
Abstract
Diagnosing lung cancer from indeterminate pulmonary nodules (IPLs) remains challenging. In this multi-institutional study involving 2032 participants with IPLs, we integrate the clinical, radiomic with circulating cell-free DNA fragmentomic features in 5-methylcytosine (5mC)-enriched regions to establish a multiomics model (clinic-RadmC) for predicting the malignancy risk of IPLs. The clinic-RadmC yields an area-under-the-curve (AUC) of 0.923 on the external test set, outperforming the single-omics models, and models that only combine clinical features with radiomic, or fragmentomic features in 5mC-enriched regions (p < 0.050 for all). The superiority of the clinic-RadmC maintains well even after adjusting for clinic-radiological variables. Furthermore, the clinic-RadmC-guided strategy could reduce the unnecessary invasive procedures for benign IPLs by 10.9% ~ 35%, and avoid the delayed treatment for lung cancer by 3.1% ~ 38.8%. In summary, our study indicates that the clinic-RadmC provides a more effective and noninvasive tool for optimizing lung cancer diagnoses, thus facilitating the precision interventions.
Similar content being viewed by others


Introduction
Lung cancer remains a major health concern worldwide1,2, underscoring the urgent need for effective strategies that can reduce lung cancer-related mortality. Early diagnoses are vital for improving clinical outcomes by facilitating timely interventions3; however, the acknowledged noninvasive lung cancer diagnosis method, which uses low-dose computed tomography (LDCT) imaging4,5,6, has a high false-positive rate, limiting its clinical applicability7. Hence, there is still an unmet clinical need for noninvasive, accurate, and cost-effective tests to facilitate the early diagnoses of lung cancer.
The identifications of 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC) modifications from circulating cell-free DNA (cfDNA) have been proposed as promising methods for the early diagnoses of lung cancer8,9,10,11,12,13,14. However, most published studies evaluating the diagnostic value of 5mC and 5hmC biomarkers have limitations, including small sample size of lung cancer cases, the absence of independent external validation sets, and restricted reproducibility due to the use of various sequencing methods. Besides, the single-dimensional information would restrict the diagnostic accuracy, leading to a high lung cancer-related mortality. The potential of multiomics data in highlighting the importance of comprehensive molecular profiling of cancer has been suggested in recent studies and offers valuable insights into cancer pathogenesis and therapeutic strategies15,16,17. Hence, constructing a multiomics biomarker in multiple large, well-characterized cohorts holds promise in improving the diagnostic accuracy and clinical applicability.
Here, the plasma cfDNA end motif profiles in 5mC- and 5hmC-enriched regions were analyzed based on a multicenter, prospective, large cohort of indeterminate pulmonary nodules (IPLs). An integrated multiomics signature was then developed by integrating end motif, CT-based radiomic and clinical factors to optimize the lung cancer diagnosis process. To our knowledge, the potential value of the multiomics model for lung cancer detection is underexplored, and by conducting this study, we aimed to provide a more accurate, robust, noninvasive, and cost-effective test for diagnosing lung cancer and promoting the precision management for patients with IPLs.
Results
Participant characteristics
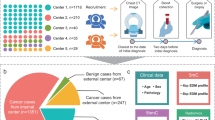
The clinical characteristics of all 2032 participants (Fig. 1) were summarized in Table 1, with detailed histopathological results listed in Supplementary Data 1. Among 1718 participants from Shanghai Pulmonary Hospital, 1019 (59.3%) were females and 699 (40.7%) were males with a mean age of 55.9 years. The majority of the IPLs manifested as subsolid nodules, including 22.4% pure-GGOs and 42.3% part-solid nodules with a mean (±SD) solid component size of 15.9 ( ± 13.5) mm. After randomization, the training, validation and internal test sets included 1030, 344, and 344 participants, respectively. The external test set included 314 participants, consisting of 197 (62.7%) females and 117 (37.3%) males with a mean age of 56.2 years. The clinical characteristics of the internal test set and the external test set were compared, and the participants on the external test set had fewer pure-solid nodules (p = 0.011) and smaller solid component sizes (p = 0.012).

A participants selecting from Shanghai Pulmonary Hospital; B participants selecting from external 4 hospitals.
