
Abstract
Cutaneous T-cell lymphoma (CTCL) is a heterogeneous non-Hodgkin lymphoma originating in the skin and invading the systemic hematopoietic system. Current treatments, including chemotherapy and monoclonal antibodies yielded limited responses with high incidence of side effects, highlighting the need for targeted therapy. Screening with small inhibitors library, herein we identify cyclin dependent kinase 9 (CDK9) as a driver of CTCL growth. Single-cell RNA-seq analysis reveals a CDK9high malignant T cell cluster with a unique actively proliferating feature. Inhibition, depletion or proteolysis targeting chimera (PROTAC)-mediated degradation of CDK9 significantly reduces CTCL cell growth in vitro and in murine models. CDK9 also promotes degradation of retinoic acid receptor α (RARα) via recruiting the E3 ligase HUWE1. Co-administration of CDK9-PROTAC (GT-02897) with all-trans retinoic acid (ATRA) leads to synergistic attenuation of tumor growth in vitro and in xenograft models, providing a potential translational treatment for complete eradication of CTCL.
Similar content being viewed by others


Introduction
Cutaneous T-cell lymphoma (CTCL) is a type of non-Hodgkin lymphoma that is primarily localized to the skin at early stage. As the disease advances, local skin malignant T cells may progress systemic dissemination to the bloodstream, lymph nodes, and visceral organs. While standard skin-directed therapies are effective for individuals with indolent early-stage disease, patients with more advanced or refractory forms require systemic treatments, which may consist of chemotherapy, retinoid analogs, interferons, HDAC inhibitors, and antibodies1,2,3,4. However, none of the treatments are considered curative and most patients suffer from relapses. Retinoids including all-trans retinoic acid (ATRA), 13-cis-retinoic acid, and the synthetic analogs isotretinoin, etretinate and acitretin, which function through nuclear retinoic acid receptor, have been employed for over three decades to treat CTCL, with bexarotene as the first retinoid approved by the US FDA for CTCL therapy. However, a series of adverse metabolic side effects namely, hyperlipidemia, hypercholesterolemia and hypothyroidism were reported5,6,7,8. Novel therapies including epigenetic modifiers such as histone deacetylase (HDAC) inhibitors yielded low responses with median survival time of patients in advanced stage less than five years9,10,11,12, highlighting the urgent needs for better understanding of the driving pathways that support malignant T cells.
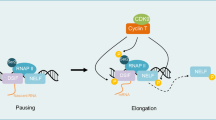
Cyclin dependent kinases (CDK) family is serine/threonine kinase whose activity relies on combination with a regulatory cyclin. Over 20 members of CDK family with specialized functions related to the cell cycle (CDK1, 2, 4, 6) or transcription regulation (CDK7, 8, 9, 12, 13, 19) have been identified13,14. In contrast to cell cycle associated CDKs, transcription associated CDKs bind with their cyclin partners constitutively, and are often recruited to chromatin as components of much larger complexes. CDK9 forms a subunit of the positive transcription elongation factor b (P-TEFb) in partnership with cyclin T, regulating gene transcription through phosphorylating the C-terminus ___domain (CTD) of RNA polymerase II (Pol II) and stimulating its release from paused phase into transcriptional elongation15,16,17. Accumulating evidences demonstrate that inhibition of CDK9 robustly induces apoptosis through transcriptional inhibition and downregulation of oncogenes (e.g., MYC) and anti-apoptotic genes (e.g., MCL1) that are pivotal for cancer proliferation and survival18,19,20. Hematopoietic cancers including acute lymphoblastic leukemia, chronic lymphocytic leukemia, diffuse large B-cell lymphoma as well as acute myeloid leukemia have been proposed to respond to CDK9-targeting strategies20,21,22,23.
Although the dependency on CDK9-related transcription is described in multiple cancer cells, clinical trials using CDK9 inhibitors yielded rather frustrating outcomes due to limited sustainable efficacy and significant toxicity19,24. Moreover, depletion of CDK9 expression displayed differential gene expression patterns compared to pharmacological inhibition of CDK9 kinase25,26, obscuring precise application of CDK9-targeting strategy. Recent studies have also highlighted the diversity of CDK9 function involved in the formation of new complexes and epigenetic alterations27,28. For instance, noncanonical cytoplasmic ___location of CDK9 was demonstrated to control messenger RNA (mRNA) translation through forming a novel mTOR-like complex, indicating a more profound role of CDK9 in carcinogenesis28. Herein, we identified CDK9 as a CTCL vulnerability and designed in vivo effective PROTAC compounds to kill CTCL. More importantly, our study unveiled a role of CDK9 in modulating RARα protein degradation through recruiting HUWE1 (HECT, UBA and WWE Domain containing E3 ubiquitin protein ligase 1) E3 ligase. Depletion of CDK9 thus leads to accumulation of RARα which endows CTCL cells with higher sensitivity to ATRA. Co-administration of CDK9 degrader with ATRA which activates RARα transcriptional activity synergistically suppressed tumor growth, providing a potential therapeutic opportunity for CTCL.
Results
Identification of a vulnerability to CDK9 inhibition of CTCL
In order to identify therapeutic target of CTCL, we performed an unbiased compound screening using an inhouse inhibitor library (L1200) in two CTCL cell lines, Hut78 and HH. Among all the inhibitors, CDK family is the top enriched category (Fig. 1a and Supplementary Data 1). Consistent with recent reports, phosphoinositide 3-kinase (PI3K) inhibitors were also enriched in our screening as the second most effective compounds in killing CTCL (Fig. 1a, b)29,30. These data suggested the dependency of CTCL cells to activity of CDKs. Further screening with a CDK inhibitors (CDKi) library including 110 compounds revealed that both CTCL cells displayed maximum sensitivity to inhibitors of transcriptional CDK subtype, particularly CDK9 and CDK7 (Fig. 1c). The top five CDK9 inhibitors displayed effective killing of CTCL cells with an IC50 between 20-1,200 nM (Fig. 1d). Furthermore, treatment of xenograft models with Flavopiridol (Fig. 1e, f) or SNS-032 (Fig. 1g, h) induced regression of HH tumors in vivo. Together, these data demonstrated inhibiting CDK9 kinase activity was effective in killing CTCL. To further verify the role of CDK9 in CTCL, we depleted CDK9 expression by stably infecting two shRNA targeting CDK9 in Hut78 and HH cell lines. We observed significant delay of tumor growth both in vitro (Fig. 1i–l) and in vivo (Fig. 1m, n), demonstrating a critical role of CDK9 in supporting CTCL growth.

a HH and Hut78 cells were treated with compounds (2 µM) from inhibitor library (L1200) and cell viability was evaluated using CCK8 assay after treatment for 48 hours. CDK Cyclin-dependent kinase, JAK/STAT Janus kinase-signal transducer and activator of transcription, PI3K Phosphoinositide 3-kinases, mTOR mammalian target of rapamycin complex. b The most effective kinase inhibitors (cell viability < 25%) identified from compound screening for HH and Hut78 cells. c Numbers of effective (red, cell viability < 25%) and ineffective (gray, cell viability\(\geq\)25%) CDK inhibitors from each different CDK subfamilies were shown. d IC50 curve of five representative CDK9 inhibitors, AT7519, Dinaciclib, Flavopiridol, SNS-032 and P276-00 in HH and Hut78 cells. Cell viability was counted by trypan blue staining. Results represent biologically independent experiments of n = 3. e–h Nude mice were subcutaneously injected with HH cells and randomly divided into Vehicle, Flavopiridol and SNS-032 groups (n = 8). Tumor volumes were measured at different time points (e, g). At 17 days after subcutaneous injection, tumors were harvested and weighed (f, h). i Western blot analysis of indicated proteins from Hut78 cells infected with lentivirus encompassing shNC, shCDK9-1 or shCDK9-2. j Growth curve of Hut78 cells upon CDK9 depletion. Cell number was counted by trypan blue. Experiments were performed in triplicate and repeated twice with similar results. k Western blot analysis of indicated proteins from HH cells infected with lentivirus encompassing shNC, shCDK9-1, or shCDK9-2. l Growth curve of HH cells upon CDK9 depletion. Cell number was counted by trypan blue. Experiments were performed in triplicate and repeated twice with similar results. m, n HH cells were infected with shNC, shCDK9-1, or shCDK9-2 lentiviruses, and subcutaneously injected into nude mice (n = 6). Tumor volumes were measured at different time points (m). At 12 days after subcutaneous injection, tumors were harvested and weighed (n). Data are presented as mean ± SEM. Unpaired, two-tailed Student’s t-test. Source data are provided as a Source Data file. i, k n = 3, independent experiments, a representative example is shown. The samples derive from the same experiment each but different gels for CDK9 and another for β-actin were processed in parallel. Band intensities were analyzed and compared using Image J. Relative densitometric values are provided below the blot images.
CDK9 is overexpressed in CTCL and defines a unique malignant T cell population
To validate the pivotal role of CDK9 in supporting CTCL growth, we measured the CDK9 expression by immunohistochemical (IHC) staining in a number of CTCL samples as well as healthy controls. Compared to healthy samples, elevated CDK9 expression was observed in CTCL (Fig. 2a and Supplementary Fig. 1). Immunofluorescent staining with CD4 and CDK9 antibodies showed overexpression of CDK9 in CD4+ T cells in CTCL tissue sections compared to normal samples (Fig. 2b). In consistency, Western blots results showed higher level of CDK9 in CTCL cell lines in comparison with CD4+ cells purified from peripheral blood mononuclear cells (PBMC) of healthy donors (Fig. 2c). Next, we analyzed public datasets of CTCL and found that compared to normal skin, CDK9 expression is remarkably upregulated in CTCL skin tumors in GSE11311331. Moreover, in patients developed aggressive large-cell transformation (LCT), level of CDK9 is significantly increased based on dataset of GSE168508 (Fig. 2d)32, indicating an adverse prognosis of CDK9.

a Immunohistochemistry (IHC) staining of CDK9 of tissues collected from healthy donors (Normal) and CTCL patients. b Immunofluorescent staining of tissue sections from Normal and CTCL patients using antibodies against CD4 and CDK9. DAPI (4’,6-diamidino-2-phenylindole) was used for nucleus staining. c Western blot analysis of samples from healthy donors (Normal) and CTCL cell lines using indicated antibodies. n = 3, independent experiments, a representative example is shown. Band intensities were analyzed and compared using Image J. Relative densitometric values are provided below the blot images. d Differential expression of CDK9 in patients with CTCL from the indicated datasets. NLCT non large-cell transformation. P values were calculated by one-tailed t test. e UMAP projection of CTCL dataset GSE165623 including cells from skin, blood and lymph node across a patient with advanced MF. f Dot plot of CTCL dataset GSE165623 showing the average expression levels and cell expression proportions of selected cell lineage markers. g Violin plots of T cell clusters in CTCL datasets GSE165623 and GSE171811 exhibiting the specific expression of CDK9 and proliferating markers. CDK9high cluster number was labeled in red. h UMAP projections of three cell cycle phases in CTCL datasets GSE165623, GSE171811, and GSE128531 respectively, T cell clusters were labeled using the bold black text and CDK9high cluster was indicated by the dotted oval (top panel). Percentage of cells in each phase of cell cycle was shown, with CDK9high cluster number labeled in red (bottom panel). i UMAP plots of T cells from GSE165623 and GSE171811 datasets, cells with the top expanded TCR α or β chain CDRs amino acid sequence per patient are colored in red (malignant), the polyclonal α or β TCRs are labeled in blue (polyclonal), and cells without detectable TCR are displayed in gray (remaining). Cell distribution was presented with contour graphs and summarized in the accompanying bar chart. The CDK9high cluster was labeled in red (right panel). j UMAP representation split by tissues of T cells from GSE165623. Malignant T cells from skin, blood, and lymph node are colored by dark cyan, red, and purple. CDK9high cluster number was labeled using the bold black text and cells were indicated by the dotted oval.
Numerous single-cell RNA sequencing (scRNA-seq) literatures have demonstrated prominent heterogeneity of CTCL cancer cells, both in lesional skin (patch/plaque/tumor) and circulation33,34,35,36,37,38,39,40. To decipher the CDK9 expression across different malignant T cell clusters, we analyzed three publicly available scRNA-seq datasets containing lesional skin or blood samples from patients with mycosis fungoides (MF) or Sézary syndrome (SS) as well as healthy controls35,37,41. We integrated samples within each dataset respectively and performed unsupervised clustering and projected cells in two dimensions using uniform manifold approximation and projection (UMAP). Major cell types based on the expression of established cell-specific marker, including T cells, B cells, macrophages, dendritic cells (DC), plasma cells, mast cells, megakaryocytes, hematopoietic stem and progenitor cells (HSPC), and skin-resident non-immune cells (fibroblasts, endothelial cells, melanocyte and sebocytes) were identified (Fig. 2e, f and Supplementary Fig. 2a, b). In all the three datasets, T cells represented the largest fraction of cells (Fig. 2e and Supplementary Fig. 2a, b). Strikingly, among all the T cell clusters, CDK9 was selectively overexpressed in one unique cluster in each dataset (cluster 11 of GSE165623, cluster 14 of GSE171811, and cluster 25 of GSE128531 respectively) (Fig. 2g and Supplementary Fig. 2c)35,37,41, which thereafter was named CDK9high T cell cluster. Importantly, these three CDK9high T cell clusters shared features of high expression of malignant-associated genes (TOX, KIR3DL2) and proliferating genes (TYMS, GINS2, MKI67, TOP2A and CDK1) (Fig. 2g and Supplementary Fig. 2c), which was reinforced by cell-cycle analysis showing elevated G2M and S-phase genes (Fig. 2h)42,43. By combining 5’ scRNA-seq with αβ T-cell receptor (TCR) sequencing, the CDK9high T cell clusters were authenticated as malignant T cells, which comprised around 30% of T-cell receptor clonality (38% in GSE165623 and 26% of GSE171811) (Fig. 2i). Intriguingly, the CDK9high T cell clusters expressed featured genes of effector T cells but not exhausted T cells (Supplementary Fig. 2d). With regards to ___location of T tumor cells, TCR sequencing revealed a comparable frequency of CDK9high T cell cluster in both skin and blood, indicating same origin of tumor cells (Fig. 2j and Supplementary Fig. 2e). Taken together, CDK9 labeled a unique actively proliferating effector T cell cluster in CTCL.
CDK9 knockdown and kinase inhibitors regulate differential sets of genes
To better understand the critical role of CDK9 in CTCL cells, we performed RNA-sequencing (RNA-Seq) to investigate the global gene expression profiles of Hut78 cells exposed to Flavopiridol or stably infected two shRNA targeting CDK9. Consistent with established role of CDK923,44,45, we observed enrichment of RNA Pol II and cell cycle pathways upon Flavopiridol treatment (Fig. 3a, Supplementary Fig. 3a, b and Supplementary Data 2). In parallel, immunoblotting showed that correlating with a decrease of Pol II phosphorylation, protein levels of known CDK9 downstream genes including apoptosis regulator myeloid cell leukemia 1 (MCL1) and the proto-oncogene MYC involved in cell growth and cell cycle progression was reduced (Supplementary Fig. 3c). Similarly, knockdown of CDK9 also induced enrichment of cell cycle pathway (Supplementary Fig. 3d). These data supported a canonical kinase-dependent mechanism of CDK9 in promoting CTCL growth and survival.

