
Abstract
The formation of \({\rm H}_{3}^{+}\) following the double ionization of small organic compounds via a roaming mechanism, which involves the generation of H2 and subsequent proton abstraction, has recently garnered significant attention. Nonetheless, a cohesive model explaining trends in the yield of \({\rm H}_{3}^{+}\) characterizing these unimolecular reactions is yet to be established. We report yield and femtosecond time-resolved measurements following the strong-field double ionization of CH3X molecules, where X = OD, Cl, NCS, CN, SCN, and I. These measurements, combined with double-ionization-potential equation-of-motion coupled-cluster ab initio calculations used to determine the geometries and energetics of CH3X2+ dications, are employed to identify the key factors governing the formation of \({\rm H}_{3}^{+}\) in certain doubly ionized CH3X species and its absence in others. We also carry out ab initio molecular dynamics simulations to obtain detailed microscopic insights into the mechanism, yields, and timescales of \({\rm H}_{3}^{+}\) production. We find that the excess relaxation energy released after double ionization of CH3X molecules combined with substantial geometrical distortion that favors H2 formation prior to proton abstraction boost the generation of \({\rm H}_{3}^{+}\). Our study provides useful guidelines for examining alternative sources of \({\rm H}_{3}^{+}\) in the universe.
Similar content being viewed by others
Introduction
The role of the trihydrogen cation (\({\rm H}^{+}_{3}\)) as a catalytic protonator of atoms and molecules, along with its capacity for radiative cooling, renders it one of the most important species in interstellar chemistry1,2,3. The formation of \({{\mbox{H}}}_{3}^{+}\) ions within the interstellar medium proceeds via the Hogness and Lunn mechanism4, \({\,{\rm{H}}}_{2}^{+}+{{{\rm{H}}}}_{2}\to {{\mbox{H}}}_{3}^{+}+{{\rm{H}}}\), where \({\rm H}_{2}^{+}\) is generated through the ionization of molecular hydrogen by cosmic rays. The resulting \({{\mbox{H}}}_{3}^{+}\) species are highly acidic and exhibit enhanced reactivity by acting as universal proton donors to other species (M) through the proton-hop reaction \({{\rm{M}}}+{{\rm{H}}}_{3}^{+}\to {{{\rm{H}}}}_{2}+{{{\rm{MH}}}}^{+}\)1. Furthermore, reactions involving \({\rm H}_{3}^{+}\) display very high rate constants2 that can be attributed to its ability to polarize and attract neutral molecules, even at large distances5. The abundance of \({\rm H}_{3}^{+}\) in the interstellar medium and its key role as the initiator of ion-molecule reactions highlight the importance of understanding the mechanisms, timescales, and yields of \({\rm H}_{3}^{+}\) production.
In recent years, various small molecules, including CH3X, where X is a halogen or pseudohalogen, have been identified as potential sources of \({\rm H}_{3}^{+}\) in interstellar and star-forming environments6. Upon double ionization, the three C–H bonds in CH3X may be cleaved and three new bonds between the detached H atoms may form, leading to the formation of \({\rm H}_{3}^{+}\). Given that equal numbers of bonds are broken and formed, this unimolecular reaction was initially thought to proceed via a concerted pathway in which the three H atoms in CH3X2+ coalesce and \({\rm H}_{3}^{+}\) is ejected by Coulombic repulsion7,8. However, it was observed that in methanol, the mechanism of \({\rm H}_{3}^{+}\) formation involves a three-step process, where the double ionization of CH3OH generates a neutral H2 which then roams the CHOH2+ dication before abstracting a proton from it9. In this case, femtosecond time-resolved strong-field ionization and photoion-photoion coincidence measurements on CH3OH and its isotopologues, accompanied by ab initio molecular dynamics (AIMD) simulations, revealed two distinct routes for the production of \({\rm H}_{3}^{+}\) depending on which proton is abstracted from CHOH2+ by H2, including a faster ∼ 100 fs process, in which the H atom attached to carbon is attacked, and a slower ∼ 250 fs one, when the abstracted proton originates from the hydroxyl group9 (for the follow-up studies of \({\rm H}_{3}^{+}\) formation from CH3OH and other alcohols, see refs. 10,11,12). It was also demonstrated that the faster pathway, corresponding to the deprotonation of carbon by the roaming H2, as shown in the lower half of Fig. 1, is the dominant process in CH3OH. In this study, we argue that a similar process applies to the \({\rm H}_{3}^{+}\)-producing doubly ionized CH3X molecules in which X lacks hydrogen atoms. It should also be noted that this way of generating \({\rm H}_{3}^{+}\) from CH3X molecules closely resembles the Hogness and Lunn mechanism, as both involve the abstraction of a proton by a neutral H2 molecule. In support of this observation, a recent experiment applying a two-color femtosecond laser field to D2—D2 dimers confirmed a 139 fs timescale for \({{\rm{{D}}}}_{3}^{+}\) production13, which matches the timeframe of \({\rm H}_{3}^{+}\) formation from the doubly ionized methanol via the pathway shown in Fig. 1, suggesting the possibility of a similar roaming mechanism in the interstellar space.

The upper half of the figure illustrates the process of generating a molecular dication by doubly ionizing methanol via electron rescattering. The electric field of the ultrafast laser pulse (represented by the dashed line) bends methanol’s potential (shown in yellow), resulting in tunnel ionization which creates a high-energy electron that accelerates back toward the molecule when the field reverses its direction. The returning electron has sufficiently high kinetic energy to remove the second electron from the molecule within the same optical cycle, producing the CH3OH2+ dication. Given the short pulse duration (∼ 35 fs), the dynamics of the CH3OH2+ species, shown in the lower half of the figure, are not influenced by the electric field of the laser. The dication in its lowest singlet state ejects a neutral H2 fragment consisting of the hydrogen atoms labeled as H(2) and H(3), which then roams the remaining CHOH2+ moiety until it abstracts the proton corresponding to the hydrogen atom H(1) bound to the carbon, leading to the formation of \({\rm H}_{3}^{+}\).
Another CH3X species that has been identified as a source of \({\rm H}_{3}^{+}\) in the interstellar clouds is methyl chloride14. Previous experiments on the strong-field ionization of CH3Cl7,15 have focused on the kinetic energy release and anisotropy of various fragmentation channels of the CH3Cl2+ dication, but insights into the timescale and mechanism of \({\rm H}_{3}^{+}\) formation from methyl chloride remain largely unexplored. Computational studies have predicted a variety of pathways for producing the trihydrogen cation from CH3Cl, including the mechanism in which \({\rm H}_{3}^{+}\) emerges from ionization of the Rydberg state of CH3Cl+ based on the lower appearance energy of the CCl+/\({\rm H}_{3}^{+}\) pair compared to the double ionization of methyl chloride16 or the concerted process without involving the roaming of H28, but the most recent calculations combining high-level quantum chemistry and molecular dynamics17 suggest the roaming mechanism similar to Fig. 1. Thus, there is a growing need to understand the intimate details of the molecular picture behind \({\rm H}_{3}^{+}\) formation following the double ionization of CH3Cl and other methyl halides and pseudohalides. A number of other observations are worth exploring as well. For example, complementary cross-section measurements at 70 eV electron ionization of the CH3X molecules with X = F, Br, and I have shown that similarly to CH3Cl, \({\rm H}_{3}^{+}\) forms readily from CH3Br, but only trace amounts of the trihydrogen cation have been found in the case of the doubly ionized CH3F and CH3I species18. This pattern, which remains consistent across different experimental studies, including two-color laser field ionization19, suggests that one has to look for factors other than electronegativities of the halogen atoms to explain \({\rm H}_{3}^{+}\) formation from methyl halogens. The behavior of methyl pseudohalides upon double ionization is not well understood either. For instance, as shown in the early photoionization and electron impact mass spectrometry measurements involving several small organic molecules20, the doubly ionized methyl cyanide produces negligible amounts of \({\rm H}_{3}^{+}\) despite the high electronegativity of the CN group. A comprehensive explanation of the varied yields of \({\rm H}_{3}^{+}\) from methyl halogens and pseudohalogens remains elusive to this date.
In this article, we report femtosecond time-resolved measurements following the strong-field double ionization of several CH3X molecules, where X is a halogen (Cl and I) or a pseudohalogen (OH, represented by OD, NCS, CN, and SCN). In the ultrafast experiments carried out in this work, the double ionization is triggered by electron rescattering21, as opposed to being induced by radiation or collision with high-energy light nuclei and electrons, which are the dominant processes in the interstellar medium. In electron rescattering, a departing electron, after tunnel-ionization of the molecule by the electric field of the laser, accelerates back toward the molecule when the field reverses its direction. The returning electron can gain tens of eV of kinetic energy, sufficient to remove the second electron from the molecule within the optical cycle of the laser pulse (∼ 2.66 fs). In principle, the returning electron can populate several electronic states of the resulting CH3X2+ dications, but the previous studies on the doubly ionized methanol, reported in refs. 11, 22, suggest that the excited manifolds of the CH3X2+ species, including singlet as well as triplet states, do not significantly impact the \({\rm H}_{3}^{+}\) formation dynamics investigated in this work. The results reported in the present article point to the same conclusion. While we chose tunnel-ionization and electron rescattering to generate CH3X2+ dications in our experiments, which are illustrated in the upper half of Fig. 1 using methanol as an example, the dynamics of \({\rm H}_{3}^{+}\) formation following the double ionization of CH3X species via a roaming mechanism examined in the present study are not influenced by the electric field of the laser pulse once H2 is formed. This also means that they do not depend on the specific method by which the CH3X2+ dications are created. To gain further insights into the yields and timescales of \({\rm H}_{3}^{+}\) formation from the various methyl halides and pseudohalides under investigation and to obtain information about the low-lying electronic states of the CH3X2+ dications involved, we augment the ultrafast time-resolved measurements reported in this study with the high-level double-ionization-potential (DIP) equation-of-motion (EOM) coupled-cluster (CC) ab initio computations with up to 3-hole–1-particle (3h-1p)23,24,25,26,27,28,29 and 4-hole–2-particle (4h-2p)28,29 correlations on top of the CC treatment30,31,32 with singles and doubles (CCSD)33,34 of the parent CH3X species, abbreviated as DIP-EOMCC(3h-1p) and DIP-EOMCC(4h-2p), respectively, and the AIMD simulations employing the complete-active-space self-consistent-field (CASSCF) approach35 calibrated to the DIP-EOMCC data. While our experimental measurements and theoretical models, despite their state-of-the-art nature, have inherent limitations, their complementary blend provides a detailed understanding of the microscopic factors that explain the formation of \({\rm H}_{3}^{+}\) by some doubly ionized CH3X species and its absence in the others, alongside the underlying molecular mechanisms, timescales, and yields, which may offer guidelines for examining small organic compounds as alternative sources of the trihydrogen cation in the interstellar medium.
