
Abstract
Metabolic reprogramming is a hallmark of cancer, enabling tumor cells to meet the high energy and biosynthetic demands required for their proliferation. High mobility group A1 (HMGA1) is a structural transcription factor and frequently overexpressed in human colorectal cancer (CRC). Here, we show that HMGA1 promotes CRC progression by driving lipid synthesis in a AOM/DSS-induced CRC mouse model. Using conditional knockout (Hmga1△IEC) and knock-in (Hmga1IEC-OE/+) mouse models, we demonstrate that HMGA1 enhances CRC cell proliferation and accelerates tumor development by upregulating fatty acid synthase (FASN). Mechanistically, HMGA1 increases the transcriptional activity of sterol regulatory element-binding protein 1 (SREBP1) on the FASN promoter, leading to increased lipid accumulation in intestinal epithelial cells. Moreover, a high-fat diet exacerbates CRC progression in Hmga1△IEC mice, while pharmacological inhibition of FASN by orlistat reduces tumor growth in Hmga1IEC-OE/+ mice. Our findings suggest that targeting lipid metabolism could offer a promising therapeutic strategy for CRC.
Similar content being viewed by others


Introduction
Metabolic reprogramming is a hallmark of malignant tumors, enabling cancer cells to adapt their metabolism to meet the increased demand for macromolecules and energy necessary for growth, proliferation, and metastasis1. As one of the three major classes of macromolecules, lipids play a critical role in cell structure and function, being key components of cellular membranes and mediators of intracellular signaling under both physiological and pathological conditions2. Abnormal lipid metabolism provides energy and biosynthetic precursors to support the rapid proliferation of tumor cells, and influences various stages of tumor progression by modulating oncogenic signaling pathways3. A defining characteristic for dysregulated lipid metabolism in tumor cells is the increased synthesis and uptake of fatty acids, alongside a reduction in fatty acid oxidation (FAO)4. In normal physiology, de novo fatty acid synthesis is predominantly confined to the liver and adipose tissue. However, unlike normal cells, tumor cells can synthesize fatty acids de novo even in the presence of sufficient exogenous fatty acids5. This is driven by the upregulation of key lipogenic enzymes, which promote lipid accumulation in cancer cells6. Understanding the mechanisms underlying the lipid metabolism may offer new therapeutic opportunities for targeting cancer metabolism.
Colorectal cancer (CRC) is the third most common malignancy worldwide, characterized by high incidence and mortality rates7. Several risk factors contribute to the majority of CRC cases, including gene mutation, smoking, obesity, poor dietary habits, excessive alcohol consumption, and diabetes8,9. Increasing evidence suggests that dysregulated lipid metabolism plays a crucial role in the initiation and progression of CRC and is closely linked to clinical outcomes and prognosis10,11. For instance, fatty acid-binding protein 5 (FABP5), an intracellular transporter of fatty acids that regulates lipid metabolism and cell growth, inhibits the ubiquitination of fatty acid synthase (FASN), resulting in FASN upregulation and enhanced lipid accumulation, which accelerates CRC progression12. Additionally, the circCAPRIN1/STAT2 complex promotes CRC development by enhancing lipid synthesis through an ACC1-dependent mechanism13. Furthermore, the silencing of protein tyrosine phosphatase receptor type O (PTPRO), a member of the R3 subtype family of receptor-type protein tyrosine phosphatases, stimulates de novo lipogenesis by activating the AKT/mTOR/SREBP1/ACC1 pathway and reduces fatty acid oxidation via the MAPK/PPARα/ACOX1 axis, thereby promoting CRC growth and metastasis14. Collectively, these findings underscore the critical role of lipid accumulation in CRC progression.
High mobility group A1 (HMGA1) is a non-histone chromosomal protein involved in crucial processes such as DNA transcription, replication, recombination, and repair15. Structurally, HMGA1 contains three AT-hook DNA binding domains and an acidic carboxyl tail. The AT-hook domains bind to A/T-rich regions of DNA, modulating the chromatin structure and thereby regulating the transcription of downstream genes16,17. Due to its role in modifying chromatin architecture, HMGA1 is often referred to as a structural transcription factor. While HMGA1 expression is significantly reduced in most adult tissues compared to embryonic cells, its levels are abnormally elevated in a wide range of tumors, including breast, gastric, pancreatic, thyroid cancers, and colorectal cancer (CRC)18,19,20,21. As a result, HMGA1 is classified as an oncofetal protein, given its re-expression during tumorigenesis. HMGA1 contributes to cancer progression by interacting with oncogenic factors through both transcriptional regulation and protein-protein interactions22. Beyond its role in transformation, HMGA1 has been implicated in regulating genes related to glucose metabolism. For instance, the insulin receptor (INSR), a critical regulator of insulin signaling and glucose homeostasis, requires HMGA1 for its normal expression. Interestingly, reduced HMGA1 expression has been observed in insulin resistance and diabetes in both humans and mice23. In a study by Williams et al.24 elevated levels of HMGA1 in the small intestine and colon were linked to abnormal cell proliferation and neoplastic transformation, alongside metabolic alterations in intestinal epithelial cells (IECs). Notably, metabolites associated with fatty acid metabolism were found to be upregulated in IECs with high HMGA1 expression, though the molecular mechanisms underlying HMGA1’s regulation of fatty acid synthesis in these cells remain unclear.
In this study, we demonstrate that HMGA1 is significantly upregulated in colorectal cancer (CRC), where it enhances the transcription of fatty acid synthase (FASN), leading to increased lipid synthesis. This elevation in lipid levels, driven by HMGA1 overexpression, promotes the proliferation of CRC cells and accelerates CRC progression in HMGA1 knock-in mice. Conversely, the knockout of HMGA1 (Hmga1△IEC) in IECs reduces lipid accumulation, thereby inhibiting the initiation and progression of CRC. Notably, a high-fat diet (HFD) exacerbates CRC progression in Hmga1△IEC mice and counteracts the suppressive effects of HMGA1 depletion on tumor development. Additionally, treatment with orlistat, a potent inhibitor of FASN-mediated lipid synthesis, significantly reduces lipid accumulation in the IECs of HMGA1 knock-in (Hmga1IEC-OE/+) mice and decreases CRC tumorigenesis. Overall, our findings suggest that HMGA1 promotes CRC development by upregulating FASN-mediated de novo fatty acid synthesis. These results provide compelling evidence for targeting lipid accumulation as a therapeutic strategy for the prevention and treatment of CRC.
Results
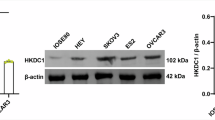
HMGA1 is highly expressed in CRC
HMGA1 is up-regulated in multiple cancers and known as an oncogene (http://gepia.cancer-pku.cn/detail.php?gene=HMGA1) (Fig. 1a). To determine the expression of HMGA1 in CRC, we firstly analyzed HMGA1 in a human CRC cohort in GEPIA database (http://gepia.cancer-pku.cn/detail.php?gene=HMGA1&clicktag=expdiy) (Fig. 1b) and GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE44076) (Fig. 1c). The results showed that the mRNA level of HMGA1 in human CRC tissues was significantly higher than that in adjacent normal colorectal tissues (Fig. 1a-c). Moreover, we identified that expression of HMGA1 was continuously up-regulated during the development of human CRC (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE41657) (Fig. 1d), and the disease-specific survival rate decreased in CRC patients with high HMGA1 expression (P = 0.022) (Fig. 1e). To further detect the expression of HMGA1 across different cellular subpopulations in colorectal tumors, we reanalyzed single-cell data from the GEO database (GSE201348). We selected single-cell sequencing results from 5 colon cancer tissues (all CRC tissue-related information contained in the uploaded dataset) and 5 normal control tissues. The data were subjected to dimensionality reduction clustering using t-distributed stochastic neighbor embedding (t-SNE) method, and different subpopulations of cells were annotated using specific markers (Supplementary Fig. 1a). Subsequently, the cells were categorized into 10 subgroups (Fig. 1f). We observed a higher proportion of intestinal epithelial cells (including Ki67+ cells) and stem cells in tumor tissues, whereas the cellular subpopulations in normal tissues appeared more homogeneous and diverse, such as goblet cells, fibroblasts, and natural killer cells (Fig. 1f and Supplementary Fig. 1b, c). We focused on the expression of HMGA1 and found that the level of HMGA1 was elevated in tumor tissues, particularly in Ki67+ IECs, as compared to that in normal tissues (Fig. 1g). These results suggest a potential association between HMGA1 and IEC proliferation. In addition, our IHC staining demonstrated that HMGA1 was up-regulated in tumor tissues of CRC patients (Fig. 1h, i).

a Relative expression of HMGA1 in various cancers from GEPIA database. N, Normal; T, Tumor. b, c Relative expression of HMGA1 in human CRC from GEPIA and GEO database. N = 349, T = 275 in (b) and N = 98, T = 98 in (c). (c) In normal group, the box plots showed minima (2.4625), maxima (5.2734), center (3.26415), lower bound (3.1050), upper bound (3.9518). Whiskers showed minima to maxima. d Relative expression of HMGA1 in different stages of carcinogenesis of human colorectum from GEO database. Normal (n = 12), low-grade dyplasia (n = 21), high-grade dyplasia (n = 30) and adenocarcinoma (n = 25). e Survival curve of patients with or without HMGA1 elevation. Patients’ data from the Study of Colorectal Adenocarcinoma (TCGA, PanCancer Atlas) with 594 patients were analyzed. The level of HMGA1 mRNA above the average + S.D. was defined as ‘high HMGA1’ (HMGA1: EXP > 1 as a search definition). p = 0.0221 with the log-rank test for the survival rate. f, g The analysis of HMGA1 level in CRC scRNA-seq dataset #GSE201348 from GEO database. T-distributed stochastic neighbor embedding (t-SNE) plot of ten main cell types identified in 5 CRC lesions and 5 normal control tissues (f). Dotplot represented the average level and the percentage of HMGA1 expression in different cell types in tumor and normal colorectal tissues of CRC patients (g). h, i Representative IHC staining of HMGA1 (h) and quantitative analysis of HMGA1 positive cells per crypt (i) in human colorectal (CRC) tissues. Data are presented as mean ± SD (c, d, i). Statistical significance was determined by two-tailed unpaired t-test (d) or two-tailed paired t-test (c, i). GEPIA: Gene Expression Profiling Interactive Analysis; GEO: Gene Expression Omnibus; scRNA-seq: single cell RNA-seq. Source data are provided as a Source Data file.
To further validate the expression of HMGA1 in CRC, we treated C57BL/6 J mice with AOM/DSS to induce CRC and detected the expression of HMGA1 in the colorectal tissues of mice (Supplementary Fig. 2a). At the end of the induction with AOM/DSS, the colorectum of mice was shortened (Supplementary Fig. 2b). Most of visible tumors were located at the distal colorectum, with the size ranging from 2 to 4 mm2 (Supplementary Fig. 2c). Colon glands in AOM/DSS-induced mice were irregularly arranged and structurally disordered in HE staining (Supplementary Fig. 2d). 45% tumors were identified as low-grade dysplastic adenomas (LGD) with infiltration of inflammatory cells (Supplementary Fig. 2d). HMGA1 mRNA and protein levels were up-regulated in CRC tissues of mice (Supplementary Fig. 2e-g). Taken together, our results demonstrate that HMGA1 is highly expressed in CRC.