a RNA-seq analysis of Hut78 cells treated with Flavopiridol or knocked down CDK9 expression with shCDK9. The Venn diagram showed the genes differentially expressed in each group. b MOLT-4 and HH cells were treated with THAL-SNS-032 and western blots were conducted. c HH cells were treated with DMSO or candidate PROTACs. Cell viability was measured using the CCK8. d Chemical structure of 23 (GT-02897). e Cell viability of Hut78 cells treated with DMSO or 19, 23, 24, 25, 26, and 27. Viability was counted by trypan blue. P value was calculated by two-tailed Student’s t-test. f Western blot analysis of indicated proteins in Hut78 cells treated with DMSO or 23, 24, 25, and 26. g, Western blot analysis of CDK9 in Hut78 cells upon THAL-SNS-032 or 23 (GT-02897) treatment. h 293T cells were treated with DMSO or 23 (GT-02897). Viability was counted by trypan blue. i Hut78 were treated with 23 (GT-02897) in the presence or absence of MG132 followed by Western blot analysis. j Hut78 cells were pre-exposed to SNS-032 or PHA-767491 followed by treatment with 23 (GT-02897). Then CDK9 protein was analyzed by Western blot. k Quantitative proteomic analysis and volcano plot of proteins that were differently regulated in Hut78 cells upon 23 (GT-02897) treatment. CDK9 was labeled. P value was derived by Wilcoxon rank-sum test. l Hut78 cells were treated with 23 (GT-02897) and Q-PCR analysis was performed for CDK9 (55 kDa) and CDK9 (42 kDa). P value was calculated by two-tailed Student’s t-test. m The regulated proteins in (k) were annotated and analyzed for the most significantly enriched pathways using Metascape. n–r Western blot analysis of indicated proteins in Hut78 cells upon 23 (GT-02897) treatment. s Hut78 cells were treated with different doses of 23 (GT-02897) for 10 h and DC50 was calculated. Results in (e, h, s) represent biologically independent experiments of n = 3. Results in (l), n = 4 for CDK9(55 kDa) and n = 3 for CDK9(42 kDa). Data are presented as mean ± SEM. Source data are provided as a Source Data file. b, f, g, i, j, n–r n = 3, independent experiments, a representative example is shown. The samples in (f) derive from the same experiment but different gels for CDK9, β-actin, another for CDK7, another for CDK2 and another for CDK6 were processed in parallel. Band intensities were analyzed and compared using ImageJ. Relative densitometric values are provided below the blot images.
Interestingly, in addition to pathways regulated by known kinase activity, we observed 2043 and 1200 significantly regulated genes (P < 0.05) in Flavopiridol-treated and CDK9-deleted Hut78 cells respectively, with only 295 genes being co-modulated in both groups (Fig. 3a). This result, in agreement with previous reports showing differential effects of dominant negative CDK9 (kinase dead mutant of CDK9) with Flavopiridol on gene expression and recent observation of CDK9 forming novel cytoplasmic complex beyond nuclear Pol II25,26,28, suggested a regulatory mechanism of CDK9 beyond phosphorylating Pol II as a kinase and prompted us to explore the feasibility of the proteolysis targeting chimeras (PROTACs) strategy, known for its application in chemically knocking down a protein of interest (POI) at the protein level.
CDK9-targeting PROTACs induce on-target degradation of CDK9 and growth arrest of CTCL cells in vitro and in vivo
We first took use of published CDK9-targeting PROTAC THAL-SNS-032, a potent degrader for CDK9 in leukemia cells. We found that while it induced significant loss of CDK9 in acute lymphoblastic leukemia cell line MOLT-4 as reported (Fig. 3b)46, it did not disturb CDK9 protein (Fig. 3b), nor did it display effective killing in CTCL cells (Supplementary Fig. 4a). We then designed a series of bivalent molecules by bridging SNS-032 and S-substituted thalidomide, a novel cereblon (CRBN) ligand developed by Yang group with alkyl or PEG (polyethylene glycol) linkers of different lengths47,48. The killing effects of these twenty-two CDK9 degraders on CTCL cells were tested in parallel with the parent compound SNS-032. We found compound 9 and 19 displayed significant killing effects below 500 nM (Fig. 3c) with the IC50 338.9 nM and 403.8 nM respectively (Supplementary Fig. 4b, c). Importantly, compound 19 presented high selectivity towards CDK9 as confirmed by immunoblotting showing that other CDK members except CDK7 were not affected (Supplementary Fig. 4d).
To further optimize the selectivity of 19, we designed additionally a series of compounds by modifying the linker, and obtained five compounds (23, 24, 25, 26, 27) displaying significantly higher killing ability than 19 (Fig. 3d, e), with IC50 between 50-165 nM (Supplementary Fig. 4e). Immunoblots confirmed the strong degradation of CDK9 by 23, 24, 25, 26 without interfering protein levels of CDK2, CDK6 and CDK7 (Fig. 3f). Next, IC50 of these compounds were tested in Hut78 and HH cell lines and 23 displayed a lower IC50 in both cell lines (49.73 nM in Hut78 and 66.23 nM in HH, Supplementary Fig. 4e, f). We then treated Hut78 cells with 23 (GT-02897) or THAL-SNS-032 at different concentrations and observed a significant loss of CDK9 after 23 (GT-02897) treatment rather than THAL-SNS-032 (Fig. 3g). In addition, 23 (GT-02897) displayed minimal killing effects on 293T cell (Fig. 3h), indicating low toxicity to normal cells. Therefore, 23 (GT-02897) was chosen for further experiments.
Next, we validated that the reduction of CDK9 by GT-02897 was through the expected degradation mechanism of the ubiquitin-proteasome system (UPS). As shown in Fig. 3i, the use of MG132 reverted the expression of CDK9 protein induced by GT-02897. Then, the on-target effect of GT-02897 was validated in vitro. Firstly, pre-treatment of Hut78 cells with the warhead SNS-032 or another CDK9 kinase inhibitor PHA-767491(IC50 is 925.8 nM in Hut78 cells) (Supplementary Fig. 4g) significantly dampened the degradation of CDK9 by GT-02897 (Fig. 3j)49. Secondly, to clarify the CDK9-engagment of GT-02897 more broadly, we performed unbiased quantitative mass-spectrometry (MS)-based proteomics analysis on Hut78 cells 6 h after GT-02897 treatment, a time point at which any changes in protein abundance would be primarily a result of compound-induced degradation. This unbiased experiment identified CDK9 as one of the most significantly downregulated proteins out of 5,689 proteins based on quantification of two or more peptides (Fig. 3k and Supplementary Data 3). Q-PCR analysis confirmed that the mRNA expression of CDK9 (42 kDa) and CDK9 (55 kDa) isoforms were not affected upon GT-02897 treatment (Fig. 3l)50. GO analysis of regulated proteins identified enrichment of CDK9-downstream pathways including RNA Pol II and cell cycle (Fig. 3m). These data further confirmed the on-target effect and selectivity of GT-02897.
Then, we investigated the dose and time effect of GT-02897 in CTCL cells. Dose dependent degradation of CDK9 by GT-02897 was observed in both low (0–100 nM) and high (0–1,000 nM) concentration ranges, and more strikingly, at concentration as low as 0.5 nM after 24 h of treatment (Fig. 3n–p). Time-dependent effect was also verified under treatment at 100 nM and 10 nM of GT-02897 (Fig. 3q, r). Similar degradation effect of GT-02897 was validated in another T lymphoma cell line EL4 (Supplementary Fig. 4h, i). In addition, the degradation efficiency of GT-02897 was measured and it displayed DC50 of 2.845 nM for CDK9 in Hut78 cells (Fig. 3s)51.
Furthermore, we evaluated the anticancer effects of GT-02897 in xenograft models. NSG mice harboring Hut78 xenografts were subjected to GT-02897 every other day by intraperitoneal (Fig. 4a–c) or subcutaneous injection (Fig. 4d, e) for 14 days. Both routes of administration of GT-02897 could significantly suppress Hut78 cell growth in vivo. Importantly, in vivo degradation of CDK9 by GT-02897 was further confirmed by IHC assays conducted on isolated tumor tissues (Fig. 4c). Meanwhile, the acute toxicities of GT-02897, through intraperitoneal or subcutaneous injection, were both measured in Sprague-Dawley rats. We observed no significant weight loss, histological damages in organs tested or increase of plasma creatinine in both treatment groups, indicating a minimal toxicity at the dose treating CTCL murine model (Supplementary Fig. 5a–c and Supplementary Fig. 6). Moreover, depletion of CDK9 expression by shRNA in Hut78 cells significantly impaired the growth inhibition effect of GT-02897, further verifying the on-target effect of this CDK9-targeting degrader in vivo (Fig. 4f–h).

a–c NSG mice were subcutaneously injected with Hut78 cells (8 × 106 cells for each mouse) and randomly divided into two groups receiving intraperitoneal injection of Vehicle or 23 (GT-02897) (n = 4). Tumor volumes were measured at different time points (a). At 14 days after subcutaneous injection, tumors were harvested and weighed (b), followed by IHC staining of human CDK9 (c). P value was calculated by two-tailed Student’s t-test. d, e NSG mice were subcutaneously injected with Hut78 cells (8 × 106 cells for each mouse) and randomly divided into two groups receiving subcutaneously injection of Vehicle or 23 (GT-02897) (n = 5). Tumor volumes were measured at different time points (d) and survival curves were shown (e). P value of tumor volumes was calculated by two-tailed Student’s t-test, P value of survival curve was calculated by log-rank test. f–h NSG mice were subcutaneously injected with Hut78 cells introduced with shNC, shCDK9-1 or shCDK9-2 (8 × 106 cells for each mouse) and treated with 23 (GT-02897) (n = 6). Tumor volumes were measured at different time points (f). At 12 days after subcutaneous injection, tumors were harvested and weighed (g, h). P value was calculated by two-tailed Student’s t-test. Data are presented as mean ± SEM. Source data are provided as a Source Data file.
CDK9 physically interacts with RARα and suppresses RARα protein expression
Notably, proteomic data revealed that RARα protein was significantly upregulated upon CDK9 degradation (Fig. 5a and Supplementary Data 4), which was validated by western blot (Fig. 5b), suggesting that CDK9 suppressed RARα expression. As ATRA has been used to treat CTCL for over 30 years, which often functions with the assistance of its receptor RARα, the accumulation of RARα protein in CDK9-null cells may have great significancy for CTCL treatment. These encouraged us to validate the association between CDK9 and RARα and explore the possible regulatory mechanism.

a Quantitative proteomic analysis and volcano plot of Hut78 cell proteins upon 23 (GT-02897) treatment, with P values derived from Wilcoxon rank-sum test. b Western blotting of indicated proteins in Hut78 under 23 (GT-02897) treatment. Western blot analysis of indicated proteins in HH and Hut78 (c) or 293T (d) cells after CDK9 depletion via lentiviruses. e A doxycycline (Dox)-inducible knockdown system was applied in HH and Hut78 cells, followed by Western blotting post Dox treatment. f Volcano plot revealing differentially expressed genes in Hut78 post CDK9 depletion, with P values derived by Wilcoxon rank-sum test. g Q-PCR assessment of CDK9 and RARA levels in Hut78 cells after CDK9 depletion. h Western blot analysis of proteins in Hut78 after CDK9 depletion, with or without MG132 treatment. i 293T cells were co-transfected with RARα and CDK9 constructs, followed by co-immunoprecipitation (Co-IP) and Western blot analysis. j GST-tagged CDK9 was incubated with HA-RARα from the WCL of 293T cells transfected with 3 × HA-tagged RARα, followed by GST pulldown and Western blot analysis. WCL: whole cell lysate. k Schematic diagram of full-length CDK9 protein (top). GST pulldown assay was performed using GST-tagged CDK9 and WCL of 293T cells transfected with 3 × HA-tagged RARα. l Lentiviral infection of Hut78 with shNC or shCDK9-1, followed by transfection of EV, 3 × Flag-tagged WT, T186A, and S347A CDK9, and Western blotting of indicated proteins. m Western blotting of indicated proteins in Hut78 post CDK9 depletion or Flavopiridol treatment. n Q-PCR analysis of RARA in Hut78 cells treated with Flavopiridol. o Schematic diagram of full-length RARα protein (top). Co-transfection of 3×Flag-tagged CDK9 with full-length or mutant RARα constructs in 293T, followed by Co-IP and immunoblotting (bottom). Results in (g, n) represent biologically independent experiments of n = 3, with P values calculated by two-tailed Student’s t-test. Data are presented as mean ± SEM, with source data provided as a Source Data file. b–e, h–m, o n = 3, independent experiments, a representative example is shown. The samples derive from the same experiment each but different gels for CDK9, β-actin and another for RARα (b, d, e, h), for RARα, β-actin and another for CDK9 (c), for HA-RARα, β-actin and another for Flag-CDK9 (i), for RARα, β-actin and another for Flag-CDK9 (l) were processed in parallel. Band intensities in (b–e, h, l, m) were analyzed and compared using Image J. Relative densitometric values are provided below the blot images.
To test the effect of CDK9 on RARα, we used distinct shRNA to disrupt CDK9 expression in multiple cell lines including HH, Hut78 and 293T cells. Western blot revealed that depletion of CDK9 caused substantial increase of RARα protein (Fig. 5c, d). Consistently, doxycycline-induced knockdown of CDK9 time-dependently induced accumulation of RARα in HH and Hut78 cell lines (Fig. 5e). The mRNA level of RARA remained stable upon CDK9 depletion in RNA-seq data (Fig. 5f and Supplementary Data 5) and was confirmed by Q-PCR (Fig. 5g), indicating a post-transcriptional regulation of RARα by CDK9. As proteasome-dependent degradation of RARα has been well documented as a key manner to maintain RARα protein homeostasis12,52,53,54, we treated cells with MG132, the proteasome inhibitor and observed abolishment of the elevation of RARα upon CDK9 depletion (Fig. 5h)12,54. Taken together, these data indicated a modulation of RARα proteasomal degradation by CDK9.
Next, we explored the potential interaction between CDK9 and RARα. Co-IP assay revealed a physical interaction between RARα with CDK9 (42 kDa) and CDK9 (55 kDa) isoforms (Fig. 5i), which was further confirmed by additional GST pull-down assay (Fig. 5j). Of note, the binding between RARα and CDK9 (42 kDa) is more potent than CDK9 (55 kDa), suggesting that the first exon might decrease the interaction. Construction of a series of CDK9 mutants, devoid of different kinase activity associated structural domains44, revealed that the kinase activity of CDK9 was dispensable for the interaction between CDK9 and RARα (Fig. 5k). Additional kinase dead (KD) mutagenesis (T186A, S347E, S347A) of CDK9 further suggested that kinase activity did not affect this interaction between CDK9 and RARα (Fig. 5k, Supplementary Fig. 7a)44,45,55. More importantly, re-expression of KD mutants in CDK9-depleted cells displayed similar effect on RARα with wild type (WT) CDK9 (Fig. 5l). In consistency, Flavopiridol treatment of Hut78 cells did not yield upregulation of RARα as shCDK9 did (Fig. 5m), nor did it affect the mRNA level of RARA (Fig. 5n). Taken together, we proposed that regulation of RARα degradation by CDK9 is through protein-protein interaction but not kinase activity.
Moreover, we mapped the CDK9-interacting domains of RARα. Deletion of AF1 (activation function 1) ___domain or ABD (ATRA binding ___domain) almost abolished the binding of RARα to CDK9, indicating that AF1 and ABD are essential for the interaction between RARα and CDK9 (Fig. 5o)56,57.
