
Abstract
Room temperature phosphorescent (RTP) hydrogels exhibit great potential but show poor mechanical performance (Tensile strengthen <1 MPa) and non-tunable RTP performance, hindering their practical applications. Here, we develop wood hydrogel (W-hydrogel) by the in situ polymerization of acrylamide in the presence of delignified wood. As a result of the molecular interactions between the components of delignified wood and polyacrylamide, the W-hydrogel exhibit a tensile strengthen of 38.4 MPa and green RTP emission with a lifetime of 32.5 ms. Moreover, the tensile strength and RTP lifetime are increased to 153.8 MPa and 69.7 ms, upon treating W-hydrogel with ethanol. Significantly, the mechanical and RTP performance of W-hydrogel is switched by alternating “ethanol and water” treatments. Additionally, W-hydrogel is used as energy donor in order to produce red afterglow emission using RhB via an energy transfer process. Taking advantage of these properties, W-hydrogel is processed into multiple hydrogel-based luminescent materials.
Similar content being viewed by others
Introduction
Organic room temperature phosphorescent (RTP) materials exhibit many advantages including structural flexibility, tunable optical properties, and relatively mild preparation conditions and as such demonstrate great potential in a wide range of applications, such as bioimaging, organic light-emitting diodes (OLED), anti-counterfeiting applications and intelligent sensors1,2,3,4,5,6,7,8. Generally, there are two methods to achieve effective RTP. One method is to facilitate the spin-orbit coupling (SOC) to enhance intersystem crossing (ISC). The other is to rigidify organic chromophores in order to protect the triplet excitons and promote radiative migration9,10,11,12,13.
Guided by these principles, multiple metal-free organic RTP materials have been developed, including, molecular crystals14, supramolecules15, polymer composites16, COF17, HOF18, and carbon dots19. Generally, these RTP materials can exist as liquids20, films21, powders22, aerogels23, hydrogels24, and structural materials25,26.
Amongst these materials, RTP hydrogels are quasi-solid materials that contain large amounts of water in their three-dimensional networks, which render the hydrogels flexible enabling their use in sensors, bio actuators, information encoding, and encryption16,27,28,29,30,31. Given that water in the hydrogel easily quenches triplet excitons for RTP emission, enhancing physical interactions via host-guest, hydrophobic, or electrostatic interactions, are generally employed to partially isolate the luminescent chromophores from water and rigidify the polymer network in order to produce effective RTP hydrogels16,24,28,31,32,33,34,35. As such the structural inhomogeneity of the gel network is enhanced on activating and enhancing RTP performance, which compromises the mechanical performance28. Therefore, the tensile strength of most RTP hydrogels is weak and less than 1 MPa. Moreover, the RTP emission and properties of the material cannot be mechanically tuned once the hydrogel has formed. Currently, it remains a challenge to prepare RTP hydrogels with a tunable/balanced RTP performance that exhibits a high tensile strength, which would broaden the scope of potential applications.
Natural cellulose and lignin have been found to be promising composites for enhancing the mechanical performance of hydrogels by way of molecular interactions with the polymer network36,37,38. Moreover, these components can generate tunable RTP emission upon appropriate processing39,40,41. Additionally, natural wood, mainly consisting of cellulose and lignin, also exhibits effective RTP emission after treatment with sugars42. Obviously, being able to produce RTP materials from natural wood resources represents an important sustainability goal. Nevertheless, the RTP of these natural components is quenched upon introducing them into hydrogels because of the high water content.
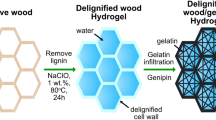
In this work, we prepare RTP hydrogel (W-hydrogel) with a lifetime of 32.5 ms and tensile strength of 38.4 MPa, from natural wood. Interestingly, both the RTP and mechanical performance of the W-hydrogel are tuned and enhanced by introducing ethanol into the hydrogel matrix. To obtain W-hydrogel, natural wood is partially delignified to increase the mechanical flexibility using H2O2 treatment. Subsequently, polymerization of an aqueous solution of acrylamide (AM) is initiated in the presence of partially delignified wood to produce W-hydrogel (Fig. 1).

