldehyde for 15 min, permeabilized with 0.1% Triton X-100 for 15 min and blocked using 3% BSA in PBS for 1 h. Subsequently, the cells were incubated with primary antibodies (1:100 dilution) for 1 h at room temperature, followed by fluorescein-conjugated secondary antibodies (1:100 dilution) for another 1 h at room temperature. Immunofluorescence was examined using an LSM 510 META laser scanning microscope (ZEISS, Germany).
PD-1 binding assay
The cells were stained with Vybrant® dil cell-labeling solution (Invitrogen) four days after exposure to radiation. Recombinant PD-1 Fc protein (R&D Systems) and Alexa 488 conjugated-secondary anti-human IgG (Invitrogen) were sequentially incubated with the cells for 1 h at room temperature. Images were acquired using a fluorescence microscope.
Sample labeling and Illumina BeadChip array hybridization
Total RNA was isolated from H460 cells using the TRIzol reagent (Invitrogen) following the manufacturer’s instructions. RNA quality was assessed by gel electrophoresis, and concentrations were measured using an Ultrospec 3100 pro spectrophotometer (Amersham Bioscience, Buckinghamshire, UK). Biotin-labeled complementary RNA (cRNA) samples were prepared according to the Illumina sample labeling protocol. Briefly, 500 ng of total RNA was used for complementary DNA (cDNA) synthesis, followed by in vitro transcription for amplification and biotin labeling using the Illumina® TotalPrep RNA Amplification Kit (Ambion Inc., Austin, TX). cRNA concentrations were determined using the RiboGreen assay (Quant-iTTM RiboGreen® RNA assay kit; Invitrogen-Molecular Probes, ON, Canada) with a Victor3 spectrophotometer (Perkin-Elmer, CT), and cRNA quality was verified on a 1% agarose gel. A total of 1.5 μg of labeled, amplified cRNA was hybridized to version 2 of the Illumina Human-6 BeadChip (48 K) following the manufacturer’s protocol (Illumina, Inc., San Diego, CA). Array signals were developed using Amersham fluorolink streptavidin-Cy3 (GE Healthcare Bio-Sciences, Little Chalfont, UK) as per the BeadChip manual. Arrays were scanned using an Illumina BeadArray Reader confocal scanner (BeadStation 500GXDW; Illumina, Inc.). Array data processing and analysis were conducted using Illumina BeadStudio software (version 3.1).
3D spheroid formation
B16F10-OVA melanoma cells were detached using trypsin/EDTA and centrifuged at 300 g for 5 min. The supernatant was discarded, and the cell pellet was resuspended in serum-free media and stained with a Vybrant® dil cell-labeled red solution (Invitrogen, C34552) and incubated for 30 min. The cells were then centrifuged at 200 g for 10 min, washed with PBS and centrifuged again at 200 g for 10 min. The supernatant was discarded, and the pellet was resuspended with complete medium and then cells were seeded at a density of 5 × 103 cells/well in 96-well round-bottom ultra-low attachment microplates (Corning B.V. Life Sciences, Amsterdam, Netherlands). The plate was then centrifuged at 135 g for 10 min and incubated at 37 °C in 5% CO2.
T-cell infiltration assay
OT- cells were stained with Vybrant® Dil cell-labeled green solution (Invitrogen, C7025) according to the manufacturer’s instruction. B16F10-OVA melanoma spheroids and OT- cells were co-cultured at a ratio of 1:5. The next day, the infiltration of OT- cells was confirmed by photographing and analyzing them using an Olympus CKX41 light microscope (Olympus, Tokyo, Japan).
T cell-mediated killing assay
B16F10-OVA melanoma cells were seeded and stained with Vybrant® dil cell-labeling solution (Invitrogen) four days after exposure to radiation. B16F10-OVA melanoma cells were treated with activated OT- cells at effector to target ratio (E: T) of 5: 1. B16F10-OVA melanoma cells and activated OT- cells were co-cultured with a fresh medium containing NucView® caspase-3 substrate (Biotium) for 48 h. GFP-positive apoptotic cells were observed by Olympus IX83 fluorescence microscopy.