CDK9 induces proteasomal degradation of RARα through recruiting HUWE1
To gain further insight into the modulation of RARα by CDK9, we applied a proteomic screening using liquid chromatography tandem mass spectrometry (LC-MS/MS) with respect to identify the CDK9-interactors that may directly degrade RARα. Given our previous data suggested the kinase independent manner of the interaction between CDK9 and RARα, we took use of the 293T cells rescuing expressed Flag-tagged WT or KD T186A or S347E mutant CDK9 respectively to recognize the binders overlapped by WT and mutant CDK9. We observed that HUWE1 is the only identified E3 ligase that could be effectively immunoprecipitated by both WT and two CDK9 KD mutants but not IgG (Fig. 6a), suggesting that HUWE1 might be the E3 ligase recruited by CDK9 to degrade RARα58,59,60. Next, GST pull-down assay was conducted using purified CDK9 and the interaction between HUWE1, CDK9, and RARα was confirmed (Fig. 6b).

a Hut78 cells were transfected with 3 × Flag-tagged WT, T186A, or S347E CDK9, followed by Co-IP with anti-Flag antibody. Mouse IgG was used as negative control. Samples were then analyzed via LC-MS/MS and E3 ligases interacting with 3 × Flag-tagged WT, T186A, or S347E CDK9 were listed. b Bacterially expressed GST-tagged CDK9 was incubated with Hut78 WCL, followed by GST pulldown and Western blot analysis. WCL: whole cell lysate. c–e Western blot analysis of indicated proteins in Hut78 (c) and 293T (d) cells with HUWE1 depletion, as well as in wild-type (WT) and HUWE1 knockout (KO) MEF cells (e). f Hut78 cells overexpressing RARα were infected with shNC or shHUWE1 lentiviruses. After 6 h of treatment with MG132 (10 µM) or DMSO, Co-IP with anti-RARα or mouse IgG was conducted, followed by immunoblotting. g In 293T cells transfected with 3 × HA-tagged RARα, Myc-tagged ubiquitin, and HUWE1, Co-IP and immunoblotting were conducted after MG132 (10 µM) or DMSO treatment. h Schematic of RARα ubiquitination K sites (left) and protein structure of RARα ligand binding ___domain (LBD) (in white) bound to ATRA (in blue) (PDB ID: 3A9E) (right). i Tandem mass spectrum of a RARα-derived peptide confirmed ubiquitin conjugation at residue K360. j Myc-tagged ubiquitin was co-transfected with wild-type or mutant RARα (HA-RARαWT, HA-RARαK244R, and HA-RARαK360R) into 293T cells with or without infection of HUWE1. Following 6 h of MG132 (10 µM) treatment, Co-IP and immunoblotting were performed. k, Myc-tagged ubiquitin was co-transfected with wild-type or mutant RARα with deletion of the Hinge ___domain (HA-ΔH-RARαWT, HA-ΔH-RARαK244R, and HA-ΔH-RARαK360R) into 293T cells with or without infection of HUWE1. Following 6 h of MG132 (10 µM) treatment, Co-IP and immunoblotting were performed. l Western blot analysis of indicated proteins in wild-type (WT) and HUWE1 knockout (KO) MEF cells with or without infection of 3×Flag-tagged CDK9. b–g and j–l n = 3, independent experiments, a representative example is shown. The samples derive from the same experiment each but different gels for RARα, β-actin, another for HUWE1 and another for GST-CDK9, GST (b), for RARα, β-actin and another for HUWE1 (c–e), for RARα, β-actin and another for Ub (f), for HA-RARα, β-actin, another for HUWE1 and another for Myc (g, j, k), for RARα, β-actin, another for Flag-CDK9 and another for HUWE1 (l) were processed in parallel. Band intensities in (c–e, l) were analyzed and compared using Image J. Relative densitometric values are provided below the blot images.
To validate whether HUWE1 is the E3 ligase for RARα, we knocked down HUWE1 in Hut78 and 293T cells and observed significant increase of RARα protein (Fig. 6c, d). In addition, RARα protein was significantly elevated in HUWE1 knockout (KO) Mouse Embryonic Fibroblast (MEF) cells compared to WT cells, in which MEF provide insights into how HUWE1-RARα interaction functioning in a more differentiated and tissue-specific environment(Fig. 6e)61. Endogenous ubiquitination of RARα, which could be detected by anti-ubiquitin antibody, was significantly attenuated upon depletion of HUWE1 (Fig. 6f). On the contrary, ectopic expression of HUWE1 increased the ubiquitination of RARα (Fig. 6g). Taken together, these results demonstrated that HUWE1 degraded RARα as an E3 ligase.
To determine which lysine site of RARα were involved in the ubiquitination regulation by HUWE1, we constructed a series of lysine to arginine mutations of RARα (Fig. 6h) on plasmids expressing WT RARα or ΔH-RARα which lacks hinge ___domain but maintains interaction with CDK9 (Fig. 5o). We found that HUWE1 primarily ubiquitinated the K360 site of RARα (Fig. 6i–k). Taken together, HUWE1 interacted with RARα and ubiquitinated RARα at the K360 site. Importantly, overexpression of CDK9 did not lead to suppression of RARα in HUWE1-KO MEF cells, further demonstrating an essential role of HUWE1 in mediating CDK9-induced degradation of RARα (Fig. 6l).
In addition, we carried out quantitative phosphoproteomics profiling in Hut78 cell overexpressing CDK9 or treated the cells with Flavopiridol. Comparison of quantified phosphopeptides would enable identification of those proteins whose phosphorylation are highly associated with CDK9 and could possibly be kinase substrates of CDK9. As expected, phosphorylation of a significant portion of proteins (222 proteins) was upregulated in CDK9-overexpressing group and inhibited upon Flavopiridol exposure (Supplementary Data 6). GO analysis revealed enrichment of known CDK9 activity associated pathways including RNA metabolism, 7SK snRNA binding and protein kinase binding (Supplementary Fig. 7b). However, no phosphopeptides from HUWE1 or RARα were identified (Supplementary Data 6), indicating the interaction between CDK9 and HUWE1 or RARα is not directly affected by the kinase activity.
RARα accumulation sensitizes CTCL cells to ATRA which induces CTCL differentiation to Th1 phenotype and regulatory T phenotype
While retinoids analog including ATRA have been used to treat CTCLs for decades, their mechanism of action and the role of RA-RARα signaling remain poorly understood62. Thus, we explored the effect of RARα on CTCL cells in the absence and presence of retinoid respectively. We constructed a RARα overexpressed system and a doxycycline-inducible knockdown of RARα system in Hut78 cells and observed little change in proliferation upon RARα overexpression or doxycycline treatment (Fig. 7a–d). Cell growth was also nearly unchanged upon depletion of RARα in shCDK9 Hut78 cells when compared with shCDK9 cells (Fig. 7e, f). These data demonstrated that RARα had almost no influence on CTCL cell growth in the absence of its ligand.

a Western blotting in Hut78 cells upon HA-RARα overexpression. b Growth curve of Hut78 cells infected with EV or HA-RARα lentivirus. Cell number was counted by trypan blue. c A doxycycline (Dox)-inducible knockdown system (shNC or shRARα) was introduced into Hut78 cells via lentiviruses, followed by Dox administration and Western blot analysis. d Growth curves of Hut78 cells with Dox-inducible shNC or shRARα post-Dox treatment, with cell counts via trypan blue. e Western blot analysis of CDK9 and RARα in CDK9-depleted Hut78 cells with or without RARα depletion. f Growth curve of control and CDK9-depleted Hut78 cells infected with or without shRARα lentivirus. Cell number was counted by trypan blue. g Hut78 cells were treated with ATRA or DMSO. Viability and cell number were measured by trypan blue. h RNA-seq analysis of HH and Hut78 cells upon ATRA treatment, with a Venn diagram showing common differentially expressed genes (left), and enriched pathways analyzed via Metascape (right). i Heatmap of regulated genes upon ATRA treatment in HH and Hut78 cells. j Schematic of CTCL cell differentiation into T cell lineages after ATRA treatment. k Q-PCR analysis of indicated genes in Hut78 cells after ATRA treatment. l Flow cytometry analysis of CD25, CD5 and CD7 expression on HH (top) and Hut78 (bottom) cells upon ATRA or DMSO treatment. m Growth curves of Hut78 cells infected with shNC or shCDK9 lentiviruses upon ATRA or DMSO treatment. Cell number was counted by trypan blue. n Hut78 cells were treated with ATRA and GT-02897 alone or combined and the cell number was counted by trypan blue. o–q NSG mice were subcutaneously injected with Hut78 cells and randomly divided into four groups (Vehicle, GT-02897, ATRA or ATRA + GT-02897, n = 10). Tumor volumes were measured over time (o).Tumors were harvested at 21 days, weighed (p), and analyzed by IHC for human CD25 (q). Results in (b, d, f, g, k, m, n) represent biologically independent experiments of n = 3, with P values calculated by two-tailed Student’s t-test. Data are presented as mean ± SEM. Source data are provided as a Source Data file. a, c, e n = 3, independent experiments, a representative example is shown. The samples in (e) derive from the same experiment but different gels for CDK9, β-actin, and another for RARα were processed in parallel. Band intensities in (a, c, e) were analyzed and compared using Image J. Relative densitometric values are provided below the blot images.
Next, we treated CTCL cells with ATRA and observed significant inhibition of cell growth without cell apoptosis (Fig. 7g). RNA-seq analysis revealed 456 regulated genes in both HH and Hut78 cells upon ATRA treatment (Fig. 7h and Supplementary Data 7). In parallel, we treated acute promyelocytic leukemia (APL) cell line NB4 with ATRA followed by RNA-seq analysis and observed shared pathways including cell adhesion molecules by three ATRA-treated cell lines, suggesting a common cellular behavior between T and myeloid-originated cancer in response to ATRA (Supplementary Fig. 8a). Notably, in addition to the well-known ATRA regulated pathways including proliferation or cell cycle, Th17, Th1 and Th2 cell differentiation pathways were significantly enriched (Fig. 7h), suggesting that ATRA may induce cell differentiation of CTCL cells63,64. Previous investigations showed that CTCL cells may display a regulatory T (Treg) phenotype expressing FOXP3 and secreting interleukin-10 (IL-10)65, while other studies showed that as the CTCL disease progressed, the secretion of the Th1 polarizing cytokines (such as IFN-γ) was decreased, accompanied by an increased level of the Th2 associated transcription factors (such as GATA-3) and cytokines (such as interleukin-4 and interleukin-13)66,67,68. We then attempted to clarify the T cell phenotype in response to ATRA treatment. Interestingly, RNA-seq data showed that genes associated with the Th2-cell subset such as GATA-3 and STAT6 were downregulated while those associated with the Th1-cell subset such as TBX21 and STAT4 were upregulated upon ATRA treatment, indicating a shift from Th2 to Th1-cell phenotype (Fig. 7i, j). These findings were confirmed by the followed Q-PCR analysis (Fig. 7k). Of note, IL-2 receptor gene α, β, and γ were also upregulated (Fig. 7i, k). Flow cytometry analysis demonstrated the absence of CD25 (IL-2 receptor gene α) on CTCL cell lines but emerging expression of CD25 on both HH and Hut78 cells, with CD5 and CD7 level unchanged upon ATRA treatment (Fig. 7l and Supplementary Fig. 9a), demonstrating a differentiation to Treg phenotype (Fig. 7j). Expression of Foxp3, however, was not changed upon ATRA treatment (Supplementary Fig. 8b and Supplementary Fig. 9b). Collectively, these results showed that ATRA inhibited CTCL by inducing cell differentiation and growth arrest.
Given that shCDK9 could accumulate RARα, we speculated that shCDK9 may enhance the sensitivity of CTCL cells to ATRA. Indeed, treatment of ATRA of shCDK9-infected Hut78 cells induced more pronounced inhibition of cell growth (Fig. 7m), strongly suggesting a synergistic therapy of co-targeting CDK9 and RARα in CTCL.
PROTAC GT-02897 displays synergistic effect with ATRA
As RARα accumulation sensitizes CTCL cells to ATRA, we proposed a synergistic therapy combing GT-02897 and low dose ATRA in the treatment of CTCL. In vitro co-administration of GT-02897 with ATRA induced a more significant inhibition of CTCL cells (Fig. 7n). The synergy score and most synergetic area were calculated using the ZIP method. GT-02897 showed a synergy score of 1.942 with ATRA (Supplementary Fig. 10)69. This effect was further validated in xenograft mice model and we observed a more significant inhibition of CTCL tumors in vivo induced by co-administration of GT-02897 with ATRA (Fig. 7o, p), demonstrating a promising translational synergistic therapy potential. Notably, in vivo upregulation of CD25 expression upon ATRA treatment was further confirmed by IHC assays conducted on isolated tumor tissues (Fig. 7q). Taken together, these data further validated the modulation of CDK9 towards RARα and proposed a rational for combined administration of GT-02897 with ATRA.
Discussion
There is urgent clinical need to develop targeted therapy for CTCL. However, due to a lack of understanding of the underlying mechanism and heterogeneity of the disease, targeted treatment strategies have not yet been developed3. In this study, we identified CDK9 as a potent driver of CTCL cells that promotes cell growth. We also unveiled the kinase-independent modulation of CDK9 towards RARα. By revealing this CDK9-RARα regulatory relationship, we have also addressed the long-standing question regarding the mechanism of ATRA in the context of CTCL. These findings expand the roles of both CDK9 and RARα in cellular signaling and cancer development.
CDK9 has been found to be overexpressed in various types of cancer including leukemia and lymphoma, breast, lung and prostate cancer in which CDK9 regulates transcription of genes involved in cell survival and drug resistance, leading to uncontrolled cell growth and progression of cancer15,44,70. Inhibition of CDK9 kinase activity holds promise as a potential therapeutic strategy for cancer treatment13,70. However, several CDK9 inhibitors under clinical investigations in leukemias yielded rather frustrating outcomes showing limited clinical activity or low tolerability71,72,which is probably due to low selectivity between CDK members or more importantly, unrecognized functions of CDK9. It is well established that CDK9 in complex with cyclin T is recruited to promoters where it stimulates transcriptional elongation by phosphorylating the CTD of RNA Pol II and negative elongation factors. However, disputes remain concerning whether CDK9 is required for transcription of global genes or alternatively restricted subsets of genes. On the other hand, it has been shown that in contrast to Flavopiridol, inhibition of CDK9 activity with dominant negative CDK9 leads to a distinctive pattern of changes in gene expression, strongly suggested kinase-independent role of CDK9. Indeed, the expression of many short-lived transcripts downregulated by Flavopiridol is not modulated by dnCDK926. In addition, multiple Pol II independent roles of CDK9 elicited outside nuclear which also play important role in cancer progress28. In this study, we report that CDK9 controls RARα protein degradation in a kinase-independent manner by recruiting HUWE1 E3 ligase. These data, along with the reports demonstrating novel plasma complex that CDK9 organized20,28, strongly suggested that simple inhibition of kinase activity of CDK9 may not be sufficient to achieve therapeutic efficacy. PROTAC, a powerful strategy to degrade target proteins, has the advantage of completely abolishing the biological function of CDK9. Unexpectedly, previously reported THAL-SNS-032 could not initiate significant degradation of CDK9, nor could it induce death of CTCL cells, which probably the results of different cell context. Thus we took efforts to design a series of SNS-032 based PROTACs by modifying CRBN ligand and linkers and obtained a powerful degrader GT-02897 which displayed efficient on-target degradation and cell killing. To optimize the selectivity of the degrader with more specified compounds, e.g.,PHA-767491, further investigation is warranted. We provided here a molecule that targets a therapeutic vulnerability that might be exploitable in the therapy for CTCL.
