
Abstract
Abundant residual heat from industrial emissions may provide energy resource for CO2 conversion, which relies on H2 gas and cannot be accomplished at low temperatures. Here, we report an approach to store electrons and hydrogen atoms in catalysts using sunlight and water, which can be released for CO2 reduction in dark at relatively low temperatures (150−300 °C), enabling on-demand CO2 conversion. As a proof of concept, a model catalyst is developed by loading single Cu sites on hexagonal tungsten trioxide (Cu/WO3). Under light illumination, hydrogen atoms are generated through photocatalytic water splitting and stored together with electrons in Cu/WO3, forming a metastable intermediate (Cu/HxWO3). Subsequent activation of Cu/HxWO3 through low-temperature heating releases the stored electrons and hydrogen atoms, reducing CO2 into valuable products. Furthermore, we demonstrate the practical feasibility of utilizing natural sunlight to drive the process, opening an avenue for harnessing intermittent solar energy for CO2 utilization.
Similar content being viewed by others


Introduction
Upcycling CO2 emissions into valuable chemicals is a highly important process toward carbon neutrality1,2,3,4,5,6. Despite the development of various techniques, energy input and hydrogen resources are two factors limiting their ultimate applications. Considering conversion rates, thermal catalysis using H2 gas is the most effective approach to CO2 conversion7,8,9, which is, however, limited by these two factors. To this end, abundant residual heat from industrial emissions is a potential energy resource for thermally driving CO2 conversion. Such an approach encounters the limitation that CO2 hydrogenation can hardly be accomplished when the industrial residual heat is within the low-temperature range10. Photocatalysis and electrocatalysis provide alternative approaches to utilize renewable energy for CO2 conversion using water as a hydrogen source11,12,13,14. However, the intermittent feature of solar energy and other renewable energy cannot sustain continuous chemical transformations, forcing the process to be coupled with energy storage devices, which inevitably results in a tremendous increase in costs15. For this reason, it is imperative to develop an approach to store intermittent solar energy as well as use readily available and cost-effective water as a hydrogen source, which can be compatible with the thermal catalysis system driven by low-temperature heat to complete the overall CO2 conversion process.
Natural photosynthesis, which provides a paradigm for separating light absorption and CO2 reduction, gives us important inspiration. Within the intricate mechanism of photosynthesis, particularly during the light-dependent reactions occurring in chloroplasts, the synthesis of reduced equivalents takes place16,17,18. These reduced equivalents play a pivotal role in the generation of reduced nicotinamide adenine dinucleotide phosphate (NADPH), a crucial molecule that serves as a potent reducing agent essential for subsequent CO2 assimilation pathways. Through this process, solar energy is efficiently captured and converted into chemical energy, facilitating the transformation of CO2 into organic molecules essential for sustaining life processes. Thus, the fundamental principles underlying natural photosynthesis provide valuable insights for the development of efficient solar-driven CO2 utilization technologies. To simulate natural photosynthesis, state-of-the-art industrial photosynthesis has proposed a multi-step CO2 reduction pathway mediated by H2, which involves the generation of H2 through photocatalysis followed by CO2 hydrogenation9,17. However, the H2 generated from photocatalysis is mixed with O2, necessitating further separation and purification before being utilized in conventional CO2 hydrogenation reaction systems19,20,21, which significantly augments the purification costs associated with the end product. Moreover, the dissociation of H2 during the CO2 hydrogenation process requires extra energy input, the magnitude of which relies on the type of catalyst employed8,22. This scenario underscores compelling opportunities for the refinement of catalytic methodologies, aiming at optimizing CO2 conversion for practical implementation.
Here, we demonstrate an approach for tandem solar energy harvesting and CO2 conversion utilizing a catalyst with the capability of storing electrons and hydrogen atoms, as shown in Fig. 1. In the presence of solar irradiation, hydrogen atoms are generated through photocatalytic water splitting and subsequently stored alongside electrons within the catalyst. In scenarios where CO2 reduction is needed, the stored electrons and hydrogen atoms are utilized to reduce CO2 into value-added products with low-temperature heat. This approach is experimentally validated using Cu-loaded tungsten trioxide (Cu/WO3) as a model catalyst. The Cu/WO3 material functions akin to a “reservoir”, effectively storing hydrogen atoms and electrons generated during photocatalytic water splitting under solar irradiation. This storage process results in the formation of a Cu/HxWO3 intermediate. When CO2 conversion is required, heating the Cu/HxWO3 intermediate allows the hydrogen atoms and electrons to be released to the catalyst surface for CO2 reduction. This work opens an avenue for the controllable acquisition of solar energy for CO2 utilization.

Diagram illustrating the integration of intermittent sunlight and residual heat for on-demand CO2 conversion with water.