Fragmentomic model establishment
The median final numbers of mapped fragments (paired reads) derived from the 5mC- and 5hmC-sequencing data were 12.7 million and 14.1 million, respectively, and no systematic bias or obvious batch effect induced by hospital regions were detected both for the 5mC- and 5hmC- sequencing data (Supplementary Fig. 1). The 4-mer (bp) end-motifs profiles were first identified from 5mC- and 5hmC- sequencing data, and then, extended to 6 bp end motifs profiles (Fig. 2A). After feature dimensionality reduction and selection, 27 4 bp end motifs and 11 6 bp end motifs from the 5mC-sequencing data, 11 4 bp end motifs and 11 6 bp end motif from the 5hmC-sequencing data were identified. The numbers of remaining features and the feature names observed after the processing steps are detailed in Supplementary Table 1. Hierarchical clustering analyses of the end motifs selected from 5mC-sequencing data is illustrated in Supplementary Fig. 2A for the training set and Fig. 2B for the rest sets, while the hierarchical clustering analyses of the end motifs selected from 5hmC-sequencing data is illustrated in Supplementary Fig. 2B, C.

A Schematic of the process for determining the first 6-nucleotide sequence (i.e., a 6-mer end motif) on each 5’ fragment end of cfDNA relative to the hg19 reference genome; B Hierarchical clustering analyses of the selected 6 bp end motifs derived from 5mC-sequencing data; Receiver operating characteristic analyses of the epigenomic models on the validation set C internal test set D and external test set (E); F Bar charts showing the TFs identified by the 6-mer end motifs selected from 5mC-sequencing data; G Bar charts showing the regulatory target genes by these identified TFs; H The top 15 most enriched GO terms based on the target genes. The area under the receiver operating characteristic curves are compared via the DeLong’s test. All statistical tests were two-sided, with p < 0.05 indicative of a statistically significant difference. 4bp-5mC, the model established by the 4-mer end motifs selected from 5mC-sequencing data; 6bp-5mC, the model established by the 6-mer end motifs selected 5mC-sequencing data; 4bp-5hmC, the model established by the 4-mer end motifs selected from 5hmC-sequencing data; 6bp-5hmC, the model established by the 6-mer end motifs selected from 5hmC-sequencing data; 5mC, 5-methylcytosine; 5hmC, 5-hydroxymethylcytosine; ROCs, receiver operating characteristic curve; AUC area under the ROCs curve, CI confidence interval, Sens sensitivity, Spec specificity, PPV positive predictive value, NPV negative predictive value, Accur accuracy, TFs transcription factors, GO gene ontology. Source data are provided as a Source Data file.
The predictive models were then established accordingly, with hyperparameter results summarized in Supplementary Table 2. As illustrated in Fig. 2C–E, for 5mC-sequencing data, the model developed by 6 bp end motifs (6bp-5mC model) produced higher AUCs of 0.805, 0.826 on the validation set and external test set, respectively, outperforming the model developed by 4 bp end motifs (4bp-5mC model), although no significant differences (p > 0.05 for all). The AUC of the 6bp-5mC model on the internal test set was slightly lower than that of the 4bp-5mC model but still did not significantly differ (p = 0.100). Also, the 6bp-5mC model maintained higher specificity and PPV than those of the 4bp-5mC model across all datasets, and higher accuracy on the external test set. For 5hmC-sequencing data, the predictive model developed with the 6 bp end motifs (6bp-5hmC model) achieved greater AUCs than did the model developed with the 4 bp end motifs (4bp-5hmC model) on the external test set (p = 0.399; Fig. 2E). The 6bp-5hmC model also exhibited greater accuracy. Hence, we selected the 6bp-5mC model and the 6bp-5hmC model for subsequent analyses. Upon downsampling the fragments to 25%, 50%, and 75%, the AUC values gradually increased, and the 6 bp end motifs-based models maintained the best performance and robustness (Supplementary Fig. 2D, E), revealing their appropriate applications. The hyperparameter of models derived from 25%, 50%, and 75% sequencing fragments are summarized in Supplementary Table 2.