HMGA1 promotes the occurrence and development of CRC
To characterize the role of HMGA1 in the growth of CRC cells, we transduced HMGA1 into human CRC cell line, HT-29 (Supplementary Fig. 3a, b), and determined cell proliferation. Enforced expression of HMGA1 led to a marked increase in cell growth, proliferation, and colony formation (Supplementary Fig. 3c-e). A similar result was obtained in HCT116 and LoVo cells with HMGA1 overexpression (Supplementary Fig. 3f-o). To determine the role of HMGA1 in the occurrence and development of CRC, we generated intestinal epithelial cell Hmga conditional knockout (Hmga1△IEC) and knock-in mice. Systemic knockout of Hmga1 gene leads to cardiac hypertrophy and type 2 diabetes in mice, and body weight and length of mice are decreased whereas Hmga1/Hmga2 double knock-out mice display a “superpygmy” phenotype25. Thus, we generated Hmga1flox/flox mice and Rosa26Hmga1/+ mice by CRISPR-Cas9 system, which were further hybridized with Villin-cre mice to produce intestinal epithelium conditional knockout (Hmga1△IEC) and knock-in HMGA1 (Hmga1IEC-OE/+) mice. IECs from Hmga1△IEC and Hmga1IEC-OE/+ mice were isolated and detected with qPCR and WB for the validation of HMGA1 manipulation. As expected, HMGA1 was successfully knocked out in the intestine of Hmga1∆IEC mice, and strikingly increased in the intestine of Hmga1IEC-OE/+ mice (Supplementary Fig. 4).
The Rosa26Hmga1/+ (control for the knock-in) mice and Hmga1IEC-OE/+ mice were treated with AOM/DSS for the induction of CRC (Fig. 2a). Compared with mice treated with mock induction (drinking water), AOM/DSS-treated Rosa26Hmga1/+ mice bore a decreased survival and a shortened colorectum, with an average of 6 tumors per colorectum, and about 50% of the tumors were 2 − 4 mm2 at the end of the induction (Fig. 2b-e). Histological examination showed that 45% of tumors in Rosa26Hmga1/+ mice induced with AOM/DSS were low-grade dysplastic (LGD) adenomas, 15% were high-grade dysplastic (HGD) adenomas, and 10% were carcinomas (Fig. 2f and Supplementary Fig. 5a). IHC staining showed HMGA1 was upregulated in CRC tissues in Rosa26Hmga1/+ mice as compared with that in normal colorectum (Fig. 2g and Supplementary Fig. 5b). About 40% of the tumor cells were Ki67 positive in the murine CRC tissues, which was much higher than that in normal colorectum of mice (Fig. 2g and Supplementary Fig. 5b). In stark contrast, Hmga1IEC-OE/+ (Hmga1 knock-in) mice treated with AOM/DSS possessed a lower survival rate and severely shortened colorectum (Fig. 2b, e). Colorectal tumor number and size were also significantly larger in AOM/DSS-induced Hmga1IEC-OE/+ mice than those in AOM/DSS-induced Rosa26Hmga1/+mice (Fig. 2c, d). Intestinal structures in Hmga1IEC-OE/+ mice without induction were largely normal under the light microscopy and there was no obvious alteration in the HE staining of intestine (Fig. 2f and Supplementary Fig. 5a). However, the proportions of HGD and carcinomas defined by histological examination were substantially increased in the colorectum of AOM/DSS-induced Hmga1IEC-OE/+ mice (Fig. 2f and Supplementary Fig. 5a). IHC staining showed that expression of HMGA1 and the number of Ki67 positive cells were increased markedly (Fig. 2g and Supplementary Fig. 5b). Together, our data suggest that HMGA1 promotes the malignant phenotype of CRC cells and enhances CRC tumor growth.

a Schematic diagram of AOM/DSS induction in Rosa26Hmga1/+ and Hmga1IEC-OE/+ mice. b Representative macroscopic views of colorectum from control and CRC mice. The colorectal length of mouse was measured. c Representative images of CRC. d The number and size of tumors in the entire colorectum per mouse were measured. e Survival curve of the Rosa26Hmga1/+ (control) mice and the Hmga1IEC-OE/+ mice with the induction of CRC. f HE staining of murine colorectum from AOM/DSS-induced Rosa26Hmga1/+ and Hmga1IEC-OE/+ mice (Scale bar = 20 μm. Representative images from 3 independent experiments). Percentages of LGD, HGD, and carcinoma in the tissues were determined. g Representative images of IHC staining for HMGA1 and Ki67 in normal colorectum and CRC (Scale bar = 20 μm). Data are presented as mean ± SD. Statistical significance was determined by two-tailed unpaired t-test (b, d, g) or log-rank test (e). n = 12. LGD: low-grade dysplasia; HGD: high-grade dysplasia; AOM: azoxymethane; DSS: dextran sulfate sodium. Source data are provided as a Source Data file.
Deletion of HMGA1 inhibits the occurrence and development of CRC
To further determine the role of HMGA1 in the maintenance of malignant phenotype of human CRC cells, we established HMGA1 knockdown cell lines in HT29, HCT116, and LoVo cells (Supplementary Fig. 6). Depletion of HMGA1 suppressed cell growth, proliferation, and colony formation (Supplementary Fig. 6). To further validate the role of HMGA1 in the tumorigenesis of CRC, we exposed Hmga1△IEC (Hmga1 conditional knockout in IECs) mice and Hmga1flox/flox (control for the knockout) mice to AOM/DSS for the induction of CRC (Fig. 3a). Conditional knockout of Hmga1 in IECs (Hmga1△IEC) showed no impact on the colorectum in the absence of AOM/DSS. Consistent with previous findings in Rosa26Hmga1/+ mice (Fig. 2b-d), AOM/DSS induction led to Hmga1flox/flox (control) mice having a lower survival rate and shorter colorectum with an average of 6 tumors in 2 − 4 mm2 per colorectum (Fig. 3b-e). Histological examination showed that pathological alterations in the colorectum of these mice included LGD and HGD adenomas and adenocarcinomas (Fig. 3f and Supplementary Fig. 7a). Ki67 staining was strikingly positive in the murine CRC tissues (Fig. 3g and Supplementary Fig. 7b). In stark contrast, Hmga1△IEC (Hmga1 conditional knockout in IECs) mice induced with AOM/DSS displayed a minimal reduction in colorectal length as compared with that in mice without induction (Fig. 3b). The survival rate was increased and the tumor number and size in colorectum were significantly decreased in Hmga1△IEC mice exposed to AOM/DSS as compared with those in Hmga1flox/flox (control) mice with a similar treatment (Fig. 3c-e). Histologic examination of colon sections demonstrated reduced proportions of adenocarcinoma, HGD, and LGD in AOM/DSS-induced Hmga1△IEC mice compared with those in AOM/DSS-induced Hmga1flox/flox (control) mice (Fig. 3f and Supplementary Fig. 7a). IHC staining showed a decrease in Ki67 positive cells in colorectum of Hmga1△IEC mice with AOM/DSS induction (Fig. 3g and Supplementary Fig. 7b). Taken together, our data suggest that HMGA1 deficiency inhibits the proliferation and tumorigenesis of CRC.

a Schematic diagram of AOM/DSS induction in Hmga1flox/flox and Hmga1△IEC mice. b Representative macroscopic views of colorectum from control and CRC mice. The colorectal length of mouse was measured. c Representative images of CRC. d The number and size of tumors in the entire colorectum per mouse were measured. e Survival curve of the Hmga1flox/flox (control) and Hmga1△IEC mice with the induction of CRC. f HE staining of colorectum from control and AOM/DSS-induced Hmga1flox/flox and Hmga1△IEC mice (Scale bar = 20 μm. Representative images from 3 independent experiments). Percentages of LGD, HGD, and carcinoma in the tissues were determined. g Representative images of IHC staining for HMGA1 and Ki67 in normal colorectum and CRC (Scale bar = 20 μm). Data are presented as mean ± SD. Statistical significance was determined by two-tailed unpaired t-test (b, d, g) or log-rank test (e). n = 12. LGD: low-grade dysplasia; HGD: high-grade dysplasia. Source data are provided as a Source Data file.
HMGA1 promotes lipid accumulation
Dysregulations of cell metabolism, such as elevated aerobic glycolysis and increased fatty acid metabolism, play a critical role in the tumorigenesis of CRC26,27,28. To determine whether HMGA1 promotes CRC through regulating cell metabolism, we performed an untargeted metabolomics using IECs from Hmga1flox/flox and Hmga1△IEC mice. In comparison with the Hmga1flox/flox mice, the Hmga1△IEC mice displayed 187 up-regulated metabolites and 120 down-regulated metabolites in IECs (Supplementary Data 1). Enrichment analysis of the metabolites showed that these differential metabolites mainly enriched in 20 pathways (Fig. 4a). Fatty acid synthesis was the most significantly deregulated signal pathway in HMGA1 deficient IECs (Fig. 4a). We separated the altered metabolites into up-regulated and down-regulated groups, and showed the top five classes of metabolites in up-regulated and down-regulated groups in heatmaps (Fig. 4b). The results showed that fatty acyl metabolites were mainly down-regulated in HMGA1-deficient IECs (Fig. 4b). Further, untargeted metabolomics analysis on tumors derived from the Hmga1IEC-OE/+ mice also showed that fatty acyl metabolites were mainly up-regulated by HMGA1 overexpression (Supplementary Fig. 8a, Supplementary Data 2). FA is a major component of structurally complex lipids and is one of the most basic categories of bio-lipids. We analyzed all lipids-related metabolites and found the free fatty acids were significantly up-regulated in tumors derived from the Hmga1IEC-OE/+ mice (Supplementary Fig. 8b). Thus, untargeted metabolomics measurement of IECs (CRCs) from both Hmga1 conditional knockout (Hmga1△IEC) and knock-in (Hmga1IEC-OE/+) mice demonstrates a positive correlation between expression of HMGA1 and activation of fatty acid synthesis.

a Pathway analysis of differentially enriched metabolites in IECs from Hmga1flox/flox mice as compared with those in IECs from Hmga1△IEC mice. b Untargeted metabolomics was used to identify metabolites in IECs from Hmga1△IEC and Hmga1flox/flox mice. The heatmaps showed significantly up-regulated metabolites (left panel) and down-regulated metabolites (right panel) in IECs from Hmga1△IEC mice. c Neutral lipids was assessed by oil red O staining in HMGA1-overexpressed HT-29 cells (Scale bar = 20 μm). d The relative levels of triglycerides, total cholesterol, and free fatty acids in HMGA1-overexpressed HT-29 cells. e Nile red staining of HMGA1-overexpressed HT-29 cells (Scale bar = 20 μm). n = 3. f, g Lipogenesis in the colorectum (CRC) from control and AOM/DSS-induced Rosa26Hmga1/+ and Hmga1IEC-OE/+ mice was determined by oil red O staining (f) (Scale bar = 20 μm) and nile red staining (g) (Scale bar = 50 μm) using frozen sections. n = 12. h HMGA1 overexpression and control HT-29 cells were cultured in glucose free DMEM in the presence of [U-13C] glucose (2 g/L) for 6 h. Saturated and monounsaturated FFAs labeled by 13C were analyzed using UHPLC-QTOF-MS-based nontargeted metabolomics. Data are presented as mean ± SD. Statistical significance was determined by two-tailed unpaired t-test (d, e, f, h) or hypergeometric test (a). Representative data from 3 independent experiments. IECs: intestinal epithelial cells. Source data are provided as a Source Data file.
To define the impact of HMGA1 on fatty acid metabolism in CRC, we detected the lipid contents in HMGA1-manipulated CRC cells. We used oil red O to stain lipid droplets in cells and found that there were much more lipid droplets in HMGA1-overexpressed HT-29 cells than those in control HT-29 cells (Fig. 4c), indicating a lipid accumulation under HMGA1-overexpression. Levels of triglycerides (TG), total cholesterol (TC) and free fatty acids (FFAs) were increased in HT-29 cells with HMGA1 overexpression (Fig. 4d). Further, application of nile red to detect intracellular lipid droplets also showed an elevated accumulation of lipids in HMGA1-overexpressed HT-29 cells (Fig. 4e). We validated the findings in HMGA1-overexpressed HCT116 and LoVo cells and demonstrated a similar increase in intracellular lipids in these cells (Supplementary Fig. 9). Moreover, oil red O staining and nile red staining revealed a marked increase in lipid contents in colorectum of Hmga1IEC-OE/+ mice (Fig. 4f, g).