Results
Synthesis and characterization of model catalyst
According to the literature report, the hexagonal WO3 was synthesized via a one-step hydrothermal process23. Then, Cu atoms were introduced to the catalyst surface under dark conditions by photo-reduced W species with a low-valence state, forming a Cu/WO3 catalyst. The crystal structure of the obtained catalyst was examined by X-ray diffraction (XRD), as shown in Fig. 2a. The Cu/WO3 displays clear diffraction peaks of hexagonal WO3, which aligns with previous reports23,24,25. No diffraction peaks related to Cu crystals were observed despite the Cu content of 0.14 wt% determined by inductively coupled plasma mass spectrometry (ICP-MS). We further employed X-ray photoelectron spectroscopy (XPS) to detect elements in the sample. As illustrated in Fig. 2b, the high-resolution Cu 2p XPS spectrum of Cu/WO3 confirms the presence of the Cu element on the surface of Cu/WO3. In addition, the signals of W and O elements can be seen from the XPS spectra (Supplementary Fig. 1). To further characterize the W–O structure, Raman spectra were collected (Fig. 2c). The band at 953 cm−1 can be assigned to the stretching vibration of the terminal W=O groups, while the peaks at 645 cm−1 and 820 cm−1 are ascribed to the O−W−O stretching vibration, and the peak at 230 cm−1 is attributed to W–O–W stretching mode24,26. There is no obvious change in the intensity and position of the peaks after Cu introduction, which indicates that Cu does not affect the crystal structure of the sample.

a XRD patterns, (b) high-resolution Cu 2p XPS spectra and (c) Raman spectra of WO3 and Cu/WO3. d HAADF-STEM image of Cu/WO3. e The structure of hexagonal WO3. The structural models in this figure were created using VESTA47. f EDS mapping for Cu, W, and O elements of Cu/WO3. g Normalized Cu K-edge XANES spectra of Cu/WO3, Cu foil, and CuO. h Cu K-edge EXAFS spectra of Cu/WO3, Cu foil, and CuO. i H2-TPR spectra of WO3 and Cu/WO3.
We further characterized the morphology of Cu/WO3 using scanning electron microscopy (SEM) and transmission electron microscopy (TEM). SEM and TEM images show that Cu/WO3 is composed of irregular nanosheets (Supplementary Figs. 2, 3). To look into the atomic structure, aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was used to characterize the catalyst. As shown in Fig. 2d, e, the arrangement of WO6 octahedra sharing corners forms a structure with stacked tunnels along the c axis, which is favorable for the insertion and extraction of hydrogen atoms23. In addition, the steep increase in adsorbed volume at low relative pressure also indicates the presence of microporosity (Supplementary Fig. 4). Due to the light weight of Cu element relative to W element, no Cu-related species can be seen in the HAADF-STEM image, but the corresponding energy-dispersive spectroscopy (EDS) mapping of Cu/WO3 clearly proves the uniform dispersion of Cu elements on the catalyst (Fig. 2f).
To analyze the local structure of Cu in Cu/WO3, X-ray absorption fine structure (XAFS) spectroscopy was performed. The Cu K-edge absorption edge position of Cu/WO3 in the X-ray absorption near edge structure (XANES) spectra is closer to that of CuO than Cu foil (Fig. 2g), indicating that the Cu element carries a positive charge and has a valence state approaching +227. Fourier-transformed (FT) extended X-ray absorption fine structure (EXAFS) spectra were used to determine the local structures of the Cu element (Fig. 2h). The Cu/WO3 does not show the peak of Cu–Cu bond at 2.2 Å in reference to standard Cu foil, revealing the absence of Cu clusters or particles in the catalyst28,29,30. According to the EXAFS fitting results (Supplementary Fig. 5 and Supplementary Table 1), the Cu atoms most likely are located in the hexagonal holes of WO3 through coordination with 4 O atoms. Such an introduction of Cu can slightly affect the photoelectric properties of WO3 (Supplementary Figs. 6, 7). We also characterized the interaction of samples with hydrogen atoms using H2 temperature programmed reduction (H2-TPR) measurement. The H2-TPR peaks shift to low temperatures after Cu is deposited onto WO3 (Fig. 2i), indicating the spillover of hydrogen atoms from Cu sites to the WO3 support31. This feature will promote the storage of H atoms rather than their release as H2. Taken together, the characterizations suggest that the well-designed Cu/WO3 catalyst has the potential to serve as a reservoir for storing hydrogen atoms and electrons.
Storage of electrons and hydrogen atoms in the catalyst
Upon gaining the structural information of the Cu/WO3 catalyst, we further assessed its capacity for storing hydrogen atoms and electrons. To this end, we dispersed the catalyst in water and subjected it to illumination, with the resulting sample named Cu/HxWO3 (Fig. 3a). Following the illumination process, the catalyst undergoes a discernible color transformation, transitioning from gray to deep blue (Supplementary Fig. 8). Compared to Cu/WO3, Cu/HxWO3 exhibits significantly enhanced absorption across the visible light spectrum, as evidenced by ultraviolet-visible diffuse reflectance spectroscopy (UV-Vis DRS) measurements (Supplementary Fig. 9). According to the literature, WO3 can form polarons due to electron localization at the d-states of tungsten, resulting in the conversion of W(6 + ) to W(5 + )32,33. Visible or near-infrared light can excite polaronic transitions, which prompts the migration of polarons from W(5 + ) to neighboring W(6 + ) sites, consequently engendering light absorption. To further validate this phenomenon, we employed electron paramagnetic resonance (EPR) spectroscopy to examine the W(5 + ) species in the catalyst during light irradiation (Fig. 3b). The EPR signal at approximately 3720 G (g value = 1.895) corresponding to W(5 + ) exhibits variation with the duration of illumination, reaching maximal intensity after 10 min23,34. This observation suggests an increase in the W(5 + ) content within the catalyst during the irradiation process. In addition, the deconvoluted W 4 f XPS spectra for Cu/WO3 and Cu/HxWO3 can be fitted into two oxidation states, W(6 + ) (4f7/2, 35.82 eV) and W( 5 + ) (4f7/2, 34.81 eV) (Supplementary Fig. 10)35. The W(5 + ) content in Cu/WO3 experiences a notable increase, rising from 0.7% to 4.5% in Cu/HxWO3. This shift implies that a portion of W(6 + ) undergoes reduction to W(5 + ) during the illumination process. The transformation from W(6 + ) to W(5 + ) under illumination signifies the successful storage of photoinduced electrons in the catalyst.