Although retinoids have been used in CTCL treatment for decades, the modes of action of RA in complex with its receptor RARα is largely unknown. In the well-known APL models, ATRA induced granulocyte differentiation via activating RARα thus initiating the transcription of a series of lineage specific genes. In the absence of its ligand, RARα rather plays an oncogenic role by blocking differentiation and leading to immortalization of myeloid progenitors73. In the context of normal T cell, ATRA-RARα controls CD4+ T cell differentiation by activating Th1 specific genes while repressing Th17 cell genes63,64. In CTCL cells, we observed little influence of RARα on tumor growth in the absence of ligand. However, in the presence of ATRA, accumulated RARα protein induced by shCDK9 sensitized tumor cells to ATRA, which clearly induced a more differentiated Th1-cell phenotype with elevated transcription of genes associated with Th1 polarizing and Treg-cell phenotype of CTCL cells by increasing the expression of CD25. Therefore, elevated level of RARα results from CDK9 depletion offers an opportunity of more effective therapeutic approaches for ATRA in CTCL.
In summary, our study reveals a role of CDK9 beyond phosphorylating RNA Pol II, which induces the degradation of RARα through CDK9-HUWE1-RARα axis. In addition, we propose a synergistic therapy combining CDK9-PROTAC with ATRA in the treatment of CTCL, providing an option of great translational perspective for clinical radical cure for CTCL.
Methods
Ethics
This research complies with all relevant ethical regulations approved by the committee for humane treatment of animals at Shanghai Jiao Tong University School of Medicine.
Reagents and antibodies
Antibodies against the following proteins were used: β-actin (MBL, PM053-7), 1:4000 diluted; RARα (Cell Signaling Technology, 62294; Santa Cruz, sc-515796), CDK6 (Cell Signaling Technology, 13331) and Myc (Cell Signaling Technology, 2276; Cell Signaling Technology, 2278), 1:500 diluted; CDK9 (Cell Signaling Technology, 2316), CDK7 (Proteintech, 27027-1-AP), CDK2 (Cell Signaling Technology, 2546), CDK4 (Cell Signaling Technology, 12790), Flag (Sigma-Aldric, F1804), HA (Cell Signaling Technology, 3724), HUWE1 (Abcam, ab70161), Ubiquitin (Proteintech, 10201-2-AP), RNA Pol II (Cell Signaling Technology, 14958), Phospho-RNA Pol II CTD Ser2 (Cell Signaling Technology, 13499), MYC (Cell Signaling Technology, 18583) and MCL1 (Cell Signaling Technology, 4572) 1:1000 diluted; CD25 (Invitrogen, 25-0259-42; Beyotime, AF7224), CD5 (Invitrogen, 12-0059-42), CD7 (Invitrogen, 11-0078-42), Foxp3 (Invitrogen, 12-5773-80) and CD4 (Abcam, ab133616), 1:100 diluted. Flavopiridol, SNS-032, THAL-SNS-032, Retinoic acid, MG-132, PHA-767491 and PR-619 were from MedChemExpress (HY-10006, HY-10008, HY-123937, HY-14649, HY-13259, HY-13461, HY-13814). Doxycycline, Puromycin, Hygromycin B, and Geneticin (G418 Sulfate) were from Selleckchem (S4163, S7417, S2908, and S3028).
Cell culture
Human CTCL cell lines (HH, Hut78, and MJ), mouse T cell lymphoma cell line EL4, human acute lymphoblastic leukemia cell line MOLT-4 and human embryonal kidney cell line HEK293T(293T) were purchased from ATCC. Mouse embryonic fibroblasts (MEF) were gifted by Professor Zhangjing61. HH, Hut78, MJ, EL4, and MOLT-4 cells were cultured in complete RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS, Sigma) and maintained in a humidified atmosphere of 5% CO2 at 37 °C. 293T and MEF cells were cultured in complete DMEM supplemented with 10% FBS (Sigma).
Statistics and reproducibility
The statistical analyses are described in the figure legends. The tests used included unpaired Student’s t-test, Pearson’s χ2 test, two-way analysis of variance (ANOVA), Mann-Whitney U-test and log-rank test, using Microsoft Excel and GraphPad Prism 9 (GraphPad Software). Differences were considered significant at P < 0.05. Three different biological replicates were tested. Each experiment was repeated independently at least three times unless indicated differently.
Compound library screening
The small molecular inhibitors library (Selleckchem L1200) and the CDK inhibitors library (MedChemExpress, HYCPK9807, HYCPK9808, HYCPK9809, HYCPK9810) were applied. Cells were seeded into 96-well plates at 1 × 105 cells/ml and inhibitors were added at 2 μM final concentration. Cell viability was measured 48 h post treatment using the CCK8 kit (CK04, Dojindo).
Patient samples
Primary samples were collected from CTCL patients or healthy donors at Department of Hematology of the Second Hospital of Dalian Medical University. All patients and healthy donors provided written consent to protocols that were approved by the Institutional Review Board and Medical Science Ethic Committee of Dalian Medical University in accordance with the Declaration of Helsinki, which enabled identified research use of biospecimens. Human Peripheral Blood Mononuclear Cells (PBMCs) were prepared from healthy donors’ blood using Ficoll Hypaque (1114547, Axis-Shield) density centrifugation. Primary T cells were isolated from the PBMCs using an EasySep Human CD4 Positive Selection Kit (17852, STEMCELL Technologies, Canada) according to the manufacturer’s instructions.
IC50 (half-maximal inhibitory concentration) assay
The IC50 was defined as the concentration of an inhibitor that reduced cell survival to 50% of the untreated control, with the highest viability (no inhibitor) defined as 100%, and the lowest viability defined as 0%. HH and Hut78 cells were seeded (1 × 105 cells/ml) in 6-well plates. Cell number and viability were determined after 48-h treatment with different compounds at indicated concentrations by trypan blue exclusion count and compared with untreated control. Mean ± SD of 3 biological replicates. Calculation of IC50 values was performed using GraphPad Prism version 9.3.1. (GraphPad Software, Inc.).
Cell proliferation assays
Cells were counted on day 0 by trypan blue exclusion and plated in 6-well plates at 1 × 105 cells/ml. Cell counts were then determined by trypan blue exclusion every day and normalized to value of day 0.
Western blotting
Protein extracts were equally loaded on SDS-polyacrylamide gel, transferred to nitrocellulose membrane (HATF00010, Merck Millipore), blocked by 2% (w/v) BSA, and immunoblotted with the indicated antibodies. Post incubation with horseradish peroxidase (HRP)-linked second antibody (Cell Signaling Technology, Beverly, MA) at room temperature for 1 h, projection was performed using Immobilon Western Chemiluminescent HRP substrate kit (WBKLS0500, Merck Millipore) according to the manufacturer’s instructions.
Co-Immunoprecipitation (Co-IP)
Cells were harvested and lysed with NP-40 lysis buffer (P0013F, Beyotime) containing 1 mM PMSF and 1× protease inhibitor cocktail (L-1009, BioTNT). After sonication, the supernatants (whole cell lysates) were collected by centrifugation at 20,000 × g for 10 min at 4 °C and then incubated with anti-Flag M2 Affinity Isolated Antibody (M8823, Sigma-Aldrich) or anti-HA Magnetic Agarose Beads (L-1009, BioTNT) for immunoprecipitation of Flag or HA-tagged proteins. The tubes were slightly rotated overnight at 4 °C, followed by separating the supernatants with a magnetic separator. For Co-IP of RARα, cells were harvested and lysed with NP-40 lysis buffer (P0013F, Beyotime) containing 1 mM PMSF, 1× protease inhibitor cocktail (L-1009, BioTNT) and 50 µM PR-619 (HY-13814, MedChemExpress), after pre-cleared with protein A/G-agarose beads (sc-2003, Santa Cruz, CA), supernatants were incubated with RARα antibody overnight and protein A/G-agarose beads for 4 h at 4 °C. All precipitates were washed three times with NP-40 lysis buffer and boiled in 1 × loading buffer before Western blot analysis.
GST pulldown assay
The GST, GST-CDK9, and CDK9 truncation mutant constructs were used for pulldown assays. GST and GST-CDK9 recombined plasmids were expressed in E. coli (Transetta DE3). At OD600 = 0.6, protein expression was induced with 0.5 mM IPTG (Solarbio) overnight at 18 °C. Bacteria were lysed and sonicated in lysis buffer containing 50 mM Tris-HCl (pH 7.4), 300 mM NaCl, 1% Triton X-100, PMSF, DTT, and lysozyme at 4 °C. After centrifugation at 12,000 × g for 15 min at 4 °C, clarified lysates were incubated with prepared glutathione-Sepharose (Merck) in GST binding buffer (PBS supplemented with 1% Triton X-100 and 1 mM PMSF) overnight at 4 °C. The GST and GST fusion proteins bound to beads were washed three times with binding buffer. Proteins of Hut78 or 293T cells were extracted using lysis buffer (50 mM Tris (pH7.4), 150 mM NaCl, 1% NP-40, and 1 mM PMSF). After centrifugation at 12,000 × g for 15 min, the supernatants were incubated with GST and GST-CDK9 bound to glutathione-Sepharose beads. After gentle shaking overnight at 4 °C, the beads were centrifuged at 500 × g for 5 min and washed three times with PBS to remove the unbound proteins. Beads were then boiled for 5 min at 100 °C in SDS loading buffer and centrifuged. The supernatant was subjected to western blot analysis.
Denaturing ubiquitination assay
293T cells were treated with 10 µM of the proteasome inhibitor MG132 for 6 h, harvested, and lysed with denature lysis buffer (denatured IP buffer 50 mM Tris-HCl, pH 6.8, 2% SDS) by boiling at 100 °C for 20 min to dissociate protein-protein interactions. After centrifugation at 20,000 × g for 10 min at 10 °C, 140 µl of supernatants was diluted by 1.26 ml NP-40 lysis buffer containing 1 mM PMSF and 1× protease inhibitor cocktail (L-1009, BioTNT) and subjected to immunoprecipitation with anti-HA Magnetic Agarose Beads (BioTNT), followed by Western blot analysis to visualize polyubiquitylated protein bands.
DC50 (half-maximal degradation concentration) assay
The DC50 was defined as the concentration of a degrader that induced the protein level of the targeted protein to 50% of the untreated control value74,75, with the highest protein level (no degrader) defined as 100%, and the lowest protein level defined as 0%. Hut78 cells were seeded (5 × 105 cells/ml) in 6 well plate and treated with 23 (GT-02897) for 10 h at indicated concentrations. The protein level of CDK9 was analyzed by Western blot and quantified by Image J 1.53a. Mean ± SD of 3 biological replicates. Calculation of DC50 values were performed using GraphPad Prism version 9.3.1. (GraphPad Software, Inc.).
Real-time quantitative reverse transcription-polymerase chain reaction (Q-PCR)
Total RNA was extracted from HH and Hut78 cells by TRIzol reagent (Invitrogen), and reverse transcription was performed by FastKing RT Kit (TIANGEN, KR116) following the manufacturer’s instructions.
The human ACTB housekeeping gene was used as an internal control (F: 5′-CATCCTCACCCTGAAGTACCC-3′, R: 5′-AGCCTGGATAGCAACGTACATG-3′). Real-time PCR was performed using SYBR Selected Master Mix (Thermo Fisher Scientific, 447308) on QuantStudio 5 instrument (Applied Biosystems) using a primer-specific standard curve with denaturation (95 °C for 10 min), amplification repeated 40 times (95 °C for 15 s, 60 °C for 1 min). For each sample, ddCt (crossing point) values were calculated as the Ct of the target gene minus the Ct of the ACTB gene.
Human CDK9 (42 kDa)-F: 5′-CCAGAAGCGGAAGGTGAA-3′,
Human CDK9 (42 kDa)-R: 5′-CCAGAAGAAGTCGTGGTTGAG-3′.
Human CDK9 (55 kDa)-F: 5′- GGGCCGCCGCTAGTAGTG-3′,
Human CDK9 (55 kDa)-R: 5′- CCTGGGACTTGTAGTTCCCG-3′.
Human RARA-F: 5′-AAGCCCGAGTGCTCTGAGA-3′,
Human RARA-R: 5′-TTCGTAGTGTATTTGCCCAGC-3′.
Human STAT4-F: 5′-TGTTGGCCCAATGGATTGAAA-3′,
Human STAT4-R: 5′-GGAAACACGACCTAACTGTTCAT-3′.
Human IL12RB1-F: 5′-AGCTGCGTATGGAGTGGGA-3′,
Human IL12RB1-R: 5′-GAGGCAGGACTCAGTATCATCA-3′.
Human LTA-F: 5′-CATCTACTTCGTCTACTCCCAGG-3′,
Human LTA-R: 5′-CCCCGTGGTACATCGAGTG-3′.
Human IFNG-F: 5′-TCGGTAACTGACTTGAATGTCCA-3′,
Human IFNG-R: 5′-TCGCTTCCCTGTTTTAGCTGC-3′.
Human GATA3-F: 5′-GCCCCTCATTAAGCCCAAG-3′,
Human GATA3-R: 5′-TTGTGGTGGTCTGACAGTTCG-3′.
Human CCR4-F: 5′-AGAAGGCATCAAGGCATTTGG-3′,
Human CCR4-R: 5′-ACACATCAGTCATGGACCTGAG-3′.
Human IL4-F: 5′-TGAAACGGCTCGACAGGAAC-3′,
Human IL4-R: 5′-TGAATGGGTTGACCAAGGGTG-3′.
Human IL2RA-F: 5′-GTGGGGACTGCTCACGTTC-3′,
Human IL2RA-R: 5′-CCCGCTTTTTATTCTGCGGAA-3′.
Human IL2RG-F: 5′-GTGCAGCCACTATCTATTCTCTG-3′,
Human IL2RG-R: 5′-GTGAAGTGTTAGGTTCTCTGGAG-3′.
Human TGFB1-F: 5′-CAATTCCTGGCGATACCTCAG-3′,
Human TGFB1-R: 5′-GCACAACTCCGGTGACATCAA-3′.
Cell line derived xenograft (CDX) models
HH cells (3 × 106 cells for each mouse) or Hut78 cells (8 × 106 cells for each mouse) were inoculated subcutaneously into 4- to 6-week-old female nude mice (Shanghai Laboratory of Animal Center, Chinese Academy of Sciences) or 4- to 6-week-old female NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice (The Jackson Laboratory) respectively and tumors were measured with caliper every other day. Tumor size was estimated using the following formula: volume (mm3) = (length × width2)/2. Mice were euthanized when individual tumor sizes reached 1000 mm3 for nude mice or 2000 mm3 for NSG mice or when mice exhibited severe weight loss. According to animal care and enforcement, the maximum allowable diameter for subcutaneous tumors in mice is 20 mm. This limit was not exceeded at any point. All animals used in this research were housed in a specific pathogen-free (SPF) environment with a 12 h dark/light cycle (lighting from 7:00 to 19:00), constant and appropriate room temperature (22–25 °C), and had free access to food and water. Animal care and experiments were performed in strict accordance with the “Guide for the Care and Use of Laboratory Animals” and the “Principles for the Utilization and Care of Vertebrate Animals” and were approved by the committee for humane treatment of animals at Shanghai Jiao Tong University School of Medicine. The study was compliant with all of the relevant ethical regulations regarding animal research.
In vivo GT-02897 treatment
The NSG mice were randomly divided into three groups, subcutaneously injected with Hut78 cells introduced with shNC, shCDK9-1 or shCDK9-2 (8 × 106 cells for each mouse). Afterwards each group was divided into two subgroups receiving intraperitoneal injection of Vehicle or GT-02897. Tumor volumes were measured at different time points. At 12 days after subcutaneous injection, tumors were harvested and weighed.
Hematoxylin-eosin (HE) and Immunohistochemistry (IHC)
For histology, samples were fixed in 4% paraformaldehyde overnight before standard processing for paraffin-embedded tissues. Then, 3-μm sections were stained with HE/IHC. The paraffin sections of human skin tissues and mice tumor tissues were dewaxed, antigen retrieval and blocked with 5% BSA as per standard protocols. Sections were stained using the polyclonal antibodies CDK9 (Cell Signaling Technology, 2316) or CD25 (AF7224, Beyotime).