Results
Experimental
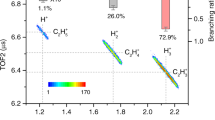
The \({\rm H}_{3}^{+}\) regions of the time-of-flight (TOF) mass spectra of all CH3X compounds with X = OD, Cl, NCS, CN, SCN, and I are shown in Fig. 2. For the CH3OD, CH3Cl, and CH3NCS molecules, which after double ionization produce the trihydrogen cation, the \({\rm H}_{3}^{+}\) signal in the mass spectrum appears as a doublet due to the forward and backward trajectories of the \({\rm H}_{3}^{+}\) ion resulting from the Coulomb repulsion of the \({{{\rm{CH}}}}_{3}{{{\rm{X}}}}^{2+}\to {{{\rm{CX}}}}^{+}+{{{\rm{H}}}}_{3}^{+}\) reaction products. This is because out of a set of randomly oriented CH3X molecules interacting with a laser, those with the dipole moments aligned parallel to the electric field of the laser are preferentially ionized, resulting in \({\rm H}_{3}^{+}\) ions that move toward or away from the detector. The \({\rm H}_{3}^{+}\) ions moving directly toward the detector arrive earlier, whereas those moving away arrive with a delay, after being turned around by the static electric field of the mass spectrometer. The forward and backward \({\rm H}_{3}^{+}\) peaks seen in Fig. 2 are sharp because the \({{{\rm{CH}}}}_{3}{{{\rm{X}}}}^{2+}\to {{{\rm{CX}}}}^{+}+{{{\rm{H}}}}_{3}^{+}\) dissociation processes that produce them are faster than rotations of the CH3X2+ dications (see Results: Timescales of \({\rm H}_{3}^{+}\) formation for further details). The presence of separate peaks due to the forward and backward trajectories of \({\rm H}_{3}^{+}\) ions resulting from the Coulomb repulsion of the positively charged CX+ and \({\rm H}_{3}^{+}\) species in our TOF mass spectra for the CH3OD, CH3Cl, and CH3NCS compounds implies that \({\rm H}_{3}^{+}\) formation in our strong-field ionization experiments originates from the dication states of the parent CH3X molecules rather than from their singly ionized counterparts. We used the deuterated form of methanol, CH3OD, where the 1H atom in the OH group is replaced by its 2H isotope, to ensure that the trihydrogen cation produced in our double ionization experiments originated from the three hydrogens of the methyl group. This gives rise to the HD+ signal from singly ionized methanol, which appears at m/z = 3 in Fig. 2 between the \({{{\rm{H}}}}_{3}^{+}\) forward and backward peaks. As shown in refs. 9, 36, the contribution of HD+ to the forward and backward peaks for CH3OD is expected to be small. We confirmed this by performing separate measurements on CD3OH which demonstrated that the contribution of HD+ to the forward and backward peaks, determined by forming a ratio of the HD+ yield normalized to all dication channels in CD3OH to the \({\rm H}_{3}^{+}\) yield normalized to all dication channels in CH3OD, is ∼ 9%.

The provided spectra correspond to CH3OD (red), CH3Cl (blue), CH3NCS (green), CH3CN (magenta), CH3SCN (brown), and CH3I (orange). In the case of the CH3OD, CH3Cl, and CH3NCS species, which produce the trihydrogen cation, the signal from \({\rm H}_{3}^{+}\) appears as a doublet labeled as fwd and bwd peaks, which are associated with the forward and backward trajectories of the \({\rm H}_{3}^{+}\) ion resulting from the Coulomb repulsion of the \({{{\rm{CH}}}}_{3}{{{\rm{X}}}}^{2+}\to {{{\rm{CX}}}}^{+}+{{{\rm{H}}}}_{3}^{+}\) reaction products. For CH3OD, we also see a central HD+ peak from singly ionized methanol between the fwd and bwd peaks of the \({\rm H}_{3}^{+}\) doublet since \({\rm H}_{3}^{+}\) and HD+ have the same m/z ratio. Based on the previous isotopologue studies involving methanol9,36 and our additional measurements on CD3OH discussed in Results: Experimental, the contribution of HD+ to the fwd and bwd peaks for CH3OD is expected to be small, having no significant effect on the reported \({\rm H}_{3}^{+}\) yields. For each studied compound, the single pulse intensity used for ionization was 5 × 1014 W/cm2. Each mass spectrum has been normalized by dividing the signal by the area of all peaks attributed to dication channels. The shaded portions of the \({\rm H}_{3}^{+}\) peaks indicate the regions that were integrated and subsequently doubled to calculate the yields of the trihydrogen cation, which are displayed as a bar graph on the right wall of the plot. In addition, to improve the visibility of the less intense signals, all mass spectra other than CH3OD have been scaled up by the numerical factors shown at the edges of the respective traces. CH3CN, CH3SCN, and CH3I do not produce appreciable \({\rm H}_{3}^{+}\) signals that would be distinguishable from the C4+ ion, which has the same m/z ratio as \({\rm H}_{3}^{+}\). All data are included in the Source Data folder.
To determine the \({\rm H}_{3}^{+}\) yield for each of the three CH3X compounds that produce it, shown in Table 1, we first normalized the corresponding mass spectrum to the integral of all ion signals attributed to dication channels. We then fit each of the two peaks in the \({\rm H}_{3}^{+}\) doublet to a Gaussian, integrated the outer halves of both peaks (the shaded regions in Fig. 2), and doubled the outcome. We only integrated the outer halves of each doublet to avoid contamination of the measured \({\rm H}_{3}^{+}\) yields by low kinetic energy features. We normalized the yields to all ion signals attributed to dication channels to correct for slight changes in pressure and/or intensity that may occur when going from one molecule to another, although these variables were carefully controlled in our experiments. In presenting the \({\rm H}_{3}^{+}\) yields resulting from the above procedure, we normalized them to the maximum one obtained for CH3OD, which was set to 1.0. This provided us with the relative \({\rm H}_{3}^{+}\) yields used in our later theoretical analysis. Furthermore, by normalizing the \({\rm H}_{3}^{+}\) yields relative to all dication channels to CH3OD, we counterbalanced the impact of the less controllable factors, such as the relative population levels of the singlet and triplet manifolds of the CH3X2+ species produced in our double ionization experiments or the competing fragmentation channels, on the \({\rm H}_{3}^{+}\) yields reported in Table 1, making them and, most importantly, the trends they follow less sensitive to all such factors. To illustrate the effect of the number of ion channels on yield determination, we also show in Table 1 the relative yields of \({\rm H}_{3}^{+}\) normalized to all positive ion channels. We only included cations in normalizing the \({\rm H}_{3}^{+}\) yields since our TOF system is configured for the detection of positively charged species. Our relative \({\rm H}_{3}^{+}\) yields match the results of the previous measurements using 70 eV electron impact ionization18.
Ab initio electronic structure calculations
To provide insights into the molecular mechanisms leading to the observed trends in \({\rm H}_{3}^{+}\) yields and the geometrical and energetic factors that affect them, we performed high-level ab initio quantum chemistry calculations for the low-lying electronic states of the neutral and doubly ionized CH3X species examined in this work, details of which are provided in Methods: Electronic structure calculations. We did not consider higher-lying states since, as shown in ref. 11 using methanol as an example, the probability of \({\rm H}_{3}^{+}\) formation is the highest when a given dicationic species is in its lowest singlet state. The selected bond lengths, which are crucial for our analysis, characterizing the equilibrium geometries of the CH3X molecules with X = OH, Cl, NCS, CN, SCN, and I in their ground electronic states and the minima on the lowest singlet potential energy surfaces of the corresponding CH3X2+ dications resulting from our most accurate CC and DIP-EOMCC computations, are shown in Table 2. The complete information about the geometries of the CH3X molecules included in our experiments and the CH3F species, which was only considered in computations, and the low-lying states of the corresponding CH3X2+ dications obtained in all of our CC and DIP-EOMCC calculations, along with the associated electronic energies, can be found in Supplementary Information (see Supplementary Data). For the three H atoms bonded to the carbon in the CH3X and CH3X2+ species, H(2) and H(3) designate hydrogens that may coalesce in the CH3X2+ dication and subsequently approach the remaining H atom on carbon, labeled as H(1), to form \({\rm H}_{3}^{+}\) (see Fig. 1). The vertical and adiabatic double ionization energies associated with the low-lying states of the CH3X2+ dications relevant to our discussion, obtained in our most accurate DIP-EOMCC calculations, are provided in Fig. 3. Our CC and DIP-EOMCC geometries of the CH3Cl, CH3Cl2+, CH3F, and CH3F2+ species and double ionization energies corresponding to the lowest singlet states of CH3Cl2+ and CH3F2+ are in very good agreement with the CASSCF-based second-order multireference perturbation theory calculations reported in ref. 17.

The vertical and adiabatic double ionization energies, in eV, of (a) CH3OH, (b) CH3Cl, (c) CH3NCS, (d) CH3CN, (e) CH3SCN, and (f) CH3I molecules associated with the lowest singlet (all systems) and triplet (CH3Cl, CH3CN, and CH3I) states of the respective dications resulting from the most accurate DIP-EOMCC computations performed in the present study (see Methods: Electronic structure calculations for the details) are shown. In each panel, the multiplicities, irreducible representations, and point group symmetries of the states of interest are highlighted in green and the adiabatic relaxation energies ΔErlx, obtained by subtracting the adiabatic double ionization energies from their vertical counterparts corresponding to the lowest singlet states of the CH3X2+ dications, Eq. (1), are emphasized using blue font. All data used to create the figure are included within it.
As shown in Fig. 3, the neutral CH3OH, CH3NCS, and CH3SCN molecules in their ground electronic states are characterized by the Cs, C1, and Cs symmetries, respectively, while the remaining CH3Cl, CH3CN, and CH3I species are all C3v-symmetric. Our computations demonstrate that the doubly ionized CH3OH, CH3NCS, and CH3SCN molecules in their lowest-energy singlet states relax to the respective adiabatic minima, adopting a Cs symmetry. As can be seen in Table 2, in the case of CH3OH2+ and CH3NCS2+, the resulting geometrical reorganization elongates the C–H(2) and C–H(3) bonds while bringing the H(2) and H(3) atoms close together, so that the distances between them are almost as small as in the isolated H2 molecule (0.741 Å37). This should be contrasted with the behavior of CH3SCN, where the H(2)–H(3), C–H(2), and C–H(3) distances do not change much when the geometries of the ground-state CH3SCN molecule and the CH3SCN2+ dication in its lowest singlet state are compared with each other.