a Schematic diagram of the process of storing electrons and hydrogen atoms. The structural models were created using VESTA47. b EPR spectra of Cu/WO3 after different times of light irradiation. c 1H NMR spectra of Cu/WO3 and Cu/HxWO3. d FTIR spectra of Cu/WO3, Cu/HxWO3 and Cu/DxWO3. e Wavelength-dependent AQE of H2O to electron−hydrogen pairs. f Normalized W L3-edge XANES spectra of Cu/WO3, Cu/HxWO3 and W foil. g W L3-edge EXAFS spectra of Cu/WO3 and Cu/HxWO3.
To maintain charge balance, the storage of electrons may be accompanied by the participation of cations. In fact, the H2 and O2 produced during the photocatalytic process do not match stoichiometrically (Supplementary Fig. 11)36. To investigate whether hydrogen atoms are stored in the catalyst, we collected 1H solid-state nuclear magnetic resonance (NMR) spectra from Cu/WO3 and Cu/HxWO3. The intensification of the peaks for Cu/HxWO3 clearly indicates the enhancement of surface terminal hydroxyl groups (1.6 ppm) and bridging hydroxyl groups (4.4 ppm) (Fig. 3c)37, signifying the successful incorporation of hydrogen atoms into the catalyst following exposure to light. This conclusion gains further support from the enhanced peaks corresponding to O–H species in the Fourier-transform infrared (FTIR) spectra, specifically in the range of 3500–3700 cm−1 (Fig. 3d). We further carried out the measurements by replacing water (H2O) with deuterium oxide (D2O) to gain a deeper understanding of the origin of hydrogen atoms. The resulting product, denoted as Cu/DxWO3, exhibits an FTIR spectrum where the peak associated with O–D species appears in the range of 2500–2700 cm−1 after light irradiation38. Remarkably, the peaks related to O−H species remains consistent with those of Cu/WO3, indicating that the hydrogen atoms inserted into the catalyst indeed originate from water splitting. Furthermore, we assessed the energy storage efficiency by analyzing the wavelength-dependent apparent quantum efficiency (AQE) of H2O to electron−hydrogen pairs (Fig. 3e). Notably, the maximum AQE was observed at 320 nm, reaching 0.66%, underscoring the excitation of catalyst by ultraviolet light. In addition, we assessed the storage of hydrogen atoms under intermittent illumination conditions (Supplementary Fig. 12). This observation underscores the capability of our designed system to mitigate the challenges posed by intermittent supply of solar energy.
To explore the impact of storing electronic and hydrogen atoms in catalysts, we conducted tests on the W L3-edge XAFS spectra of Cu/WO3 and Cu/HxWO3. From the XANES spectra (Fig. 3f), it can be observed that the absorption edge position of Cu/HxWO3 is slightly lower compared to Cu/WO3, but both are higher than that of W foil. This suggests a slight reduction in average valence state of W element, primarily attributed to an increase in W(5 + ) content, which is consistent with the findings from EPR and XPS results. In addition, the EXAFS spectra show that the intensity of W−O coordination at 1.35 Å slightly decreases, primarily due to insertion of hydrogen atom (Fig. 3g). No significant changes can be found in XRD patterns and Raman spectra, indicating that the overall crystal structure of the catalyst does not change obviously after the storage of electrons and hydrogen atoms (Supplementary Fig. 13). Taken together, the above results indicate that the storage of hydrogen atoms and electrons does not lead to the collapse of crystalline structure, which is crucial for the recyclability of catalyst.
Catalytic CO2 reduction performance
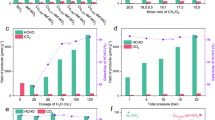
After confirming the ability to store hydrogen atoms and electrons in the catalyst using light and water, we further explored the utilization of low-temperature residual heat to thermally activate Cu/HxWO3 for CO2 conversion. To this end, the catalytic CO2 reduction performance by the stored hydrogen atoms and electrons was evaluated under heated conditions in a homemade reactor (Supplementary Fig. 14, see the experiment section for details). Specifically, a gas-solid phase reaction was implemented, wherein only gaseous products—such as CH4, CO, H2, and C2H4—are detected to assess the efficacy of catalyst39. It turns out that the CO2 conversion can be achieved at a temperature as low as 90 °C, but the production rate is very limited. For this reason, we chose a temperature above 150 °C for the demonstration (Supplementary Fig. 15). When CO2 reacts with Cu/HxWO3 at 150 °C, a small amount of CO (0.13 mL/g) and CH4 (0.11 mL/g) are generated (Fig. 4a). As the reaction temperature is elevated to 180 °C, C2H4 becomes detectable (0.07 mL/g). Remarkably, by increasing reaction temperature, product selectivity for CO2 conversion gradually rises—from 11.4% at 150 °C to 91.8% at 300 °C (Fig. 4b). At 270 °C, a variety of products are yielded at relatively high levels (Supplementary Table 2), with the stored electrons and hydrogen atoms contributing to the formation of C2H4. As the formation of C2H4 requires the transfer of 12 electrons and 12 hydrogen atoms, this observation underscores that the elevated temperatures can facilitate the provide additional electrons and protons for the sites of CO2 activation. To better understand this aspect, we determined the conversion rates of hydrogen atoms and electrons by analyzing the yield of final products, which show a positive correlation with the temperatures (Supplementary Fig. 16). This suggests that thermal energy input enhances the migration and release of electrons and hydrogen atoms. Our further exploration was fixed at 270 °C to uphold a relative balance between efficiency and electron/hydrogen utilization.