Immunofluorescent (IF)
Immunofluorescent (IF) staining was performed to examine the expression of CDK9 and CD4 in human skin tissues. Briefly, slides were deparaffinized, rehydrated and subjected to antigen retrieval by heating the sample with citrate buffer (pH 6.0) in a microwave oven. After blocking, sections were incubated with CDK9 (Cell Signaling Technology, 2316) and CD4 (Abcam, ab133616) primary antibodies at 4 °C overnight. The slides were then incubated with fluorochrome-conjugated secondary antibodies for 1 h at room temperature. Finally, DAPI was applied for nuclear staining, and images were viewed using confocal fluorescence microscope (Leica).
Integrated analysis of public CTCL scRNA-seq datasets
Count matrices from three studies including GSE165623 (n = 3), GSE171811 (n = 14), and GSE128531 (n = 9) were downloaded from the Gene Expression Omnibus and re-analyzed respectively. Doublets or multiple cells, and low quality barcodes (including dead or dying cells) were initially removed if having less than 200 expressed genes or more than 40% mitochondrial transcripts. Genes that were detected in fewer than 60 cells were also filtered out. Seurat (v3.0.2) anchor-based integration using canonical correlational analysis and mutual nearest neighbors was performed to eliminate the biological and technical batch effects of the samples and scRNA-seq libraries76. The count data were normalized by a scale factor (10,000) followed by a natural-log transformation. The top 3000 highly variable genes (HVGs) which were detected using the “FindVariableGenes” function were subsequently used to identify the integration anchors based on the first 30 dimensions by employing the “IntegrateData” function. Principal component analysis (PCA) was performed using the “RunPCA” function to project the cells in a two-dimensional space. A graph-based Louvain clustering and dimensional reduction to form the Uniform Manifold Approximation and Projection (UMAP) plots used the top 30 calculated dimensions following a resolution of 0.6 for GSE165623, 0.4 for GSE171811 and 0.9 for GSE128531 respectively.
Cell type labels were assigned to clusters based on the inference from scHCL package (v0.1.1)77 and the expression of signature genes. Cell cycle state of T cells was evaluated by Seurat “CellCycleScoring” function based on a list of canonical markers delineating G1, S, and G2/M phases described in ref. 78. Clonotype annotation files were obtained from GSE165623 and GSE171811. Three types of T cells were assigned as follows: (1) malignant cells: cells with the highest CDR3 sequence frequency for TCR β, (2) polyclonal cells: all T cells that possess non-malignant clonotype based on the CDR3 sequence, (3) remaining cells: T cell barcodes without TCR detected.
Bulk RNA-seq data processing
For the gene expression, we mapped the sequencing data to reference genome (hg38) using HISAT, and defined transcript coordinates according to the gene annotation format file (GTF file) from GENCODE (Release 27, GRCh38). The Gene abundances are given as Reads Per Kilobase per Million mapped reads (RPKM) with “cuffnorm” command using the Cufflinks package.
MS data processing
All MS/MS ion spectra were analyzed using PEAKS X (Bioinformatics Solutions) for processing, de novo sequencing, database searching and label-free quantification. Resulting sequences were searched against the UniProt Human Proteome database (downloaded 5 May 2018) with mass error tolerances of 10 ppm and 0.02 Da for parent and fragment, respectively, the digestion enzyme semiTrypsin allowed for two missed tryptic cleavages, Carbamidomethyl of cysteine specified as a fixed modification, and Oxidation of methionine, acetyl of the N-terminus and phosphorylation of tyrosine, serine, and threonine as variable modifications. FDR estimation was enabled. Peptides were filtered for -Log10 P ≥ 15, and proteins were filtered for −Log10 P ≥ 15 and one unique peptide. For all experiments, this gave an FDR of <1% at the peptide-spectrum match level. Proteins sharing significant peptide evidence were grouped.
Intracellular cytokine staining
HH cells or Hut 78 cells were treated with ATRA (1 µM) or DMSO for 48 h. Thoroughly resuspend cells were fixed and permeabilized in 250 µl of BD Cytofix/Cytoperm solution (BD Biosciences, 51-2090 KZ) and incubated for 20 min at 4 °C. Wash cells two times in BD Perm/Wash buffer (BD Biosciences, 51-2091 KZ). Thoroughly resuspend fixed/permeabilized cells in 50 µl of BD Perm/Wash buffer (BD Biosciences, 51-2091 KZ) containing 0.5 µl of Foxp3 Monoclonal Antibody (Invitrogen, 12577380) or 0.5 µl of IgG Mouse-PE Isotype Control (Beckman, IM0670U). Incubate at 4 °C for 30 min in the dark. Flow cytometric data were acquired using flow cytometer (Cytoflex s, Beckman). Data were analyzed using FlowJo software.
Flow cytometry
HH cells or Hut 78 cells were treated with ATRA (2 µM) or DMSO for 48 hours, followed by staining with antibodies against CD25 (Invitrogen, 25-0259-42), CD5 (Invitrogen, 12-0059-42) and CD7 (Invitrogen, 11-0078-42). The immunophenotyping was performed on flow cytometry (Cytoflex s, Beckman).
Compound competition assay
Hut78 cells were seeded (5 × 105 cells/ml) in 6-well plates and treated with PHA-767491 (10 μM) or SNS-032 (10 μM) for 2 h prior to 6-h of 23 (GT-02897) (100 nM) treatment. Afterwards cells were harvested and lysed for Western blot analysis using the CDK9 antibody.
Acute toxicity assay
Sprague-Dawley rats were randomly allocated into four groups, including two Vehicle groups (Vehicle for intraperitoneal injection is 2% DMSO + 30% PEG-300 + 68% saline; Vehicle for subcutaneous injection is corn oil) and two treatment (intraperitoneal: 0.625 mg/kg, and subcutaneous: 2.5 mg/kg) groups. 3 males and 3 females in each group. A single injection of GT-02897 was administered to each group followed by a 2-week observation. Body weights were measured on days 0, 1, 2, 3, 4, 7, and 14 after administration79. On day 15, rats were sacrificed for evaluating their organs including morphological and histopathology appearances. The hearts, livers, spleens, kidneys, and lungs were harvested for hematoxylin-eosin (HE) staining80. In addition, plasma creatinine was measured by creatine oxidase method.
Detection of plasma creatinine
Plasma levels of creatinine were measured using creatinine quantification kit (Jiangsu Aidisheng Biological Technology Co., Ltd, ADS-W-FM034), according to the manufacturer’s instructions. Creatinine was first converted into creatine by creatinine amidohydrolase. Creatine was then sequentially oxidized by creatinase and sarcosine oxidase, producing hydrogen peroxide (H2O2). The generated H2O2 reacted with a chromogen to form a purple-colored compound, which exhibited a maximum absorption peak at 546 nm. Optical density was determined at 546 nm absorbance using a microplate reader.
Plasmids, virus construction and infection
Information of all plasmids were provided in Supplementary Table 1. Information of the shRNA sequences were provided in Supplementary Table 2. Lentivirus was produced by co-transfecting 293T cells with the lentiviral construct pCMV-dR8.91 (Δ8.9) plasmid, containing the genes gag, pol, and rev, and the pMDG envelope-expressing plasmid, using Lipofectamine 2000 (Invitrogen, 11668500). Viral supernatant was harvested at 48 h post-transfection, passed through a 0.45 μm filter, and infected the indicated CTCL cells using polybrene (Sigma–Aldrich).
Chemical synthesis
General methods
Reagents and solvents were purchased from commercial sources without further purification, unless otherwise indicated. Starting materials of S2 and S3 were synthesized according to the patent WO2019196812. The progress of reactions was monitored by thin-layer chromatography (TLC) and/or LC-MS. The final compounds were purified by prepared HPLC. NMR spectra were obtained from an Ascend 400 MHz Bruker spectrometer (operating at 400 MHz for 1H NMR). Multiplicities of signals are described as follows: s—singlet, br. s—broad singlet, d—doublet, t—triplet, m—multiple. High Resolution Mass spectra were recorded on Agilent-6125B ESI mass spectrometer with acetonitrile and water as solvents.
Scheme S1. Synthesis of compound 1-22

General procedure
To a mixture of N-[5-({[5-(2-methylprop-2-yl)-1,3-oxazol-2-yl]methyl}sulfanyl)-1,3-thiazol-2-yl]hexahydropyridine-4-carboxamide S1 (20 mg, 0.053 mmol), S2 (0.053 mmol, 1.0 eq), EDCI (20.3 mg, 0.106 mmol, 2.0 eq), HOAt (14.4 mg, 0.106 mmol, 2.0 eq) and DMF (5 mL) were added DIEA (43.8 μL, 0.265 mmol, 5.0 eq). The mixture was stirred for 2 h at room-temperature. LC-MS showed compound S1 was consumed and the desired product was major. The mixture was purified by prep-HPLC (0.05% HCl aq./acetonitrile) and lyophilized to give the desired product.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(2-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)thio)acetyl)piperidine-4-carboxamide (1). Light yellow solid (18.5 mg, yield 50%).1H NMR (400 MHz, DMSO-d6) δ 12.33 (s, 1H), 11.00 (d, J = 3.2 Hz, 1H), 7.72 (t, J = 6.1 Hz, 1H), 7.56 (dd, J = 15.8, 9.5 Hz, 2H), 7.40 (d, J = 5.4 Hz, 1H), 6.72 (s, 1H), 5.14 (dd, J = 13.3, 4.6 Hz, 1H), 4.41–4.30 (m, 2H), 4.21–4.11 (m, 2H), 4.06 (s, 2H), 4.01 (d, J = 13.3 Hz, 1H), 3.18–3.02 (m, 2H), 2.94 (dd, J = 22.1, 8.7 Hz, 1H), 2.80–2.66 (m, 2H), 2.61 (d, J = 18.3 Hz, 1H), 2.48 (s, 1H), 2.06–1.98 (m, 1H), 1.83 (s, 2H), 1.68 (d, J = 12.4 Hz, 1H), 1.43 (s, 1H), 1.18 (d, J = 3.1 Hz, 9H). MS (ESI) m/z: calcd. for C32H37N6O6S3+ [M + H]+, 697.19; found 697.3.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(3-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)thio)propanoyl)piperidine-4-carboxamide (2). Light yellow solid (18.8 mg, yield 52%). 1H NMR (400 MHz, DMSO-d6) δ 12.30 (s, 1H), 10.99 (s, 1H), 7.67 (d, J = 7.2 Hz, 1H), 7.60–7.52 (m, 2H), 7.40 (d, J = 8.0 Hz, 1H), 6.76–6.68 (m, 1H), 5.13 (d, J = 13.1 Hz, 1H), 4.40–4.33 (m, 2H), 4.23 (dd, J = 17.4, 5.8 Hz, 1H), 4.06 (s, 2H), 3.83 (s, 1H), 3.27 (d, J = 6.6 Hz, 2H), 3.00 (t, J = 11.8 Hz, 1H), 2.95–2.86 (m, 1H), 2.73 (dd, J = 14.2, 8.3 Hz, 3H), 2.64–2.57 (m, 2H), 2.46 (s, 1H), 2.07–1.97 (m, 1H), 1.80 (d, J = 12.7 Hz, 2H), 1.62–1.49 (m, 1H), 1.42 (d, J = 12.7 Hz, 1H), 1.18 (d, J = 4.6 Hz, 9H). MS (ESI) m/z: calcd. for C33H39N6O6S3+ [M + H]+, 711.21; found 711.2.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(4-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)thio)butanoyl)piperidine-4-carboxamide (3). Light yellow solid (17.7 mg, yield 46%). 