The situation with the CH3Cl, CH3CN, and CH3I molecules is more complex. These three species, in their neutral forms, possess C3v-symmetric ground electronic states in which all C–H bonds have the same length. Following vertical double ionization, prior to any geometric relaxation, the three lowest electronic states of each of the resulting dications include a spatially nondegenerate triplet of the A2(C3v) symmetry and a doubly degenerate singlet that belongs to the E irreducible representation of C3v (see Fig. 3). The two components of the doubly degenerate singlet consist of a multi-configurational state mixing two closed-shell determinants, in which one of the two highest-occupied molecular orbitals (HOMOs) of the neutral species remains occupied by two electrons and the other one is empty, and the open-shell singlet state where each of the two degenerate HOMOs of the neutral system is singly occupied. The two degenerate HOMOs are also singly occupied in the lowest triplet state. Upon the geometric relaxation after double ionization, the lowest-energy triplet states of CH3Cl2+, CH3CN2+, and CH3I2+ remain C3v-symmetric, having structures similar to their neutral counterparts. In particular, the C–H bond lengths do not change much and the hydrogen atoms remain separated by rather large distances (see Supplementary Data, especially Supplementary Table 1). However, the lowest doubly degenerate singlets of these dications undergo Jahn–Teller (JT) distortion that removes the degeneracy and lowers their symmetry to Cs, splitting the 1E(C3v) singlets into the \({}^{1}{{{\rm{A}}}}^{{\prime} }({{{\rm{C}}}}_{{{\rm{s}}}})\) and \({}^{1}{{{\rm{A}}}}^{{\prime\prime} }({{{\rm{C}}}}_{{{\rm{s}}}})\) states. While the geometrical distortions and the JT splitting in CH3I2+ are minimal, so that the \({}^{1}{{{\rm{A}}}}^{{\prime} }({{{\rm{C}}}}_{{{\rm{s}}}})\) and \({}^{1}{{{\rm{A}}}}^{{\prime\prime} }({{{\rm{C}}}}_{{{\rm{s}}}})\) states, having three practically equidistant C–H bonds and large H–H separations, remain nearly degenerate and the A2(C3v)-symmetric triplet remains the ground state, the analogous changes in the CH3Cl2+ and CH3CN2+ species are massive. Indeed, as shown in Fig. 3, the \({}^{1}{{{\rm{A}}}}^{{\prime} }({{{\rm{C}}}}_{{{\rm{s}}}})\) states of the CH3Cl2+ and CH3CN2+ dications lower their energies so much, compared to the multi-configurational closed-shell components of the respective E(C3v)-symmetric singlets from which they originate, that they become the nondegenerate, predominantly single-reference, ground states. The \({}^{1}{{{\rm{A}}}}^{{\prime\prime} }({{{\rm{C}}}}_{{{\rm{s}}}})\) states resulting from the JT splitting of the E(C3v)-symmetric singlets of CH3Cl2+ and CH3CN2+, which retain the open-shell singlet character upon geometrical relaxation, lower their energies too, but not enough to go below the A2(C3v)-symmetric triplet states. Similarly to the triplet states, they do not undergo significant structural changes compared to the neutral CH3Cl and CH3CN molecules either (see Supplementary Data and Supplementary Table 1). As a result, the ordering of the adiabatic minima on the lowest potential energy surfaces of the CH3Cl2+ and CH3CN2+ dications becomes \({}^{1}{{{\rm{A}}}}^{{\prime} }({{{\rm{C}}}}_{{{\rm{s}}}}){ < \,}^{3}{{{\rm{A}}}}_{2}({{{\rm{C}}}}_{3{{\rm{v}}}}){ < \,}^{1}{{{\rm{A}}}}^{{\prime\prime} }({{{\rm{C}}}}_{{{\rm{s}}}})\). The stabilization of the Cs-symmetric structures of CH3Cl2+ and CH3CN2+ in their ground electronic states and the distortion in nuclear geometries compared to their neutral parents are so significant that the main features of the ground-state geometries of these two dications relevant to \({\rm H}_{3}^{+}\) production are similar to those characterizing the lowest singlet states of CH3OH2+ and CH3NCS2+. Indeed, as shown in Table 2, and in analogy to the lowest singlet states of CH3OH2+ and CH3NCS2+, the H(2)–H(3) distances in the ground-state CH3Cl2+ and CH3CN2+ structures are close to that in the isolated H2 molecule and the C–H(2) and C–H(3) bonds are considerably elongated compared to the neutral CH3Cl and CH3CN species. This should be contrasted with CH3I2+, where the H–H distances in the lowest singlet and triplet states are similar to those in neutral CH3I, preventing two of the three hydrogen atoms from coalescing and disabling the roaming mechanism of \({\rm H}_{3}^{+}\) production examined in this study.
One might think that the CH3X systems that undergo significant structural changes upon double ionization, which bring the H(2) and H(3) atoms bonded to the carbon close together while elongating the C–H(2) and C–H(3) bonds, favor \({\rm H}_{3}^{+}\) generation via the roaming mechanism shown in Fig. 1, but these structural changes are not the only factors influencing the yield of \({\rm H}_{3}^{+}\). For example, as shown in Table 2, the H(2)–H(3) distance in the lowest singlet state of CH3CN2+ is quite close to that in the isolated H2 (and much shorter than in neutral CH3CN) and the C–H(2) and C–H(3) bonds substantially elongate upon the double ionization of CH3CN, and yet we do not see an appreciable \({\rm H}_{3}^{+}\) signal in our experiments involving CH3CN (cf. Fig. 2 and Table 1). As noted in the Introduction, only trace amounts of the trihydrogen cation were found in the double ionization experiments involving CH3F18 despite the fact that CH3F2+ has a lot in common with its CH3Cl2+ analog, which readily forms \({\rm H}_{3}^{+}\) (as pointed out in Discussion, our AIMD simulations show no evidence for \({\rm H}_{3}^{+}\) production from the CH3F2+ species either). Indeed, the effects of JT distortion in the CH3F2+ and CH3Cl2+ species are similar, resulting in the \({}^{1}{{{\rm{A}}}}^{{\prime} }({{{\rm{C}}}}_{{{\rm{s}}}})\)-symmetric ground states and short, nearly identical, H(2)–H(3) distances (see the geometry of the CH3F2+ dication in its lowest-energy \({}^{1}{{{\rm{A}}}}^{{\prime} }({{{\rm{C}}}}_{{{\rm{s}}}})\) state and the electronic state ordering in CH3F2+ in Supplementary Data, especially Supplementary Fig. 1 and Supplementary Tables 1 and 2), but unlike in CH3Cl2+, no \({\rm H}_{3}^{+}\) is produced from CH3F2+. For all these reasons, in addition to examining the relationship between the geometrical changes in the CH3X systems upon double ionization and the observed yield of \({\rm H}_{3}^{+}\), we also considered other factors that might affect the production of trihydrogen cation from the CH3X2+ species. Thus, as further elaborated on in Methods: Electronic structure calculations, we computed the adiabatic relaxation energies
where ΔEvert and ΔEadiab are the vertical and adiabatic double ionization energies corresponding to the lowest singlet states of the CH3X2+ dications, and the dissociation energies associated with the H2 formation from the doubly ionized CH3X molecules, defined as
where E(CHX2+) and E(CH3X2+) are the energies of the CHX2+ and CH3X2+ species in their lowest singlet states, respectively, and E(H2) is the ground-state energy of a neutral H2. ΔErlx provides a measure of the excess energy available to the CH3X2+ dications at their adiabatic minima, which may drive the separation of a neutral H2 from the CHX2+ moiety and facilitate \({\rm H}_{3}^{+}\) generation via a roaming mechanism shown in Fig. 1. ΔEdiss estimates the energy required to liberate H2 from the parent CH3X2+ ion. We also computed potential energy profiles for the \({{{\rm{CH}}}}_{3}{{{\rm{X}}}}^{2+}\to {{{\rm{CX}}}}^{+}+{{{\rm{H}}}}_{3}^{+}\) reactions initiated by double ionization of the CH3X species with X = OH, Cl, and NCS that produce \({\rm H}_{3}^{+}\) in our experiments, finding out that the adiabatic relaxation energies ΔErlx characterizing the CH3OH2+, CH3Cl2+, and CH3NCS2+ dications are sufficiently large to overcome the barriers associated with the transition states on their lowest singlet potentials and release neutral H2 molecules that can subsequently roam their CHX2+ companions, abstracting protons from them to form \({\rm H}_{3}^{+}\) (see Supplementary Discussion and Supplementary Fig. 4). As shown in Discussion, the H(2)–H(3) distances combined with the ΔErlx and ΔEdiss values characterizing the doubly ionized CH3X species and information about the \({{{\rm{CH}}}}_{3}{{{\rm{X}}}}^{2+}\to {{{\rm{CX}}}}^{+}+{{{\rm{H}}}}_{3}^{+}\) reaction profiles, including barrier heights associated with the final \({{{\rm{CHX}}}}^{2+}+{{{\rm{H}}}}_{2}\to {{{\rm{CX}}}}^{+}+{{{\rm{H}}}}_{3}^{+}\) proton abstraction steps, can be used to explain why some CH3X species form \({\rm H}_{3}^{+}\) while others do not and rationalize the observed \({\rm H}_{3}^{+}\) ion yields.
Timescales of \({\rm H}_{3}^{+}\) formation
For each compound, we also studied the \({\rm H}_{3}^{+}\) formation time using a technique called disruptive probing, as outlined in Methods: Experimental details38. In short, we split the laser pulse into a strong pump (5 × 1014 W/cm2) and a weak probe (9.5 × 1013 W/cm2). By changing the delay between these two pulses with a variable delay stage, the weak probe can disrupt the formation of some ions in the mass spectra, leading to an exponential decay and recovery in the ion yields as a function of pump-probe delay. The timescale of formation of each ion seen in the mass spectrum, including \({\rm H}_{3}^{+}\), can be determined by fitting the corresponding pump-probe delay-dependent ion yield to the function
where
and t is a pump-probe time delay. The first term on the right-hand side of Eq. (3) describes the dependence of the ion yield on the Gaussian pump pulse, whereas the second and third terms are solutions of the differential rate equation described in ref. 38 that accounts for changes in the populations of ion states by Gaussian pump pulses. In the case of the pump-probe experiments reported in the present study, we need two types of such solutions, P1(t, τ1) characterized by time constant τ1 for a decay (τdecay) of the ion signal and P2(t, τ2) using time constant τ2 for the signal’s rise (τrise). The parameters A in Eq. (3) and A1 and A2 in Eq. (4) are the respective amplitude factors and s is related to the full width at half maximum pulse duration τFWHM through the expression \({\tau }_{{{\rm{FWHM}}}}=2s\sqrt{\ln 2}\).