Schematic illustration of the preparation of room temperature phosphorescent W-hydrogel from natural wood.
Results
Mechanical and RTP performance of W-hydrogel
Initially, natural wood was delignified to produce delignified wood. Importantly, the delignified wood maintained the skeleton structure of natural wood (Supplementary Fig. 1). W-hydrogel was prepared via in situ gelation of acrylamide in partially delignified wood, consisting of holocellulose (cellulose and hemicellulose) and lignin ca. 98.3% (88.1% cellulose and 10.2% hemicellulose) and 1.7%, respectively (Fig. 2a)43,44. SEM images of W-hydrogel indicated that acrylamide was evenly polymerized throughout the porous structure of the delignified wood (Supplementary Fig. 2).

a Digital images of W-hydrogel. b Tensile stress of W-hydrogel (red line) and PAM gel (black line). c Fluorescence (black line) and phosphorescence spectra (red line) of W-hydrogel upon excitation of 290 nm. (measurement conditions: room temperature, delay time = 10 ms). d Changes of phosphorescence spectrum of W-hydrogel (black line) and W-hydrogel-E (red line). e Radar images of W-hydrogel, W-hydrogel-E, and W-hydrogel-E-W on the RTP lifetime, tensile strength, Young’s modulus, impact resistance, and specific puncture absorption energy. f Digital images of W-hydrogel and W-hydrogel-E loaded with a weight.
The water content in W-hydrogel was ~47.8% w/w. The as-obtained W-hydrogel was flexible and exhibited a tensile strengthen of 38.4 MPa, which was much higher than the hydrogel (PAM gel) prepared by initiating the polymerization of acrylamide in the absence of partially delignified wood (Fig. 2b). Moreover, the tensile strength of W-hydrogel was stronger than most reported hydrogels (Supplementary Fig. 3). Subsequently, the optical properties of W-hydrogel were investigated. W-hydrogel exhibited fluorescence centered at 390 nm upon 290 nm-light excitation and exhibited delayed phosphorescence emission centered at 490 nm after switching off the excitation source (Fig. 2c and Supplementary Movie 1). However, the introduction of pre-polymerized polyacrylamide into delignified wood did not trigger effective RTP emission (Supplementary Fig. 4). Notably, W-hydrogel with RTP emission can also be obtained using different monomers including acrylic acid (AA), N-Isopropyl acrylamide (NIAPM) and N.N’-Dimethylacrylamide (NDEAM) (Supplementary Fig. 5). Excitation-dependent emission of W-hydrogel was also observed. RTP emission shifted from 490 nm to 510 nm when the excitation wavelength went from 260 nm to 340 nm (Supplementary Fig. 6).
Time-resolved spectroscopy indicated that the W-hydrogel exhibited a long-lasting and stable afterglow emission (Supplementary Fig. 7). The lifetime of W-hydrogel was 32.5 ms. The lifetime of W-hydrogel was comparable to most of the reported RTP hydrogels (Supplementary Fig. 8). Notably, the lifetime of W-hydrogel was further increased to 102.1 ms using external heavy atoms via adding chloride ions to the W-hydrogel (Supplementary Fig. 9). Additionally, the lifetime was also enhanced by removing water from W-hydrogel. The lifetime increased to ~75.7 ms when the water content decreased from 47.8% to 13.8% in W-hydrogel (Supplementary Fig. 10). Interestingly, the RTP intensity of W-hydrogel was also enhanced upon treatment with ethanol (Fig. 2d). The RTP lifetime increased from 32.5 ms to 69.7 ms (Fig. 2e). Interestingly, the lifetime change was reversible and as such by immersing of the ethanol-treated W-hydrogel in water resulted in a decreased lifetime (Fig. 2e and Supplementary Fig. 11). The tensile strengthen of W-hydrogel was also enhanced after immersion in ethanol for 3 h. The tensile strength of the as-obtained W-hydrogel-E increased from 38.4 MPa to 153.8 MPa (Fig. 2e). Additionally, the impact resistance and specific puncture absorption energy also increased from 90.04 J m−1 and 128.63 J m−1 (g cm−3)−1 to 246.63 J m−1 and 269.59 J m−1 (g cm−3)−1, respectively (Fig. 2e, f and Supplementary Movie 2). Moreover, the whole process was reversible. W-hydrogel-E-W, which was obtained via immersing W-hydrogel-E into water, exhibited similar performance as W-hydrogel.