For the functional analysis of 6 bp end motifs incorporated into the 6bp-5mC model, 131 transcription factors (TFs) were identified (Fig. 2F), with the top 20 TFs illustrated in Fig. 2G. The most enriched biological processes were cancer-associated pathways (Fig. 2H), such as the epithelial cell proliferation. The similar analysis was performed for 6 bp end motifs incorporated into the 6bp-5hmC model (Supplementary Fig. 2F, G), while the most enriched pathways illustrated in Supplementary Fig. 2H. The impacts of each end motif on the 6bp-5mC model and 6bp-5hmC model predictions for malignancy risks of IPLs are visualized in Supplementary Fig. 3.
Radiomic model establishment
The construction schemes of the radiomic models are illustrated in Fig. 3A, B. The trainable parameters of the DL-radiomics model are described in Supplementary Method 5, 6, while the hyperparameter results for the C-radiomics model are listed in Supplementary Table 2. The DL-radiomics model achieved AUCs of 0.842, 0.842 and 0.855 on the validation set (Fig. 3C), internal test set (Fig. 3D) and external test set (Fig. 3E), respectively, outperformed the C-radiomics model, especially on the external test set (p = 0.012), revealing the superiority of the deep learning technology. The attribution scores of all 64 deep learning-based radiomics features obtained from the training set is illustrated in Fig. 3F, and the DL-feature 07, 52, and 25 had the largest contributions to the DL-radiomics model’s output (Fig. 3G). Nevertheless, the definite meanings of DL-features are still difficult to explain. To further enhance the interpretability of the DL-radiomics model, the biological basis of it was evaluated. A total of 116 patients from Radiogenomics dataset were identified, and classified as low-risk (DL-radiomics model score<47.8, the cutoff value calculated based on the maximum Youden index on the training set) or high-risk (DL-radiomics model score≥47.8). As illustrated in Fig. 3H, 218 differentially expressed genes, consisting of 89 upregulated genes and 129 downregulated genes, were found between the low and high-risk subgroups. The tumorigenesis-related pathways, such as the HIF-1 signaling pathways and transcriptional misregulation in cancer, were significantly upregulated (Fig. 3I), whereas the pathways related to tumor immunity, such as the B-cell receptor signaling pathway and the T-cell receptor signaling pathway, were significantly downregulated (Fig. 3J). Further, high-risk patients classified by the DL-radiomics model were tend to show fewer activated CD8 T cells, fewer myeloid-derived suppressor cells (MDSCs), and fewer T follicular helper cells in their tumor microenvironments, although the difference were not significant (Fig. 3K).

Construction procedures utilized for the DL-radiomics model A and the C-radiomics model B; Receiver operating characteristics analyses of the DL-radiomics model and C-radiomics model on the validation set C, the internal test set D, and the external test set E; F, The attribution score distribution of each deep learning-based radiomics feature on the training set; G The impact of the deep learning-based radiomics features included in the DL-radiomics models on the risk probabilities output for the training set; H Volcano plot for differentially expressed genes between the low-risk and high-risk subgroups classified by the DL-radiomics model; I GSEA conducted for upregulated genes; J GSEA conducted for downregulated genes; K ssGSEA conducted for the low-risk and high-risk subgroups classified by the DL-radiomics model, The center of the box denotes the 50th percentile, the bounds of the box contain the 25th to 75th percentiles, the whiskers mark the maximum and minimum values, and values beyond these upper and lower whiskers are considered outliers and are marked with dots. n = 116 biologically independent samples were analyzed. All statistical tests were two-sided, with p < 0.05 indicative of a statistically significant difference. DL-radiomics, the deep learning-based radiomics model score, while C-radiomics referred to the classic radiomics model. ROCs, receiver operating characteristic analyses; AUC, area under the ROCs curve; CI, confidence interval; DL, deep learning; KEGG, Kyoto Encyclopedia of Genes and Genomes; MDSC, myeloid derived suppressor cell; GSEA, gene set enrichment analyses; ssGSEA, single sample gene set enrichment analyses. Source data are provided as a Source Data file.