1H NMR (400 MHz, DMSO-d6) δ 12.30 (s, 1H), 10.99 (s, 1H), 7.69 (dt, J = 11.0, 5.5 Hz, 1H), 7.58–7.48 (m, 2H), 7.40 (d, J = 7.0 Hz, 1H), 6.76–6.65 (m, 1H), 5.13 (dd, J = 13.3, 5.1 Hz, 1H), 4.30 (dd, J = 58.2, 17.4 Hz, 3H), 4.06 (s, 2H), 3.87 (d, J = 11.7 Hz, 1H), 3.15–3.09 (m, 2H), 3.01 (t, J = 12.3 Hz, 1H), 2.95–2.86 (m, 1H), 2.75–2.54 (m, 4H), 2.49–2.37 (m, 2H), 2.05–1.98 (m, 1H), 1.82 (dd, J = 14.1, 7.2 Hz, 4H), 1.53 (d, J = 12.5 Hz, 1H), 1.40 (d, J = 9.4 Hz, 1H), 1.18 (d, J = 3.6 Hz, 9H). MS (ESI) m/z: calcd. for C34H41N6O6S3+ [M + H]+, 725.22; found 725.3.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(5-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)thio)pentanoyl)piperidine-4-carboxamide (4). Light yellow solid (16.1 mg, yield 41%). 1H NMR (400 MHz, DMSO-d6) δ 12.30 (s, 1H), 10.99 (s, 1H), 7.65 (dd, J = 7.3, 1.3 Hz, 1H), 7.60–7.47 (m, 2H), 7.40 (d, J = 7.6 Hz, 1H), 6.72 (s, 1H), 5.13 (dd, J = 13.3, 5.1 Hz, 1H), 4.30 (dd, J = 57.6, 17.4 Hz, 3H), 4.06 (s, 2H), 3.92 (t, J = 14.5 Hz, 1H), 3.11 (s, 2H), 3.01 (t, J = 11.7 Hz, 1H), 2.98–2.85 (m, 1H), 2.72 (dd, J = 13.1, 9.5 Hz, 1H), 2.64–2.54 (m, 2H), 2.49–2.27 (m, 3H), 2.07–1.94 (m, 1H), 1.80 (s, 2H), 1.64 (d, J = 3.1 Hz, 4H), 1.54 (d, J = 12.1 Hz, 1H), 1.38 (dd, J = 23.0, 13.7 Hz, 1H), 1.18 (d, J = 4.8 Hz, 9H). MS (ESI) m/z: calcd. for C35H43N6O6S3+ [M + H]+, 739.24; found 739.3.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(6-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)thio)hexanoyl)piperidine-4-carboxamide (5). Light yellow solid (21.9 mg, yield 55%). 1H NMR (400 MHz, DMSO-d6) δ 12.30 (s, 1H), 10.99 (s, 1H), 7.63 (t, J = 6.7 Hz, 1H), 7.59–7.52 (m, 2H), 7.41 (d, J = 7.1 Hz, 1H), 6.76–6.68 (m, 1H), 5.13 (dd, J = 13.3, 5.1 Hz, 1H), 4.40–4.19 (m, 3H), 4.06 (s, 2H), 3.89 (d, J = 13.2 Hz, 1H), 3.09 (t, J = 7.2 Hz, 2H), 3.00 (d, J = 11.4 Hz, 1H), 2.96–2.87 (m, 1H), 2.72 (t, J = 11.5 Hz, 1H), 2.64–2.55 (m, 2H), 2.49–2.40 (m, 1H), 2.32–2.26 (m, 2H), 2.04–1.98 (m, 1H), 1.80 (s, 2H), 1.61 (dd, J = 13.9, 7.1 Hz, 2H), 1.56–1.48 (m, 3H), 1.46–1.37 (m, 3H), 1.18 (d, J = 4.5 Hz, 9H). MS (ESI) m/z: calcd. for C36H45N6O6S3+ [M + H]+, 753.26; found 753.3.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(7-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)thio)heptanoyl)piperidine-4- carboxamide (6). Light yellow solid (18.3 mg, yield 45%). 1H NMR (400 MHz, DMSO-d6) δ 12.31 (s, 1H), 10.99 (s, 1H), 7.63 (dt, J = 6.6, 3.3 Hz, 1H), 7.59–7.52 (m, 2H), 7.40 (d, J = 6.9 Hz, 1H), 6.72 (d, J = 3.5 Hz, 1H), 5.13 (dd, J = 13.3, 5.1 Hz, 1H), 4.39–4.19 (m, 3H), 4.06 (s, 2H), 3.89 (d, J = 13.7 Hz, 1H), 3.09 (t, J = 7.2 Hz, 2H), 3.00 (d, J = 12.5 Hz, 1H), 2.94–2.86 (m, 1H), 2.73 (t, J = 11.5 Hz, 1H), 2.63–2.54 (m, 2H), 2.49–2.42 (m, 1H), 2.32–2.25 (m, 2H), 2.05–1.97 (m, 1H), 1.80 (s, 2H), 1.60 (dd, J = 14.3, 7.2 Hz, 2H), 1.46 (ddd, J = 21.9, 14.5, 7.3 Hz, 6H), 1.33–1.27 (m, 2H), 1.18 (d, J = 4.6 Hz, 9H). MS (ESI) m/z: calcd. for C37H47N6O6S3+ [M + H]+, 767.27; found 767.3.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)thio)acetyl)piperidine-4-carboxamide (7). Yellow solid (13.9 mg, 37%). 1H NMR (400 MHz, DMSO-d6) δ 12.31 (s, 1H), 10.99 (s, 1H), 7.63 (dt, J = 6.6, 3.3 Hz, 1H), 7.59–7.52 (m, 2H), 7.40 (d, J = 6.9 Hz, 1H), 6.72 (d, J = 3.5 Hz, 1H), 5.13 (dd, J = 13.3, 5.1 Hz, 1H), 4.39–4.19 (m, 3H), 4.06 (s, 2H), 3.89 (d, J = 13.7 Hz, 1H), 3.09 (t, J = 7.2 Hz, 2H), 3.00 (d, J = 12.5 Hz, 1H), 2.94–2.86 (m, 1H), 2.73 (t, J = 11.5 Hz, 1H), 2.63–2.54 (m, 2H), 2.49–2.42 (m, 1H), 2.32–2.25 (m, 2H), 2.05–1.97 (m, 1H), 1.80 (s, 2H), 1.60 (dd, J = 14.3, 7.2 Hz, 2H), 1.46 (ddd, J = 21.9, 14.5, 7.3 Hz, 6H), 1.33–1.27 (m, 2H), 1.18 (d, J = 4.6 Hz, 9H). MS (ESI) m/z: calcd. for C32H35N6O7S3 + [M + H]+, 711.17; found 711.2.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(3-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)thio)propanoyl)piperidine-4-carboxamide (8). Yellow solid (15.4 mg, 40%). 1H NMR (400 MHz, DMSO-d6) δ 12.36 (d, J = 47.6 Hz, 1H), 11.12 (s, 1H), 7.86–7.74 (m, 2H), 7.68–7.59 (m, 1H), 7.40 (d, J = 7.5 Hz, 1H), 6.76–6.65 (m, 1H), 5.12 (dd, J = 12.8, 5.4 Hz, 1H), 4.40 (d, J = 13.4 Hz, 1H), 4.10–4.02 (m, 2H), 3.88 (d, J = 14.3 Hz, 1H), 3.03 (t, J = 12.9 Hz, 1H), 2.93–2.52 (m, 9H), 2.07 (dd, J = 12.4, 7.0 Hz, 1H), 1.81 (d, J = 11.5 Hz, 2H), 1.64–1.48 (m, 1H), 1.43 (d, J = 12.6 Hz, 1H), 1.23–1.16 (m, 9H). MS (ESI) m/z: calcd. for C33H37N6O7S3+ [M + H]+, 725.19; found 725.2.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(4-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)thio)butanoyl)piperidine-4-carboxamide (9). Yellow solid (19.2 mg, 49%). 1H NMR (400 MHz, DMSO-d6) δ 12.31 (s, 1H), 11.13 (s, 1H), 7.88 (d, J = 8.2 Hz, 1H), 7.84–7.76 (m, 1H), 7.64 (d, J = 7.1 Hz, 1H), 7.40 (s, 1H), 6.73 (d, J = 4.9 Hz, 1H), 5.12 (dd, J = 12.8, 5.4 Hz, 1H), 4.41 (d, J = 12.7 Hz, 1H), 4.06 (s, 2H), 3.91 (d, J = 12.9 Hz, 1H), 3.19–3.13 (m, 2H), 3.05 (t, J = 11.8 Hz, 1H), 2.89 (td, J = 13.5, 6.4 Hz, 1H), 2.74 (t, J = 11.2 Hz, 1H), 2.68–2.52 (m, 5H), 2.11–2.02 (m, 1H), 1.92–1.77 (m, 4H), 1.63–1.51 (m, 1H), 1.48–1.37 (m, 1H), 1.18 (s, 9H). MS (ESI) m/z: calcd. for C34H39N6O7S3+ [M + H]+, 739.20; found 739.3.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(5-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)thio)pentanoyl)piperidine-4-carboxamide (10). Yellow solid (21.5 mg, 54%). 1H NMR (400 MHz, DMSO-d6) δ 12.30 (s, 1H), 11.13 (s, 1H), 7.88–7.72 (m, 2H), 7.62 (t, J = 11.8 Hz, 1H), 7.40 (d, J = 7.1 Hz, 1H), 6.81–6.66 (m, 1H), 5.12 (dd, J = 12.7, 5.4 Hz, 1H), 4.39 (d, J = 12.7 Hz, 1H), 4.04 (d, J = 14.3 Hz, 2H), 3.93 (d, J = 13.1 Hz, 1H), 3.16 (d, J = 6.4 Hz, 2H), 3.03 (t, J = 12.2 Hz, 1H), 2.93–2.84 (m, 1H), 2.72 (d, J = 11.2 Hz, 1H), 2.66–2.53 (m, 3H), 2.39 (d, J = 5.0 Hz, 2H), 2.07 (dd, J = 12.1, 6.8 Hz, 1H), 1.81 (s, 2H), 1.69 (d, J = 6.0 Hz, 4H), 1.55 (d, J = 9.2 Hz, 1H), 1.44–1.31 (m, 1H), 1.18 (d, J = 4.4 Hz, 9H). MS (ESI) m/z: calcd. for C35H41N6O7S3+ [M + H]+, 753.22; found 753.3.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(6-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)thio)hexanoyl)piperidine-4-carboxamide (11). Yellow solid (19.5 mg, 48%).1H NMR (400 MHz, DMSO-d6) δ 12.30 (s, 1H), 11.12 (s, 1H), 7.85–7.71 (m, 2H), 7.62 (t, J = 8.8 Hz, 1H), 7.40 (d, J = 6.6 Hz, 1H), 6.79–6.66 (m, 1H), 5.12 (dd, J = 12.7, 5.4 Hz, 1H), 4.39 (d, J = 13.5 Hz, 1H), 4.05 (d, J = 6.0 Hz, 2H), 3.92 (d, J = 13.7 Hz, 1H), 3.14 (t, J = 7.2 Hz, 2H), 3.03 (t, J = 12.4 Hz, 1H), 2.94–2.84 (m, 1H), 2.73 (t, J = 11.2 Hz, 1H), 2.65–2.52 (m, 3H), 2.37–2.28 (m, 2H), 2.06 (dd, J = 12.4, 6.9 Hz, 1H), 1.81 (s, 2H), 1.73–1.65 (m, 2H), 1.60–1.36 (m, 6H), 1.24–1.16 (m, 9H). MS (ESI) m/z: calcd. for C36H43N6O7S3+ [M + H]+, 767.23; found 767.3.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(7-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)thio)heptanoyl)piperidine-4-carboxamide (12). Yellow solid (17.8 mg, 43%).1H NMR (400 MHz, DMSO-d6) δ 12.30 (s, 1H), 11.12 (s, 1H), 7.85–7.71 (m, 2H), 7.67–7.57 (m, 1H), 7.40 (d, J = 7.0 Hz, 1H), 6.77–6.66 (m, 1H), 5.12 (dd, J = 12.8, 5.4 Hz, 1H), 4.39 (d, J = 12.2 Hz, 1H), 4.05 (s, 2H), 3.91 (d, J = 13.7 Hz, 1H), 3.13 (t, J = 7.3 Hz, 2H), 3.03 (t, J = 12.0 Hz, 1H), 2.95–2.82 (m, 1H), 2.72 (dd, J = 21.2, 10.0 Hz, 1H), 2.57 (dd, J = 25.4, 13.1 Hz, 3H), 2.38–2.26 (m, 2H), 2.06 (dd, J = 10.7, 5.4 Hz, 1H), 1.81 (s, 2H), 1.67 (dd, J = 14.3, 7.2 Hz, 2H), 1.56–1.30 (m, 8H), 1.23–1.14 (m, 9H). MS (ESI) m/z: calcd. for C37H45N6O7S3+ [M + H]+, 781.25; found 781.3.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)thio)ethoxy)acetyl)piperidine-4-carboxamide (13). Light yellow solid (17.7 mg, 45%). 1H NMR (400 MHz, DMSO-d6) δ 12.31 (s, 1H), 10.99 (s, 1H), 7.69 (s, 1H), 7.60–7.54 (m, 2H), 7.40 (t, J = 3.3 Hz, 1H), 6.72 (d, J = 4.5 Hz, 1H), 5.13 (dd, J = 13.1, 5.0 Hz, 1H), 4.44–4.22 (m, 3H), 4.18 (d, J = 15.7 Hz, 1H), 4.05 (d, J = 6.8 Hz, 2H), 3.77 (s, 1H), 3.68 (d, J = 6.0 Hz, 2H), 3.28 (s, 2H), 3.03–2.96 (m, 1H), 2.93–2.87 (m, 1H), 2.73 (s, 1H), 2.68–2.53 (m, 3H), 2.49–2.42 (m, 1H), 2.01 (d, J = 11.8 Hz, 1H), 1.80 (d, J = 12.0 Hz, 2H), 1.60 (s, 1H), 1.46–1.32 (m, 1H), 1.18 (d, J = 6.2 Hz, 9H). MS (ESI) m/z: calcd. for C34H41N6O7S3+ [M + H]+, 741.22; found 741.3.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)thio)ethoxy)ethoxy)acetyl)piperidine-4-carboxamide (14). Light yellow solid (19.6 mg, 47%). 1H NMR (400 MHz, DMSO-d6) δ 12.23 (s, 1H), 10.92 (s, 1H), 7.62 (d, J = 7.5 Hz, 1H), 7.54–7.40 (m, 2H), 7.33 (d, J = 8.4 Hz, 1H), 6.65 (d, J = 5.1 Hz, 1H), 5.06 (dd, J = 13.2, 5.0 Hz, 1H), 4.35–4.13 (m, 3H), 4.06 (d, J = 19.6 Hz, 1H), 3.98 (s, 2H), 3.75 (d, J = 11.9 Hz, 1H), 3.56 (t, J = 6.4 Hz, 2H), 3.47 (s, 4H), 3.19 (t, J = 6.4 Hz, 2H), 2.97–2.88 (m, 1H), 2.82 (dd, J = 17.5, 5.5 Hz, 1H), 2.64 (d, J = 11.3 Hz, 1H), 2.59–2.45 (m, 3H), 2.40–2.30 (m, 1H), 1.98–1.89 (m, 1H), 1.73 (d, J = 11.5 Hz, 2H), 1.52 (d, J = 11.2 Hz, 1H), 1.35 (d, J = 9.8 Hz, 1H), 1.11 (d, J = 5.7 Hz, 9H). MS (ESI) m/z: calcd. for C36H45N6O8S3+ [M + H]+, 785.25; found 785.3.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(2-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)thio)ethoxy)ethoxy)ethoxy)acetyl)piperidine-4-carboxamide (15). Light yellow solid (18.0 mg, 41%). 1H NMR (400 MHz, DMSO-d6) δ 12.23 (s, 1H), 10.92 (s, 1H), 7.61 (d, J = 7.4 Hz, 1H), 7.53–7.39 (m, 2H), 7.33 (d, J = 8.7 Hz, 1H), 6.65 (d, J = 4.8 Hz, 1H), 5.06 (dd, J = 13.3, 5.0 Hz, 1H), 4.24 (dt, J = 34.4, 17.5 Hz, 3H), 4.07 (d, J = 20.2 Hz, 1H), 3.96 (d, J = 14.9 Hz, 2H), 3.76 (d, J = 12.4 Hz, 1H), 3.55 (t, J = 6.4 Hz, 2H), 3.51–3.42 (m, 8H), 3.17 (d, J = 6.4 Hz, 2H), 2.91 (d, J = 14.8 Hz, 1H), 2.85–2.77 (m, 1H), 2.64 (d, J = 11.6 Hz, 1H), 2.62–2.45 (m, 3H), 2.37 (d, J = 13.4 Hz, 1H), 1.98–1.89 (m, 1H), 1.73 (d, J = 11.6 Hz, 2H), 1.51 (d, J = 11.2 Hz, 1H), 1.34 (d, J = 13.0 Hz, 1H), 1.11 (d, J = 5.2 Hz, 9H). MS (ESI) m/z: calcd. for C38H49N6O9S3+ [M + H]+, 829.27; found 829.3.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(14-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)thio)-3,6,9,12-tetraoxatetradecanoyl)piperidine-4-carboxamide (16). Light yellow solid (16.2 mg, 35%). 1H NMR (400 MHz, DMSO-d6) δ 12.23 (s, 1H), 10.92 (s, 1H), 7.61 (d, J = 7.6 Hz, 1H), 7.54–7.43 (m, 2H), 7.33 (d, J = 8.3 Hz, 1H), 6.64 (d, J = 4.3 Hz, 1H), 5.06 (dd, J = 13.2, 5.1 Hz, 1H), 4.36–4.14 (m, 3H), 4.12–4.00 (m, 2H), 3.98 (d, J = 4.7 Hz, 2H), 3.48–3.36 (m, 16H), 3.18 (t, J = 6.0 Hz, 2H), 2.87–2.79 (m, 1H), 2.66 (t, J = 11.1 Hz, 1H), 2.57–2.46 (m, 2H), 2.42–2.31 (m, 1H), 1.97–1.90 (m, 1H), 1.74 (d, J = 10.5 Hz, 2H), 1.51 (d, J = 9.5 Hz, 1H), 1.36 (d, J = 11.9 Hz, 1H), 1.11 (d, J = 4.7 Hz, 9H). MS (ESI) m/z: calcd. for C40H53N6O10S3+ [M + H]+, 873.30; found 873.3.