Mechanism
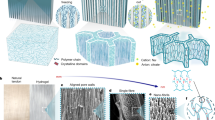
To understand the mechanism of RTP emission, PAM hydrogel was evaluated for RTP performance and did not exhibit any RTP emission (Fig. 3a). Additionally, partially delignified wood treated with water (DW in water) and partially delignified wood treated with a solution of acrylamide (DW in AM) also did not exhibit any RTP emission. These results confirm that the interactions between the components of delignified wood and polyacrylamide in W-hydrogel are crucial for the RTP emission properties. To further understand these interactions, a theoretical simulation was conducted (Supplementary Fig. 12). The calculations indicated that the adsorption force between monomer acrylamide and polysaccharide, the main component in delignified wood, was −191 kJ mol−1. As a comparison, the interaction force between polysaccharide and polyacrylamide increased to −223 kJ mol−1 (Fig. 3b). As such oxygen-incorporated moieties, such as hydroxyl moieties/carbonyl moieties of the polysaccharide and polyacrylamide, easily formed molecular clusters via hydrogen bonding, which are beneficial for the RTP emission. In addition, the strength of interaction between monomer acrylamide and lignin was determined to be −74 kJ mol−1, which was lower than the interaction force between lignin and polyacrylamide (−159 kJ mol−1) (Fig. 3c). Such enhanced interactions between lignin and polyacrylamide facilitate the confinement of lignin and trigger the radiative migration of triplet excitons. All these results confirmed that the interactions between the components of delignified wood and polyacrylamide triggered formation of molecular clusters and confined aromatic units, which contributed to the RTP emission of W-hydrogel. Moreover, adding 98.3% cellulose and 1.7% lignin, or adding 10.2% hemicellulose (xylan), which is similar to the components of delignified wood to PAM hydrogel did not trigger RTP emission of the hydrogel (Supplementary Figs. 13, 14). This result further illustrates that the orderly arrangement of the components in the delignified wood is crucial for effective RTP emission. Subsequently, the enhanced emission of W-hydrogel after treatment with ethanol was evaluated. 2D WAXS indicated that both the microfibrils of the W-hydrogel and W-hydrogel-E exhibited bright birefringence along the wood growth direction, indicating excellent alignment and crystallinity (Fig. 3d)45. Additionally, comparison of WAXS spectra between PAM hydrogel, W-hydrogel, W-hydrogel-E and delignified wood indicated that W-hydrogel and W-hydrogel-E exhibited similar crystal structure as delignified wood (Fig. 3e and Supplementary Fig. 15). However, the crystallinity index (CI) calculated by the Segal method indicated that W-hydrogel exhibited a lower crystallinity (46.98%) than W-hydrogel-E (49.70%). Further analysis indicated that the (200) diffraction peaks of W-hydrogel and W-hydrogel-E were at 22.44° and 22.56°, respectively. The signals shifted towards higher angles for W-hydrogel-E indicating a decrease in the crystallite spacing (d200) (Fig. 3e)46,47. After that, the crystal sizes were studied. There were four peak types (1-10), (110), (102) and (200) obtained via Gaussian fitting in the range of 5°−30° (Supplementary Fig. 16) from one-dimensional scattering profiles of 2D WAXS. The crystal sizes of W-hydrogel in (1-10), (110), and (200) directions increased from 0.67, 0.60, and 0.71 nm to 0.85, 0.81, and 0.82 nm, respectively, and the crystal width decreased from 7.63, 5.66, and 3.97 nm to 5.96, 5.08, and 3.93 nm, respectively after ethanol treatment. These results indicate that the crystal size increased and the crystal spacing decreased. All these results indicate that W-hydrogel-E exhibits a more crystalline and rigid environment than W-hydrogel, which is beneficial for stabilizing the triplet excitons and RTP emission18,28,31,34. Significantly the signals for hydroxyl moieties shifted from 3333 to 3326 cm−1 in the FT-IR spectra after treating the W-hydrogel with ethanol, indicating more intensive hydrogen bonding formed upon addition of ethanol to the W-hydrogel (Fig. 3f)48. Such enhanced orientation and hydrogen bonding enhance the mechanical performance and RTP emission of the W-hydrogel.