To further validate the role of HMGA1 in the accumulation of intracellular lipids in CRC, we detected the lipid content in HMGA1 knocked down HT-29, HCT116, and LoVo cells. It showed that depletion of HMGA1 reduced the levels of lipid droplets in the oil red O staining and nile red fluorescence staining and decreased the contents of TG, TC and FFAs in CRC cells (Supplementary Fig. 10). Lipid accumulation in IECs of Hmga1ΔIEC mice was much weaker than that in IECs of Hmga1flox/flox mice regardless of the induction for CRC with AOM/DSS (Supplementary Fig. 11). These results suggest that HMGA1 upregulates lipid accumulation in IECs and in CRC cells.
To further determine the role of HMGA1 in de novo lipogenesis, we used [U-13C] glucose to trace newly synthesized lipids in HMGA1-manipulated HT-29 cells (Fig. 4h, Supplementary Fig. 12). Since de novo lipogenesis mainly produces saturated fatty acids and monounsaturated fatty acids29, 13C-labeled saturated fatty acids and monounsaturated fatty acids were analyzed by UHPLC-QTOF-MS. It showed that 13C-labeled newly synthesized fatty acids were increased in HMGA1-overexpression cells (Fig. 4h) and decreased in HMGA1-knockdown cells compared to control cells (Supplementary Fig. 12). Taken together, these data suggest that HMGA1 promotes de novo lipogenesis in CRC cells.
Lipid accumulation mediates the oncogenic activity of HMGA1
To determine the role of lipid accumulation in HMGA1-involved oncogenic activities, we first exposed HMGA1-depleted HT-29 cells to palmitic acid (PA) and determined cellular oncogenic phenotypes. PA, generated by the condensation of seven malonyl-CoA and one acetyl-CoA catalyzed by FASN, is the main product of de novo fatty acid synthesis. The expression of FASN was not affected by the addition of PA (Supplementary Fig. 13a, b). However, supplement of PA reversed HMGA1 knockdown-induced decrease of intracellular neutral lipids (Supplementary Fig. 13c-e) and restored the proliferation and colony formation of HMGA1 depleted cells (Supplementary Fig. 13f–h).
Next, we exposed HMGA1-overexpressed HT-29 cells to orlistat, an inhibitor for de novo fatty acid synthesis12,30. Application of orlistat reduced lipid accumulation (Supplementary Fig. 14a-c) and mitigated HMGA1 overexpression-induced CRC cell proliferation and colony formation (Supplementary Fig. 14d-f). Collectively, these results support our notion that lipid accumulation mediates HMGA1 oncogenic activity in CRC cells.
HMGA1 upregulates FASN
To explore the mechanism by which HMGA1 promotes lipid accumulation in CRC, we performed an RNA-seq of control and HMGA1-knocked down HT-29 cells. Consistent with findings in the untargeted metabolomic analysis (Fig. 4a), the transcriptome assay revealed an enrichment in genes in fatty acid biosynthesis in HMGA1 deficient cells (Fig. 5a, b). We analyzed the correlation between HMGA1 and key molecules involved in fatty acid synthesis, such as ACLY, FASN, MLYCD, SCD1, ACC1, ACSL1, ACSL3, ACSL4, ACSL5, ACSL6, FABP4, and FABP5, and fatty acid oxidation, such as ACOX1 and CPT1A (Fig. 5a). It showed that depletion of HMGA1 mainly reduced the expression of fatty acid synthesis genes (Fig. 5a). We validated the changes in lipid metabolism genes identified by the RNA-seq analysis with qPCR. It showed that HMGA1 overexpression upregulated and HMGA1 depletion downregulated genes associated with de novo fatty acid synthesis, such as FASN, ACLY, and ACC1, but not fatty acid β-oxidation genes, such as ACSL4, ACOX1, and CPT1A in HT-29 cells (Supplementary Fig. 15a, b). Consistently, validations with HMGA1-manipulated HCT116 and LoVo cells also demonstrated that HMGA1 mainly regulated genes in fatty acid synthesis, but not genes in fatty acid β-oxidation (Supplementary Fig. 15c-f).

a Differentially expressed genes in lipid metabolism in HMGA1-knockdown CRC cells compared with control CRC cells from RNA-seq. b GSEA plot of fatty acid synthesis genes identified in RNA-seq in HMGA1-silenced HT-29 cells. NES, normalized enrichment score. c Saturated and monounsaturated FFAs with 13C-labeled in control and HMGA1-overexpressed cells treated with orlistat. Control and HMGA1-overexpressed HT29 cells were treated with 20 µM orlistat for 18 h. Cell culture medium was then replaced with glucose free DMEM in the presence of [U-13C] glucose (2 g/L) and orlistat for 6 h. Each dot represents a sample (n = 3). d The samples derive from the same experiment but different gels for HMGA1, FASN, another for CPT1A and another for β-actin were processed in parallel. β-actin served as loading control (The quantification provided under the blots is for the representative blot from 3 independent experiments). e The correlation between HMGA1 and FASN in CRC patients from the TCGA database. f Representative IHC staining of HMGA1 and FASN in human colorectal and CRC tissues (Scale bar = 20 μm). g Representative images of IHC staining for FASN in the coloretum (CRC) from Rosa26Hmga1/+ and Hmga1IEC-OE/+ mice induced with or without AOM/DSS (Scale bar = 20 μm) (n = 12 mice per group). h Representative images of IHC staining for FASN in the colorectum (CRC) from Hmga1flox/flox and Hmga1△IEC mice induced with or without AOM/DSS (Scale bar = 20 μm) (n = 12 mice per group). Data are presented as mean ± SD. Statistical significance was determined by two-tailed unpaired t-test (c, d, g, h), two-tailed paired t-test (f), permutation test (b), or Pearson correlation coefficient (e). GSEA: gene set enrichment analysis, TCGA: The Cancer Genome Atlas. Source data are provided as a Source Data file.
We found that FASN was consistently the most significantly changed gene in fatty acid synthesis in all three HMGA1-manipulated cells (Supplementary Fig. 15). FASN is a key gene in de novo fatty acid synthesis. 13C-labeled newly synthesized FFAs in HMGA1 overexpression cells were increased, which was reversed by FASN inhibitor, orlistat (Fig. 5c), indicating that FASN may mediate HMGA1-induced intracellular de novo lipogenesis. Western blot detection demonstrated that expression of FASN was compromised in HMGA1-knocked down cells and elevated in HMGA1-transduced cells (Fig. 5d). We also found that ACC1 and ACLY were only marginally affected by the expression of HMGA1 in the Western blot assay (Supplementary Fig. 16). To further exclude the potential impact of ACC1 and ACLY on HMGA1-induced de novo lipogenesis, we detected the outcome of ACC1 and ACLY inhibition on HMGA1-induced lipid droplet formation and lipid synthesis and found that inhibition of ACC1 with TOFA (RMI14514) and suppression of ACLY with ETC-1002 (bempedoic acid) could not affect HMGA1-induced lipid accumulation (Supplementary Fig. 17).
We analyzed the expression of FASN in human CRC in TCGA and found that FASN was positively correlated with HMGA1 at the mRNA level (Fig. 5e) and at the protein level analyzed with reverse-phase protein array (RPPA) (Supplementary Fig. 18). Our IHC staining showed that the expression of FASN was very low in human normal colon tissues, and increased markedly in human CRC tissues (Fig. 5f), which displayed a similar pattern with the expression of HMGA1 in the CRC tissues (Fig. 5f). In addition, we detected the expression of FASN in intestinal epithelia (CRC) from HMGA1 genetically engineered mice treated with or without AOM/DSS for the induction of CRC. A weak staining of FASN was observed in normal colon tissue of mice without the induction of CRC. Dysplastic adenomas and adenocarcinomas in the colorectum from wild-type mice treated with AOM/DSS displayed a strong staining of FASN (Fig. 5g, h and Supplementary Fig. 19). More importantly, CRC tumors generated in Hmga1 conditional knock-in mice (Hmga1IEC-OE/+) bore a much stronger staining of FASN (Fig. 5g and Supplementary Fig. 19a). Whereas IHC staining of FASN in the intestinal epithelia of Hmga1 conditional knock-out (Hmga1△IEC) mice was much weaker than that in Hmga1flox/flox mice regardless the presence or absence of AOM/DSS induction (Fig. 5h and Supplementary Fig. 19b). Taken together, our current data suggest that HMGA1 upregulates FASN, which leads to the increased de novo fatty acid synthesis in CRC.
FASN mediates HMGA1-upregulated fatty acid accumulation
To determine the role of FASN in HMGA1-promoted accumulation of fatty acids, we transduced FASN into HMGA1-knockdown cells. Depletion of HMGA1 led to a reduction in intracellular neutral lipids (Supplementary Fig. 20a, b). Intracellular TG, TC, and FFAs were decreased due to the depletion of HMGA1 (Supplementary Fig. 20c). Transduction of FASN reversed the reduction in intracellular lipids and recovered the levels of TG, TC, and FFAs in HMGA1 knockdown cells (Supplementary Fig. 20). In contrast, silencing FASN abrogated HMGA1 overexpression-induced accumulation of neutral lipids and elevation of TG, TC, and FFAs in cells (Supplementary Fig. 21). Further, application of orlistat, an inhibitor of FASN, also reduced HMGA1 overexpression-induced lipid accumulation and mitigated intracellular contents of TG, TC, and FFAs (Supplementary Fig. 14). Collectively, these results suggest that FASN mediates HMGA1-induced intracellular lipid accumulation.
FASN inhibition has been shown to affect multiple signaling pathways as FASN is a central metabolic hub involved in the generation of bio-active lipids, many of which possess growth factor properties. FASN inhibition may also disturb the NADP/NADPH balance resulting in high levels of ROS, and affects acetyl-CoA levels potentially impacting protein acetylation31. We detected the ratio of NADP/NADPH and the level of acetyl-CoA in HMGA1 overexpression or knockdown cells and found that alterations of HMGA1 were unable to affect NADP/NADPH ratio and acetyl-CoA levels compared with control cells in our experimental settings (Supplementary Fig. 22).
HMGA1 promotes the transcription of FASN by enhancing the activity of SREBP1
HMGA1 has previously been shown to play a role in the regulation of PI3K/AKT/mTOR signaling, which is known to increase de novo lipid synthesis through multiple pathways, including activation of SREBP1 and upregulation of FASN14. In addition, HMGA1 has also been shown to regulate c-Myc expression32 and FASN is an important effector downstream of mTORC1 and c-Myc in hepatocellular carcinoma33. Thus we detected the effects of HMGA1 overexpression on PI3K/AKT/mTORC1 and c-Myc. It showed that HMGA1-manipulations did not affect the expression of PI3K/AKT, mTORC1, and c-Myc in HT-29 cells (Supplementary Fig. 23). c-Myc and mTORC1 were also not affected by the expression of HMGA1 in the intestinal epithelia (CRC) of Hmga1 conditional knock-in or knockout mice (Supplementary Fig. 24). None of the observed phenotypes on lipid synthesis induced by HMGA1 overexpression were rescued by inhibitor of AKT (MK-2206), c-Myc (10058-F4), and mTORC1 (rapamycin) in cells (Supplementary Fig. 25). In addition, inhibition of mTORC1 with rapamycin or c-Myc with 10058-F4 was unable to affect the expression of FASN in HMGA1-overexpression HT-29 cells (Supplementary Fig. 26). Taken together, our data suggest that HMGA1-upregulated FASN is independent of the activation of PI3K/AKT, mTORC1, and c-Myc in our current experimental settings.