To obtain information about the time-resolved dynamics of \({\rm H}_{3}^{+}\) formation for each of the three CH3X compounds that produce the trihydrogen cation in measurable amounts, i.e., CH3OD, CH3Cl, and CH3NCS, the peak area of the doublet assigned to \({\rm H}_{3}^{+}\) in the corresponding mass spectrum was integrated as a function of pump-probe delay. The resulting time-resolved yields of \({\rm H}_{3}^{+}\) from CH3OD, CH3Cl, and CH3NCS, shown in Fig. 4, were normalized such that the ion yield at negative pump-probe delays is 1.0 and the minimum is 0.0. We could not perform the analogous analysis for the remaining CH3X species examined in this study since they do not generate enough \({\rm H}_{3}^{+}\) to obtain time-resolved data with sufficient signal-to-noise ratio (cf. Fig. 2 and Table 1).

The \({\rm H}_{3}^{+}\) yields are shown for CH3OD (red), CH3Cl (green), and CH3NCS (blue). Black lines indicate fits of the experimental data to Eq. (3). The resulting values of parameter τrise, displayed in the panels, indicate the times of formation of \({\rm H}_{3}^{+}\) from the three molecules. The \({\rm H}_{3}^{+}\) yields for the CH3OD2+ and CH3Cl2+ transients level off below the baseline (ion yield = 1), indicating fragmentation of the \({\rm H}_{3}^{+}\) ion while it is still confined within the focal volume. In the case of the CH3NCS2+ transient, the long pump-probe delay asymptote is at a value above the baseline, which suggests that the pump pulse has populated an excited state of the corresponding monocation that has subsequently been ionized by the weak probe to the dication state, resulting in \({\rm H}_{3}^{+}\) production from an additional channel. All data are included in the Source Data folder.
From the time-resolved \({\rm H}_{3}^{+}\) yields in Fig. 4, we see a large spike at the pump-probe delay t = 0. The presence of this spike is a consequence of the intensities of the pump and probe pulses combining to give an increased ionization efficiency within the focal volume. Shortly after the double ionization of the CH3X compounds by the pump pulse, the resulting CH3X2+ dications are far from equilibrium and can easily be disrupted by the probe pulse. This leads to a depletion of the \({\rm H}_{3}^{+}\) ion yield at short positive pump-probe delay times t because, as explained in ref. 38, the probe pulse can promote the transient CH3X2+ system to some other electronic state that results in a reduced \({\rm H}_{3}^{+}\) yield or does not produce \({\rm H}_{3}^{+}\) at all. For example, the probe may suppress the return of the roaming H2 to the CHX2+ moiety, preventing \({\rm H}_{3}^{+}\) formation. For each CH3X2+ molecule, a resonant single or multiphoton transition induced by the probe has a complicated relationship with the reaction coordinate as the H2 moiety changes orientation and distance from the CHX2+ species during the roaming process. Averaging over the ensemble of CH3X2+ dications excited by the pump pulse washes out these specific details, leaving only a measure of the fraction of the ensemble that has yet to form \({\rm H}_{3}^{+}\). As the pump-probe delay becomes larger and the fraction of the roaming ensemble that has undergone the \({{{\rm{CHX}}}}^{2+}+{{{\rm{H}}}}_{2}\to {{{\rm{CX}}}}^{+}+{{{\rm{H}}}}_{3}^{+}\) reaction increases, the yield of \({\rm H}_{3}^{+}\) ions begins to grow, eventually leveling off because the \({\rm H}_{3}^{+}\) ions that have already been formed are not appreciably disturbed by the probe pulse. Averaging over the ensemble’s stochastic roaming dynamics results in an exponential signal dependence proportional to the reaction time, as observed here and in other direct measurements of roaming reactions9,10,39,40. Interestingly, the doubly ionized CH3OD generates the most \({\rm H}_{3}^{+}\) (Table 1), while having the shortest time for \({\rm H}_{3}^{+}\) formation when compared to CH3Cl and CH3NCS (Fig. 4).
Ab initio molecular dynamics simulations
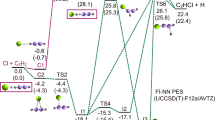
To further investigate the timescale and mechanism of \({\rm H}_{3}^{+}\) formation, AIMD simulations were conducted for a few representative CH3X2+ species in their lowest singlet states using the CASSCF approach to generate electronic energies and energy gradients (see Methods: Ab initio molecular dynamics simulations for the details). Exploring single-state dynamics on the lowest-energy singlet potentials of the CH3X2+ species is sufficient to provide useful insights that help us understand the outcomes of our experiments since, as already alluded to above (cf. the beginning of Results: Ab initio electronic structure calculations), the probability of \({\rm H}_{3}^{+}\) formation from the doubly ionized methanol is the highest in its lowest singlet state, rapidly dropping down when higher excited states are considered11. At the same time, states belonging to triplet manifolds of the CH3X2+ dications are not conducive to the release of a neutral H2 that could subsequently roam and abstract a proton from the CHX2+ moiety to form \({\rm H}_{3}^{+}\) 22. Of all the molecules that we examined with AIMD, \({\rm H}_{3}^{+}\) only forms from the doubly ionized CH3OH and CH3Cl, and, to a much lesser extent, CH3CN. Consistent with the experiment, from the trajectories obtained in our AIMD calculations, the doubly ionized CH3OH produced the most \({\rm H}_{3}^{+}\) (9% of the trajectories, counting only those where all three hydrogens in \({\rm H}_{3}^{+}\) originate from the methyl group), followed by CH3Cl (6% of the trajectories). In the case of CH3CN2+, the calculated \({\rm H}_{3}^{+}\) yield was much smaller than those obtained for CH3OH2+ and CH3Cl2+ (only 1% of the trajectories). This matches our experimental findings too, as the doubly ionized CH3CN molecule does not produce an appreciable \({\rm H}_{3}^{+}\) signal in the mass spectrum under the conditions of our ionization experiments (cf. Fig. 2). Furthermore, formation of the H2 precursor, which in the roaming mechanism explored in this work leads to \({\rm H}_{3}^{+}\), rarely occurred in CH3CN2+ trajectories. In agreement with our experiments, none of the trajectories obtained for CH3SCN2+ produced \({\rm H}_{3}^{+}\). Consistent with the earlier experimental18 and computational17 studies, no trajectories obtained for the doubly ionized CH3F molecule resulted in the formation of \({\rm H}_{3}^{+}\) either.
To understand the different timescales of \({\rm H}_{3}^{+}\) formation, trajectories that resulted in \({\rm H}_{3}^{+}\) were further analyzed. On average, \({\rm H}_{3}^{+}\) formed faster from CH3OH2+ (∼ 208 fs) than from CH3Cl2+ (∼ 343 fs), which agrees with previous AIMD simulations9,11,17 and our experimental observations. Interestingly, in a majority of the trajectories that led to \({\rm H}_{3}^{+}\), the neutral H2 fragment formed very fast (in ∼ 30 fs) and then roamed the CHX2+ moiety for most of the remaining time prior to proton abstraction. Snapshots of one of the AIMD trajectories resulting in the formation of \({\rm H}_{3}^{+}\) from the doubly ionized CH3Cl are shown in Fig. 5. Supplementary Movies 1 and 2 show representative trajectories leading to \({\rm H}_{3}^{+}\) formation from CH3Cl2+ and CH3OH2+. Supplementary Movie 3 shows an example of a trajectory obtained for CH3F2+, where the liberated H2 fragment becomes positively charged and subsequently dissociates, so that the final products are the CH2F+ moiety and a proton.

The trajectory from which these snapshots originate is shown in Supplementary Movie 1. The Cartesian coordinates of the molecular geometries shown in the figure, along with the corresponding AIMD trajectory, are included in the Source Data folder.
Discussion
In our ultrafast time-resolved double ionization experiments on a series of methyl halides and pseudohalides, we observed a progressive decrease in the yield of \({\rm H}_{3}^{+}\) from CH3OD to CH3Cl to CH3NCS, while no appreciable amount of this ion was detected in CH3CN, CH3SCN, and CH3I (see Table 1 and Fig. 2). Moreover, as shown in Fig. 4, the process of forming \({\rm H}_{3}^{+}\) from the doubly ionized CH3X species was the fastest for CH3OD, followed by CH3NCS and CH3Cl, with the time of \({\rm H}_{3}^{+}\) formation from CH3Cl being the longest. Based on our experiments and computations, the following three conditions must be simultaneously met in order for the doubly ionized CH3X species to have the potential of generating \({\rm H}_{3}^{+}\) via a roaming mechanism illustrated in Figs. 1 and 5:
-
(i)
Geometrical distortion following double ionization should result in two substantially elongated C–H bonds compared to the neutral CH3X molecule.
-
(ii)
The H(2)–H(3) internuclear distance characterizing the equilibrium geometry of a given CH3X2+ dication in its lowest singlet electronic state should be close to the H–H bond length in an isolated H2 molecule.
-
(iii)
The adiabatic relaxation energy ΔErlx characterizing the CH3X2+ dication, as defined by Eq. (1), should exceed the dissociation energy ΔEdiss, Eq. (2), associated with the fragmentation of the doubly ionized CH3X species into CHX2+ and H2, and be large enough to overcome the barrier(s) along the \({{{\rm{CH}}}}_{3}{{{\rm{X}}}}^{2+}\to {{{\rm{CX}}}}^{+}+{{{\rm{H}}}}_{3}^{+}\) potential energy profile, including that corresponding to the \({{{\rm{CHX}}}}^{2+}+{{{\rm{H}}}}_{2}\to {{{\rm{CX}}}}^{+}+{{{\rm{H}}}}_{3}^{+}\) proton abstraction by the roaming H2, but it cannot be so large that other reactive mechanisms and other ways of decomposing CH3X2+ become dominant.
The C–H bond lengths characterizing the neutral and doubly ionized CH3X molecules in their lowest singlet states, summarized in Table 2, and the H(2)–H(3) distances and the ΔErlx and ΔEdiss values characterizing the CH3X2+ dications, reported in Fig. 6, along with the \({{{\rm{CH}}}}_{3}{{{\rm{X}}}}^{2+}\to {{{\rm{CX}}}}^{+}+{{{\rm{H}}}}_{3}^{+}\) (X = OH, Cl, and NCS) reaction profiles shown in Supplementary Fig. 4, demonstrate that of all CH3X compounds considered in our experiments, only CH3OH, CH3Cl, and CH3NCS satisfy all three conditions listed above in their entirety. This is in perfect agreement with our observations, where CH3OD, CH3Cl, and CH3NCS are the only three molecules of the six CH3X species considered in our double ionization experiments that produce \({\rm H}_{3}^{+}\) (the experimental \({\rm H}_{3}^{+}\) yields normalized to CH3OH2+, taken from Table 1, are included in Fig. 6 as well). In fact, as shown in Fig. 6, the sequence of the experimental \({\rm H}_{3}^{+}\) yields CH3OH > CH3Cl > CH3NCS closely mirrors the difference between the adiabatic relaxation and dissociation energies, ΔErlx and ΔEdiss, respectively. This difference is the largest for CH3OH, which produces the highest amount of \({\rm H}_{3}^{+}\), smaller for CH3Cl, resulting in a correspondingly lower yield, and the smallest for CH3NCS, characterized by the least production of \({\rm H}_{3}^{+}\).