a RTP lifetime of PAM gel, partially delignified wood treated with water (DW in water), partially delignified wood treated with a solution of acrylamide (DW in AM) and W-hydrogel. b Theoretical simulation of the interaction between monomer acrylamide (6 units) and polysaccharide (upper); Theoretical simulation of the interaction between polyacrylamide (6 units) and polysaccharide (lower). c Theoretical simulation of the interaction between polyacrylamide (6 units) and lignin (upper); Theoretical simulation of the interaction between monomer acrylamide (6 units) and lignin (lower). d 2D WAXS of W-hydrogel and W-hydrogel-E. e One-dimensional scattering profiles of W-hydrogel (black line) and W-hydrogel-E (red line). f FT-IR of W-hydrogel (black line) and W-hydrogel-E (red line).
TS-FRET between W-hydrogel and RhB
Shifting the afterglow emission to the red region is required to extend the potential applications of the RTP materials33. RTP emission of W-hydrogel overlapped with the absorbance of RhB, indicating energy transfer could happen between W-hydrogel and RhB (Fig. 4a). As such W-hydrogel was evaluated for the production of red afterglow emission using a triplet to singlet Förster resonance energy transfer (TS-FRET) strategy with RhB. Therefore, RhB was loaded into W-hydrogel to prepare RhB@W-hydrogel. The as-obtained RhB@W-hydrogel exhibited red afterglow emission upon UV excitation and the afterglow emission matched the fluorescence spectra of RhB (Fig. 4b). The lifetime of the red afterglow emission at 600 nm in the RhB@W-hydrogel was 17.8 ms (Supplementary Fig. 17).

a The RTP emission of W-hydrogel (red line) and the absorbance of RhB (black line). b Fluorescence (black line) and RTP spectra (red line) of RhB@W-hydrogel. c Phosphorescence spectra of RhB@W-hydrogel at different RhB concentrations (0.002 mg mL−1 (red line), 0.005 mg mL−1 (blue line), 0.01 mg mL−1 (purple line), 0.02 mg mL−1 (orange line), 0.05 mg mL-1 (green line)) (excitation wavelength = 290 nm). d RhB@W-hydrogel energy transfer efficiency at different RhB concentrations.
The effect of the loading amount of RhB on the optical performance of W-hydrogel was further investigated. The emission spectra of RhB@W-hydrogel (λex = 290 nm) exhibited a gradual decrease of the phosphorescence emission centered at 490 nm and a concomitant enhancement of RhB fluorescence at 600 nm with an increased concentration of RhB (0.002 to 0.05 mg mL−1), which indicated an efficient energy transfer from the triplet state of the donor to the acceptor molecules (Fig. 4c). The highest energy transfer efficiency can reach 77.8% when the concentration of RhB was 0.05 mg mL−1 (Fig. 4d). Furthermore, time-resolved emission lifetime analyses of the RhB@W-hydrogel phosphorescence monitored at 490 nm (λex = 290 nm), exhibited a gradual decrease of the lifetime from 23.0 ms to 7.2 ms with higher RhB content (Supplementary Fig. 18). All these results eliminate the possibility of a simple emission-reabsorption energy transfer process, since for this situation the donor lifetime is not expected to change. Thus, the energy transfer between W-hydrogel and RhB was attributed to non-radiative FRET from the triplet state of the W-hydrogel donors to the singlet states of the RhB acceptors in the hydrogel (TS-FRET). Additionally, upon selective excitation of the acceptor at 490 nm, no afterglow emission was observed (Supplementary Fig. 19). Hence, it was evident that the long-lived triplet excitons of the donor W-hydrogel were the only source of the delayed population of the singlet state of the acceptor RhB via an efficient TS-FRET mechanism resulting in persistently delayed fluorescence.