To elucidate the mechanism by which HMAG1 regulates FASN, we performed a co-IP assay to determine whether there is a direct interaction between HMGA1 and FASN. It showed that there was no direct interaction between HMGA1 and FASN by the co-IP assay (Fig. 6a). HMGA1 is a non-histone chromatin protein, which acts as a structural transcription factor affecting the transcriptional efficiency of target genes. Our previous data showed that HMGA1 regulated FASN at both mRNA and protein levels (Fig. 5a, d and Supplementary Fig. 15). Thus, we speculated that HMGA1 might regulate FASN by enhancing its transcription. To test our hypothesis, we performed an ATAC-seq assay to determine the chromatin accessibility in cells with or without HMGA1 manipulation. We found that the transcriptional signals around the transcriptional start site (TSS) in FASN gene were strikingly reduced in HMGA1-knockdown cells in the ATAC-seq assay (Fig. 6b), indicating that depletion of HMGA1 compromised the openness of TSS of FASN and reduced the chromatin accessibility of the region to transcription factors.

a Co-IP assay to detect the interaction between HMGA1 and FASN. b IGV plots of ATAC-seq peaks at the FASN locus in HMGA1 silencing and control CRC cells. Boxed is the TSS site. c, d Luciferase reporter assay to detect the effect of HMGA1 silencing (c) or overexpression (d) on the luciferase activity of the FASN promoter in CRC cells. e Correlation between FASN and SREBP1 in CRC patients from the TCGA database. f The samples derive from the same experiment but different gels for HMGA1, SREBP1, and another for β-actin were processed in parallel. β-actin served as loading control (The quantification provided under the blots is for the representative blot from 3 independent experiments). g Co-IP assay to detect the interaction between HMGA1 and SREBP1. The samples derive from the same experiment but different gels for HMGA1, and another for SREBP1 were processed in parallel (The quantification provided under the blots is for the representative blot from 3 independent experiments). h Immunofluorescence detection of HMGA1 and SREBP1 in CRC cells (Scale bar = 10 μm). i Luciferase reporter assay to detect the effect of SREBP1- overexpression or silencing on FASN transcription in HT-29 cells with or without HMGA1 knockdown. j Luciferase reporter assay to detect the effect of SREBP1 silencing or overexpression on HMGA1-activated FASN transcription in HT-29 cells. k Schematic illustration of the mutated promoter sites in pGL3-Basic-FASN reporter. Potential binding sites of SREBP1 in FASN promoter targeted by HMGA1 were mutated. l, m Luciferase reporter assay to detect the effect of SREBP1 binding sites mutated on HMGA1-activated FASN transcription in HMGA1-knocked down (l) or HMGA1-overexpressed (m) HT-29 cells. n Chromatin immunoprecipitation-qPCR assay (ChIP-qPCR) to verify that HMGA1 facilitated the binding of SREBP1 to the promoter of FASN in HT-29 cells. Data are presented as mean ± SD from 3 independent experiments. Statistical significance was determined by two-tailed unpaired t-test (c, d, i, j, l, m, n) or Pearson correlation coefficient (e). Ns, no statistical difference. IGV: integrative genomics viewer; ATAC-seq: assay for transposase accessible chromatin using sequencing; Co-IP: co-immunoprecipitation. Source data are provided as a Source Data file.
To determine whether expression of HMGA1 affects the transcription of FASN, we cloned the promoter region of FASN from 2000 bp upstream to 500 bp downstream of the TSS (−2000 bp to +500 bp) into a luciferase report vector (pGL3-basic), transfected the vector into HMGA1-manipulated cells, and performed a dual luciferase reporter assay. As compared with empty vector-transduction, HMGA1 knockdown significantly decreased the luciferase activity of the FASN promoter (Fig. 6c), whereas overexpression of HMGA1 markedly enhanced the luciferase signal of the FASN promoter (Fig. 6d), indicating that HMGA1 could promote the transcription of FASN.
HMGA1 is an ‘architectural’ transcription factor, which does not possess an intrinsic transcriptional activity, but is able to transactivate promoters through mechanisms that facilitate the assembly and stability of a multicomponent enhancer complex, which drives gene transcription in response to extracellular and intracellular signals34. SREBP1, as a transcription factor, plays an important role in de novo fatty acid synthesis by regulating fatty acid synthesis related genes, such as FASN, ACLY and ACC135. We analyzed the expression of SREBP1 and FASN in CRC in TCGA and identified a strong positive correlation between FASN and SREBP1 (Fig. 6e). Thus, we asked whether HMGA1 affects the transcription of FASN by regulating the binding of SREBP1 to FASN promoter region. Manipulations of HMGA1 did not affect the expression of SREBP1 (Fig. 6f), but indeed there was a direct interaction between HMGA1 and SREBP1 (Fig. 6g). Immunofluorescence staining showed that HMGA1 and SREBP1 were co-located in the nucleus of CRC cells (Fig. 6h). Depletion of HMGA1 substantially reduced the promoter activity of FASN, which was restored by the enforced expression of SREBP1 and further suppressed by the silencing of SREBP1 (Fig. 6i). In contrast, enforced expression of HMGA1 significantly increased the promoter activity of FASN, which was suppressed by the silencing of SREBP1 and further promoted by the overexpression of SREBP1 (Fig. 6j). These results suggest that HMGA1 elevates FASN by enhancing SREBP1-promoted transcription of FASN.
Next, we tried to identify the specific sites at which HMGA1 regulates the transcriptional binding of SREBP1 to FASN promoter. The existing FASN promoter with the length of 2500 bp was divided into three segments: F1 (−2000 bp ~ −1000 bp), F2 (−1050 bp ~ −500 bp), and F3 (-500 bp ~ +500 bp), and were cloned into the pGL3-basic vector respectively (Fig. 6k). The fragments of FASN promoter were then transduced into cells with or without HMGA1 manipulations. The results showed that there was no luciferase signal when the F1 was applied regardless of the presence of HMGA1 or not. However, overexpression of HMGA1 increased and knockdown of HMGA1 inhibited the luciferase activity of F2 and F3 (Fig. 6l, m).
We screened the transcriptional binding sites of SREBP1 in the FASN promoter region through the JASPAR database (http://jaspar.genereg.net/) and identified 4 high score binding sites. Among them, the binding site -1179 ~ -1170 bp was located in F1 fragment, which we previously showed no activity for the transcription of FASN. The other 3 binding sites were located in F2 and F3 fragment respectively. Because of the continuity of the two predicted fragments (-67 ~ -58 bp, −57 ~ −48 bp) located in F3 fragments, we named the two predicted site R2 (−67 ~ −48 bp) jointly, and the remaining site was named R1 (−651 ~ −642 bp) (Fig. 6k). We mutated the possible binding sites of SREBP1 in the promoter fragment sequences, cloned them into pGL3-basic vector, and performed a dual-luciferase report assay. Mutation of R2 (−67 ~ −48 bp), but not R1 (−651 ~ −642 bp), led to the decrease of the luciferase activity, which was no longer regulated by HMGA1 (Fig. 6l, m). In addition, chromatin immunoprecipitation assay (ChIP) confirmed that HMGA1 promoted the binding of SREBP1 to the FASN promoter region in HT-29 cells (Fig. 6n). R2 (−67 ~ −48 bp) was essential for the activation of FASN transcription by HMGA1, and inhibition of HMGA1 significantly reduced the binding of SREBP1 to FASN promoter in the ChIP qPCR assay (Fig. 6n). These results suggest that HMGA1 contributes to the binding of SREBP1 to FASN promoter and promotes its transcription.
HMGA1 binds to AT-rich regions in the minor groove of DNA through three basic AT-hook domains17. Next, we aimed to determine whether HMGA1 regulates the transcriptional activation of FASN through the AT-hook domains. We used a dual luciferase assay to detect the transcriptional activity of FASN promoters in cells transduced with wild-type or HMGA1 mutants containing different AT-hook ___domain deletions (Supplementary Fig. 27a). The results showed that deletions of AT-hook I (21-31aa) and AT-hook III (78-89aa) had almost no impact on the transcriptional activity of FASN promoter, whereas deletion of AT-hook II (53-62aa) significantly decreased the transcriptional activation of FASN (Supplementary Fig. 27b). Thus, we conclude that HMGA1 promotes the transcriptional activation of FASN through its AT-hook II ___domain.
Administration of high-fat diet eliminates the protection of HMGA1 depletion on Hmga1 △IEC mice from CRC tumorigenesis
In order to further clarify the role of lipid synthesis in HMGA1-involved tumorigenesis of CRC, we fed Hmga1flox/flox (control for the knockout) mice and Hmga1△IEC mice with high-fat diet (HFD) and induced murine CRC with AOM/DSS (Fig. 7a, d). HFD increased AOM/DSS-induced CRC tumorigenesis and decreased the survival of Hmga1flox/flox (control) mice (Fig. 7b, c and Supplementary Fig. 28a). The number and size of CRC in Hmga1flox/flox mice were increased due to the exposure to HFD. Hmga1△IEC mice induced with AOM/DSS bore much less and smaller CRC tumors as compared with those observed in Hmga1flox/flox (control) mice (Fig. 7b, c, e, f). However, administration of HFD eliminated the protection of HMGA1 depletion on Hmga1△IEC mice from CRC tumorigenesis and led AOM/DSS-induced CRC to the level occurred in Hmga1flox/flox mice (Fig. 7b, c, e, f and Supplementary Fig. 28b). All forms of the foci (LGD, HGD, and adenocarcinoma) in the colorectum of mice induced with AOM/DSS were increased due to the exposure of HFD regardless of HMGA1 state in the colorectum (Fig. 7g, i and Supplementary Fig. 29a). The expression of FASN was only marginally affected by HFD exposure in normal colorectal tissue (Supplementary Fig. 30a, b). However, FASN levels were markedly increased in the intestinal epithelia (CRCs) of mice treated with AOM/DSS for the induction of CRC, in particular in CRCs in mice fed with HFD (Supplementary Fig. 30). Compared to AOM/DSS-induced tumors generated in Hmga1△IEC mice fed with normal diet, tumors from mice co-treated with AOM/DSS and HFD bore a high level of Ki67 positive cells (Fig. 7h, j and Supplementary Fig. 29b). The results indicate that HFD supplementation promotes AOM/DSS-induced CRC tumorigenesis in Hmga1△IEC mice.

Hmga1flox/flox and Hmga1△IEC mice were treated with high fat diet (HFD) and AOM/DSS for the induction of CRC. a, d Schematic diagram of the induction. b, e Representative images of colorectum in the mice. c, f The number and size of tumors in the entire colorectum in each mouse were measured. g, i HE staining of the colorectum (CRC) from mice treated with control diet (CD), CD and AOM/DSS, HFD, and HFD and AOM/DSS (Scale bar = 20 μm. Representative images from 3 independent experiments). The percentage of LGD, HGD, and carcinoma was determined. LGD: low-grade dysplasia; HGD: high-grade dysplasia. h, j Representative images of IHC staining of Ki67 in murine colorectum (Scale bar = 20 μm). Data are presented as mean ± SD. Statistical significance was determined by two-tailed unpaired t-test (c, f, h, j). n = 12. Source data are provided as a Source Data file.
Inhibition of FASN-mediated lipid synthesis abrogates HMGA1-promoted tumorigenesis of CRC
Next, we detected the functional consequence of suppressing FASN-mediated lipid synthesis on HMGA1-promoted CRC. We treated AOM/DSS-induced or non-induced Hmga1IEC-OE/+ (knock-in) mice and Rosa26Hmga1/+ (control for knock-in) mice with orlistat (50 mg/kg), an inhibitor of FASN-mediated lipid synthesis, by gavage every other day (Fig. 8a, d). Surprisingly, the survival of Hmga1IEC-OE/+ mice induced with AOM/DSS was increased with the treatment of orlistat (Supplementary Fig. 31). The number and size of CRC in these mice were significantly decreased with the administration of orlistat (Fig. 8b, c, e, f). The proportions of LGD, HGD, and adenocarcinoma in these mice were all decreased (Fig. 8g, i and Supplementary Fig. 32a). The percentage of Ki67 positive cells in CRC (intestinal epithelia) from AOM/DSS-induced Hmga1IEC-OE/+ mice treated with orlistat was also reduced (Fig. 8h, j and Supplementary Fig. 32b), indicating that inhibition of FASN-mediated lipid synthesis suppresses CRC tumorigenesis in Hmga1IEC-OE/+ mice. It is worth noting that orlistat reduced CRC formation and development not only in Hmga1 gene knock-in mice (Hmga1IEC-OE/+), but also in Hmga1 gene normal background mice (Rosa26Hmga1/+) (Fig. 8), warranting an application of orlistat in the prevention and treatment of CRC. Taken all together, our data reveal that suppressing lipid synthesis could be an effective therapeutic method for CRC, in particular to those with elevated HMGA1.