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(17-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)thio)-3,6,9,12,15-pentaoxaheptadecanoyl)piperidine-4-carboxamide (17). Light yellow solid (22.4 mg, 46%). 1H NMR (400 MHz, DMSO-d6) δ 12.23 (s, 1H), 10.92 (s, 1H), 7.61 (d, J = 7.4 Hz, 1H), 7.48 (dt, J = 15.1, 7.4 Hz, 2H), 6.65 (d, J = 4.0 Hz, 1H), 5.06 (dd, J = 13.3, 5.1 Hz, 1H), 4.36–4.13 (m, 3H), 4.12–4.00 (m, 2H), 3.99–3.95 (m, 2H), 3.76 (d, J = 14.2 Hz, 1H), 3.59–3.44 (m, 18H), 3.17 (d, J = 6.3 Hz, 2H), 2.90–2.79 (m, 2H), 2.66 (t, J = 11.3 Hz, 1H), 2.53 (d, J = 20.1 Hz, 2H), 2.38 (dd, J = 13.4, 9.0 Hz, 1H), 1.97–1.91 (m, 1H), 1.74 (d, J = 10.6 Hz, 2H), 1.51 (d, J = 9.5 Hz, 1H), 1.35 (d, J = 9.0 Hz, 1H), 1.11 (d, J = 4.5 Hz, 9H). MS (ESI) m/z: calcd. for C42H57N6O11S3+ [M + H]+, 917.32; found 917.4.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)thio)ethoxy)acetyl)piperidine-4-carboxamide (18). Yellow solid (16.8 mg, 42%). 1H NMR (400 MHz, DMSO-d6) δ 12.30 (s, 1H), 11.12 (s, 1H), 7.89–7.74 (m, 2H), 7.64 (d, J = 6.7 Hz, 1H), 7.39 (s, 1H), 6.75–6.67 (m, 1H), 5.12 (dd, J = 12.8, 5.3 Hz, 1H), 4.34 (s, 1H), 4.23 (d, J = 8.0 Hz, 2H), 4.07 (d, J = 10.5 Hz, 4H), 3.39 (dd, J = 7.4, 3.6 Hz, 4H), 2.86 (s, 1H), 2.73 (s, 1H), 2.65–2.54 (m, 3H), 2.07–1.99 (m, 1H), 1.81 (d, J = 10.0 Hz, 2H), 1.60 (s, 1H), 1.43 (s, 1H), 1.23–1.10 (m, 9H). MS (ESI) m/z: calcd. for C34H39N6O8S3+ [M + H]+, 755.20; found 755.3.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)thio)ethoxy)ethoxy)acetyl)piperidine-4-carboxamide (19, GT-02865). Yellow solid (18.6 mg, 44%). 1H NMR (400 MHz, DMSO-d6) δ 12.29 (s, 1H), 11.12 (s, 1H), 7.85–7.68 (m, 2H), 7.68–7.54 (m, 1H), 7.40 (d, J = 11.1 Hz, 1H), 6.72 (d, J = 5.0 Hz, 1H), 5.12 (dd, J = 12.7, 5.3 Hz, 1H), 4.33 (d, J = 11.9 Hz, 1H), 4.23–4.09 (m, 2H), 4.04 (d, J = 11.0 Hz, 2H), 3.84 (d, J = 13.3 Hz, 1H), 3.73 (t, J = 6.2 Hz, 2H), 3.64–3.45 (m, 6H), 2.99 (d, J = 12.8 Hz, 1H), 2.87 (d, J = 13.9 Hz, 1H), 2.73 (s, 1H), 2.60 (dd, J = 35.6, 16.8 Hz, 3H), 2.05 (d, J = 12.5 Hz, 1H), 1.81 (d, J = 12.0 Hz, 2H), 1.60 (d, J = 12.3 Hz, 1H), 1.43 (d, J = 9.8 Hz, 1H), 1.18 (d, J = 6.0 Hz, 9H). MS (ESI) m/z: calcd. for C36H43N6O9S3+ [M + H]+, 799.22; found 799.3.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(2-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)thio)ethoxy)ethoxy)ethoxy)acetyl)piperidine-4-carboxamide (20). Yellow solid (22.8 mg, 51%). 1H NMR (400 MHz, DMSO-d6) δ 12.29 (d, J = 48.1 Hz, 1H), 11.05 (s, 1H), 7.81–7.66 (m, 2H), 7.56 (ddd, J = 5.9, 3.9, 2.2 Hz, 1H), 7.32 (d, J = 9.8 Hz, 1H), 6.72–6.59 (m, 1H), 5.05 (dd, J = 12.6, 5.3 Hz, 1H), 4.22–4.01 (m, 2H), 3.97 (dd, J = 12.0, 3.8 Hz, 2H), 3.75 (s, 1H), 3.64 (t, J = 6.1 Hz, 2H), 3.52–3.45 (m, 10H), 2.93–2.78 (m, 3H), 2.73–2.64 (m, 1H), 2.50 (dd, J = 23.7, 11.3 Hz, 3H), 1.99 (dd, J = 12.5, 6.9 Hz, 1H), 1.90 (d, J = 13.8 Hz, 1H), 1.67 (t, J = 43.3 Hz, 3H), 1.09 (t, J = 13.3 Hz, 9H). MS (ESI) m/z: calcd. for C38H47N6O10S3+ [M + H]+, 843.25; found 843.3.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(14-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)thio)-3,6,9,12-tetraoxatetradecanoyl)piperidine-4-carboxamide (21). Yellow solid (25.4 mg, 54%). 1H NMR (400 MHz, DMSO-d6) δ 12.22 (s, 1H), 11.05 (s, 1H), 7.77–7.66 (m, 2H), 7.56 (dd, J = 5.7, 2.2 Hz, 1H), 7.37–7.25 (m, 1H), 6.65 (d, J = 4.4 Hz, 1H), 5.05 (dd, J = 12.7, 5.4 Hz, 1H), 4.24 (d, J = 13.0 Hz, 1H), 4.07 (q, J = 13.6 Hz, 2H), 4.01–3.94 (m, 2H), 3.77 (d, J = 12.5 Hz, 1H), 3.64 (t, J = 6.3 Hz, 2H), 3.52–3.39 (m, 14H), 2.93 (t, J = 11.5 Hz, 1H), 2.87–2.78 (m, 1H), 2.64 (d, J = 12.5 Hz, 1H), 2.61–2.47 (m, 3H), 2.02–1.93 (m, 1H), 1.74 (d, J = 10.1 Hz, 2H), 1.51 (d, J = 11.8 Hz, 1H), 1.35 (d, J = 11.8 Hz, 1H), 1.16–1.05 (m, 9H). MS (ESI) m/z: calcd. for C40H51N6O11S3+ [M + H]+, 887.28; found 887.3.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(17-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)thio)-3,6,9,12,15-pentaoxaheptadecanoyl)piperidine-4-carboxamide (22). Yellow solid (19.2 mg, 39%). 1H NMR (400 MHz, DMSO-d6) δ 12.23 (s, 1H), 11.05 (s, 1H), 7.71 (dd, J = 9.0, 5.1 Hz, 2H), 7.56 (dd, J = 5.8, 2.0 Hz, 1H), 7.33 (d, J = 8.6 Hz, 1H), 6.64 (s, 1H), 5.05 (dd, J = 12.8, 5.4 Hz, 1H), 4.24 (d, J = 11.9 Hz, 1H), 4.07 (q, J = 13.6 Hz, 2H), 3.96 (d, J = 14.2 Hz, 2H), 3.76 (d, J = 14.0 Hz, 1H), 3.64 (t, J = 6.3 Hz, 2H), 3.54–3.41 (m, 18H), 2.98–2.88 (m, 1H), 2.80 (d, J = 12.1 Hz, 1H), 2.66 (s, 1H), 2.61–2.47 (m, 3H), 2.01–1.93 (m, 1H), 1.74 (d, J = 11.8 Hz, 2H), 1.52 (s, 1H), 1.35 (d, J = 9.7 Hz, 1H), 1.16–1.03 (m, 9H). MS (ESI) m/z: calcd. for C42H55N6O12S3+ [M + H]+, 931.30; found 931.3.
Scheme 2: Synthesis of compound 23–27

General procedure
To a solution of N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)piperidine-4-carboxamide S1 (26.6 mg, 0.07 mmol, 1.0 eq) in NMP (2 mL) were added bromide S3 (0.070 mmol, 1.0 eq) and DIPEA (0.210 mmol, 3.0 eq), and the reaction was stirred at 50 °C overnight. LC-MS showed the reaction was completed. The reaction mixture was purified by prep-HPLC (0.05% HCl aq./acetonitrile) and lyophilized to afford the desired product.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(5-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)thio)pentyl)piperidine-4-carboxamide (23, GT-02897). Light yellow solid (29.9 mg, 59%). 1H NMR (400 MHz, DMSO-d6) δ 12.43 (s, 1H), 11.01 (s, 1H), 9.80 (s, 1H), 7.66 (d, J = 7.4 Hz, 1H), 7.57 (dt, J = 14.7, 7.2 Hz, 2H), 7.41 (d, J = 3.7 Hz, 1H), 6.73 (s, 1H), 5.15 (dd, J = 13.3, 5.1 Hz, 1H), 4.30 (dd, J = 57.5, 17.4 Hz, 2H), 4.02 (d, J = 37.5 Hz, 2H), 3.52 (t, J = 14.5 Hz, 2H), 3.12 (t, J = 7.0 Hz, 2H), 2.99 (d, J = 16.8 Hz, 2H), 2.97–2.81 (m, 3H), 2.72 (dd, J = 23.9, 12.1 Hz, 1H), 2.60 (d, J = 17.2 Hz, 1H), 2.45 (dd, J = 13.1, 8.7 Hz, 1H), 2.03 (d, J = 12.6 Hz, 3H), 1.90 (dd, J = 23.6, 12.4 Hz, 2H), 1.74–1.56 (m, 4H), 1.45 (d, J = 7.0 Hz, 2H), 1.21 (d, J = 17.7 Hz, 9H). MS (ESI) m/z: calcd. for C35H45N6O5S3+ [M + H]+, 725.26; found 725.3.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(8-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)thio)octyl)piperidine-4-carboxamide (24). Light yellow solid (32.2 mg, 0.042 mmol, 60%). 1H NMR (400 MHz, DMSO-d6) δ 12.44 (s, 1H), 11.00 (s, 1H), 9.93 (s, 1H), 7.64 (d, J = 7.3 Hz, 1H), 7.56 (dt, J = 14.8, 6.8 Hz, 2H), 7.41 (d, J = 3.4 Hz, 1H), 6.73 (d, J = 2.5 Hz, 1H), 5.14 (dd, J = 13.2, 5.0 Hz, 1H), 4.40–4.19 (m, 2H), 4.07 (s, 2H), 3.53 (d, J = 12.0 Hz, 2H), 3.09 (t, J = 6.8 Hz, 2H), 2.99 (dd, J = 10.5, 5.0 Hz, 2H), 2.95–2.83 (m, 3H), 2.74 (t, J = 12.0 Hz, 1H), 2.60 (d, J = 17.5 Hz, 1H), 2.45 (dd, J = 13.3, 8.8 Hz, 1H), 2.02 (d, J = 12.5 Hz, 3H), 1.98–1.81 (m, 2H), 1.62 (dt, J = 14.1, 5.9 Hz, 4H), 1.41 (s, 2H), 1.28 (s, 6H), 1.19 (s, 9H). MS (ESI) m/z: calcd. for C38H51N6O5S3+ [M + H]+, 767.31; found 767.3.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(4-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)thio)butyl)piperidine-4-carboxamide (25). Yellow solid (27.9 mg, 55%). 1H NMR (400 MHz, DMSO-d6) δ 12.44 (s, 1H), 11.14 (s, 1H), 9.66 (s, 1H), 7.87–7.75 (m, 2H), 7.70–7.58 (m, 1H), 7.41 (d, J = 4.6 Hz, 1H), 6.72 (d, J = 1.8 Hz, 1H), 5.13 (dd, J = 12.7, 5.4 Hz, 1H), 4.07 (s, 2H), 3.55 (d, J = 11.6 Hz, 2H), 3.21 (t, J = 7.0 Hz, 2H), 3.11 (s, 2H), 2.99–2.84 (m, 3H), 2.74 (d, J = 12.0 Hz, 1H), 2.61 (d, J = 20.3 Hz, 1H), 2.56–2.52 (m, 1H), 2.05 (d, J = 12.4 Hz, 3H), 1.90 (dd, J = 24.6, 10.6 Hz, 4H), 1.73 (d, J = 6.8 Hz, 2H), 1.19 (s, 9H). MS (ESI) m/z: calcd. for C34H41N6O6S3+ [M + H]+, 725.22; found 725.3.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(7-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)thio)heptyl)piperidine-4-carboxamide (26). Yellow solid (33.8 mg, 63%). 1H NMR (400 MHz, DMSO-d6) δ 12.44 (s, 1H), 11.13 (s, 1H), 9.98 (s, 1H), 7.78 (dt, J = 16.5, 8.1 Hz, 2H), 7.64 (d, J = 7.0 Hz, 1H), 7.41 (d, J = 3.4 Hz, 1H), 6.73 (d, J = 6.5 Hz, 1H), 5.12 (dd, J = 12.7, 5.4 Hz, 1H), 4.04 (d, J = 19.6 Hz, 2H), 3.53 (d, J = 10.5 Hz, 2H), 3.15 (t, J = 7.1 Hz, 2H), 3.06–2.98 (m, 2H), 2.96–2.83 (m, 3H), 2.74 (dd, J = 21.0, 9.4 Hz, 1H), 2.60 (d, J = 19.4 Hz, 1H), 2.57–2.52 (m, 1H), 2.09–1.87 (m, 5H), 1.68 (d, J = 6.8 Hz, 4H), 1.47 (s, 2H), 1.33 (s, 4H), 1.21 (d, J = 17.8 Hz, 9H). MS (ESI) m/z: calcd. for C37H47N6O6S3+ [M + H]+, 767.27; found 767.3.

N-(5-(((5-(tert-butyl)oxazol-2-yl)methyl)thio)thiazol-2-yl)-1-(8-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)thio)octyl)piperidine-4-carboxamide (27). Yellow solid (36.1 mg, 66%). 1H NMR (400 MHz, DMSO-d6) δ 12.43 (s, 1H), 11.13 (s, 1H), 9.77 (s, 1H), 7.78 (dt, J = 16.4, 8.0 Hz, 2H), 7.64 (d, J = 6.9 Hz, 1H), 7.41 (d, J = 3.5 Hz, 1H), 6.73 (s, 1H), 5.12 (dd, J = 12.8, 5.4 Hz, 1H), 4.07 (s, 2H), 3.54 (d, J = 11.8 Hz, 2H), 3.14 (t, J = 7.2 Hz, 2H), 3.02 (d, J = 6.3 Hz, 2H), 2.95–2.83 (m, 3H), 2.73 (dd, J = 24.9, 13.0 Hz, 1H), 2.60 (d, J = 19.5 Hz, 1H), 2.55–2.52 (m, 1H), 2.05 (dd, J = 13.9, 9.7 Hz, 3H), 1.91 (dd, J = 23.5, 11.9 Hz, 2H), 1.68 (d, J = 6.9 Hz, 4H), 1.46 (s, 2H), 1.32 (s, 6H), 1.19 (s, 9H). MS (ESI) m/z: calcd. for C38H49N6O6S3+ [M + H]+, 781.29; found 781.3.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw RNA-seq data of cultured cells upon ATRA treatment have been deposited in the Gene Expression Omnibus database (accession number GSE262586). The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the iProX partner repository81,82 (data set identifier PXD051037). RNA-seq data of CTCL patients and healthy donors were obtained from GEO (GSE113113, GSE168508)31,32. scRNA-seq data of CTCL patients and healthy donors were obtained from GEO (GSE165623, GSE171811, GSE128531)35,37,41. Source data are provided with this paper. All the other data are available within the article and its Supplementary Information. Source data are provided with this paper.
References
Whittaker, S., Hoppe, R. & Prince, H. M. How I treat mycosis fungoides and Sezary syndrome. Blood 127, 3142–3153 (2016).
Horwitz, S. M. et al. Activity of the PI3K-delta, gamma inhibitor duvelisib in a phase 1 trial and preclinical models of T-cell lymphoma. Blood 131, 888–898 (2018).
Dummer, R. et al. Cutaneous T cell lymphoma. Nat. Rev. Dis. Prim. 7, 61 (2021).
Kamijo, H. et al. Aberrant CD137 ligand expression induced by GATA6 overexpression promotes tumor progression in cutaneous T-cell lymphoma. Blood 132, 1922–1935 (2018).