TCGA analysis
TCGA analysis was carried out using TIMER2.0, a R package that includes six cutting-edge methods, including TIMER, XCELL, MCPCOUNTER, CIBERSORT, EPIC, and QUANTISEQ40. The Gene DE module was used to assess RPN1 level between tumor and matched normal tissues across all TCGA cancers. The Gene_Corr module was used to examine the correlation between RPN1 and PD-L1 levels. The Cellular Composition Estimate module was used to create a heatmap table of Spearman’s correlations between IFN-β/HIF-1α/RPN1 expression and CD8 + T cells across all TCGA tumors.
Flow cytometry analysis
H460 cell were suspended in cell staining buffer (Biolegend, San Diego, CA, USA, 420201) and stained with PE-labeled PD-L1 antibodies (Biolegend, 329705, 1:100 dilution) or isotype control antibodies (Biolegend, 400313, 1:100 dilution) for 30 min at 4 °C while protecting from light. Flow cytometric analyzes were performed with CytoFLEX flow cytometer (A00-1-1102) from Beckman Coulter.
Generation of PD-L1 stable cells
Using a pGIPz-shPD-L1/Flag-PD-L1 dual expression construct17,24, we established H460 stable cell lines expressing Flag-PD-L1 WT and 4NQ with endogenous PD-L1 knockdown. To package lentivirus, the dual expression constructs were transfected into HEK293T cells with two packaging plasmids (VSV-G and delta 8.2). The supernatant was collected at 48 h after transfection and centrifuged for 10 minutes. The centrifuged supernatant was filtered using a 0.45 µm filter. H460 was incubated in lentivirus-containing medium with polybrene. 48 h following infection, cell was subjected to puromycin selection (InvivoGen, San Diego, CA, USA, 1 µg/ml).
Plasmid
The human PD-L1 Flag was obtained from pGIPz-shPD-L1/Flag-PD-L1 and cloned into pCI-neo vector (Promega, Madison, WI, USA, E1841). The construct was confirmed using DNA sequencing.
N- glycan release and enrichment
N-glycans were prepared as previously described65,66. Brifely, The N-glycans were enzymatically released by PNGase F from PD-L1. The isolated N-glycans were purified and enriched with Porous Graphitize Carbon-Solid Phase Extraction (PGC-SPE). All SPE fractions were vacuum-dried prior to LC/MS analysis.
Nano-LC/MS analysis
Purified N-glycans were chromatographically separated and analyzed using a nanoLC/Q-TOF MS system (Agilent 1260 and 6530, Agilent Technologies, Santa Clara, CA). The nanoLC chip consisted of a 9 × 0.075 mm i.d. enrichment column and a 43 × 0.075 mm i.d. analytical column, both packed with PGC. The glycan elution gradient was delivered at 0.4 μl/min using solutions of: (A) 3% ACN with 0.3% formic acid (v/v) in H2O and (B) 90% ACN with 0.3% formic acid (v/v) in H2O, at the following proportions and time points: 4% B, 0–2.5 min; 4–16% B, 2.5–30 min; 16–45% B, 30–55 min; 45–100% B, 55–65 min. MS spectra were obtained from m/z 500–2500 in the positive ion mode.
LC/MS Data Processing
Raw LC/MS data were processed using MassHunter Qualitative Analysis v7.0 software (Agilent Technologies). The molecular feature extractor algorithm generated the target compound list providing neutral mass values, retention times of each ion, and chromatographic peak areas. N-Glycan compositions consisting of hexose (Hex), N-acetylhexosamine (HexNAc), fucose (Fuc), N-acetylneuraminic acid (NeuAc) were identified by accurate masses (with a mass error of 10 ppm), retention times and known glycan biosynthetic correlations65,67,68. The peak areas of all ions (e.g., a single-and multiple-protonated ions) of each glycan were summed to obtain compound abundance, which was used for relative quantitation.