a Yield of CO2 conversion products for Cu/HxWO3 at different temperatures. b Products selectivity for Cu/HxWO3 at different reaction temperatures. c Product yields for WO3, Cu/WO3, HxWO3 and Cu/HxWO3 at 270 °C. Mass spectra of (d)CO, (e) CH4, and (f) C2H4 over Cu/HxWO3 with 13CO2 or 12CO2 as the reactant at 270 °C. The error bar represents the standard deviation of the measurements.
Stability is a key parameter determining the practical feasibility of a catalyst. The performance of the catalyst remains stable after six testing cycles—an encouraging sign of its endurance and potential for practical applications (Supplementary Fig. 17). Furthermore, we underscored the catalyst stability by employing characterization techniques, including XRD, Raman spectroscopy, XPS, and FTIR spectroscopy (Supplementary Fig. 18). Notably, no significant structural changes can be detected in the catalyst after undergoing cyclic reactions, indicating excellent stability of the catalyst.
To gain a comprehensive understanding of CO2 reduction by Cu/HxWO3, a series of comparative experiments were conducted. We first conducted a thorough assessment using WO3 and Cu/WO3 for CO2 conversion. Apart from a small amount of CO, no other carbonous products were detected (Fig. 4c). This product may be attributed to residual carbon impurities on the surface that have not been entirely eliminated40. Subsequently, we examined WO3 after storing electrons and hydrogen atoms (noted as HxWO3). Notably, a small number of carbonous products emerged with primarily H2 due to the scarcity of suitable active sites. When high-purity CO2 was replaced by Ar, only a negligible amount of CO was detected (0.012 mL/g) at 270 °C. To strengthen this conclusion, we conducted in-situ FTIR measurements for the formation of intermediate products. Only CO intermediate was generated in a CO2 atmosphere, indicating that the carbonous product indeed originates from CO2 reduction (Supplementary Figs. 19, 20). To trace the carbon source in the products, we performed tests using 13CO2 instead of 12CO2 and analyzed the ion fragment distribution of CO, CH4, and C2H4. The detection of 13C-associated ion fragments indicates that the product is indeed derived from CO2 reduction (Fig. 4d–f)11,41. Furthermore, during the stage of collecting electrons and hydrogen atoms, H2O was substituted with D2O, resulting in the formation of a Cu/DxWO3 catalyst. We conducted CO2 reduction reactions using Cu/DxWO3 and observed that the primary hydrocarbon products were CD4 and C2D4 (Supplementary Fig. 21). As such, we can safely attribute the carbonous products to CO2 reduction by the hydrogen atoms and electrons in the catalyst.
Catalytic mechanisms
Following the validation of the stored hydrogen atoms and electrons for CO2 reduction, the investigation of the reaction mechanism is warranted. We employed the kinetic isotope effect (KIE, H/D) to elucidate the rate-limiting step38,42. By using Cu/HxWO3 and Cu/DxWO3 for CO2 conversion reactions, based on the consumption of hydrogen atoms and electrons in product generation, the KIE (H/D) was calculated to be 5.24 (Fig. 5a). This understanding of the reaction process underscores the pivotal role of hydrogen transfer within the catalytic framework37,43.

a Release rate of stored H(D) atoms for Cu/HxWO3 and Cu/DxWO3. b Calculated free energy diagram for transfer of H to catalyst surface over WO3 and Cu/WO3. The inserted figure shows the optimized H atom adsorption structure at various sites. c Electron density difference plot of Cu/WO3 surface. Electron accumulation and depletion are indicated in yellow and blue, respectively. The structural models in (b) and the differential charge density plot in (c) were visualized using VESTA47.