The H(2)–H(3) distances, in Å, in the CH3X2+ dications, where X = OH, Cl, NCS, CN, SCN, and I (blue numbers, circles, and line), the adiabatic relaxation energies ΔErlx, Eq. (1), in eV, corresponding to the lowest singlet states of the doubly ionized CH3X molecules (red numbers, squares, and line), and the dissociation energies ΔEdiss, Eq. (2), also in eV, associated with the fragmentation of the CH3X2+ species into H2 and CHX2+ (green numbers, diamonds, and line) resulting from the most accurate DIP-EOMCC computations carried out in the present study described in Methods: Electronic structure calculations are shown alongside the normalized yields of \({\rm H}_{3}^{+}\) relative to all dication channels taken from Table 1 for X = OD, Cl, and NCS (magenta numbers and bars) and assumed to be zero for the remaining species that do not generate appreciable \({\rm H}_{3}^{+}\) amounts. The H–H bond length of an isolated H2 molecule is highlighted in gray. All data used to create the figure are included within it.
None of the remaining CH3X compounds included in our experiments satisfy all three conditions (i)–(iii), which means that they should not generate \({\rm H}_{3}^{+}\) upon double ionization, and, as shown in Fig. 2, they do not. The CH3SCN and CH3I systems violate each of the three conditions. CH3CN, which meets conditions (i) and (ii), violates condition (iii), which states that ΔErlx must be greater than ΔEdiss. Clearly, the energy released by the geometrical relaxation of the CH3X2+ dication, measured by the difference of the vertical and adiabatic double ionization energies defining ΔErlx, must be large enough to liberate a neutral H2, without which the roaming and proton abstraction mechanism of \({\rm H}_{3}^{+}\) production shown in Figs. 1 and 5 cannot be initiated, and CH3CN2+ does not satisfy this requirement. This demonstrates that the elongation of two of the three C–H bonds and two of the three hydrogens bonded to the carbon in CH3X2+ coming close, while important, do not suffice; one also has to meet the condition ΔErlx > ΔEdiss. Interestingly, despite satisfying conditions (i) and (ii) and the ΔErlx > ΔEdiss requirement, which is part of condition (iii) (see Supplementary Tables 1 and 2 for the C–H and H(2)–H(3) distances characterizing the neutral and doubly ionized CH3F molecule and the ΔErlx and ΔEdiss values obtained in our best DIP-EOMCC calculations for CH3F2+), the CH3F species that we studied only computationally does not form \({\rm H}_{3}^{+}\) upon double ionization. As mentioned in Results: Ab initio molecular dynamics simulations, and in agreement with the older experimental18 and more recent computational17 studies, none of the trajectories obtained in our AIMD simulations for CH3F2+ produced \({\rm H}_{3}^{+}\). In this case, as shown in Supplementary Fig. 3, the adiabatic relaxation energy ΔErlx, i.e., the energy released by the geometrical relaxation of CH3F2+ after double ionization of CH3F, is much higher than in the remaining CH3X2+ species included in our experiments and, as a result, redistributed very differently than in the CH3OH2+, CH3Cl2+, and CH3NCS2+ dications that form \({\rm H}_{3}^{+}\). This is illustrated by Supplementary Movie 3, which shows a typical trajectory obtained in our AIMD simulations for CH3F2+, where the liberated, positively charged, H2 fragment dissociates and the final products are CH2F+ and a proton. It may be worth noticing that the adiabatic relaxation energy ΔErlx resulting from our most accurate DIP-EOMCC computations for the CH3F2+ dication, reported in Supplementary Table 2 and Supplementary Fig. 3, is large enough to dissociate both H2 and \({{{\rm{H}}}}_{2}^{+}\), and we plan to study the significance of this finding further in the future work, but we can already observe that CH3F2+ is the only CH3X2+ species considered in the present study which has this property. In a small number of trajectories, the liberated H2 fragment, having sufficiently large energy available to it, escaped the system, preventing \({\rm H}_{3}^{+}\) formation as well. Once again, this is consistent with condition (iii), which allows such a scenario if the adiabatic relaxation energy ΔErlx is high enough, which is the case when CH3F2+ is examined.
As explained in Results and Methods, the mechanism of \({\rm H}_{3}^{+}\) formation from the doubly ionized CH3X species that emerges from the above conditions (i)–(iii), in which the neutral H2 fragment resulting from a geometrical distortion of the CH3X2+ dication roams CHX2+, abstracting a proton from it, was confirmed and further examined using AIMD simulations. AIMD simulations have limitations, which in our case are the relatively small number and limited duration of the computed trajectories and the limited quality of the CASSCF potential surfaces on which we had to rely (see Methods: Ab initio molecular dynamics simulations). It is also very difficult to precisely model the stochastic nature of the roaming dynamics examined in our experiments, discussed in Results: Timescales of \({\rm H}_{3}^{+}\) formation. Nevertheless, our calculated yields and timescales of \({\rm H}_{3}^{+}\) formation are in qualitative agreement with the experimental observations. The analysis of the AIMD trajectories shows that the timescale of \({\rm H}_{3}^{+}\) formation is related to the ability of the roaming H2 to appropriately align itself relative to the CHX2+ fragment. From the trajectories resulting from our AIMD simulations, such as that shown in Fig. 5 or those visualized by Supplementary Movies 1 and 2, for a substantial portion of time prior to the formation of \({\rm H}_{3}^{+}\), a neutral H2 roams the CHX2+ moiety before orienting itself in a suitable edge-on configuration facing the hydrogen of CHX2+, which facilitates the completion of the \({{{\rm{CHX}}}}^{2+}+{{{\rm{H}}}}_{2}\to {{{\rm{CX}}}}^{+}+{{{\rm{H}}}}_{3}^{+}\) proton abstraction step (molecular structures corresponding to these edge-on configurations can also be seen in the \({{{\rm{CH}}}}_{3}{{{\rm{X}}}}^{2+}\to {{{\rm{CX}}}}^{+}+{{{\rm{H}}}}_{3}^{+}\) reaction profiles shown in Supplementary Fig. 4). The easier it is for the H2 and CHX2+ fragments to find this configuration, the faster the timescale of \({\rm H}_{3}^{+}\) formation becomes. This is reflected in our AIMD computations which demonstrate that the neutral H2 roaming the CHCl2+ fragment in the doubly ionized CH3Cl species explores a much larger portion of the phase space, when searching for a suitable alignment with CHCl2+, than the H2 roaming CHOH2+ in CH3OH2+. As a result, the average timescale of \({\rm H}_{3}^{+}\) formation from the doubly ionized CH3Cl species seen in our experiments and AIMD simulations is longer than in the CH3OH case (see Fig. 4, Results: Ab initio molecular dynamics simulations, and Supplementary Movies 1 and 2). Based on the above observation that the H2 and CHX2+ fragments have to align in a very specific configuration to enable \({\rm H}_{3}^{+}\) formation, one might speculate that larger adiabatic relaxation energy of a given doubly ionized CH3X molecule should lead to a faster exploration of the phase space and, thus, a shorter time needed to produce \({\rm H}_{3}^{+}\), but the results in Figs. 4 and 6 show that there is no direct correlation between the ΔErlx values characterizing the CH3X2+ dications and the timescales of \({\rm H}_{3}^{+}\) formation from these ions. This agrees with the recent computational study of the CH3X systems with X = F, Cl, and Br17.
Finally, an interesting pattern emerges when we compare the molecular orbitals obtained in the restricted Hartree–Fock (RHF) calculations for the CH3X molecules and the corresponding CH3X2+ dications that produce \({\rm H}_{3}^{+}\). This is illustrated in Fig. 7 using the neutral CH3OH and CH3Cl species and the corresponding dications at the respective equilibrium geometries as examples. In the neutral CH3Cl molecule, the two degenerate HOMOs exhibit bonding and antibonding characteristics with respect to the H(2) and H(3) atoms involved in H2 formation. The same is true when the nondegenerate HOMO − 1 and HOMO of the neutral CH3OH are examined. According to our DIP-EOMCC computations, the dominant process defining the double ionization of the CH3OH and CH3Cl species is the removal of two electrons from the highest-energy occupied orbital that has the antibonding H2 character. This makes the orbital that binds the two hydrogens of H2 the HOMO of the respective dication, bringing the H(2) and H(3) atoms close together, almost as in the isolated H2 molecule, facilitating its liberation and initiating the roaming mechanism shown in Figs. 1 and 5.

The RHF/cc-pVTZ orbitals describe the HOMO − 1 and HOMO(s) of the neutral CH3OH and CH3Cl molecules and the HOMOs of the corresponding dications at their respective equilibrium geometries. The GAMESS input and output files and information about the software used to plot the orbitals are provided in the Source Data folder.
In conclusion, we reported highly accurate yield and femtosecond time-resolved measurements following the strong-field double ionization of the CH3X molecules with X = OD, Cl, NCS, CN, SCN, and I, along with the ab initio quantum chemistry and AIMD computations. The goal was to obtain a detailed understanding of the microscopic factors that govern the production of \({\rm H}_{3}^{+}\) via a roaming mechanism in which a neutral H2 liberated from the CH3X2+ dication abstracts a proton from the CHX2+ moiety. By combining state-of-the-art experiments and computations, we provided deep insights into the molecular mechanism of \({\rm H}_{3}^{+}\) formation from the doubly ionized CH3X species, which allowed us to explain the observed yields and timescales and determine geometric and energetic conditions that must be met in order for \({\rm H}_{3}^{+}\) to form. The results reported in this work may be used to predict the behavior of methyl halides and pseudohalides upon strong-field double ionization and help future studies of alternative sources of \({\rm H}_{3}^{+}\) in the universe. They also lead to interesting new questions. One such question is the generality of the geometrical and energetic conditions (i)–(iii) laid down in Discussion, i.e., to what extent these three conditions apply to other organic compounds of astrophysical significance, such as hydrocarbons41,42,43,44 and longer-chain alcohols10,45, which may produce \({\rm H}_{3}^{+}\) as well. Our preliminary examination of the doubly ionized ethane and ethene using the same CC and DIP-EOMCC methodologies as those employed in our calculations for the CH3X systems, which are analyzed in greater detail in the Supplementary Discussion section of Supplementary Information, and our earlier studies of ethanol10,45 suggest that our conditions (i)–(iii) may apply to organic compounds outside the CH3X family, although additional factors may have to be considered as well. We will investigate to what extent these three conditions developed in the context of examining \({\rm H}_{3}^{+}\) formation from the doubly ionized methyl halides and pseudohalides apply to other organic compounds in future work.