Animal experiments
In Fig. 7a–j and Fig. 8a–d, CT26 cells (1 × 106) transfected with Control Si (Cont si) or RPN1 si were injected subcutaneously into the lateral hind leg of 6-week-old male BALB/c mice (n = 6 mice per group). On days 10, the tumors were exposed to 12 Gy of IR. Over the course of 40 days, tumors were injected with Cont si or RPN1 si 14 additional times using in vivo-jetPEI® (Polyplus transfection;Illkirch, France). Mice were sacrificed 40 days after inoculation and tumors were removed for further analysis. In Fig. 7k and l, CT26 cells (1 × 106) transfected with Cont si or RPN1 si were injected subcutaneously into the lateral hind leg of 6-week-old male BALB/c mice (n = 6 mice per group). On days 10, the tumors were exposed to 12 Gy of IR. From day 8 for a total of 50 days, tumors were administered 250 μg of anti-CD8 (clone 2.43; Bio X cell) via IP injection, and Cont si or RPN1 si was administered via intratumoral injection every 3 days. Over a total of 110 days, tumor size and survival rate were measured. In Fig. 8e–j and Supplementary Fig. 12, CT26 cells (1 × 106) were subcutaneously injected into the lateral hind leg of 6-week-old male BALB/c mice (n = 9 mice per group). On days 13, the tumors were irradiated with 12 Gy of IR. On days 14 and 16, the mice received injections of anti-mouse PD-1 (BE0146; Bio X cell) In Fig. 8k and l, CT26 cancer cells were inoculated into BALB/c male mice on day 1 (n = 9 mice per group). Mice were treated 250 ug anti-CD8 antibody on day 10, once every three days for a total of four times. Mice were irradiated with 12 Gy of IR on day 11 and treated with 200 µg anti-PD-1 antibody per mouse through IP injection. Tumor size was measured at the specified time. Survival data for each group were recorded over 120 days. The average tumor volume was calculated using the formula (L x W2)/2, where the tumor length (L) and width (W) were determined using calipers. Seventeen days after the tumor injection, mice were sacrificed, and their tumors were harvested for further analysis. BALB/c mice were purchased from the Orient Bio (Gyeonggi-do, Korea) and were maintained with in a 12 h light–12 h dark cycle. The ambient temperature was 20–24 °C, with 45% humidity.
Fluorescent immunohistochemistry
Tissues were cryosectioned at 20-μm thickness, fixed with 3.7% formaldehyde and permeabilized with 0.01% Triton X-100 for 15 min. The samples were then blocked with 3% BSA for 1 h at room temperature and incubated with primary antibody diluted (1:50) in blocking solution overnight at 4 °C. Sections were washed in PBS and incubated with secondary antibodies diluted (1:50) in blocking solution for 1 h at room temperature. The sections were washed three times with PBS and mounted on microscopy slides.
Extraction of genomic DNA
The KAPA Mouse Genotyping Kit (KK7301) was used to extract and amplify DNA from mouse ear. One-millimeter pieces of mouse ear were placed in PCR tubes containing 100 μL of KAPA Express Extraction Buffer and Enzyme. The samples were then heated to 75 °C for 15 min to release genomic DNA, and then the enzyme activity was stopped at 95 °C for 5 min. After the tubes were cooled to 4 °C, the sample was centrifuged to pellet the tissue debris, and the supernatant was immediately used for PCR.
Genotyping PCR
PCR samples consisted of 2 μL of genomic DNA, 12.5 μL of 2X KAPA2G Fast Genotyping Mix with dye and 5 μL of mixed primers (0.5 μM primers were mixed in a volume ratio of 1:1:1:1:1). The target DNA was amplified in a thermocycler machine (T100 thermal cycler, BIO-RAD). An initial denaturation at 94 °C for 2 min, followed by touchdown program for 10 cycles of 94 °C for 20 s, 65 °C for 15 s, 68 °C for 10 s. Then the subsequent 28 cycles of 94 °C for 15 s, 60 °C for 15 s, 72 °C for 10 s. The final extension step was performed at 72 °C for 2 min. The PCR products were electrophoresed on a 1.5% agarose gel.
OT-1 specific TCR expression by FACS
Mouse blood was added to 1.5 ml microcentrifuge tubes containing 100 μL FACS buffer. Cells were stained with the cell surface markers (1:100) PerCP-labeled CD8α (BioLegend, 100732), PE-labeled Vα2 TCR (BD Pharmingen, 553289), FITC-labeled Vβ5 TCR (BD Pharmingen, 553189) for 30 min. Add 1 ml red blood cell lysis buffer and incubate until the blood becomes clear. Centrifuge the tube at 848 g for 3 min and remove the supernatant. After washing, cells were analyzed using CytoFLEX (Beckman Coulter) with CytExpert software (version 2.3 Beckman).