Processability and applications
Utilizing the ethanol-sensitive mechanical performance, a flexible flat-shaped W-hydrogel was processed into a self-standing 3D “roll” with RTP emission using ethanol (Fig. 5a). The as-obtained 3D shapes could be further processed into soft and flexible shapes by subjecting them to water treatment. The “ethanol and water” processing was reversible and could be repeated several times. Moreover, the soft and flexible W-hydrogel or RhB@W-hydrogel can also be processed into complicated 3D structures with RTP emission, which become tough and self-standing after treatment with ethanol (Fig. 5b). W-hydrogel or RhB@W-hydrogel were also cut into RTP threads using scissors (Fig. 5c). The as-obtained hydrogel threads of W-hydrogel or RhB@W-hydrogel were mechanically flexible and could be used as building blocks for producing textiles with multiple-color afterglow emission (Fig. 5d). Notably, these as-obtained textiles were also sensitive to ethanol. They can be converted into self-standing and tough shapes after treatment with ethanol (Fig. 5e). However, the textiles become soft and flexible after they are again immersed in the water. The whole process was also switchable and could be recycled several times. Utilizing these properties, W-hydrogel was used as a building block for producing functional RTP materials (Fig. 5f). To this end, a section of the W-hydrogel was pre-folded and treated with ethanol. As a result, two different mechanical performances were observed in one W-hydrogel. The untreated part was flexible and the treated part was tough and maintained its shape even after loading with a weight. Considering the potential of luminescent sutures for accurate ___location during surgical introduction and removal49, the as-obtained RhB@W-hydrogel threads were used for suturing tissue. The W-hydrogel and RhB@W-hydrogel threads exhibited blue/red emission upon UV excitation (Fig. 5g and Supplementary Fig. 20). Green and red afterglow emission was observed from these threads after removing the excitation sources. Such afterglow emission effectively avoids background emission from the tissue and provides the possibility for imaging the sutures in a biological environment.

a RTP image of W-hydrogel after processing into different shapes. b Afterglow images of 3D shapes from W-hydrogel and RhB@W-hydrogel. c Digital, fluorescent, and afterglow images of threads made from W-hydrogel and RhB@W-hydrogel. d Digital, fluorescent, and afterglow images of textiles made from W-hydrogel and RhB@W-hydrogel threads. e RTP images of textiles made from W-hydrogel threads reversibly treated by ethanol and water. f Digital, fluorescent, and afterglow images of treated W-hydrogel threads loaded with weight. g Digital, fluorescent, and afterglow images of pork tissues sutured using RhB@W-hydrogel threads.
Discussion
In summary, we have developed W-hydrogel using delignified wood as the structural skeleton. Driven by the interaction between the polyacrylamide and components of delignified wood, our as-obtained W-hydrogel exhibited RTP emission with lifetimes of 32.5 ms and a tensile strengthen up to 38.4 MPa. Moreover, the mechanical and RTP performances were further enhanced upon treating the W-hydrogel with ethanol. These mechanical and RTP changes were reversible via repeated “ethanol and water” treatments. Additionally, W-hydrogel was used as energy donor for producing red afterglow emission at 600 nm together with RhB via a TS-FRET energy transfer process with efficiency up to 77.8%. Our as-obtained W-hydrogel and RhB@W-hydrogel with green/red afterglow emission exhibited good processing flexibility. As such we used the processing flexibility induced by ethanol treatment, to generate self-standing 3D emissive structures, threads, and textiles from W-hydrogel and RhB@W-hydrogel. Moreover, W-hydrogel and RhB@W-hydrogel threads exhibited great potential as luminescent sutures.