Methods
Experimental details
We employed the disruptive probing technique for the synchronous monitoring of the ultrafast time-resolved product yields induced by strong-field ionization38. Our experimental setup features a regeneratively amplified Ti:sapphire laser system as the ionization source, which produces 35 fs pulses at a central wavelength of ∼ 800 nm and operates at a repetition rate of 1 kHz. The pulses were compressed using a Multiphoton Intrapulse Interference Phase Scan (MIIPS)46, which measures all orders of spectral phase distortions and compensates them using a pulse shaper (MIIPS-HD) with a spatial light modulator (640 × 800, Hamamatsu). Following compression, the pulses were split into a strong pump and a weak probe, with intensities of 5 × 1014 W/cm2 and 9.5 × 1013 W/cm2, respectively, using a beam splitter. The pump pulses used in this work are sufficiently intense to doubly ionize the CH3X molecules, and they are somewhat more intense than those employed in ref. 38, potentially populating several electronic states of the CH3X2+ dications, but this does not have a significant effect on the outcome of our experiments. The \({\rm H}_{3}^{+}\) formation dynamics via a roaming mechanism considered in the present study proceeds mainly on the lowest singlet states of the CH3X2+ species11, which means that any pump pulse providing enough energy to doubly ionize the molecules of interest suffices. For yield measurements, the probe pulse was blocked, allowing only the pump pulse to interact with the sample. The time delay between the pump and probe pulses was controlled using an optical delay line equipped with a programmable translation stage capable of nanometer-level precision control. The laser pulses were focused inside a Wiley-McLaren TOF mass spectrometer by a gold-coated concave focusing mirror with f = 300 mm. At the focus, the pump and probe pulses reached their peak intensities, which were calibrated by measuring the Ar2+/Ar+ ion yield ratios47.
The ionization potentials of the compounds under investigation span a range of approximately 9–12 eV, which corresponds to an average Keldysh parameter γ of 0.4 under our laser conditions48. It is well-known that γ < 1 suggests tunnel ionization as the dominant process, where the timescale of classical ionization is faster than the optical cycle. On the other hand, γ > 1 signifies that the ionization spans multiple optical cycles, indicative of multiphoton ionization. The average γ value of 0.4 in our experiments points to the tunneling regime, implying that the electric field is strong enough to ionize the molecules within a single optical cycle.
The CH3OD, CH3Cl, CH3NCS, CH3CN, CH3SCN, and CH3I samples, used without further purification, were degassed and loaded into the TOF chamber as a room-temperature effusive beam through a needle valve. The static pressure inside the vacuum chamber was maintained below 1 × 10−5 Torr during data acquisition. Upon closing the needle valve, the vacuum pressure quickly dropped to the baseline pressure of 5 × 10−8 Torr, which ensures fast sample refresh. The positively charged ions formed in the interaction volume were sent to a Chevron dual microchannel plate detector (RM Jordan) by a + 2168 V repeller plate and a + 1080 V extractor plate separated by 10 mm. Ion signals generated at the microchannel plate detector were digitized using an oscilloscope (LeCroy WaveRunner 610Zi, 1 GHz) and transferred to a computer for further processing and analysis.
Electronic structure calculations
To enhance our understanding of the experimental findings, we performed a series of high-level ab initio quantum chemistry calculations which shed light on the geometrical, energetic, and other factors that influence the mechanisms and yields of \({\rm H}_{3}^{+}\) formation following the strong-field double ionization of the methyl halides and pseudohalides examined in this work. As is normally done, all of our electronic structure calculations, including those for methanol, which in our experiments was represented by its deuterated CH3OD form, were performed using masses of atomic nuclei that correspond to the most abundant isotopes since isotopic substitution has no effect on electronic properties. We started by optimizing the ground-state equilibrium geometries of the closed-shell neutral CH3X (X = OH, Cl, NCS, CN, SCN, I, and F) molecules using the CCSD approach33,34. Then, in order to obtain the desired information about the electronic states of the CH3X2+ dications involved in our strong-field double ionization experiments and, to enrich our discussion of the behavior of the CH3X systems upon double ionization, of CH3F2+, we computed vertical double ionization energies of all studied CH3X species at their respective CCSD-optimized geometries using high-level methods belonging to the DIP-EOMCC hierarchy23,24,25,26,27,28,29. For the CH3X systems with the smaller X = OH, Cl, and F groups, we used both the state-of-the-art DIP-EOMCC(4h-2p) approach28,29 and its more economical DIP-EOMCC(3h-1p) counterpart23,24,25,26,28,29. In the case of the remaining CH3X molecules with the larger X = NCS, CN, SCN, and I fragments, we utilized DIP-EOMCC(3h-1p). The same DIP-EOMCC methods were applied to determine the minima on the ground and low-lying excited-state potential energy surfaces of the CH3X2+ species, which allowed us to obtain information about the adiabatic double ionization energies associated with the respective dicationic states and, more importantly, the energies released upon geometrical relaxation of the doubly ionized CH3X molecules to their respective equilibrium structures. We chose the particle-nonconserving DIP-EOMCC methodology in our computations since it is particularly well suited for an accurate description of the many-electron systems that are formally obtained by removing two electrons from their closed-shell parents (which themselves were treated by a highly correlated CCSD level) while relaxing the remaining electrons after double ionization. Furthermore, the DIP-EOMCC methods, including the DIP-EOMCC(3h-1p) and DIP-EOMCC(4h-2p) approaches used in our calculations, offer several advantages over the conventional particle-conserving CC30,31,32 and EOMCC49,50,51,52 treatments of open-shell species employing unrestricted or restricted open-shell reference determinants, such as rigorous spin and symmetry adaptation of the calculated electronic states and the ability to describe singlet and triplet manifolds of the doubly ionized species in a well-balanced manner.
The DIP-EOMCC computations for the CH3X2+, CHX2+, and CX+ ions and the preceding CCSD calculations for their closed-shell CH3X, CHX, and CX− parents, needed to set up the respective DIP-EOMCC eigenvalue problems, in addition to the aforementioned geometry optimizations for the CH3X molecules, were performed using the routines described in refs. 28,29,53, 54, which are part of the GAMESS software package55,56,57. In constructing the relevant wave functions, all CCSD and DIP-EOMCC computations relied on the RHF molecular orbitals of the CH3X, CHX, and CX− species and the correlation-consistent basis sets, including the cc-pVDZ and cc-pVTZ bases for H, C, N, F, O, S, and Cl atoms58,59, as implemented in GAMESS, with the additional tight d functions for S and Cl taken from ref. 60, and cc-pVTZ-PP61,62, taken from the EMSL Basis Set Exchange63, for iodine. In all post-RHF calculations, the core orbitals correlating with the 1s shells of the C, N, F, and O atoms and the 1s, 2s, and 2p shells of the S and Cl atoms were kept frozen. For the heavier I atom, the 28 inner-shell electrons ([Ar]3d10) were replaced by the relativistic effective core potential of Peterson et al.61,62 and the remaining 25 valence electrons were described by the cc-pVTZ-PP basis set. All of our geometry optimizations were initially carried out without imposing any symmetry constraints, but, once the minima on the relevant potential energy surfaces were found and a given species had symmetry higher than C1, which was the case for all systems examined in this work other than CH3NCS, we reoptimized the resulting structures using the appropriately chosen symmetrized Cartesian coordinates adapted to the point group symmetries of the molecules of interest. To confirm that all of our optimized geometries are minima on the respective potential energy surfaces, we determined the corresponding harmonic vibrational frequencies, which were real in each studied case.
The Cartesian coordinates defining all geometries of the CH3X and CH3X2+ species optimized in our CCSD (all CH3X molecules), DIP-EOMCC(3h-1p) (all CH3X2+ dications), and DIP-EOMCC(4h-2p) (CH3OH2+, CH3Cl2+, and CH3F2+) calculations, along with the corresponding total electronic energies, can be found in Supplementary Data. In presenting the selected bond lengths in Table 2, which are important for the material included in Results and the follow-up analysis in Discussion, characterizing the optimized geometries of the CH3X molecules with X = OH, Cl, NCS, CN, SCN, and I in their ground electronic states and the minima on the lowest singlet potential energy surfaces of the corresponding CH3X2+ dications, we use the data obtained in the most accurate CCSD (neutral CH3X molecules), DIP-EOMCC(4h-2p) (CH3OH2+ and CH3Cl2+), and DIP-EOMCC(3h-1p) (CH3NCS2+, CH3CN2+, CH3SCN2+, and CH3I2+) calculations performed in this work in which we utilized the cc-pVTZ basis set for H, C, N, O, S, and Cl (with additional tight d functions on S and Cl) and the cc-pVTZ-PP basis for iodine. The same applies to the vertical and adiabatic double ionization energies of the CH3OH, CH3Cl, CH3NCS, CH3CN, CH3SCN, and CH3I molecules reported in Fig. 3 and the geometries of the lowest singlet and triplet states of the CH3X2+ species with X = F, Cl, CN, and I and their ground-state CH3X parents shown in Supplementary Table 1 (the computational methodologies applied to CH3F and CH3F2+ were exactly the same as those used for the neutral and doubly ionized CH3OH and CH3Cl molecules).
Among the quantities that are useful in our theoretical analyses (other than the geometries and energies of the neutral and doubly ionized CH3X molecules) are the adiabatic relaxation energies ΔErlx characterizing the CH3X2+ dications, shown in Figs. 3 and 6, Supplementary Figs. 1–4, and (for X = F only) Supplementary Table 2 and defined by Eq. (1), and the dissociation energies ΔEdiss associated with the fragmentation of the doubly ionized CH3X species into CHX2+ and H2 prior to \({\rm H}_{3}^{+}\) formation, Eq. (2), reported in Fig. 6, Supplementary Fig. 3, and Supplementary Table 2. In our most accurate calculations presented in Figs. 3 and 6, Supplementary Fig. 3, and Supplementary Table 2, the vertical and adiabatic double ionization energies entering the definition of ΔErlx were calculated at the DIP-EOMCC(4h-2p)/cc-pVTZ level of theory for X = OH, Cl, and F and the DIP-EOMCC(3h-1p)/cc-pVTZ approach (using cc-pVTZ-PP for I atom) for the remaining CH3X2+ dications. The energies of the CHX2+ and CH3X2+ species in their lowest singlet states entering the definition of ΔEdiss were determined using the DIP-EOMCC(3h-1p)/cc-pVDZ theory level for all CH3X2+ and CHX2+ dications except CH3I2+ and CHI2+, which were treated with DIP-EOMCC(3h-1p)/cc-pVTZ (cc-pVTZ-PP for I atom). To be consistent in our calculations of the energies that appear on the right-hand side of Eq. (2), the energy of the H2 molecule entering the definition of ΔEdiss was computed using the CCSD/cc-pVDZ approach when the X = OH, Cl, NCS, CN, SCN, and F systems were considered and CCSD/cc-pVTZ in the X = I case (determining the equilibrium H–H distance in H2 at the respective CCSD/cc-pVDZ or CCSD/cc-pVTZ level using GAMESS). We note that the CCSD calculation for H2, which has only two electrons, is equivalent to an exact, full configuration interaction, computation.