To look into the hydrogen transfer process from the carrier material (WO3) to the active site (Cu sites), we conducted density functional theory (DFT) calculations to discern the energy barrier associated with hydrogen transfer. We scrutinized the sites within the WO3 carrier material where hydrogen atoms reside and discovered a substantial number of catalyst sites capable of adsorbing hydrogen atoms (Supplementary Fig. 22). This raises a key question: how do hydrogen atoms reach the catalyst surface to participate in the reaction? The Gibbs free energy variation at each stage of hydrogen transfer—from the interior of WO3 to the surface sites—was meticulously computed (Fig. 5b). The hydrogen adsorption shows a remarkable weakness in the vicinity of the six-octahedron tunnels of WO3 (sites 2 and 4), while it shows a considerate strength along the four-octahedron channels (sites 1 and 5), indicating that the latter is more favorable for hydrogen adsorption. The migration of a hydrogen atom from the inner WO layer to the outer WO layer requires crossing the intermediate O layer (site 3). The computed Gibbs free energy difference associated with adsorbed hydrogen between site 2 and site 3 is 0.93 eV, highlighting the inherent difficulty of hydrogen transfer from the WO3 interior to the surface sites. Interestingly, the Gibbs free energy difference of adsorbed hydrogen between site 2 and site 3 on Cu/WO3 is lower than that on WO3, with a barrier of 0.60 eV. This outcome suggests that the hydrogen transfer process is significantly facilitated on Cu/WO3. Furthermore, we observed stronger hydrogen adsorption on the Cu site (site 5) of the Cu/WO3 surface compared to the six octahedron channels (site 4), thereby favorably influencing the CO2 reduction reaction. To unravel the intricacies of this facilitated hydrogen transfer process, we examined the charge density difference and revealed distinct electron accumulation regions on the surface layer of Cu/WO3 upon the introduction of a Cu atom (Fig. 5c). The change in the electron distribution can affect the trapped hydrogen atom by interacting with unsaturated electrons in the H 1 s orbital of the hydrogen atom44, which can account for the change in the free energy of the hydrogen transfer process.
Demonstration of solar-driven application
To validate the practicality of our concept, we designed a process to integrate utilization of intermittent sunlight and residual heat for on-demand CO2 conversion (Fig. 6a). In addition, we investigated the solar energy harvesting process through a demonstration experiment under cloudy weather (Supplementary Fig. 23). As depicted in Fig. 6a, the initial sample exhibits a subdued gray hue prior to solar exposure (at 6:00). When exposed to sunlight, the color of catalyst gradually changes (at 10:00). Despite fluctuations in sunlight intensity due to passing clouds wherein irradiance nears 0 W/m², the catalyst consistently accumulates solar energy, reaching saturation around 16:00 (Supplementary Table 3). Subsequently, we used the catalyst to perform CO2 reduction tests. Remarkably, the amount of hydrogen stored in the catalyst using both natural sunlight and simulated sunlight is comparable (Fig. 6b), confirming the successful use of solar energy. These results demonstrate the feasibility of collecting hydrogen atoms directly from atmospheric water vapor.

a Proposed block flow diagram to achieve on-demand CO2 conversion based on electron-hydrogen storage in catalysts with sunlight and water and their release with residual heat. The images on the left show the catalyst under natural light, demonstrating the process of collecting hydrogen atoms and electrons. b Product yields for Cu/HxWO3 prepared under natural sunlight and simulated sunlight. c Product yields for Cu/HxWO3 after being stored in the dark for 15 days and 90 days. d Product yields for Cu/HxWO3 with different catalyst dosages. The error bar represents the standard deviation of the measurements.
To verify that the catalyst can perform CO2 reduction on demand, we tested the catalyst after 15 days and 90 days of storage. Although the catalytic performance slightly decreases, it still maintains over 80% of the stored hydrogen atoms and electrons (Fig. 6c). Moreover, the consumption of the stored electrons and hydrogen atoms is mainly in the initial stage (0–15 days), with very little change over the subsequent extended period, indicating that long-term storage is feasible. The reduced charge loss during storage suggests that this method holds promise for mitigating the uneven seasonal distribution of solar energy. In addition, the catalyst used for CO2 conversion was scaled up by two orders of magnitude, with a corresponding increase in product yield (Fig. 6d and Supplementary Fig. 24). The results have confirmed the theoretical feasibility of large-scale application. It should be acknowledged that the purpose of the aforementioned experiments is mainly to verify the feasibility of integrable utilization of intermittent sunlight and residual heat for on-demand CO2 conversion with water, without targeted optimization of material costs and reaction devices. Achieving large-scale applications necessitates substantial efforts, particularly in catalyst preparation and reactor design, which are the focal points of our future research. Overall, our results have confirmed the practicality of directly collecting, storing, and deploying catalysts when CO2 conversion is required, significantly increasing the flexibility of catalytic CO2 reduction.
Discussion
In summary, we have proposed a conceptual approach inspired by natural photosynthesis, to achieve on-demand CO2 conversion utilizing an electron−hydrogen reservoir. To validate the feasibility of this concept, we prepared a model catalyst of Cu-loaded WO3. This catalyst exhibits abundant mesoporous structures that serve as storage sites for hydrogen atoms. Indeed, under simulated sunlight, it efficiently collects electrons and hydrogen atoms, leading to the formation of an electron/hydrogen-stored intermediate, Cu/HxWO3. The Cu/HxWO3 demonstrates the capability to convert CO2 into high-value products under heating conditions ranging from 150 °C to 300 °C. Of particular note is its ability to produce ethylene (C2H4) as a high-value product at temperatures above 180 °C. Our investigation revealed that the reaction rate is significantly affected by the migration of stored hydrogen atoms to the surface, and the introduction of Cu sites can accelerate this migration process. To assess the practical application of the catalyst, we conducted experiments on cloudy days when the solar supply is unstable, with the aim of driving the reaction using natural sunlight. The results demonstrate that varying solar irradiance does not impede the collection of electrons and hydrogen atoms. More importantly, the electron/hydrogen-stored intermediates can be preserved for over three months, facilitating further scale-up of experiments. By harnessing unstable sunlight and residual heat, this study opens an avenue for efficient solar-driven CO2 conversion.