Statistical analysis
Statistical analyzes were performed using GraphPad Prism v9.5.1 software. Data are expressed as the mean ± SD from at least three independent experimental replicates/groups. Statistical analyzes were conducted using two-tailed unpaired t-tests and one-way or two-way ANOVA test, followed by Tukey’s post hoc analysis to determine the significance levels, which were reported as p-values. Statistical tests used are detailed in respective figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The microarray data generated in this study have been deposited in the NCBI Gene Expression Omnibus (GEO) under accession code GSE280729. The raw LC-MS data of PD-L1 N-glycans generated in this study have been deposited in the GlycoPOST database under accession code GPST000489. The analysis of public data from TCGA was conducted using the TIMER 2.0 portal (http://timer.cistrome.org/). ‘The remaining data are available within the Article, Supplementary Information or Source Data file. Source data are provided with this paper.
References
Wang, L., Lankhorst, L. & Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 22, 340–355 (2022).
Park, S. S., Choi, Y. W., Kim, J. H., Kim, H. S. & Park, T. J. Senescent tumor cells: an overlooked adversary in the battle against cancer. Exp. Mol. Med 53, 1834–1841 (2021).
Wang, B., Kohli, J. & Demaria, M. Senescent cells in cancer therapy: friends or foes? Trends Cancer 6, 838–857 (2020).
Coppe, J. P., Desprez, P. Y., Krtolica, A. & Campisi, J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev. Pathol. 5, 99–118 (2010).
Takasugi, M., Yoshida, Y. & Ohtani, N. Cellular senescence and the tumour microenvironment. Mol. Oncol. 16, 3333–3351 (2022).
Yang, J., Liu, M., Hong, D., Zeng, M. & Zhang, X. The paradoxical role of cellular senescence in cancer. Front Cell Dev. Biol. 9, 722205 (2021).
Saleh, T. et al. Clearance of therapy-induced senescent tumor cells by the senolytic ABT-263 via interference with BCL-X(L) -BAX interaction. Mol. Oncol. 14, 2504–2519 (2020).
Sieben, C. J., Sturmlechner, I., van de Sluis, B. & van Deursen, J. M. Two-step senescence-focused cancer therapies. Trends Cell Biol. 28, 723–737 (2018).
Onorati, A. et al. Upregulation of PD-L1 in senescence and aging. Mol. Cell Biol. 42, e0017122 (2022).
Wang, T. W. et al. Blocking PD-L1-PD-1 improves senescence surveillance and ageing phenotypes. Nature 611, 358–364 (2022).
Francisco, L. M., Sage, P. T. & Sharpe, A. H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 236, 219–242 (2010).
Qin, W. et al. The diverse function of PD-1/PD-L pathway beyond cancer. Front Immunol. 10, 2298 (2019).
Yamaguchi, H., Hsu, J. M., Yang, W. H. & Hung, M. C. Mechanisms regulating PD-L1 expression in cancers and associated opportunities for novel small-molecule therapeutics. Nat. Rev. Clin. Oncol. 19, 287–305 (2022).
Cha, J. H., Chan, L. C., Li, C. W., Hsu, J. L. & Hung, M. C. Mechanisms controlling PD-L1 expression in cancer. Mol. Cell 76, 359–370 (2019).
Naimi, A. et al. Tumor immunotherapies by immune checkpoint inhibitors (ICIs); the pros and cons. Cell Commun. Signal 20, 44 (2022).
Schjoldager, K. T., Narimatsu, Y., Joshi, H. J. & Clausen, H. Global view of human protein glycosylation pathways and functions. Nat. Rev. Mol. Cell Biol. 21, 729–749 (2020).
Li, C. W. et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 7, 12632 (2016).
Li, S. M. et al. Recent Findings in the Posttranslational Modifications of PD-L1. J. Oncol. 2020, 5497015 (2020).
Hsu, J. M. et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 9, 1908 (2018).
Harada, Y., Ohkawa, Y., Kizuka, Y. & Taniguchi, N. Oligosaccharyltransferase: A Gatekeeper of Health and Tumor Progression. Int J Mol Sci 20 https://doi.org/10.3390/ijms20236074 (2019).
Cherepanova, N. A., Venev, S. V., Leszyk, J. D., Shaffer, S. A. & Gilmore, R. Quantitative glycoproteomics reveals new classes of STT3A- and STT3B-dependent N-glycosylation sites. J. Cell Biol. 218, 2782–2796 (2019).
Wilson, C. M. & High, S. Ribophorin I acts as a substrate-specific facilitator of N-glycosylation. J. cell Sci. 120, 648–657 (2007).
Qin, S. Y. et al. Malectin forms a complex with ribophorin I for enhanced association with misfolded glycoproteins. J. Biol. Chem. 287, 38080–38089 (2012).