Rosa26Hmga1/+ and Hmga1IEC-OE/+ mice were treated with AOM/DSS for the induction of CRC. Orlistat (50 mg/kg) was applied to the mice by gavage every other day for the entire duration in the CRC induction. a, d Schematic diagram of the induction and orlistat treatment. b, e Representative images of colorectum in the mice. c, f The number and size of tumors in the entire colorectum of each mouse were measured. g, i HE staining of colorectum (CRC) from mice treated with control (water), AOM/DSS, orlistat, and AOM/DSS and orlistat (Scale bar = 20 μm. Representative images from 3 independent experiments). The percentage of LGD, HGD, and carcinoma was determined. LGD, low-grade dysplasia; HGD, high-grade dysplasia. h, j Representative images of IHC staining of Ki67 in colorectum (CRC) from the mice (Scale bar = 20 μm). Data are presented as mean ± SD. Statistical significance was determined by two-tailed unpaired t-test (c, f, h, j). n = 12. Source data are provided as a Source Data file.
Discussion
Metabolic remodeling is a critical hallmark of tumors, with abnormal lipid metabolism supplying the energy required for malignant progression and accelerating CRC proliferation. Therefore, understanding the molecular mechanisms underlying dysregulated lipid metabolism in CRC may offer valuable therapeutic strategies36,37. In this study, we found HMGA1 was overexpressed in patients with CRC. HMGA1 promoted tumourigenesis in AOM/DSS-treated mouse CRC models although it did not induce neoplasias by itself but promoted tumourigenesis once mice were treated with the carcinogen AOM/DSS. We employed the AOM/DSS mouse model of CRC and found that HMGA1 drives CRC progression by activating FASN-mediated lipid synthesis. Our results demonstrated that HMGA1 is overexpressed in CRC and increases lipid accumulation within CRC cells. Genetically engineered HMGA1 mice further confirmed that HMGA1 promotes both the initiation and progression of CRC. We also identified that HMGA1 enhances de novo fatty acid synthesis in CRC cells by upregulating FASN transcription (Supplementary Fig. 33). These findings highlight the critical role of HMGA1 in reprogramming lipid metabolism in CRC and suggest it as a potential therapeutic target.
Belton et al. previously reported that HMGA1 transgenic mice develop polyposis, characterized by increased proliferation of IECs, though no neoplasia was detected, with only hyperplasia noted in their findings38. The level of HMGA1 overexpression observed in that study appears comparable to our current work. While Belton et al. demonstrated that HMGA1 promotes polyp formation—often considered an early stage of precancerous lesions—the study did not show progression to neoplasia. It is crucial to recognize that chronic colonic inflammation is a well-established risk factor for CRC, particularly in individuals with inflammatory bowel disease (IBD), who face a significantly increased risk of developing CRC39,40. In our study, the emergence of a neoplastic phenotype following DSS-induced colitis more accurately reflects the inflammation-driven carcinogenesis process seen in humans. The DSS treatment models chronic inflammation, and in combination with HMGA1 overexpression, likely drives the transition from hyperplasia to neoplasia, closely mirroring the inflammatory environment that fosters CRC development in humans.
HMGA1 regulates the transcription of key genes involved in glucose metabolism and plays a critical role in maintaining glucose homeostasis41. In gastric cancer, elevated HMGA1 levels promote glycolysis by directly upregulating c-Myc, a key transcription factor involved in aerobic glycolysis32. In CRC, caveolin-1, a scaffolding protein that is the primary component of caveolae plasma membranes, facilitates the translocation of HMGA1 from the cytoplasm to the nucleus, thereby enhancing aerobic glycolysis by stimulating GLUT3 transcription42. Williams et al.24,43 conducted metabolomics and bioinformatics analyzes, revealing that fatty acid content was elevated in fecal samples and highly proliferative colorectal polyps with HMGA1 overexpression, suggesting that HMGA1 may regulate fatty acid synthesis. To further investigate the mechanisms by which HMGA1 regulates fatty acid metabolism, we performed a metabolomics analysis on the intestinal epithelium of Hmga1△IEC and Hmga1flox/flox mice, identifying significant alterations in lipid metabolism-related pathways. RNA-seq analysis demonstrated a marked reduction in FASN, a key enzyme in de novo fatty acid synthesis, in the absence of HMGA1, supporting our hypothesis that HMGA1 promotes lipid accumulation by enhancing fatty acid synthesis.
De novo fatty acid synthesis is a critical step in lipid metabolism, essential for maintaining cellular functions. While most normal human tissues rely on exogenous (dietary) lipids to generate structural lipids, endogenous fatty acid synthesis is typically suppressed44. However, in tumor cells, even when exogenous lipids are available, the de novo fatty acid synthesis pathway is activated to meet the bioenergetic demands of rapid cell division45. These newly synthesized fatty acids support signal transduction, energy storage, and membrane synthesis46,47,48. Thus, HMGA1-driven de novo fatty acid synthesis may play a broad role in CRC tumorigenesis and progression.
To explore how HMGA1 upregulates lipid content in CRC cells, we performed RNA-seq analysis to identify lipid metabolism-related genes deregulated by HMGA1. A cluster of genes encoding key enzymes in de novo fatty acid synthesis, including ACLY, ACC1, FASN, and SCD, was significantly downregulated following HMGA1 knockdown. Among these, FASN exhibited the largest fold change (Fig. 5a). As a key biosynthetic enzyme, FASN catalyzes the production of fatty acids, driving their accumulation within cells. Previous studies have shown that FASN expression in CRC is regulated by various factors, and increased fatty acid synthesis promotes CRC development12,49,50,51. In this study, we demonstrated that HMGA1 is a critical factor for FASN upregulation, thereby enhancing lipid accumulation and contributing to CRC progression. This suggests that a detailed understanding of the signaling pathway through which HMGA1 influences CRC tumorigenesis is necessary.
Like HMGA1, FASN plays an essential role in embryonic development. Fasn-/- mutant mouse embryos die at early developmental stages, underscoring the enzyme’s importance. In post-embryonic life, FASN expression is relatively low in most tissues, except in the liver, adipose tissue, and lactating mammary glands, where it is highly expressed52,53. However, FASN is notably overexpressed in various tumors54. To investigate the molecular mechanisms by which HMGA1 regulates FASN, we first confirmed through co-immunoprecipitation (co-IP) assays that there is no direct interaction between HMGA1 and FASN at the protein level. However, dual-luciferase assays revealed that HMGA1 enhances FASN promoter activity, indicating transcriptional regulation. FASN transcription is regulated by various factors, with SREBP1 being a well-established transcription factor in lipid synthesis regulation. SREBP1 is synthesized in the endoplasmic reticulum, transported to the Golgi apparatus, and cleaved at the N-terminal to produce its active form, which then translocates to the nucleus to initiate the transcription of lipid synthesis genes35. Our co-IP assay showed a direct interaction between HMGA1 and SREBP1. Previous work by Zhu et al.55 identified that apelin induces HMGA1 deamidation, enhancing the binding affinity of the apelin-HMGA1-SREBP1 complex to regulate lipid metabolism and cell growth in non-small cell lung cancer (NSCLC). In our study, silencing SREBP1 abrogated HMGA1-enhanced FASN promoter activity, supporting the idea that SREBP1 mediates HMGA1’s regulation of FASN and subsequent lipid accumulation.
Orlistat, an FDA-approved drug for obesity prevention, irreversibly inhibits the thioesterase (TE) ___domain of FASN, thereby blocking its activity56. In our study, orlistat reversed HMGA1-driven CRC progression in murine models. Notably, orlistat not only reduced CRC formation and progression in HMGA1-knockin mice (Hmga1IEC-OE/+) but also significantly suppressed CRC tumorigenesis in HMGA1-normal mice (Rosa26Hmga1/+) (Fig. 8). These findings suggest that orlistat could have broader applications in CRC treatment. Future studies should explore the therapeutic potential of orlistat in CRC patients, particularly those at high risk, such as individuals with polyps exhibiting elevated HMGA1 expression. Expanding the use of orlistat for CRC prevention in these high-risk populations is also warranted.
Despite significant progress in understanding the mechanisms by which HMGA1 drives malignancy, the upstream regulators governing its activation remain largely unknown. Bush et al.57 previously reported that HMGA1 is markedly elevated in intestinal tumors of adenomatous polyposis coli (APCMin/+) mice compared to normal intestinal mucosa. Inhibition of Wnt signaling through enforced APC expression led to the downregulation of HMGA1 in CRC cells. Since APC mutations are linked to the mobilization of the beta-catenin/TCF-4 transcriptional complex and the subsequent activation of downstream oncogenic targets, the authors analyzed the 5’-flanking region of HMGA1 and identified two specific regions that bind the beta-catenin/TCF-4 complex both in vitro and in vivo. This established HMGA1 as a direct target of the beta-catenin/TCF-4 signaling pathway in CRC. These findings strongly implicate Wnt/beta-catenin/TCF-4 signaling in the regulation of HMGA1. Additionally, Wnt/beta-catenin/TCF-4 signaling also activates the transcription of c-Myc, a known transcriptional activator of HMGA1, raising the possibility that HMGA1 activation by the Wnt/beta-catenin/TCF-4 pathway may be, in part, mediated through c-Myc. This notion is supported by Akaboshi et al., who demonstrated that HMGA1 expression is reduced in gastric tissue upon c-Myc knockdown58. Thus, it is plausible that APC mutations, at least partially, drive HMGA1 upregulation.
In colon cancer, APC inactivation and deregulation of associated transcription factors are among the most common disease drivers. APC mutations initiate the transition of cells into a hyperproliferative state, and ~80% of CRCs arise from APC inactivation due to either germline or sporadic mutations59. The new classification system categorizes CRC into four consensus molecular subtypes (CMSs). Among these, CMS2, CMS3, and CMS4 exhibit a higher frequency of APC mutations (over 50%) compared to CMS1, making them more commonly associated with APC-mutated CRC. Therefore, we hypothesize that HMGA1 is likely to be more highly expressed in CMS2, CMS3, and CMS4 CRCs.
Chromosomal imbalances are critical drivers of malignant transformation and cancer progression. DNA copy number aberrations, including the formation of isochromosomes, oncogene overexpression, and tumor suppressor loss, are frequently observed in tumors. For instance, chromosomal translocations have been identified as causative factors for the misexpression of HMGA2, another member of the HMGA family, in mesenchymal tumors60,61,62. Similarly, chromosomal aberrations at 6p21-23, where HMGA1 (HMGI-Y) is located, have been implicated in benign mesenchymal tumors63,64, suggesting that DNA copy number aberrations may contribute to HMGA-driven tumorigenesis. Copy number gains affecting chromosome 6p have also been detected in various cancers and are associated with more aggressive or metastatic disease, poor prognosis, and venous invasion in colorectal, bladder, ovarian, and hepatocellular carcinomas, as well as in large B-cell lymphoma and melanoma65. Notably, most of these gains map to the 6p21-p23 region, where HMGA1 resides (6p21.31). Although the specific role of HMGA1 in CRC predisposition due to chromosome 6p alterations is not yet fully established, increased copy numbers of 6p have been linked to advanced CRC stages (e.g., Dukes’ stage D) and metastasis. A comparative genomic hybridization (CGH) study revealed that 41% of CRC patients with liver metastasis exhibit gains at 6p2165. These findings suggest that chromosome 6p gains may contribute to CRC progression, and further characterization of genes in this region, such as HMGA1, could offer valuable insights into CRC tumorigenesis mechanisms.