Methods
Materials preparation
1.5 g of Na2WO4·2H2O was added to 30 mL of deionized (DI) water and stirred for 30 min. Subsequently, 265 µL of hydrazine hydrate solution (50%) was added and stirred continuously for 2 h. Next, a certain amount of HCl was added to adjust the pH to 0.5, and the solution was stirred for another 12 h. Then, the solution underwent a hydrothermal reaction for 12 h at 120 °C. The final precipitation was collected centrifugally, then washed alternately three times with deionized water and absolute ethanol, and finally dried under vacuum at 70 °C. The dried sample was then calcinated in air at a temperature of 320 °C for 12 h, followed by an additional calcination in high-purity N2 for another 12 h, to obtain WO3.
To prepare the Cu/WO3, 1 g of WO3 catalyst was dispersed in 30 mL solution (5 mL of absolute ethanol and 25 mL of DI water), followed by ultrasonic treatment for 6 h at 20 °C. Then, the solution was irradiated by a 300 W xenon lamp for 1 min. Subsequently, a certain amount of Cu(NO3)2·3H2O solution (the Cu content is 0.5 wt% of the WO3 content) was added dropwise, and the mixture was stirred for 12 h under dark. The resulting sample was collected centrifugally, then washed alternately three times with deionized water and absolute ethanol, and finally dried under vacuum at 70 °C. After drying, the samples were calcinated at 320 °C for 12 h, first in air and then in high-purity N2 to obtain Cu/WO3 catalyst. All samples were stored in the dark for further experiments.
Collection of electrons and hydrogen atoms from simulated sunlight
To verify the feasibility of sunlight harvesting, 0.5 g of as-synthesized catalyst was dispersed in 30 mL of DI water and subjected to 10 min of ultrasonic treatment to ensure uniform dispersion. The sample was then irradiated by a 300 W xenon lamp with an AM1.5 filter for 10 min under continuous stirring. The optical power of the light source was recorded by the optical power meter (PM100D, Thorlabs, USA), and the corresponding optical power density was 0.45 W/cm2. The wavelength distribution information of the light source is shown in Supplementary Fig. 25. Subsequently, the sample was centrifuged and vacuum-dried for 24 h at 55 °C. According to the different catalysts added, the products are noted as HxWO3 (added WO3) and Cu/HxWO3 (added Cu/WO3). Wavelength-dependent AQE of H2O to electron−hydrogen pairs was calculated by: AQE (%) = (Number of electron−hydrogen pairs) / (Number of incident photons) × 100%. Number of electron−hydrogen pairs was calculated by the number of H atoms obtained by ion exchange using an excess of K+ in water23.
Evaluation of CO2 conversion performance
The synthesized samples were used for CO2 reduction reactions in a homemade reactor consisting of a temperature-controlled heater and a double-sealed quartz tube. The sample was placed in the middle of the quartz tube, and gas was injected or extracted through sampling ports located at the end of the quartz tube. The reactor was purged with high-purity CO2 (99.999%) ten times before reaction. Then, 90 mL of high-purity CO2 was injected into the reactor to keep the pressure at 0.1 MPa. The temperature of the samples was regulated by the heater and monitored using a thermocouple. After a certain period of reaction, 2 ml of gas was extracted from the reactor to measure the product yield. The products were detected by gas chromatography (7890B, Agilent). Gaseous products such as CH4, C2H4, CO, and CO2 were detected using a flame ionization detector (FID), while O2 and H2 were detected using a thermal conductivity detector (TCD). The amount of the CO2 reduction product was determined by comparing the peak area with the standard curve of the gas with known concentration (Supplementary Fig. 26). For the isotope-labeling experiment, high-purity 13CO2 was used as the reactant instead of 12CO2 for the reaction, and the products were analyzed using gas chromatography-mass spectrometry (GC-MS, 7890 A and 5975 C, Agilent). The volume of the products has been converted to the volume under standard temperature and pressure (STP), which corresponds to a temperature of 0 °C and a pressure of 1 atmosphere.
Cyclic stability test
The cyclic stability test of the catalyst comprises two stages: the collection of electrons and hydrogen atoms under simulated sunlight illumination, and the thermal reaction at 270 °C in a high-purity CO2 atmosphere. The specific procedures for these stages are detailed in the above sections. A cycle was considered complete when the product yield ceased to increase under heating conditions. Subsequently, the catalyst was irradiated by simulated sunlight illumination to initiate the next cycle.