As outlined in the introduction, RTP hydrogels exhibit great potential as sensors, bio actuators, and for information encryption because of the semi-solid nature, combining the unique advantages of solid and solution states. However, the weak mechanical performance and non-tunable RTP performance hindered the realization of their full potential. Thus, this research aims to build a practical solution to the challenges associated with RTP hydrogels. Moreover, considering the convenient manipulation, low price, and sustainability of W-hydrogel, we anticipate that this research will also provide a practical method for producing high-performance RTP hydrogels on a large scale.
Methods
Preparation of partially delignified wood
The basswood was immersed in a mixed aqueous solution (2.5 M NaOH and 0.4 M Na2SO3) and boiled for 8 h to remove part of the hemicellulose and lignin. After that, the treated wood was washed with hot deionized water 4 times. Then, the wood samples were coated with hydrogen peroxide (30% w/w) and exposed to a Xenon lamp (400 mW cm−2) for further bleaching the lignin for 210 min. The resulting partially delignified wood was then washed 5 times with hot deionized water and ethanol.
Preparation of W-hydrogel
The wet partially delignified wood was soaked in 17 mL of acrylamide solution (0.34 g mL−1) for 2.5 h, and the samples were then transferred to a mold composed of two glass plates (15 cm × 15 cm) and a silicone frame (thickness: 1 mm) and placed in an oven at 60 °C for 2 h for initiating the gelation process.
Preparation of RhB@W-hydrogel
A certain amount of RhB was weighed and dissolved in water (10 mL) to prepare aqueous solutions of RhB with different concentrations. The partially delignified wood was soaked in 17 mL of acrylamide (0.34 g mL−1) and a RhB solution with different concentrations for 2.5 h, and the samples were then transferred to a mold composed of two glass plates (15 cm × 15 cm) and a silicone frame (thickness: 1 mm) and placed in an oven at 60 °C for 2 h for initiating the gelation. The as-obtained sample was RhB@W-hydrogel.
Data availability
All data are included in this article and its Supplementary Information files, including Supplementary Information, Supplementary Movie 1, and Supplementary Movie 2. Source data are provided with this paper. All data underlying this study are available from the corresponding author Zhijun Chen upon request. Source data are provided with this paper.
References
Luo, X. et al. Room-temperature phosphorescent materials derived from natural resources. Nat. Rev. Chem. 7, 800–812 (2023).
Guo, J., Yang, C. & Zhao, Y. Long-lived organic room-temperature phosphorescence from amorphous polymer systems. Acc. Chem. Res. 55, 1160–1170 (2022).
Wang, J. et al. A facile strategy for realizing room temperature phosphorescence and single molecule white light emission. Nat. Commun. 9, 2963 (2018).
Zhang, T. et al. Molecular engineering for metal-free amorphous materials with room-temperature phosphorescence. Angew. Chem. Int. Ed. 59, 11206 (2020).
Louis, M. et al. Blue-light-absorbing thin films showing ultralong room-temperature phosphorescence. Adv. Mater. 31, e1807887 (2019).
Guo, H. et al. Photocured room temperature phosphorescent materials from lignosulfonate. Nat. Commun. 15, 1590 (2024).
Wang, K., Qu, L. & Yang, C. Long-lived dynamic room temperature phosphorescence from carbon dots based materials. Small 19, e2206429 (2023).
Li, D., Yang, J., Fang, M., Tang, B. Z. & Li, Z. Stimulus-responsive room temperature phosphorescence materials with full-color tunability from pure organic amorphous polymers. Sci. Adv. 8, eabl8392 (2022).
Zhao, W., He, Z. & Tang, B. Z. Room-temperature phosphorescence from organic aggregates. Nat. Rev. Mater. 5, 869–885 (2020).
Ma, X., Wang, J. & Tian, H. Assembling-induced emission: an efficient approach for amorphous metal-free organic emitting materials with room-temperature phosphorescence. Acc. Chem. Res. 52, 738–748 (2019).