Cancer cells may revert to a progenitor-like state through dedifferentiation, evading the typical process of differentiation and shedding during CRC development. This dedifferentiation can occur when partially differentiated cells revert to a less differentiated state or when the differentiation of colon stem cells is blocked, as both cell types have the potential to give rise to colon cancer. In human embryonic stem cells, HMGA1 maintains a dedifferentiated state by regulating genes associated with stemness and is crucial for reprogramming somatic cells into induced pluripotent stem cells (iPSCs)66,67,68. While no comprehensive studies have specifically examined HMGA1’s role in the dedifferentiation of colorectal epithelial cells in CRC, existing evidence strongly supports HMGA1’s involvement in promoting stem-like characteristics in poorly differentiated tumors and stem cells68,69. HMGA1 is aberrantly expressed in many cancers, where it drives tumor progression and worsens prognosis by activating stem cell transcription networks. It promotes the expression of key pluripotency genes, such as SOX2 and cMYC, and its knockdown reduces the expression of these genes69. Additionally, HMGA1 interacts with other key transcriptional regulators, such as STAT3, to maintain cancer stem cell properties, highlighting its dual role in both normal development and cancer progression70.
In summary, our study demonstrated that HMGA1 is upregulated in CRC, where its elevated expression accelerates CRC progression by promoting lipid synthesis. FASN was identified as a key gene regulated by HMGA1 in this process, with HMGA1 enhancing FASN expression through SREBP1-mediated transcription. Inhibition of FASN reversed HMGA1-induced lipid accumulation and cell proliferation, ultimately slowing CRC development. These findings suggest that targeting the HMGA1-FASN axis could be a promising therapeutic strategy for the prevention and treatment of CRC.
Methods
Ethics statement
All animal protocols in this study were approved by the Institutional Animal Care and Use Committee (IACUC) at Henan University, China, and were conducted in accordance with institutional guidelines and the Laboratory Animal Center of Henan University. The study complies with ethical regulations for research involving animals and human subjects. The collection and use of clinical materials and human samples were approved by the Institutional Research Ethics Committee of the People’s Hospital of Anyang City.
Clinical samples
CRC and adjacent colorectal tissues were obtained from patients at the People’s Hospital of Anyang City, who had not undergone radiotherapy or chemotherapy prior to tumor resection. Normal tissues adjacent to the CRCs were collected at least 5 cm from the tumor margin, following surgical guidelines. The tissue samples were fixed in formalin and embedded in paraffin for subsequent experiments. Pathological diagnoses were confirmed by two pathologists. Low-grade dysplasia (LGD) was defined as low-grade adenoma/intraepithelial neoplasia, while high-grade dysplasia (HGD) included high-grade adenoma/intraepithelial neoplasia, carcinoma in situ, suspicion of invasive carcinoma, and intramucosal carcinoma. Adenocarcinoma was characterized by submucosal tumor invasion71. We used de-identified CRC tissue samples solely for immunohistochemistry (IHC) staining, and no patient data, including demographic or clinical information, was collected or used. As such, no compensation was provided to participants, and no sex-based stratification or analysis was performed, as the study only focused on the tissues and not on patient-specific information.
This study was approved by the Human Medical Ethics Committee of Henan University, and informed consent was obtained from all patients for the use of their tissue samples.
HMGA1 genetically engineered mice
HMGA1KI/KIPVillin-cre+ (Hmga1IEC-OE/+) conditional knock-in and HMGA1flox/floxPVillin-Cre (Hmga1ΔIEC) conditional knockout mice were C57BL/6 J background and originally ordered from Cyagen (Suzhou, China) and maintained in the laboratory for crossbreeding.
Induction of CRC in mice
Experiments involving mice were conducted using the C57BL/6 J genetic background. All mice were housed in an animal barrier facility at Henan University under a 12-hour light/dark cycle, with food and water provided ad libitum. Male and female mice (6–8 weeks old, n = 6 per sex) were selected for the induction of CRC using azoxymethane/dextran sulfate sodium (AOM/DSS). Mice received an intraperitoneal injection of AOM (Sigma-Aldrich, St. Louis, MO, USA) at 10 mg/kg body weight. One week later, they were given 2% (w/v) DSS (Meilunbio, Dalian, China) in drinking water for 5 days, followed by regular water for 14 days. This sequence, termed a DSS cycle, was repeated three times. Body weight and stool were continuously monitored until euthanasia at week 12. For the survival assay, 12 mice (6 males and 6 females) in each group were treated with AOM/DSS (water as control) and observed for 5 months post-treatment. Since orthotopic colorectal tumors induced by AOM/DSS were not directly visible during the mice’s lifetime, we determined the timing of sacrifice based on weight loss and behavioral observations. Mice were euthanized when they lost 20% of their pre-experiment body weight, showed signs of severe debilitation, or were on the verge of death, such as being unable to move, and/or body condition scoring (BCS)72 reached 2.0. Distal colon tissues were collected, and visible tumors on the colorectal mucosa were counted. The weight and length of the colorectum were measured, photographed, and fixed in 4% formalin for histological analysis.
High-fat diet (HFD, Cat# XTHF60, Jiangsu Xietong Pharmaceutical Bio-engineering Co., Ltd., Nanjing, China) and control diet (Cat# AIN-93M) were used. The HFD contained 60% kcal from fat, 20% kcal from protein, and 20% kcal from carbohydrates, while the control diet consisted of 9.4% kcal from fat, 14.7% kcal from protein, and 75.9% kcal from carbohydrates73. Weight and food intake were recorded every three days.
Cell culture
The human colon cancer cell lines HT-29, HCT116, and LoVo were derived from the American Type Culture Collection (ATCC) and cultured in RPMI1640 (Procell, Wuhan, China) with 10% fetal bovine serum (FBS, Lonza, Basel, Switzerland) and 1% penicillin / streptomycin (Solarbio, Beijing, China). Cells were maintained in the cell incubator with 5% CO2 and a humidified atmosphere at 37 °C.
Lentiviral transfection
shHMGA1 plasmids were purchased from Genepharma (Shanghai, China). LentiCRISPRv3-HA-FLAG-HMGA1 plasmid containing HMGA1 cDNA full-length was constructed in the lab. Target plasmids were mixed with the packaging plasmid psPAX.2 and the envelope plasmid pMD2.G in a ratio of 4:3:1 and transfected into HEK-293T cells with Lipofectamine 2000 (Invitrogen, Carlsad, CA, USA). After 48 h, the conditioned medium containing viral particles was collected and filtered through 0.45 µm filter. HT-29, HCT116, and LoVo cells were incubated with the conditioned medium containing virus particles and polybrene (8 μg / mL) (Solarbio, Beijing, China) for 24 h, and then selected with puromycin (2.5 μg / mL) (Solarbio, Beijing, China) for 2 weeks for the establishment of stable cell lines. qRT-PCR and Western blot were used for confirming the knockdown or overexpression efficiency.
Real-time PCR assay
Total RNAs were extracted using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA) and measured on a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). cDNA synthesis was performed with HiScript III RT SuperMix Kit (Vazyme, Nanjing, China). Real-time quantitative PCR was carried out using ChamQ Universal SYBR qPCR Master Mix (Vazyme, Nanjing, China) on a Lightcycler 480 II Detection System (Roche, Mannheim, Germany). After normalizing with β-actin gene as internal control, the relative mRNA was quantified by 2−∆∆Ct method74. Primer sequences for the qPCR were listed in Supplementary Table 1.
Western blotting
Total proteins were extracted using RIPA buffer containing protease inhibitors and phosphatase inhibitors. Protein concentration was measured with a BCA kit (Solarbio, Beijing, China). The total proteins were separated by electrophoresis using SDS-PAGE gel and transferred to PVDF membrane (Millipore, Temecula, CA, USA). Specific antibodies were used for the detection of target proteins. ECL chemiluminescence kit (Biosharp, Beijing, China) was applied for the development of the membrane. The antibodies used in this study were listed on Supplementary Table 1.
Co-immunoprecipitation assay
The cell lysates (500 μg total proteins) were mixed with antibody (3 μg) or corresponding IgG (negative control) and incubated on a shaker at 4 °C for overnight. On the next day, 20 μL protein A/G agarose beads (Santa Cruz Biotech, Santa Cruz, CA, USA) were added to the mixture and incubated at 4 °C for 4 h. Agarose beads were collected by centrifuging at 4 °C with 13,500 × g for 1 min and washed with washing buffer (1/3 lysis buffer and 2/3 PBS) for 4 times with 5 min for each time. 1 × SDS-PAGE protein sample loading buffer was then added to the precipitation and boiled at 100 °C for 5 min for the following immunoblot analysis.
Cell proliferation assay
CRC cells were inoculated into 96-well plates (2000 cells per well) and treated differently. Cell count kit-8 (CCK8, Biosharp, Beijing, China) was used to detect the proliferation of CRC cells.
Colony formation assay
CRC cells were inoculated into 6-well plates at a density of 600 cells per well. The cells were cultured for 2 weeks. Cells were then fixed with 4% paraformaldehyde for 30 min, and then stained with 0.1% crystal violet (Solarbio, Beijing, China). Colonies with 50 or more cells were counted.
The detection of triglycerides, total cholesterol and free fatty acids
CRC cells with different treatments were collected and counted. 1 × 106 cells were suspended in 200 μL of PBS and treated with the ultrasound. The homogenate obtained was used for subsequent detection of cellular lipids. The triglycerides, total cholesterol, and free fatty acids were then measured with corresponding assay kits from Solarbio Science & Technology (Beijing, China) according to the manufacturer’s instructions. The protein concentration in the homogenate was detected using a BCA Protein Assay Kit (Solarbio, Beijing, China) and applied for the normalization of the lipids.
Oil red O staining
CRC cells were seeded into 6-well plates and cultured for overnight. Cells were washed with freshly prepared PBS and fixed with 4% paraformaldehyde. The cells were then stained with oil red O working solution (Solarbio, Beijing, China) for 30 min at room temperature. After staining, the cells were washed with PBS for three times. Frozen sections from murine orthotopic CRC were also stained with oil red O solutions as described above.
Nile red staining
CRC cells were fixed with 4% paraformaldehyde, and then stained with nile red dye (Solarbio, Beijing, China) at a final concentration of 1 μM. DAPI (UElandy, Suzhou, China) was used to counterstain the nucleus. The images were captured using a confocal laser scanning system (Carl Zeiss, Oberkochene, Germany).
[U-13C] glucose labeled lipid mass spectrometry
HMGA1-overexpression, HMAG1-knockdown and control HT-29 cells were cultured in complete medium for 24 h. Standard complete media were replaced by glucose free DMEM, which [U-13C6] glucose (2 g/L) were added in. Cells were cultured for 6 h to allow the isotope to be incorporated into metabolites. The medium was then discarded and cells were washed with 2 mL PBS. 1 mL pre-cooled mixture (400 µL carbinol, 400 µL acetonitrile and 200 µL H2O) was added, and cells were scraped off from the cell culture dish. The samples were applied for the mass spectrometry analysis75 conducted by Shanghai Biotree Biotechnology Co., LTD. (Shanghai, China).