Calculation details
All calculations were carried out using the Vienna Ab Initio Simulation Package (VASP)45. The exchange-correlation interaction was described by Perdew−Burke−Ernzerhof exchange-correlation functional (GGA-PBE)46. To prevent interactions between periodic structures, a vacuum of 20 Å was introduced during the construction of surface models. The energy cutoff and Monkhorst-Pack k-point mesh were set to 450 eV and 3 × 3 × 1, respectively. The convergence criteria for geometry optimization was set as 1 × 10−5 eV/atom for energy and 0.03 eV/Å for force. The Gibbs free energy was determined under reaction conditions using the following equation: \({{{\rm{G}}}}={{{{\rm{E}}}}}_{{{{\rm{DFT}}}}}-{{{{\rm{E}}}}}_{{{{\rm{ZPE}}}}}+{{{{\rm{E}}}}}_{{{{\rm{enthalpy}}}}}-{{{\rm{TS}}}}\). Here, \({{{{\rm{E}}}}}_{{{{\rm{DFT}}}}}\) represents the static electron energy of the system at 0 K, \({{{{\rm{E}}}}}_{{{{\rm{ZPE}}}}}\) is zero-point energy, \({{{{\rm{E}}}}}_{{{{\rm{enthalpy}}}}}\) is the enthalpy contribution, and TS is the entropy contribution. All the structural model visualization is achieved using VESTA47.
Data availability
Source data are provided in this paper.
References
Hepburn, C. et al. The technological and economic prospects for CO2 utilization and removal. Nature 575, 87–97 (2019).
Jiang, Z. et al. Filling metal-organic framework mesopores with TiO2 for CO2 photoreduction. Nature 586, 549–554 (2020).
Diercks, C. S., Liu, Y., Cordova, K. E. & Yaghi, O. M. The role of reticular chemistry in the design of CO2 reduction catalysts. Nat. Mater. 17, 301–307 (2018).
Yu, J. et al. Artificial spherical chromatophore nanomicelles for selective CO2 reduction in water. Nat. Catal. 6, 464–475 (2023).
Schappi, R. et al. Drop-in fuels from sunlight and air. Nature 601, 63–68 (2022).
Zhou, B. et al. Light-driven synthesis of C2H6 from CO2 and H2O on a bimetallic AuIr composite supported on InGaN nanowires. Nat. Catal. 6, 987–995 (2023).
Feng, Z. et al. Asymmetric sites on the ZnZrOx catalyst for promoting formate formation and transformation in CO2 hydrogenation. J. Am. Chem. Soc. 145, 12663–12672 (2023).
Vogt, C. et al. Unravelling structure sensitivity in CO2 hydrogenation over nickel. Nat. Catal. 1, 127–134 (2018).
Shih, C. F., Zhang, T., Li, J. & Bai, C. Powering the future with liquid sunshine. Joule 2, 1925–1949 (2018).
Jiang, X., Nie, X., Guo, X., Song, C. & Chen, J. G. Recent advances in carbon dioxide hydrogenation to methanol via heterogeneous catalysis. Chem. Rev. 120, 7984–8034 (2020).
Yu, Y., He, Y., Yan, P., Wang, S. & Dong, F. Boosted C-C coupling with Cu-Ag alloy sub-nanoclusters for CO2-to-C2H4 photosynthesis. Proc. Natl. Acad. Sci. USA 120, e2307320120 (2023).
Ma, W. et al. Synergistic interplay of dual‐active‐sites on metallic Ni‐MOFs loaded with Pt for thermal‐photocatalytic conversion of atmospheric CO2 under infrared light irradiation. Angew. Chem. Int. Ed. 62, e202313784 (2023).
Rashid, R. T. et al. Tunable green syngas generation from CO2 and H2O with sunlight as the only energy input. Proc. Natl. Acad. Sci. USA 119, e2121174119 (2022).
Wang, Y. et al. Direct and indirect Z-scheme heterostructure-coupled photosystem enabling cooperation of CO2 reduction and H2O oxidation. Nat. Commun. 11, 3043 (2020).
Amthor, S. et al. A photosensitizer-polyoxometalate dyad that enables the decoupling of light and dark reactions for delayed on-demand solar hydrogen production. Nat. Chem. 14, 321–327 (2022).
Wang, Y. et al. Mimicking natural photosynthesis: solar to renewable H2 fuel synthesis by Z-scheme water splitting systems. Chem. Rev. 118, 5201–5241 (2018).
Ulmer, U. et al. Fundamentals and applications of photocatalytic CO2 methanation. Nat. Commun. 10, 3169 (2019).
Johnson, M. P. Photosynthesis-An overview of photosynthesis. Essays Bioche 60, 255–273 (2016).
Zhang, Y. et al. Internal quantum efficiency higher than 100% achieved by combining doping and quantum effects for photocatalytic overall water splitting. Nat. Energy 8, 504–514 (2023).
Che, W. et al. Fast photoelectron transfer in (cring)-C3N4 plane heterostructural nanosheets for overall water splitting. J. Am. Chem. Soc. 139, 3021–3026 (2017).
Chowdhury, F. A., Trudeau, M. L., Guo, H. & Mi, Z. A photochemical diode artificial photosynthesis system for unassisted high efficiency overall pure water splitting. Nat. Commun. 9, 1707 (2018).
Sordakis, K. et al. Homogeneous catalysis for sustainable hydrogen storage in formic acid and alcohols. Chem. Rev. 118, 372–433 (2018).
Besnardiere, J. et al. Structure and electrochromism of two-dimensional octahedral molecular sieve h’-WO3. Nat. Commun. 10, 327 (2019).
Wang, H. P. et al. Photocatalytic CO2 reduction over platinum modified hexagonal tungsten oxide: Effects of platinum on forward and back reactions. Appl. Catal. B Environ. 263, 118331 (2020).