Multiomics model establishment
The multivariable logistic regression analyses revealed that age, radiological solid component size, the DL-radiomics model score, the 6bp-5mC model score, and the 6bp-5hmC model score were significant predictors for malignancy risk of IPLs (p < 0.001 for all; Table 2). All single-omics model score (DL-radiomics, 6bp-5mC, and 6bp-5hmC) showed weak correlations with clinical factors, with the absolute value of Pearson correlation coefficients≤0.10 for all (Fig. 4A–C), suggesting that both radiomic and fragmentomic features provide additional information to clinical data for lung cancer detection. A clinical model and 4 multiomics models were then established, with the parameter information detailed in Supplementary Table 2. The clinic-RadmC model, which integrated the clinical factors, DL-radiomics model with the 6bp-5mC model, yielded significantly improved AUCs of 0.883 on the validation set (Fig. 4D), 0.889 on the internal test set (Fig. 4E), and 0.923 on the external test set (Fig. 4F), outperforming the models who only integrated clinical factors with the DL-radiomics model, or with the 6bp-5mC model (p < 0.050 for all; Supplementary Table 3). Interestingly, we found that after adding the diagnostic information from the 5hmC-sequencing data, the clinic-Rad(h)mC model did not significantly outperform the clinic-RadmC model (p > 0.050 for all; Supplementary Table 3). Based on the cutoff value of 78.0, 77.9, 80.5, 82.7, and 82.7 for the clinical, clinic-Radiomics, clinic-mC, clinic-RadmC and clinic-Rad(h)mC model, comparable performance metrics were observed between the clinic-RadmC model and the clinic-Rad(h)mC model (Fig. 4D–F). A calibration curve analysis revealed that the predicted probabilities of the clinic-RadmC model and the actual malignancy risk probabilities had good agreement (Hosmer-Lemeshow test, p > 0.050 for all datasets; Supplementary Fig. 4A–C). DCA suggested that the clinic-RadmC model achieved better net clinical benefits than did the clinic-Radiomics, clinic-mC and clinical model, but similar clinical benefits with the clinic-Rad(h)mC model (Supplementary Fig. 4D–F).

Pearson correlation coefficients analyses for the features included in the multiomics model on validation set A, internal test set B, and external test set C. Receiver operating characteristic analyses and performance metrics for the models on validation set D, internal test set E, and external test set F. All statistical tests were two-sided, with p < 0.05 indicative of a statistically significant difference, and *** denotes p < 0.001; ** denotes p < 0.01; * denotes p < 0.05. Size, the radiological solid component size of pulmonary nodules; Radiomics, the deep learning-based radiomics model score; 6bp-5mC, the model score established by the 6-mer end motifs selected from 5mC-equencing data; 6bp-5hmC, the model score established by the 6-mer end motifs selected from 5hmC-sequencing data; clinical, the model established by the age and radiological solid component size of pulmonary nodule; clinic-Radiomics, the model established by combining clinical variables with the DL-radiomics model sore; clinic-mC, the model established by combining clinical variables with the 6bp-5mC model score; clinic-RadmC, the model established by combining clinical variables, DL-radiomics model score with the 6bp-5mC model score; clinic-Rad(h)mC, the model established by combining clinical variables, the DL-radiomics model score, the 6bp-5mC model score with the 6bp-5hmC model score. ROCs, receiver operating characteristics analyses; AUC area under the ROCs curve, Sens sensitivity, Spec specificity, PPV positive predictive value, NPV negative predictive value, Accur accuracy. Source data are provided as a Source Data file.
In further comparisons with single-omics models, the clinic-RadmC outperformed the 6 bp end motifs-based model did (p < 0.010 for all datasets; Supplementary Fig. 5A–C). It kept the better performance than the DL-radiomics model, especially on the external test set (p < 0.001; Supplementary Fig. 5C). DCA suggested that the net clinical benefits derived by the clinic-RadmC model was better than all single-omics models (Supplementary Fig. 5D–F), as well as the IDI and NRI analyses (Supplementary Table 4). The impact of each feature on the malignancy probability output by the clinic-RadmC model is illustrated in Fig. 5A–C, with the DL-radiomics model score harboring the largest contribution, followed by the 6bp-5mC score. Additionally, 2 example participants with similar radiological characteristics, the SHAP maps of which are illustrated in Fig. 5D, E, could be classified into different subgroups with distinct malignancy risk levels based on the probability output by the clinic-RadmC model, enabling to select high-risk participants for early intervention.