Baryshnikov, G., Minaev, B. & Ågren, H. Theory and calculation of the phosphorescence phenomenon. Chem. Rev. 117, 6500–6537 (2017).
An, Z. et al. Stabilizing triplet excited states for ultralong organic phosphorescence. Nat. Mater. 14, 685–690 (2015).
Wu, W. & Liu, B. Aggregation-induced emission: challenges and opportunities. Natl Sci. Rev. 8, nwaa222 (2021).
Gu, L. et al. Colour-tunable ultra-long organic phosphorescence of a single-component molecular crystal. Nat. Photonics 13, 406–411 (2019).
Zhou, X., Zhao, X., Bai, X., Cheng, Q. & Liu, Y. Thermal activated reversible phosphorescence behavior of solid supramolecule mediated by β-cyclodextrin. Adv. Funct. Mater. 34, 2400898 (2024).
Ju, H. et al. Polymerization-induced crystallization of dopant molecules: an efficient strategy for room-temperature phosphorescence of hydrogels. J. Am. Chem. Soc. 145, 3763–3773 (2023).
Hamzehpoor, E. et al. Efficient room-temperature phosphorescence of covalent organic frameworks through covalent halogen doping. Nat. Chem. 15, 83–90 (2023).
Luo, W. et al. Hydrogen-bonded organic frameworks enabling highly robust aqueous phase ultralong room-temperature phosphorescence. Adv. Funct. Mater. 34, 2401728 (2024).
Wang, P. et al. Confinement of sustainable carbon dots results in long afterglow emitters and photocatalyst for radical photopolymerization. Angew. Chem. Int. Ed. 63, e202402915 (2024).
Xu, Z. et al. Supercooled liquids with dynamic room temperature phosphorescence using terminal hydroxyl engineering. Angew. Chem. Int. Ed. 62, e202301564 (2023).
Liu, R. et al. Producing a room temperature phosphorescent film from natural wood using a top-down approach. Adv. Funct. Mater. 34, 2312254 (2024).
Lv, B. et al. Natural hybrid-mediated long-lived room temperature phosphorescence of milk powder. J. Mater. Chem. C. 10, 15629–15637 (2022).
Cao, M. et al. Producing naturally degradable room-temperature phosphorescent materials by covalently attaching lignin to natural polymers. Cell. Rep. Phys. Sci. 5, 101811 (2024).
Hou, L. et al. Intrinsic anti-freezing and unique phosphorescence of glassy hydrogels with ultrahigh stiffness and toughness at low temperatures. Adv. Mater. 35, e2300244 (2023).
Wan, K. et al. Structural materials with afterglow room temperature phosphorescence activated by lignin oxidation. Nat. Commun. 13, 5508 (2022).
Shi, W. et al. Time-dependent phosphorescence color of carbon dots in binary salt matrices through activations by structural confinement and defects for dynamic information encryption. Angew. Chem. Int. Ed. 62, e202303063 (2023).
Liu, D. et al. Tough, transparent, and slippery PVA hydrogel led by syneresis. Small 19, e2206819 (2023).
Deng, J. et al. Room-temperature phosphorescent tough hydrogels based on ionically crosslinked nonaromatic polymers. Adv. Funct. Mater. 34, 2308420 (2024).
Su, G. et al. Information-storage expansion enabled by a resilient aggregation-induced-emission-active nanocomposite hydrogel. Adv. Mater. 34, e2207212 (2022).
Zhou, W. et al. Ultralong purely organic aqueous phosphorescence supramolecular polymer for targeted tumor cell imaging. Nat. Commun. 11, 4655 (2020).
Cao, Y. et al. Multi-functional integration of phosphor, initiator, and crosslinker for the photo-polymerization of flexible phosphorescent polymer gels. Angew. Chem. Int. Ed. 63, e202401331 (2024).
Deng, J. et al. Metal cation-responsive and excitation-dependent nontraditional multicolor fluorescent hydrogels for multidimensional information encryption. ACS Appl. Mater. Interfaces 13, 39967–39975 (2021).