Zeng, D. et al. Single-atom copper modified hexagonal tungsten oxide for efficient photocatalytic CO2 reduction to acetic acid. Chem. Eng. J. 451, 138801 (2023).
Daniel, M. F., Desbat, B., Lassegues, J. C., Gerand, B. & Figlarz, M. Infrared and Raman study of WO3 tungsten trioxides and WO3·xH2O tungsten trioxide tydrates. J. Solid State Chem. 67, 235–247 (1987).
Xie, W., Li, K., Liu, X. H., Zhang, X. & Huang, H. P-mediated Cu-N4 sites in carbon nitride realizing CO2 photoreduction to C2H4 with selectivity modulation. Adv. Mater. 35, e2208132 (2023).
Li, X. G. et al. Single-atom Pt as co-catalyst for enhanced photocatalytic H2 evolution. Adv. Mater. 28, 2427–2431 (2016).
Gao, C. et al. Heterogeneous single-atom catalyst for visible-light-driven high-turnover CO2 reduction: The role of electron transfer. Adv. Mater. 30, 1704624 (2018).
Xiao, X. D. et al. A promoted charge separation/transfer system from Cu single atoms and C3N4 layers for efficient photocatalysis. Adv. Mater. 32, 2003082 (2020).
Xiong, M., Gao, Z. & Qin, Y. Spillover in heterogeneous catalysis: new insights and opportunities. ACS Catal. 11, 3159–3172 (2021).
Bondarenko, N., Eriksson, O. & Skorodumova, N. V. Polaron mobility in oxygen-deficient and lithium-doped tungsten trioxide. Phys. Rev. B 92, 165119 (2015).
Shi, X. et al. Sustainable all-weather CO2 utilization by mimicking natural photosynthesis in a single material. Natl. Sci. Rev. 11, nwad275 (2024).
Poulos, A. S. et al. Photochromic hybrid organic-inorganic liquid-crystalline materials built from nonionic surfactants and polyoxometalates: Elaboration and structural study. Langmuir 24, 6285–6291 (2008).
Park, J. et al. Investigation of the support effect in atomically dispersed Pt on WO3-x for utilization of Pt in the hydrogen evolution reaction. Angew. Chem. Int. Ed. 58, 16038–16042 (2019).
Li, Y. et al. Boosting thermo-photocatalytic CO2 conversion activity by using photosynthesis-inspired electron-proton-transfer mediators. Nat. Commun. 12, 123 (2021).
Zheng, X. et al. Tailoring a local acid-like microenvironment for efficient neutral hydrogen evolution. Nat. Commun. 14, 4209 (2023).
Yin, S. et al. Elucidating protonation pathways in CO2 photoreduction using the kinetic isotope effect. Nat. Commun. 15, 437 (2024).
Chai, Y. et al. Metal to non-metal sites of metallic sulfides switching products from CO to CH4 for photocatalytic CO2 reduction. Nat. Commun. 14, 6168 (2023).
Dong, C. et al. Size-dependent activity and selectivity of carbon dioxide photocatalytic reduction over platinum nanoparticles. Nat. Commun. 9, 1252 (2018).
Wang, Y. et al. Visible-light driven overall conversion of CO2 and H2O to CH4 and O2 on 3D-SiC@2D-MoS2 heterostructure. J. Am. Chem. Soc. 140, 14595–14598 (2018).
Gunnemann, C., Bahnemann, D. W. & Robertson, P. K. J. Isotope effects in photocatalysis: An underexplored issue. ACS Omega 6, 11113–11121 (2021).
Liu, P. et al. Synergy between palladium single atoms and nanoparticles via hydrogen spillover for enhancing CO2 photoreduction to CH4. Adv. Mater. 34, e2200057 (2022).
Chen, J. et al. Reversible hydrogen spillover in Ru-WO3-x enhances hydrogen evolution activity in neutral pH water splitting. Nat. Commun. 13, 5382 (2022).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 78, 1396–1396 (1997).
Momma, K. & Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276 (2011).
Acknowledgements
This work was supported by the Youth Cross Team Scientific Research Project of the Chinese Academy of Sciences (Grant No. JCTD-2022-17 to Y.H.), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA23010300 to Y.H.) and the National Natural Science Foundation of China (Grant No. 22232003 and Grant No. 52261135635 to Y.X.).
Author information
Authors and Affiliations
Contributions
Y.H., J.C. and Y.X. conceived and designed the experiments. Y.H. and Y.X. supervised the project. X.S., W.P., Y.F., Z.W., L.Y., Z.Z. and F.R. synthesized catalysts and analyzed experimental data. F.R. and G.Z. carried out HAADF-STEM measurements. X.S., W.P., Y.H., C.G., J.C., G.Z., S.L. and Y.X. co-wrote the manuscript. All the authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shi, X., Peng, W., Huang, Y. et al. Integrable utilization of intermittent sunlight and residual heat for on-demand CO2 conversion with water. Nat Commun 15, 10135 (2024). https://doi.org/10.1038/s41467-024-54587-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-54587-2
This article is cited by
-
CO2 conversion by solar photothermo co-catalysis on montmorillonite-based catalysts
Discover Applied Sciences (2025)