Isolation and culture of colonic epithelial cells from HMGA1 genetically engineered mice
The mice were euthanized, washed with pre-cooled PBS, and dissected longitudinally. Colon tissues of the mice were isolated and cut into 0.5 - 1.0 cm fragments and flushed 15–20 times with pre-cooled PBS followed by incubation in 20 mL HBSS containing 2% fetal bovine serum, 1 mM EDTA, and 1 mM DTT in a 50 mL centrifugal tube with constant shaking at 200 rpm for 20 min at 37 °C. The washed colon tissue pieces were transferred into a new tube containing 20 mL HBSS buffer and incubated for another 20 min. The colon tissues were cleaned again with 5 mL DMEM medium containing 10% FBS and then transfered to a tube with 5 mL DMEM containing 10% FBS, 50 U/mL collagenase type VIII, and 50 U/mL DNase I, and incubated at 37 °C for 50 min with constant shaking at 200 rpm. The digested tissue fragments were swirled for 30 s and the undigested tissue was removed through a 40 μm filter. The filtered cell suspension was centrifugated at 410 × g for 5 min at 4 °C and then re-suspended in 10 mL DMEM containing 15% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin (Thermo Fisher Scientific) for further culture76.
Untargeted metabolomics analysis
IECs isolated from Hmga1flox/flox (control for the knockout) and Hmga1△IEC (knockout) mice and CRCs from Rosa26Hmga1/+ (control for the knock-in) and Hmga1IEC-OE/+ (knock-in) mice induced with AOM/DSS were used for the metabolomics and MS spectra. Three biologically independent samples were applied in each group. Both the metabolomics and the MS spectra were processed by Metware Biotechnology Co., Ltd (Wuhan, China). Briefly, Acquit Ultra Performance LC I-class system (Waters Corp. Milford, MA) connected to a Waters Synapt G2-Si QTOF MS was applied for UHPLC-QTOF/MS assay. Sample was separated using a Waters Acquity premier HSS T3 Column (1.8 μm, 2.1 mm × 100 mm) maintained at 40 °C, a flow rate of 0.4 mL/min, and the injection volume of 2 μL. The raw data acquired by LC-MS were converted into mzXML format by ProteoWizard software77. All data were analyzed using XCMS program for peak picking, alignment, and normalization. Identified metabolites were annotated using KEGG Compound database (http://www.kegg.jp/kegg/compound/). Annotated metabolites were then mapped to KEGG pathway database (http://www.kegg.jp/kegg/pathway.html).
Assay for transposase accessible chromatin with high-throughput sequencing
HMGA1 silencing and control CRC cells were used for the ATAC sequencing by Frasergen Biotechnology Co., Ltd (Wuhan, China). Sequencing depth, sequencing error rate, and fragment length distribution were analyzed with the FastQC tool for validating the quality of ATAC-seq data. The raw sequencing data was preprocessed to remove low-quality sequences and duplicated reads. The ATAC-seq data were then analyzed as follows. The preprocessed reads were compared with the reference genome using the alignment tool Bowtie2. The peak calling algorithms (MACS2) was used to identify open chromatin regions from sequencing data, which represented accessible DNA regions. The genomic characterization of the identified open chromatin regions was annotated using ChIPseeker, contributing to determine the region’s association with genes and potential functional roles. The enrichment of specific functions was determined by enrichment analysis. The Visualization Tool (IGV 2.5.3) was used to visualize the results of the ATAC-seq data, showing peak heights, the distribution of open chromatin regions, and other relevant information78.
Dual-luciferase report assay
CRC cells (7 × 103/well) were planted into 96-well plates and transfected with 50 ng pGL3 control or target plasmid and 50 ng Renilla A plasmid per well for 48 h. The dual luciferase reporter gene activity was determined by the Duo-Lite Luciferase Assay System (Vazyme, Nanjing, China). The Renilla luciferase luminescence was used as a quantitation control.
Chromatin immunoprecipitation assay
Proliferating cells were cross-linked with 1% formaldehyde at 37 °C for 10 min, and the glycine solution was added to a final concentration of 0.125 M to terminate the cross-linking. Cells were then collected and lysed with SDS lysis buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA, 1% SDS). The crosslinked DNAs were broken into 200–500 bp fragments by ultrasound. Protein A/G agarose beads were then added to the pre-cleared lysates at 4 °C for 2 h. SREBP1 antibody or IgG was added to the lysates and incubated for overnight on a 4 °C shaker. After eluting the complex of immunoprecipitation and reversing the crosslinking, the purified DNA was quantified with qPCR. The PCR primers are listed on the Supplementary Table 1.
Haematoxylin - eosin (HE) staining and immunohistochemistry
The mice colorectal tissues were fixed in formalin and embedded in paraffin, then cut into 5 μm-thick sections. The histological morphology was observed by HE staining. Pathologic criteria used to define LGD versus HGD are as follows. Normal means normal colonic mucosa. LGD is characterized by simple glandular architecture. The neoplastic crypt cells are elongated and dense, with deeply stained nuclei, but maintain polarity relative to the basement membrane. HGD is characterized by more pronounced nuclear atypia, with complex glandular structures such as a cribriform appearance, and loss of epithelial nuclear polarity. Carcinomas are malignant tumors of the glandular epithelium consisting of tubular and/or villous structures that penetrate the mucous membrane of the muscularis79,80.
For immunohistochemical staining, the tissue sections were dewaxed and hydrated, and the antigen was repaired by 0.01 mol/L citric acid antigen repairing solution. The tissue slides were blocked by 1% BSA at room temperature for 1 h and incubated overnight with specific primary antibody at 4 °C. The Dako Envision HRP / DAB system (Dako, Heverlee, Belgium) was used to detect the slices according to the manufacturer’s instructions. Finally, the nucleus was stained with hematoxylin. The stained sections were photographed with AxioScopeA1 microscope (Zeiss, Germany) at 40× magnification. The staining intensity was defined as: negative (intensity score, 0), weak (1), medium (2), and strong (3), and the sum of staining scores per 100 colonic epithelial cells was calculated79.
Immunofluorescence staining
CRC cells were cultured on cover slides and fixed with 4% paraformaldehyde at room temperature for 30 min. The cells were blocked with 1% BSA at room temperature for 1 h and then incubated with mouse anti-HMGA1 monoclonal antibody (1: 200) and rabbit anti-SREBP1 polyclonal antibody (1: 200) at 4 °C for overnight. The cells were incubated with goat anti-mouse Alexa Fluor 647 antibody (Abcam, Cambridge, UK) and goat anti-rabbit Alexa Fluor 488 (Abcam, Cambridge, UK) at room temperature in darkness for 1 h. The nucleus was re-stained with DAPI dye. For immunofluorescence staining of tissues, paraffin-embedded tissues were dewaxed and hydrated, and then the antigen was repaired with 0.01 mol/L citric acid antigen repairing solution. These slides were blocked with 1% BSA at room temperature for 1 h, and the subsequent antibody incubation steps were carried out to complete the immunofluorescent experiment. The fluorescence microscope images were captured by confocal laser scanning microscope system (CarlZeiss, Oberkochene, Germany) and analyzed by ZEN (version 2012).
Statistics and reproducibility
Statistical analyzes were performed by using Student’s t-test, one-way ANOVA or two-way ANOVA with GraphPad Prism 8.0 software. The experimental data were presented as mean ± SD. Survival curves were plotted using the Kaplan-Meier method and compared by log-rank test. P < 0.05 was statistically significant. RNA sequencing and metabolic profiling assays were conducted in three biological replicates. All experiments requiring statistical analysis were repeated at least three times with similar results. All repeated data presented are from the biological replicates with 3 technical replicates in each biological replicate. No statistical method was used to predetermine sample size. No data were excluded from the analyzes. The experiments were not randomized, except that mice were randomly grouped before different treatments. Western blot and fluorescence images are shown as representatives of three or more repeated experiments with similar results. Uncropped versions of western blots are presented in the Source Data file.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq data generated in this study have been deposited in GEO under the accession number GSE247894. GEPIA was used for HMGA1 expression analysis. RNA-seq data in GEO under the accession number GSE44076 and GSE41657 were analyzed for the expression of HMGA1. Metabolomics data were included in Supplementary Data 1 and Supplementary Data 2, and deposited in the MetaboLights database81 under accession code MTBLS11471 and MTBLS11495. All data supporting current study are provided in the article, Supplementary Information, and Source data. Source data are provided with this paper.
References
Xia, L. et al. The cancer metabolic reprogramming and immune response. Mol. Cancer 20, 28 (2021).
Li, C. et al. Association between abnormal lipid metabolism and tumor. Front. Endocrinol. (Lausanne) 14, 1134154 (2023).
Shuvalov, O., Daks, A., Fedorova, O., Petukhov, A. & Barlev, N. Linking metabolic reprogramming, plasticity and tumor progression. Cancers (Basel) 13, 762 (2021).
Ye, L., Li, Y., Zhang, S., Wang, J. & Lei, B. Exosomes-regulated lipid metabolism in tumorigenesis and cancer progression. Cytokine Growth Factor Rev. 73, 27–39 (2023).
Wang, W., Bai, L., Li, W. & Cui, J. The lipid metabolic landscape of cancers and new therapeutic perspectives. Front Oncol. 10, 605154 (2020).
Broadfield, L. A., Pane, A. A., Talebi, A., Swinnen, J. V. & Fendt, S. M. Lipid metabolism in cancer: New perspectives and emerging mechanisms. Dev. Cell 56, 1363–1393 (2021).
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74, 229–263 (2024).
Low, E. E. et al. Risk factors for early-onset colorectal cancer. Gastroenterology 159, 492–501.e497 (2020).
Kastrinos, F., Kupfer, S. S. & Gupta, S. Colorectal cancer risk assessment and precision approaches to screening: brave new world or worlds apart? Gastroenterology 164, 812–827 (2023).
Bian, X. et al. Lipid metabolism and cancer. J. Exp. Med. 218, e20201606 (2021).
Ecker, J. et al. The colorectal cancer lipidome: identification of a robust tumor-specific lipid species signature. Gastroenterology 161, 910–923.e919 (2021).
Ye, M. et al. FABP5 suppresses colorectal cancer progression via mTOR-mediated autophagy by decreasing FASN expression. Int. J. Biol. Sci. 19, 3115–3127 (2023).
Yang, Y. et al. circCAPRIN1 interacts with STAT2 to promote tumor progression and lipid synthesis via upregulating ACC1 expression in colorectal cancer. Cancer Commun. (Lond.) 43, 100–122 (2023).
Dai, W. et al. PTPRO represses colorectal cancer tumorigenesis and progression by reprogramming fatty acid metabolism. Cancer Commun. (Lond.) 42, 848–867 (2022).
Saed, L. & Jeleń, A. Prognostic significance of HMGA1 expression in lung cancer based on bioinformatics analysis. Int. J. Mol. Sci. 23, 6933 (2022).
Vignali, R. & Marracci, S. HMGA genes and proteins in development and evolution. Int. J. Mol. Sci. 21, 654 (2020).
De Martino, M. & Esposito, F. Critical role of the high mobility group a proteins in hematological malignancies. Hematol. Oncol. 40, 2–10 (2022).
Sgarra, R. et al. High mobility group A (HMGA) proteins: molecular instigators of breast cancer onset and progression. Biochim. Biophys. Acta Rev. Cancer 1869, 216–229 (2018). 19.
Zanin, R. et al. HMGA1 promotes breast cancer angiogenesis supporting the stability, nuclear localization and transcriptional activity of FOXM1. J. Exp. Clin. Cancer Res. 38, 313 (2019).
Chia, L. et al. HMGA1 induces FGF19 to drive pancreatic carcinogenesis and stroma formation. J. Clin. Invest. 133, e151601 (2023).
Zhong, J. et al. TGF-β1 induces HMGA1 expression: The role of HMGA1 in thyroid cancer proliferation and invasion. Int. J. Oncol. 50, 1567–1578 (2017).
Pallante, P., Sepe, R., Puca, F. & Fusco, A. High mobility group a proteins as tumor markers. Front. Med. (Lausanne) 2, 15 (2015).
Foti, D. et al. Lack of the architectural factor HMGA1 causes insulin resistance and diabetes in human and mice. Nat. Med. 7, 765–773 (2005).