Garain, S., Garain, B., Eswaramoorthy, M., Pati, S. & George, S. Light-harvesting supramolecular phosphors: highly efficient room temperature phosphorescence in solution and hydrogels. Angew. Chem. Int. Ed. 60, 19720–19724 (2021).
Yu, S. et al. Room-temperature phosphorescence hydrogel with multiple color based on salt-induced aggregation. Adv. Opt. Mater. 12, 2303330 (2024).
Mao, Q. et al. Advanced marine antifouling hydrogels based on 7-amino-4-methylcoumarin fluorescence driven by rare-earth Phosphorescence. ACS Appl. Mater. Interfaces 15, 57582–57592 (2023).
Gan, D. et al. Plant-inspired adhesive and tough hydrogel based on Ag-Lignin nanoparticles-triggered dynamic redox catechol chemistry. Nat. Commun. 10, 1487 (2019).
Hu, D., Zeng, M., Sun, Y., Yuan, J. & Wei, Y. Cellulose-based hydrogels regulated by supramolecular chemistry. SusMat 1, 266–284 (2021).
Zhong, Y. et al. Antimicrobial/biocompatible hydrogels dual-reinforced by cellulose as ultrastretchable and rapid self-healing wound dressing. Biomacromolecules 22, 1654–1663 (2021).
You, J. et al. Aggregation-regulated room-temperature phosphorescence materials with multi-mode emission, adjustable excitation-dependence and visible-light excitation. Nat. Commun. 14, 4163 (2023).
Gao, Q. et al. Stereospecific redox-mediated clusterization reconstruction for constructing long-lived, color-tunable, and processable phosphorescence cellulose. Chem. Eng. J. 451, 138923 (2023).
Wan, K. et al. Sustainable afterglow room-temperature phosphorescence emission materials generated using natural phenolics. Angew. Chem. Int. Ed. 61, e202202760 (2022).
Yin, W. et al. Producing sustainable room temperature phosphorescent materials using natural wood and sucrose. Cell. Rep. Phys. Sci. 5, 101792 (2024).
Zhai, Y. et al. Room temperature phosphorescence from natural wood activated by external chloride anion treatment. Nat. Commun. 14, 2614 (2023).
Zhu, L. et al. Transparent bioplastics from super-low lignin wood with abundant hydrophobic cellulose crystals. ACS Sustain. Chem. Eng. 10, 13775–13785 (2022).
Zhang, T. et al. Shape and stiffness switchable hydroplastic wood with programmability and reproducibility. ACS Nano. 17, 23524–23534 (2023).
Hill, S. J. et al. Effect of drying and rewetting of wood on cellulose molecular packing. Holzforschung 64, 421–427 (2010).
Tang, J. et al. Superstrong, sustainable, origami wood paper enabled by dual-phase nanostructure regulation in cell walls. Sci. Adv. 10, eado5142 (2024).
Gong, K., Hou, L. & Wu, P. Hydrogen-bonding affords sustainable plastics with ultrahigh robustness and water-assisted arbitrarily shape engineering. Adv. Mater. 34, e2201065 (2022).
Ma, Z. et al. Bioinspired tough gel sheath for robust and versatile surface functionalization. Sci. Adv. 7, eabc3012 (2021).
Acknowledgements
Z.C. wishes to thank the National Natural Science Foundation of China (32471803, 31890774). T.D.J. wishes to thank the University of Bath and the Open Research Fund of the School of Chemistry and Chemical Engineering, Henan Normal University (2020ZD01) for their support.
Author information
Authors and Affiliations
Contributions
Conceptualization: Z.C., S.Li., and T.D.J.; Methodology: R.L.; Investigation: R.L., and H.G.; Visualization: J.L., and S.Liu.; Supervision: Z.C., S.Li., and T.D.J.; Writing-original draft: All authors; Writing-review & editing: All authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Baozhong Lü, Jitendra Mata, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, R., Guo, H., Liu, S. et al. Room temperature phosphorescent wood hydrogel. Nat Commun 15, 10588 (2024). https://doi.org/10.1038/s41467-024-55025-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55025-z