Wang, L. et al. RARalpha/RXR synergism potentiates retinoid responsiveness in cutaneous T-cell lymphoma cell lines. Exp. Dermatol. 26, 1004–1011 (2017).
Zhang, C. & Duvic, M. Retinoids: therapeutic applications and mechanisms of action in cutaneous T-cell lymphoma. Dermatol. Ther. 16, 322–330 (2003).
Zhang, C., Hazarika, P., Ni, X., Weidner, D. A. & Duvic, M. Induction of apoptosis by bexarotene in cutaneous T-cell lymphoma cells: relevance to mechanism of therapeutic action. Clin. Cancer Res. 8, 1234–1240 (2002).
Zhang, C. & Duvic, M. Treatment of cutaneous T-cell lymphoma with retinoids. Dermatol. Ther. 19, 264–271 (2006).
Qu, K. et al. Chromatin accessibility landscape of cutaneous T cell lymphoma and dynamic response to HDAC inhibitors. Cancer Cell 32, 27–41 e24 (2017).
Tensen, C. P., Quint, K. D. & Vermeer, M. H. Genetic and epigenetic insights into cutaneous T-cell lymphoma. Blood 139, 15–33 (2022).
Duvic, M. et al. Bexarotene is effective and safe for treatment of refractory advanced-stage cutaneous T-cell lymphoma: multinational phase II-III trial results. J. Clin. Oncol. 19, 2456–2471 (2001).
Zhu, J. et al. Retinoic acid induces proteasome-dependent degradation of retinoic acid receptor alpha (RARalpha) and oncogenic RARalpha fusion proteins. Proc. Natl. Acad. Sci. USA 96, 14807–14812 (1999).
Malumbres, M. et al. Cyclin-dependent kinases: a family portrait. Nat. Cell Biol. 11, 1275–1276 (2009).
Suski, J. M., Braun, M., Strmiska, V. & Sicinski, P. Targeting cell-cycle machinery in cancer. Cancer Cell 39, 759–778 (2021).
Parua, P. K. & Fisher, R. P. Dissecting the Pol II transcription cycle and derailing cancer with CDK inhibitors. Nat. Chem. Biol. 16, 716–724 (2020).
Baumli, S. et al. The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. EMBO J. 27, 1907–1918 (2008).
Fujinaga, K., Huang, F. & Peterlin, B. M. P-TEFb: The master regulator of transcription elongation. Mol. Cell 83, 393–403 (2023).
Chen, R. et al. Mechanism of action of SNS-032, a novel cyclin-dependent kinase inhibitor, in chronic lymphocytic leukemia. Blood 113, 4637–4645 (2009).
Asghar, U., Witkiewicz, A. K., Turner, N. C. & Knudsen, E. S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 14, 130–146 (2015).
Narita, T. et al. Cyclin-dependent kinase 9 is a novel specific molecular target in adult T-cell leukemia/lymphoma. Blood 130, 1114–1124 (2017).
Thieme, E. et al. CDK9 inhibition induces epigenetic reprogramming revealing strategies to circumvent resistance in lymphoma. Mol. Cancer 22, 64 (2023).
Sher, S. et al. VIP152 is a selective CDK9 inhibitor with pre-clinical in vitro and in vivo efficacy in chronic lymphocytic leukemia. Leukemia 37, 326–338 (2023).
Phillips, D. C. et al. A novel CDK9 inhibitor increases the efficacy of venetoclax (ABT-199) in multiple models of hematologic malignancies. Leukemia 34, 1646–1657 (2020).
Fiskus, W. et al. Efficacy of CDK9 inhibition in therapy of post-myeloproliferative neoplasm (MPN) secondary (s) AML cells. Blood Cancer J. 12, 23 (2022).
Garriga, J. & Grana, X. CDK9 inhibition strategy defines distinct sets of target genes. BMC Res. Notes 7, 301 (2014).
Garriga, J., Xie, H., Obradovic, Z. & Grana, X. Selective control of gene expression by CDK9 in human cells. J. Cell Physiol. 222, 200–208 (2010).
Zhang, H. et al. Targeting CDK9 reactivates epigenetically silenced genes in cancer. Cell 175, 1244–1258 e1226 (2018).
Beauchamp, E. M. et al. Identification and targeting of novel CDK9 complexes in acute myeloid leukemia. Blood 133, 1171–1185 (2019).
Flinn, I. W. et al. Duvelisib, a novel oral dual inhibitor of PI3K-delta, gamma, is clinically active in advanced hematologic malignancies. Blood 131, 877–887 (2018).
Bresin, A. et al. Preclinical evidence for targeting PI3K/mTOR signaling with dual-inhibitors as a therapeutic strategy against cutaneous T-Cell lymphoma. J. Investig. Dermatol. 140, 1045–1053 e1046 (2020).
Querfeld, C. et al. Primary T cells from cutaneous T-cell lymphoma skin explants display an exhausted immune checkpoint profile. Cancer Immunol. Res. 6, 900–909 (2018).
Liu, F. et al. PEG10 amplification at 7q21.3 potentiates large-cell transformation in cutaneous T-cell lymphoma. Blood 139, 554–571 (2022).
Liu, X. et al. Single-cell transcriptomics links malignant T cells to the tumor immune landscape in cutaneous T cell lymphoma. Nat. Commun. 13, 1158 (2022).
Iyer, A. et al. Skin colonization by circulating neoplastic clones in cutaneous T-cell lymphoma. Blood 134, 1517–1527 (2019).
Gaydosik, A. M. et al. Single-cell lymphocyte heterogeneity in advanced cutaneous T-cell lymphoma skin tumors. Clin. Cancer Res. 25, 4443–4454 (2019).
Gluud, M. et al. Malignant T cells induce skin barrier defects through cytokine-mediated JAK/STAT signaling in cutaneous T-cell lymphoma. Blood 141, 180–193 (2023).
Herrera, A. et al. Multimodal single-cell analysis of cutaneous T-cell lymphoma reveals distinct subclonal tissue-dependent signatures. Blood 138, 1456–1464 (2021).
Buus, T. B. et al. Single-cell heterogeneity in Sezary syndrome. Blood Adv. 2, 2115–2126 (2018).
Borcherding, N. et al. Single-cell profiling of cutaneous T-cell lymphoma reveals underlying heterogeneity associated with disease progression. Clin. Cancer Res. 25, 2996–3005 (2019).
Rindler, K. et al. Single-cell RNA sequencing reveals markers of disease progression in primary cutaneous T-cell lymphoma. Mol. Cancer 20, 124 (2021).
Rindler, K. et al. Single-cell RNA sequencing reveals tissue compartment-specific plasticity of mycosis fungoides tumor cells. Front. Immunol. 12, 666935 (2021).
Yang, L. X. et al. C1Q labels a highly aggressive macrophage-like leukemia population indicating extramedullary infiltration and relapse. Blood 141, 766–786 (2023).
Zhang, X. et al. Single-cell heterogeneity and dynamic evolution of Ph-like acute lymphoblastic leukemia patient with novel TPR-PDGFRB fusion gene. Exp. Hematol. Oncol. 12, 19 (2023).
Anshabo, A. T., Milne, R., Wang, S. & Albrecht, H. CDK9: a comprehensive review of its biology, and its role as a potential target for anti-cancer agents. Front. Oncol. 11, 678559 (2021).
Egloff, S. CDK9 keeps RNA polymerase II on track. Cell Mol. Life Sci. 78, 5543–5567 (2021).
Olson, C. M. et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol. 14, 163–170 (2018).
Liu, H. et al. Discovery of novel BCR-ABL PROTACs based on the cereblon E3 ligase design, synthesis, and biological evaluation. Eur. J. Med. Chem. 223, 113645 (2021).
Liu, H. et al. Discovery and characterization of novel potent BCR-ABL degraders by conjugating allosteric inhibitor. Eur. J. Med. Chem. 244, 114810 (2022).
Morrell, R. C. et al. The BH3 Mimetic, ABT-737, Overcomes Stromal-Mediated Pro-Survival Signals and Synergizes with PHA-767491, a Dual Cdc7/CDK9 Inhibitor, In Acute Myeloid Leukaemia. Blood 116, 768–768 (2010).
Alfano, L. et al. CDK9-55 guides the anaphase-promoting complex/cyclosome (APC/C) in choosing the DNA repair pathway choice. Oncogene 43, 1263–1273 (2024).
Ji, C.H. et al. The AUTOTAC chemical biology platform for targeted protein degradation via the autophagy-lysosome system. Nat. Commun. 13, 904 (2022).
Xu, A. et al. Post-translational modification of retinoic acid receptor alpha and its roles in tumor cell differentiation. Biochem. Pharm. 171, 113696 (2020).
Lu, Y. et al. 2-Bromopalmitate targets retinoic acid receptor alpha and overcomes all-trans retinoic acid resistance of acute promyelocytic leukemia. Haematologica 104, 102–112 (2019).
Ying, M. et al. The E3 ubiquitin protein ligase MDM2 dictates all-trans retinoic acid-induced osteoblastic differentiation of osteosarcoma cells by modulating the degradation of RARα. Oncogene 35, 4358–4367 (2016).
Mandal, R., Becker, S. & Strebhardt, K. Targeting CDK9 for anti-cancer therapeutics. Cancers 13, 2181 (2021).
Melnick, A. & Licht, J. D. Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood 93, 3167–3215 (1999).
Keidel, S., Lamour, F. P. & Apfel, C. M. Mutational analysis reveals that all-trans-retinoic acid, 9-cis-retinoic acid, and antagonist interact with distinct binding determinants of RARalpha. J. Biol. Chem. 272, 18267–18272 (1997).
Grabarczyk, D. B. et al. HUWE1 employs a giant substrate-binding ring to feed and regulate its HECT E3 ___domain. Nat. Chem. Biol. 17, 1084–1092 (2021).
Hunkeler, M. et al. Solenoid architecture of HUWE1 contributes to ligase activity and substrate recognition. Mol. Cell 81, 3468–3480 e3467 (2021).
Wang, Y., Argiles-Castillo, D., Kane, E.I., Zhou, A. & Spratt, D.E. HECT E3 ubiquitin ligases—emerging insights into their biological roles and disease relevance. J. Cell Sci. 133, jcs228072 (2020).
Wu, Y. et al. Ubiquitin ligase E3 HUWE1/MULE targets transferrin receptor for degradation and suppresses ferroptosis in acute liver injury. Cell Death Differ. 29, 1705–1718 (2022).
Wang, X. et al. Retinoic acid receptor alpha drives cell cycle progression and is associated with increased sensitivity to retinoids in T-cell lymphoma. Oncotarget 8, 26245–26255 (2017).
Brown, C. C. et al. Retinoic acid is essential for Th1 cell lineage stability and prevents transition to a Th17 cell program. Immunity 42, 499–511 (2015).
Pino-Lagos, K. et al. A retinoic acid-dependent checkpoint in the development of CD4+ T cell-mediated immunity. J. Exp. Med. 208, 1767–1775 (2011).
Berger, C. L. et al. Cutaneous T-cell lymphoma: malignant proliferation of T-regulatory cells. Blood 105, 1640–1647 (2005).
Kim, E. J. et al. Immunopathogenesis and therapy of cutaneous T cell lymphoma. J. Clin. Investig. 115, 798–812 (2005).
Wasik, M. A. IL-13 as a novel growth factor in CTCL. Blood 125, 2737–2738 (2015).
Geskin, L. J., Viragova, S., Stolz, D. B. & Fuschiotti, P. Interleukin-13 is overexpressed in cutaneous T-cell lymphoma cells and regulates their proliferation. Blood 125, 2798–2805 (2015).
Sanjiv, K. et al. MTH1 inhibitor TH1579 induces oxidative DNA damage and mitotic arrest in acute myeloid leukemia. Cancer Res 81, 5733–5744 (2021).
Chou, J., Quigley, D. A., Robinson, T. M., Feng, F. Y. & Ashworth, A. Transcription-associated cyclin-dependent kinases as targets and biomarkers for cancer therapy. Cancer Discov. 10, 351–370 (2020).
Tong, W. G. et al. Phase I and pharmacologic study of SNS-032, a potent and selective Cdk2, 7, and 9 inhibitor, in patients with advanced chronic lymphocytic leukemia and multiple myeloma. J. Clin. Oncol. 28, 3015–3022 (2010).
Byrd, J. C. et al. Treatment of relapsed chronic lymphocytic leukemia by 72-hour continuous infusion or 1-hour bolus infusion of flavopiridol: results from Cancer and Leukemia Group B study 19805. Clin. Cancer Res 11, 4176–4181 (2005).
Du, C., Redner, R. L., Cooke, M. P. & Lavau, C. Overexpression of wild-type retinoic acid receptor alpha (RARalpha) recapitulates retinoic acid-sensitive transformation of primary myeloid progenitors by acute promyelocytic leukemia RARalpha-fusion genes. Blood 94, 793–802 (1999).
Weng, G. et al. PROTAC-DB: an online database of PROTACs. Nucleic Acids Res. 49, D1381–D1387 (2021).
Ji, C. H. et al. The AUTOTAC chemical biology platform for targeted protein degradation via the autophagy-lysosome system. Nat. Commun. 13, 904 (2022).
Stuart, T. et al. Comprehensive Integration of single-cell data. Cell 177, 1888–1902 e1821 (2019).
Han, X. et al. Construction of a human cell landscape at single-cell level. Nature 581, 303–309 (2020).
Tirosh, I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016).
Wang, Y. et al. Acute, subchronic toxicity and genotoxicity studies of JointAlive, a traditional Chinese medicine formulation for knee osteoarthritis. PLoS One 18, e0292937 (2023).
Bello, I. et al. Acute and sub-acute toxicity evaluation of the methanolic extract of Alstonia scholaris stem bark. Med. Sci. 4, 4 (2016).
Ma, J. et al. iProX: an integrated proteome resource. Nucleic Acids Res. 47, D1211–D1217 (2019).
Chen, T. et al. iProX in 2021: connecting proteomics data sharing with big data. Nucleic Acids Res. 50, D1522–D1527 (2022).
Acknowledgements
This work was supported by the National Natural Science Foundation (82170179, 81970131, 81770146 to Y.L., 82293661, 92253303 to G.Q.C, and 32171431 to L.X.), Foundation from Science and Technology Commission of Shanghai Municipality (22S11900400 to Y.L.), Foundation from the National Key R&D Program of China (2020YFA0803403 to G.Q.C), Foundation from the Shanghai Scientific and the Technological Innovation Action Plan (20JC1410100 to Y.L.). This work was supported by grants from the National Key Research and Development Program of China (2022YFC3401500 to X.B.Y).
Author information
Authors and Affiliations
Contributions
Y.L., X.B.Y., and G.Q.C. designed the research. C.H.L., L.H.H., L.X., J.Y.L., and M.Y.Y. performed the experiments. R.H.S., G.Q.C., and X.B.Y. contributed the new reagents. C.H.L., L.H.H., J.Y.L., X.H.Z., and M.Y.Y. collected the samples and managed the subject information. X.H.Z. carried out the bioinformatic analysis. C.C.L., X.Q., and B.J. contribute to compound synthesis. C.H.L., Y.L., M.Y.Y., G.Q.C., L.X., and L.Z. performed other data analysis. Y.L. and G.Q.C. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Antonio Giordano, Ying Lu, Yang Wang, and Andrew Yen for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Luo, CH., Hu, LH., Liu, JY. et al. CDK9 recruits HUWE1 to degrade RARα and offers therapeutic opportunities for cutaneous T-cell lymphoma. Nat Commun 15, 10594 (2024). https://doi.org/10.1038/s41467-024-54354-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-54354-3