Williams, M. D. et al. HMGA1 drives metabolic reprogramming of intestinal epithelium during hyperproliferation, polyposis, and colorectal carcinogenesis. J. Proteome Res. 14, 1420–1431 (2015).
Federico, A. et al. Hmga1/Hmga2 double knock-out mice display a “superpygmy” phenotype. Biol. Open 3, 372–378 (2014).
Zhou, M. et al. N(6)-methyladenosine modification of REG1α facilitates colorectal cancer progression via β-catenin/MYC/LDHA axis mediated glycolytic reprogramming. Cell Death Dis. 14, 557 (2023).
Tracz-Gaszewska, Z., Sowka, A. & Dobrzyn, P. Stearoyl-CoA desaturase 1 inhibition impairs triacylglycerol accumulation and lipid droplet formation in colorectal cancer cells. J. Cell Physiol. 238, 2888–2903 (2023).
Xu, C. & Gu, L. FADS1-arachidonic acid axis enhances arachidonic acid metabolism by altering intestinal microecology in colorectal cancer. Nat. Commun. 14, 2042 (2023).
Yao, Z. et al. Upregulation of WDR6 drives hepatic de novo lipogenesis in insulin resistance in mice. Nat. Metab. 5, 1706–1725 (2023).
Shang, C. et al. The prognostic miR-532-5p-correlated ceRNA-mediated lipid droplet accumulation drives nodal metastasis of cervical cancer. J. Adv. Res. 37, 169–184 (2022).
Bartolacci, C. et al. Targeting de novo lipogenesis and the Lands cycle induces ferroptosis in KRAS-mutant lung cancer. Nat. Commun. 13, 4327 (2022).
Cao, X. P., Cao, Y., Zhao, H., Yin, J. & Hou, P. HMGA1 promoting gastric cancer oncogenic and glycolytic phenotypes by regulating c-myc expression. Biochem. Biophys. Res. Commun. 516, 457–465 (2019).
Jia, J. et al. Pivotal Role of Fatty acid synthase in c-MYC driven hepatocarcinogenesis. Int. J. Mol. Sci. 21, 8467 (2020).
Chiefari, E. et al. HMGA1 is a novel downstream nuclear target of the insulin receptor signaling pathway. Sci. Rep. 2, 251 (2012).
Zhao, Q., Lin, X. & Wang, G. Targeting SREBP-1-mediated lipogenesis as potential strategies for cancer. Front. Oncol. 12, 952371 (2022).
Aguirre-Portolés, C., Fernández, L. P. & Ramírez de Molina, A. Precision nutrition for targeting lipid metabolism in colorectal cancer. Nutrients 9, 1076 (2017).
Salita, T. & Rustam, Y. H. Reprogrammed lipid metabolism and the lipid-associated hallmarks of colorectal cancer. Cancers (Basel) 14, 3714 (2022).
Belton, A. et al. HMGA1 induces intestinal polyposis in transgenic mice and drives tumor progression and stem cell properties in colon cancer cells. PLoS One 7, e30034 (2012).
Chatila, W. K. et al. Integrated clinical and genomic analysis identifies driver events and molecular evolution of colitis-associated cancers. Nat. Commun. 14, 110 (2023).
Lee, M. et al. Protein stabilization of ITF2 by NF-κB prevents colitis-associated cancer development. Nat. Commun. 14, 2363 (2023).
Chiefari, E. et al. Cross-talk among HMGA1 and FoxO1 in control of nuclear insulin signaling. Sci. Rep. 8, 8540 (2018).
Ha, T. K. et al. Caveolin-1 increases aerobic glycolysis in colorectal cancers by stimulating HMGA1-mediated GLUT3 transcription. Cancer Res. 72, 4097–4109 (2012).
Williams, M. D. et al. Fecal metabolome in Hmga1 transgenic mice with polyposis: evidence for potential screen for early detection of precursor lesions in colorectal cancer. J. Proteome Res. 15, 4176–4187 (2016).
Zhang, J., Zou, S. & Fang, L. Metabolic reprogramming in colorectal cancer: regulatory networks and therapy. Cell Biosci. 13, 25 (2023).
Mashima, T., Seimiya, H. & Tsuruo, T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br. J. Cancer 100, 1369–1372 (2009).
Sena, L. A. & Denmeade, S. R. Fatty acid synthesis in prostate cancer: vulnerability or epiphenomenon? Cancer Res. 81, 4385–4393 (2021).
Koundouros, N. & Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 122, 4–22 (2020).
Yoon, H., Shaw, J. L., Haigis, M. C. & Greka, A. Lipid metabolism in sickness and in health: emerging regulators of lipotoxicity. Mol. Cell 81, 3708–3730 (2021).
Wei, W. et al. FBXW7β loss-of-function enhances FASN-mediated lipogenesis and promotes colorectal cancer growth. Signal Transduct. Target Ther. 8, 187 (2023).
Han, Z. et al. Derepression of the USP22-FASN axis by p53 loss under oxidative stress drives lipogenesis and tumorigenesis. Cell Death Discov. 8, 445 (2022).
Wang, H. et al. The lncRNA ZFAS1 regulates lipogenesis in colorectal cancer by binding polyadenylate-binding protein 2 to stabilize SREBP1 mRNA. Mol. Ther. Nucleic Acids 27, 363–374 (2022).
Chirala, S. S. et al. Fatty acid synthesis is essential in embryonic development: fatty acid synthase null mutants and most of the heterozygotes die in utero. Proc. Natl Acad. Sci. USA 100, 6358–6363 (2003).
Fhu, C. W. & Ali, A. Fatty acid synthase: an emerging target in cancer. Molecules 25, 3935 (2020).
Buckley, D. et al. Fatty acid synthase - Modern tumor cell biology insights into a classical oncology target. Pharmacol. Ther. 177, 23–31 (2017).
Zhu, Y. et al. Apelin-mediated deamidation of HMGA1 promotes tumorigenesis by enhancing SREBP1 activity and lipid synthesis. Cancer Sci. 113, 3722–3734 (2022).
Schcolnik-Cabrera, A. et al. Orlistat as a FASN inhibitor and multitargeted agent for cancer therapy. Expert Opin. Investig. Drugs 27, 475–489 (2018).
Bush, B. M., Brock, A. T., Deng, J. A., Nelson, R. A. & Sumter, T. F. The Wnt/β-catenin/T-cell factor 4 pathway up-regulates high-mobility group A1 expression in colon cancer. Cell Biochem. Funct. 31, 228–236 (2013).
Akaboshi, S. et al. HMGA1 is induced by Wnt/beta-catenin pathway and maintains cell proliferation in gastric cancer. Am. J. Pathol. 175, 1675–1685 (2009).
Adebayo, A. S., Agbaje, K., Adesina, S. K. & Olajubutu, O. Colorectal cancer: disease process, current treatment options, and future perspectives. Pharmaceutics 15, 2620 (2023).
Zaidi, M. R., Okada, Y. & Chada, K. K. Misexpression of full-length HMGA2 induces benign mesenchymal tumors in mice. Cancer Res. 66, 7453–7459 (2006).
Panagopoulos, I., Gorunova, L., Bjerkehagen, B., Lobmaier, I. & Heim, S. The recurrent chromosomal translocation t(12;18)(q14~15;q12~21) causes the fusion gene HMGA2-SETBP1 and HMGA2 expression in lipoma and Osteochondrolipoma. Int. J. Oncol. 47, 884–890 (2015).
Bartuma, H. et al. Expression levels of HMGA2 in adipocytic tumors correlate with morphologic and cytogenetic subgroups. Mol. Cancer 8, 36 (2009).
Tkachenko, A., Ashar, H. R., Meloni, A. M., Sandberg, A. A. & Chada, K. K. Misexpression of disrupted HMGI architectural factors activates alternative pathways of tumorigenesis. Cancer Res. 57, 2276–2280 (1997).
Kazmierczak, B. et al. HMGIY is the target of 6p21.3 rearrangements in various benign mesenchymal tumors. Genes Chromosomes Cancer 23, 279–285 (1998).
Santos, G. C., Zielenska, M., Prasad, M. & Squire, J. A. Chromosome 6p amplification and cancer progression. J. Clin. Pathol. 60, 1–7 (2007).
Li, L. et al. HMGA1 chromatin regulators induce transcriptional networks involved in GATA2 and proliferation during MPN progression. Blood 139, 2797–2815 (2022).
Parisi, S., Piscitelli, S., Passaro, F. & Russo, T. HMGA proteins in stemness and differentiation of embryonic and adult stem cells. Int. J. Mol. Sci. 21, 362 (2020).
Xian, L. et al. HMGA1 amplifies Wnt signalling and expands the intestinal stem cell compartment and Paneth cell niche. Nat. Commun. 8, 15008 (2017).
Shah, S. N. et al. HMGA1 reprograms somatic cells into pluripotent stem cells by inducing stem cell transcriptional networks. PLoS One 7, e48533 (2012).
Belton, A. et al. STAT3 inhibitor has potent antitumor activity in B-lineage acute lymphoblastic leukemia cells overexpressing the high mobility group A1 (HMGA1)-STAT3 pathway. Leuk. Lymphoma 57, 2681–2684 (2016).
Stolte, M. The new Vienna classification of epithelial neoplasia of the gastrointestinal tract: advantages and disadvantages. Virchows Arch. 442, 99–106 (2003).
Ullman-Culleré, M. H. & Foltz, C. J. Body condition scoring: a rapid and accurate method for assessing health status in mice. Lab Anim. Sci. 49, 319–323 (1999).
Sun, L. et al. Bile salt hydrolase in non-enterotoxigenic Bacteroides potentiates colorectal cancer. Nat. Commun. 14, 755 (2023).
Zhao, X. et al. Sulfonated perylene as three-in-one STING agonist for cancer chemo-immunotherapy. Angew. Chem. Int. Ed. Engl. 63, e202318799 (2024).
Kamphorst, J. J. et al. Hypoxic and ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl Acad. Sci. USA 110, 8882–8887 (2013).
Wu, N. et al. MAP3K2-regulated intestinal stromal cells define a distinct stem cell niche. Nature 592, 606–610 (2021).
Kikuchi, K. et al. Gut microbiome-derived phenyl sulfate contributes to albuminuria in diabetic kidney disease. Nat. Commun. 10, 1835 (2019).
Aegerter, H. et al. Influenza-induced monocyte-derived alveolar macrophages confer prolonged antibacterial protection. Nat. Immunol. 21, 145–157 (2020).
Boivin, G. P. et al. Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology 124, 762–777 (2003).
Gobert, A. P. et al. Protective role of spermidine in colitis and colon carcinogenesis. Gastroenterology 162, 813–827.e818 (2022).
Yurekten, O. et al. MetaboLights: open data repository for metabolomics. Nucleic Acids Res. 52, D640–d646 (2024).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2023YFE0109800 Z.X.), the National Natural Science Foundation of China (Nos. 82020108024 Z.X., 82200596 Q.S. and 82400658 Y.J.), and the China Postdoctoral Science Foundation (No. 2022M721014 Y.J.). We thank scientists in Xu lab for technical supports and effective discussions.
Author information
Authors and Affiliations
Contributions
Y.Z. designed the study, performed the experiments, and wrote the manuscript; M.L., L.Z., Q.Y., X.L., K.H., and J.L. performed the experiments; Q.S., J.G., and J.Y. analyzed the data. Y.J. participated in organizing figures. Z.X. contributed to the conception and writing. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Peer review
Peer review information
: Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, Y., Liu, MJ., Zhang, L. et al. High mobility group A1 (HMGA1) promotes the tumorigenesis of colorectal cancer by increasing lipid synthesis. Nat Commun 15, 9909 (2024). https://doi.org/10.1038/s41467-024-54400-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-54400-0
This article is cited by
-
STING inhibits the progression of esophageal squamous cell carcinoma by suppressing CPT1A-mediated fatty acid β-oxidation
Acta Pharmacologica Sinica (2025)