Cha, J. H. et al. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol. Cell 71, 606–620.e607 (2018).
Yu, X., Li, W., Young, K. H. & Li, Y. Posttranslational Modifications in PD-L1 Turnover and Function: From Cradle to Grave. Biomedicines 9 https://doi.org/10.3390/biomedicines9111702 (2021)
Zhu, Q. et al. O-GlcNAcylation promotes tumor immune evasion by inhibiting PD-L1 lysosomal degradation. Proc. Natl Acad. Sci. USA 120, e2216796120 (2023).
Benicky, J. et al. PD-L1 glycosylation and its impact on binding to clinical antibodies. J. Proteome Res 20, 485–497 (2021).
Wang, Y. N., Lee, H. H., Hsu, J. L., Yu, D. & Hung, M. C. The impact of PD-L1 N-linked glycosylation on cancer therapy and clinical diagnosis. J. Biomed. Sci. 27, 77 (2020).
He, X. & Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res 30, 660–669 (2020).
Du, S. S. et al. Radiation therapy promotes hepatocellular carcinoma immune cloaking via PD-L1 upregulation induced by cGAS-STING activation. Int J. Radiat. Oncol. Biol. Phys. 112, 1243–1255 (2022).
Sato, H. et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 8, 1751 (2017).
Gilad, Y. et al. Drug-induced PD-L1 expression and cell stress response in breast cancer cells can be balanced by drug combination. Sci. Rep. 9, 15099 (2019).
Shahbandi, A. et al. Breast cancer cells survive chemotherapy by activating targetable immune-modulatory programs characterized by PD-L1 or CD80. Nat. Cancer 3, 1513–1533 (2022).
Scott, S. D., Joiner, M. C. & Marples, B. Optimizing radiation-responsive gene promoters for radiogenetic cancer therapy. Gene Ther. 9, 1396–1402 (2002).
Rao Gogineni, V. et al. Radiation-inducible silencing of uPA and uPAR in vitro and in vivo in meningioma. Int J. Oncol. 36, 809–816 (2010).
Kavanagh, E. L. et al. N-Linked glycosylation profiles of therapeutic induced senescent (TIS) triple negative breast cancer cells (TNBC) and their extracellular vesicle (EV) progeny. Mol. Omics 17, 72–85 (2021).
Itakura, Y. et al. N- and O-glycan cell surface protein modifications associated with cellular senescence and human aging. Cell Biosci. 6, 14 (2016).
Chan, L. C. et al. IL-6/JAK1 pathway drives PD-L1 Y112 phosphorylation to promote cancer immune evasion. J. Clin. Invest 129, 3324–3338 (2019).
Roboti, P. & High, S. The oligosaccharyltransferase subunits OST48, DAD1 and KCP2 function as ubiquitous and selective modulators of mammalian N-glycosylation. J. Cell Sci. 125, 3474–3484 (2012).
Li, T. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res 48, W509–W514 (2020).
Li, T. et al. TIMER: a web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res 77, e108–e110 (2017).
Li, B. et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. 17, 174 (2016).
Ewald, J. A., Desotelle, J. A., Wilding, G. & Jarrard, D. F. Therapy-induced senescence in cancer. J. Natl Cancer Inst. 102, 1536–1546 (2010).
Starbeck-Miller, G. R., Xue, H. H. & Harty, J. T. IL-12 and type I interferon prolong the division of activated CD8 T cells by maintaining high-affinity IL-2 signaling in vivo. J. Exp. Med 211, 105–120 (2014).
Marrack, P., Kappler, J. & Mitchell, T. Type I interferons keep activated T cells alive. J. Exp. Med 189, 521–530 (1999).
Scharping, N. E. et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 22, 205–215 (2021).
Hatfield, S. M. et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 7, 277ra230 (2015).
Wilson, C. M., Roebuck, Q. & High, S. Ribophorin I regulates substrate delivery to the oligosaccharyltransferase core. Proc. Natl Acad. Sci. USA 105, 9534–9539 (2008).
Wang, H. et al. HIP1R targets PD-L1 to lysosomal degradation to alter T cell-mediated cytotoxicity. Nat. Chem. Biol. 15, 42–50 (2019).
Burr, M. L. et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 549, 101–105 (2017).