In addition to the above characteristics of the CH3X2+ ions, we calculated potential energy profiles for the \({{{\rm{CH}}}}_{3}{{{\rm{X}}}}^{2+}\to {{{\rm{CX}}}}^{+}+{{{\rm{H}}}}_{3}^{+}\) reactions following double ionization of the CH3X molecules with X = OH, Cl, and NCS, along with the corresponding \({{{\rm{CH}}}}_{3}{{{\rm{X}}}}^{2+}\to {{{\rm{CHX}}}}^{ 2 +}+{{{\rm{H}}}}_{2}\) dissociation asymptotes relevant to the roaming mechanism examined in the present study (see Supplementary Fig. 4). In the case of the CH3X2+, CHX2+, and CX+ ions, we used the DIP-EOMCC(3h-1p)/cc-pVDZ approach, whereas the CH3X, H2, and \({\rm H}_{3}^{+}\) molecules were handled with CCSD/cc-pVDZ (again using additional tight d functions for S and Cl atoms). These calculations allowed us to demonstrate that the doubly ionized CH3OH, CH3Cl, and CH3NCS species have enough energy to liberate molecular hydrogen, which is a key step in the roaming mechanism considered in this work, and to overcome the barriers for \({\rm H}_{3}^{+}\) formation.
The details of our preliminary CC and DIP-EOMCC computations for the doubly ionized ethane and ethene species, which resulted in Supplementary Figs. 5–7 and Supplementary Tables 3 and 4 analyzed in Supplementary Discussion, can be found in Supplementary Information.
Ab initio molecular dynamics simulations
To gain further insights into the yields and timescales of \({\rm H}_{3}^{+}\) formation, we performed AIMD simulations for the CH3X2+ ions with X = OH, Cl, CN, SCN, and F by utilizing the GPU-accelerated CASSCF code implemented in the TeraChem software64,65,66,67. For each CH3X2+ species, the potential energy surface corresponding to its lowest singlet electronic state and its gradients were computed on-the-fly by using the CASSCF method with 4 electrons in 4 orbitals as an active space, abbreviated as CAS(4,4)SCF, in conjunction with the cc-pVDZ basis set. The orbital active spaces were carefully tailored to closely reproduce the adiabatic relaxation energies ΔErlx, Eq. (1), and the geometries of the CH3X2+ dications in their lowest singlet states obtained using the DIP-EOMCC(4h-2p)/cc-pVDZ approach for CH3OH2+, CH3Cl2+ and CH3F2+ and DIP-EOMCC(3h-1p)/cc-pVDZ for the remaining species (see Supplementary Fig. 2). To provide a reasonable representation of the molecular dynamics following double ionization, a total of 200 trajectories were computed for each CH3X2+ system. All trajectories were initiated and subsequently propagated on the lowest singlet states of the CH3X2+ dication species reached after vertical double ionization of their CH3X parents. The initial conditions (positions and momenta of atomic nuclei) were sampled from the Wigner distributions obtained for the neutral CH3X molecules using the B3LYP/cc-pVDZ approach68,69. All trajectories were integrated for 700 fs with a time step of 0.5 fs by using the velocity Verlet algorithm. Of the six CH3X systems included in our experiments, three, namely, CH3OH, CH3Cl, and CH3NCS, produce \({\rm H}_{3}^{+}\) after double ionization, so we wanted to perform AIMD simulations for CH3NCS2+ as well. However, the low yield of \({\rm H}_{3}^{+}\) resulting from the double ionization of CH3NCS would necessitate the determination of a much larger number of trajectories than in the CH3OH2+ and CH3Cl2+ cases to obtain sufficiently good statistics for describing the dynamics of the \({{{\rm{CH}}}}_{3}{{{\rm{NCS}}}}^{2+}\to {{{\rm{CNCS}}}}^{+}+{{{\rm{H}}}}_{3}^{+}\) process, which turned out to be prohibitively expensive for us.
Data availability
All data needed to evaluate the conclusions of this study are included in the article and Supplementary Information. Source data are provided in the Source Data folder. Any additional information related to this study may be requested from the authors.
Code availability
All ab initio DIP-EOMCC computations for the CH3X2+, CHX2+, and CX+ cations and the preceding CCSD calculations for the CH3X, CHX, and CX− parent molecules, as well as the additional computations for the neutral and doubly ionized ethane and ethene, were performed using the routines described in refs. 28, 29, 53, 54, which are part of the GAMESS software package55,56,57 that can be obtained from https://www.msg.chem.iastate.edu/gamess/download.html. The AIMD simulations for the selected CH3X2+ species, using CASSCF information calibrated to the DIP-EOMCC data, were carried out using the development version of TeraChem software64,65,66,67 provided by B.G.L.
References
Oka, T. Interstellar \({{{\rm{H}}}}_{3}^{+}\). Chem. Rev. 113, 8738–8761 (2013).
Herbst, E. & Klemperer, W. The formation and depletion of molecules in dense interstellar clouds. ApJ 185, 505–534 (1973).
Kannan, G., Chien, J. R., Benjamin, A. J., Bhatia, N. & Saykally, R. J. Rydberg states of H3 and HeH as potential coolants for primordial star formation. J. Phys. Chem. A 125, 4267–4275 (2021).
Hogness, T. & Lunn, E. The ionization potentials of hydrogen as interpreted by positive ray analysis. Proc. Natl. Acad. Sci. USA 10, 398–405 (1924).
Tennyson, J. Spectroscopy of \({{{\rm{H}}}}_{3}^{+}\) : planets, chaos and the universe. Rep. Prog. Phys. 58, 421–476 (1995).
Pilling, S. et al. Production of \({{{\rm{H}}}}_{3}^{+}\) via photodissociation of organic molecules in interstellar clouds. Mon. Not. R. Astron. Soc. 375, 1488–1494 (2007).
Sun, S. et al. Ejection of triatomic molecular ion \({{{\rm{H}}}}_{3}^{+}\) from methyl chloride in an intense femtosecond laser field. Chem. Phys. Lett. 581, 16–20 (2013).
Palaudoux, J. & Hochlaf, M. Formation of \({{{\rm{H}}}}_{3}^{+}\) through chloromethane dication fragmentation. ACS Earth Space Chem. 3, 980–985 (2019).
Ekanayake, N. et al. Mechanisms and time-resolved dynamics for trihydrogen cation (\({{{\rm{H}}}}_{3}^{+}\)) formation from organic molecules in strong laser fields. Sci. Rep. 7, 4703 (2017).
Ekanayake, N. et al. H2 roaming chemistry and the formation of \({{{\rm{H}}}}_{3}^{+}\) from organic molecules in strong laser fields. Nat. Commun. 9, 5186 (2018).
Livshits, E., Luzon, I., Gope, K., Baer, R. & Strasser, D. Time-resolving the ultrafast H2 roaming chemistry and \({{{\rm{H}}}}_{3}^{+}\) formation using extreme-ultraviolet pulses. Commun. Chem. 3, 49 (2020).
Wang, E. et al. Ultrafast proton transfer dynamics on the repulsive potential of the ethanol dication: Roaming-mediated isomerization versus coulomb explosion. J. Phys. Chem. A 124, 2785–2791 (2020).
Zhou, L. et al. Ultrafast formation dynamics of \({{{\rm{D}}}}_{3}^{+}\) from the light-driven bimolecular reaction of the D2–D2 dimer. Nat. Chem. 15, 1229–1235 (2023).
Fayolle, E. C. et al. Protostellar and cometary detections of organohalogens. Nat. Astron. 1, 703–708 (2017).
Ma, P. et al. Ultrafast proton migration and coulomb explosion of methyl chloride in intense laser fields. J. Chem. Phys. 146, 244305 (2017).
Duflot, D., Robbe, J.-M. & Flament, J.-P. Ab initio study of the chloromethane dication fragmentation. Int. J. Mass Spectrom. Ion Proc. 171, 215–230 (1997).
Matsubara, T. Dynamic effects on the ionization-induced reactions of methyl halides: Quantum mechanical and molecular dynamics studies. J. Phys. Chem. A 127, 4801–4814 (2023).
Newton, A. S., Sciamanna, A. & Thomas, G. The occurrence of the \({{{\rm{H}}}}_{3}^{+}\) ion in the mass spectra of organic compounds. Int. J. Mass Spectrom. Ion Phys. 5, 465–482 (1970).
Walt, S. G. et al. Role of multi-electron effects in the asymmetry of strong-field ionization and fragmentation of polar molecules: The methyl halide series. J. Phys. Chem. A 119, 11772–11782 (2015).
Eland, J. The origin of primary \({{{\rm{H}}}}_{3}^{+}\) ions in mass spectra. Rapid Commun. Mass Spectrom. 10, 1560–1562 (1996).
Corkum, P. Plasma perspective on strong multiphoton ionization. Phys. Rev. Lett. 71, 1994–1997 (1993).
Gope, K., Livshits, E., Bittner, D. M., Baer, R. & Strasser, D. Absence of triplets in single-photon double ionization of methanol. J. Phys. Chem. Lett. 11, 8108–8113 (2020).
Nooijen, M. & Bartlett, R. J. A new method for excited states: Similarity transformed equation-of-motion coupled-cluster theory. J. Chem. Phys. 106, 6441–6448 (1997).
Wladyslawski, M. & Nooijen, M. The photoelectron spectrum of the NO3 radical revisited: A theoretical investigation of potential energy surfaces and conical intersections. In Low-lying potential energy surfaces. ACS Symposium Series (eds. Hoffmann, M. R. & Dyall, K. G.), vol. 828, pp. 65–92 (American Chemical Society, Washington, D.C., 2002).
Nooijen, M. State selective equation of motion coupled cluster theory: Some preliminary results. Int. J. Mol. Sci. 3, 656–675 (2002).
Sattelmeyer, K. W., Schaefer III, H. F. & Stanton, J. F. Use of 2h and 3h-p-like coupled-cluster Tamm–Dancoff approaches for the equilibrium properties of ozone. Chem. Phys. Lett. 378, 42–46 (2003).
Kuś, T. & Krylov, A. I. Using the charge-stabilization technique in the double ionization potential equation-of-motion calculations with dianion references. J. Chem. Phys. 135, 084109 (2011).
Shen, J. & Piecuch, P. Doubly electron-attached and doubly ionized equation-of-motion coupled-cluster methods with 4-particle-2-hole and 4-hole-2-particle excitations and their active-space extensions. J. Chem. Phys. 138, 194102 (2013).
Shen, J. & Piecuch, P. Doubly electron-attached and doubly ionised equation-of-motion coupled-cluster methods with full and active-space treatments of 4-particle-2-hole and 4-hole-2-particle excitations: The role of orbital choices. Mol. Phys. 112, 868–885 (2014).
Čížek, J. On the correlation problem in atomic and molecular systems. Calculation of wavefunction components in Ursell-type expansion using quantum-field theoretical methods. J. Chem. Phys. 45, 4256–4266 (1966).
Čížek, J. On the use of the cluster expansion and the technique of diagrams in calculations of correlation effects in atoms and molecules. Adv. Chem. Phys. 14, 35–89 (1969).
Paldus, J., Čížek, J. & Shavitt, I. Correlation problems in atomic and molecular systems. IV. Extended coupled-pair many-electron theory and its application to the BH3 molecule. Phys. Rev. A 5, 50–67 (1972).
Purvis III, G. D. & Bartlett, R. J. A full coupled-cluster singles and doubles model: The inclusion of disconnected triples. J. Chem. Phys. 76, 1910–1918 (1982).
Cullen, J. M. & Zerner, M. C. The linked singles and doubles model: An approximate theory of electron correlation based on the coupled-cluster ansatz. J. Chem. Phys. 77, 4088–4109 (1982).
Roos, B. O., Taylor, P. R. & Siegbahn, P. E. M. A complete active space SCF method (CASSCF) using a density matrix formulated super-CI approach. Chem. Phys. 48, 157–173 (1980).
Eland, J. H. D. & Treves-Brown, B. J. The fragmentation of doubly charged methanol. Int. J. Mass Spectrom. Ion Processes 113, 167–176 (1992).
Huber, K. P. & Herzberg, G. Molecular spectra and molecular structure: IV. Constants of diatomic molecules (Van Nostrand Reinhold, New York, 1979).
Jochim, B., DeJesus, L. & Dantus, M. Ultrafast disruptive probing: Simultaneously keeping track of tens of reaction pathways. Rev. Sci. Instrum. 93, 033003 (2022).
Ekanayake, N. et al. Substituent effects on \({{{\rm{H}}}}_{3}^{+}\) formation via H2 roaming mechanisms from organic molecules under strong-field photodissociation. J. Chem. Phys. 149, 244310 (2018).
Word, M. D. et al. Ultrafast dynamics of nitro-nitrite rearrangement and dissociation in nitromethane cation. J. Phys. Chem. A 126, 879–888 (2022).
Zhang, Y. et al. Formation of \({{{\rm{H}}}}_{3}^{+}\) from ethane dication induced by electron impact. Commun. Chem. 3, 160 (2020).
Hoshina, K., Kawamura, H., Tsuge, M., Tamiya, M. & Ishiguro, M. Metastable decomposition and hydrogen migration of ethane dication produced in an intense femtosecond near-infrared laser field. J. Chem. Phys. 134, 064324 (2011).
Hoshina, K., Furukawa, Y., Okino, T. & Yamanouchi, K. Efficient ejection of \({{{\rm{H}}}}_{3}^{+}\) from hydrocarbon molecules induced by ultrashort intense laser fields. J. Chem. Phys. 129, 104302 (2008).
Zhang, Y. et al. Formation of \({{{\rm{H}}}}_{3}^{+}\) from hydrocarbon dications induced by collisions with charged particles. Phys. Rev. A 100, 052706 (2019).
Severt, T. et al. Initial-site characterization of hydrogen migration following strong-field double-ionization of ethanol. Nat. Commun. 15, 74 (2024).
Xu, B., Gunn, J. M., Dela Cruz, J. M., Lozovoy, V. V. & Dantus, M. Quantitative investigation of the multiphoton intrapulse interference phase scan method for simultaneous phase measurement and compensation of femtosecond laser pulses. J. Opt. Soc. Am. B 23, 750–759 (2006).
Guo, C., Li, M., Nibarger, J. P. & Gibson, G. N. Single and double ionization of diatomic molecules in strong laser fields. Phys. Rev. A 58, 4271–4274 (1998).
Keldysh, L. V. Ionization in the field of a strong electromagnetic wave. Soviet Phys. JETP 20, 1307–1314 (1965).
Emrich, K. An extension of the coupled cluster formalism to excited states (I). Nucl. Phys. A 351, 379–396 (1981).
Geertsen, J., Rittby, M. & Bartlett, R. J. The equation-of-motion coupled-cluster method: Excitation energies of Be and CO. Chem. Phys. Lett. 164, 57–62 (1989).
Stanton, J. F. & Bartlett, R. J. The equation of motion coupled-cluster method. A systematic biorthogonal approach to molecular excitation energies, transition probabilities, and excited state properties. J. Chem. Phys. 98, 7029–7039 (1993).
Piecuch, P. & Bartlett, R. J. EOMXCC: A new coupled-cluster method for electronic excited states. Adv. Quantum Chem. 34, 295–380 (1999).
Piecuch, P., Kucharski, S. A., Kowalski, K. & Musiał, M. Efficient computer implementation of the renormalized coupled-cluster methods: The R-CCSD[T], R-CCSD(T), CR-CCSD[T], and CR-CCSD(T) approaches. Comp. Phys. Commun. 149, 71–96 (2002).
Shen, J. & Piecuch, P. Double electron-attachment equation-of-motion coupled-cluster methods with up to 4-particle–2-hole excitations: Improved implementation and application to singlet–triplet gaps in ortho-, meta-, and para-benzyne isomers. Mol. Phys. 119, e1966534 (2021).
Schmidt, M. W. et al. General atomic and molecular electronic structure system. J. Comput. Chem. 14, 1347–1363 (1993).
Barca, G. M. J. et al. Recent developments in the general atomic and molecular electronic structure system. J. Chem. Phys. 152, 154102 (2020).
Zahariev, F. et al. The general atomic and molecular electronic structure system (GAMESS): Novel methods on novel architectures. J. Chem. Theory Comput. 19, 7031–7055 (2023).
Dunning Jr, T. H. Gaussian basis functions for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 90, 1007–1023 (1989).
Woon, D. E. & Dunning Jr, T. H. Gaussian basis functions for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 103, 1358–1371 (1993).
Dunning, J., Thom, H., Peterson, K. A. & Wilson, A. K. Gaussian basis sets for use in correlated molecular calculations. X. The atoms aluminum through argon revisited. J. Chem. Phys. 114, 9244–9253 (2001).
Peterson, K. A., Figgen, D., Goll, E., Stoll, H. & Dolg, M. Systematically convergent basis sets with relativistic pseudopotentials. II. Small-core pseudopotentials and correlation consistent basis sets for the post-d group 16-18 elements. J. Chem. Phys. 119, 11113–11123 (2003).
Peterson, K. A., Shepler, B. C., Figgen, D. & Stoll, H. On the spectroscopic and thermochemical properties of ClO, BrO, IO, and their anions. J. Phys. Chem. A 110, 13877–13883 (2006).
Pritchard, B. P., Altarawy, D., Didier, B., Gibson, T. D. & Windus, T. L. New basis set exchange: An open, up-to-date resource for the molecular sciences community. J. Chem. Inf. Model. 59, 4814–4820 (2019).
Ufimtsev, I. S. & Martínez, T. J. Quantum chemistry on graphical processing units. 3. Analytical energy gradients, geometry optimization, and first principles molecular dynamics. J. Chem. Theory Comput. 5, 2619–2628 (2009).
Fales, B. S. & Levine, B. G. Nanoscale multireference quantum chemistry: Full configuration interaction on graphical processing units. J. Chem. Theory Comput. 11, 4708–4716 (2015).
Hohenstein, E. G., Luehr, N., Ufimtsev, I. S. & Martínez, T. J. An atomic orbital-based formulation of the complete active space self-consistent field method on graphical processing units. J. Chem. Phys. 142, 224103 (2015).
Seritan, S. et al. TeraChem: A graphical processing unit-accelerated electronic structure package for large-scale ab initio molecular dynamics. WIREs Comput. Mol. Sci. 11, e1494 (2021).
Becke, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993).
Lee, C., Yang, W. & Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789 (1988).
Acknowledgements
S.K. was supported by the Air Force Office of Scientific Research under award number FA9550-21-1-0428 to M.D. The experimental work was funded by the Department of Energy, Office of Basic Energy Sciences, Atomic, Molecular, and Optical Sciences Program under award number SISGR (DE-SC0002325) to M.D. The theoretical and computational work using the CC and DIP-EOMCC methodologies was supported by the Chemical Sciences, Geosciences, and Biosciences Division, Office of Basic Energy Sciences, Office of Science, U.S. Department of Energy (Grant No. DE-FG02-01ER15228 to P.P.). The AIMD simulations were performed using the Expanse GPU system at the San Diego Supercomputer Center through allocation CHE230046 from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program, which is supported by National Science Foundation grants #2138259, #2138286, #2138307, #2137603, and #2138296. A.M. and B.G.L. gratefully acknowledge support from the National Science Foundation under award number CHE-1954519 and from the Institute for Advanced Computational Science. S.S. thanks Moaid Shaik, for his help with analyzing preliminary AIMD trajectories.
Author information
Authors and Affiliations
Contributions
M.D. conceived the project and oversaw the execution of all experiments. J. Stamm, S.K., and C.W. carried out the experiments under M.D.’s supervision, and J. Sandhu helped with the analysis of experimental data. P.P. and B.G.L. conceived the computational parts of the project. A.C., S.S.P., and J. Shen performed all ab initio electronic structure calculations using the CC and DIP-EOMCC methodologies under P.P.’s supervision. S.S. performed all AIMD simulations, consulting A.M., B.G.L., and P.P. All authors contributed to the interpretation of the results and J. Stamm, S.S.P., S.S., A.C., P.P., and M.D. led the manuscript writing, with the remaining authors offering comments and helping with respective tables and/or figures.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Stamm, J., Priyadarsini, S.S., Sandhu, S. et al. Factors governing \({\rm H}_{3}^{+}\) formation from methyl halogens and pseudohalogens. Nat Commun 16, 410 (2025). https://doi.org/10.1038/s41467-024-55065-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55065-5