Zhang, J., Jiang, Z. & Shi, A. Rab GTPases: The principal players in crafting the regulatory landscape of endosomal trafficking. Comput Struct. Biotechnol. J. 20, 4464–4472 (2022).
Fregno, I. et al. ER-to-lysosome-associated degradation of proteasome-resistant ATZ polymers occurs via receptor-mediated vesicular transport. EMBO J 37 https://doi.org/10.15252/embj.201899259 (2018).
Forrester, A. et al. A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM134B complex. EMBO J 38 https://doi.org/10.15252/embj.201899847 (2019).
Xiao, S. et al. Cellular senescence: a double-edged sword in cancer therapy. Front Oncol. 13, 1189015 (2023).
Schmitt, C. A., Wang, B. & Demaria, M. Senescence and cancer - role and therapeutic opportunities. Nat. Rev. Clin. Oncol. 19, 619–636 (2022).
Demaria, M. et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 7, 165–176 (2017).
Ruscetti, M. et al. Senescence-induced vascular remodeling creates therapeutic vulnerabilities in pancreas cancer. Cell 184, 4838–4839 (2021).
Uceda-Castro, R. et al. Re-purposing the pro-senescence properties of doxorubicin to introduce immunotherapy in breast cancer brain metastasis. Cell Rep. Med 3, 100821 (2022).
Langer, C. J. et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. 17, 1497–1508 (2016).
Reck, M. et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N. Engl. J. Med. 375, 1823–1833 (2016).
Theelen, W. et al. Effect of pembrolizumab after stereotactic body radiotherapy vs pembrolizumab alone on tumor response in patients with advanced non-small cell lung cancer: results of the PEMBRO-RT phase 2 randomized clinical trial. JAMA Oncol. 5, 1276–1282 (2019).
Sun, H. et al. Using PAMPs and DAMPs as adjuvants in cancer vaccines. Hum. Vaccin Immunother. 17, 5546–5557 (2021).
Marin, I. et al. Cellular senescence is immunogenic and promotes antitumor immunity. Cancer Discov. 13, 410–431 (2023).
Chen, H. A. et al. Senescence rewires microenvironment sensing to facilitate antitumor immunity. Cancer Discov. 13, 432–453 (2023).
Hua, S. et al. Comprehensive native glycan profiling with isomer separation and quantitation for the discovery of cancer biomarkers. Analyst 136, 3663–3671 (2011).
Seo, Y. et al. Comprehensive characterization of biotherapeutics by selective capturing of highly acidic glycans using stepwise PGC-SPE and LC/MS/MS. Anal. Chem. 91, 6064–6071 (2019).
Lee, S. H. et al. Glycomic profiling of targeted serum haptoglobin for gastric cancer using nano LC/MS and LC/MS/MS. Mol. Biosyst. 12, 3611–3621 (2016).
Abrahams, J. L., Campbell, M. P. & Packer, N. H. Building a PGC-LC-MS N-glycan retention library and elution mapping resource. Glycoconj. J. 35, 15–29 (2018).
Acknowledgements
We would like to express our gratitude to Professor Young Ju Suh (Department of Biomedical Science, College of Medicine, Inha University) for kindly supporting the biostatistical analysis. This study was supported in part by following: The National Research Foundation of Korea grant funded by the Korea government Ministry of Science and ICT (MSIT) (grant number: 2021R1A5A2031612 and 2023R1A2C2003673 to J.S.L; RS-2024-00342075 and RS-2024-00406723 to J-H.C; RS-2024-00354219 to H.J.H.).
Author information
Authors and Affiliations
Contributions
H.J.H. and D.H.K. designed and performed the experiments, analyzed data, and wrote the manuscript; J.S.S. and J.H.J. performed experiments and analyzed data; W.-H.Y. provided scientific input and performed TCGA analysis; S.K., K.H.J. and S.S.H. provided the OT-1 system. J.E.P., M.J.O. and H.J.A. conducted the analysis of the PD-L1 glycan structure. Y.-G. K. provided scientific input and analyzed data. J.-H.C. and J.-S.L. supervised the entire project, designed the experiments, analyzed data, and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Tyler Curiel and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hwang, H.J., Kang, D., Shin, J. et al. Therapy-induced senescent cancer cells contribute to cancer progression by promoting ribophorin 1-dependent PD-L1 upregulation. Nat Commun 16, 353 (2025). https://doi.org/10.1038/s41467-024-54132-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-54132-1