SHAP analyses results obtained for ranking the impact of the continuous features included in the clinic-RadmC models on the risk probabilities output for the validation set A, internal test set B, and external test set C; 2 example participants with similar clinic-radiological characteristics in the low-risk D and high-risk subgroups E, respectively. Each participant was represented by a single dot on each feature flow. The horizontal position of the dot was determined by the SHAP value of that feature, and dots were accumulated along each feature row to show density values. DL-radiomics, deep learning-based radiomic model; 6bp-5mC, the model established by the 6-mer end motifs selected from 5mC-sequencing data; solid size, the radiological solid component size of pulmonary nodules; SHAP, Shapley additive explanations. Source data are provided as a Source Data file.
Performance evaluation of the models in various clinical scenarios
The diagnostic performance of the clinic-RadmC model was further evaluated after adjusting for clinical and radiological characteristics. The clinic-RadmC model yielded significantly improved AUCs over clinical model for participants aged≤50 years on test sets (p < 0.050 for all; Fig. 6A & Supplementary Table 5). For participants aged>50 years who were identified as high-risk population for lung cancer18, the clinic-RadmC model outperformed the other models (p < 0.050 for all; p = 0.070 for vs. clinic-Radiomics on the internal test set) but performed comparably with the clinic-Rad(h)mC model (p > 0.050 for all; Fig. 6B & Supplementary Table 5).

Predictive performance achieved by the models in subgroups stratified by age A, B radiological image C–E, and nodule size F–H. Pure-GGO comprised nodules with only GGO, and part-solid nodules consisted of GGOs and solid components, whereas pure-solid nodules had only solid components without GGOs. Subcentimeter pulmonary nodules were defined as the nodules with solid component size≤10 mm, and large nodules were defined as those with 15 mm≤solid component size≤30 mm, whereas pulmonary massed were defined as those with solid component size>30 mm. 6bp-5mC, the model established by the 6-mer end motifs selected from 5mC-sequencing data; DL-radiomics, the deep learning-based radiomic model score; clinic-mC, the model established by combining clinical variables with the 6bp-5mC model score; clinic-Radiomics, the model established by combining clinical variables with the DL-radiomics model sore; clinic-Rad(h)mC, the model established by combining clinical variables, the DL-radiomics model score, the 6bp-5mC model score with the 6bp-5hmC model score; clinic-RadmC, the model established by combining clinical variables, the DL-radiomics model score with the 6bp-5mC model score. AUCs areas under the receiver operating characteristics curves; GGO ground-glass opacity. Source data are provided as a Source Data file.
For subsolid nodules, the clinic-RadmC model did not perform better in pure-GGOs (p > 0.050 for all; p = 0.005 for vs. the DL-radiomics model on the internal test set; Fig. 6C and Supplementary Table 6), but performed better than the other models on the external test set for part-solid nodules (p < 0.050 for all; Fig. 6D & Supplementary Table 6). However, the clinic-RadmC model exhibited excellent performance for pure-solid nodules (Fig. 6E), outperforming the single-omics models (p < 0.050 for all; Supplementary Table 6). Our findings revealed the increasing applicability of the clinic-RadmC model along with the increase in solid components size of IPLs.
Diagnosing subcentimeter pulmonary nodules remains challenging19,20 in clinical practice, and compared with the clinical model, the clinic-RadmC model yielded higher AUCs of 0.776, and 0.937 for predicting the malignancy risks of subcentimeter IPLs on test sets (p < 0.001; Fig. 6F & Supplementary Table 7). In addition, the current guidelines still rely on the clinical risk models, and large IPLs (size≥15 mm) satisfy the 10% threshold for investigation, whereas the British Thoracic Society Guidelines suggest a broad range of options. The clinic-RadmC model achieved improved AUCs of 0.988, and 0.936 on the internal and the external test set, when compared with the 6bp-5mC model (p = 0.002 for all), the clinic-mC model (p = 0.003 for all), and the clinical model did (p < 0.001 for all; Fig. 6G & Supplementary Table 7). Similar trends concerning the superiority of the clinic-RadmC model were observed for pulmonary massed with size>30 mm (Fig. 6H & Supplementary Table 7).
Support for guiding individual management process
The impacts of the models on the malignancy risk reclassification for IPLs were evaluated on the combined test sets (n = 658 consisting of 137 benign and 521 malignant samples). Based on the cutoff value of 82.7 on the training set, the clinic-RadmC model correctly classified 112 samples as low-risk and 456 samples as high-risk, leading to a 78.2% reduction in unnecessary invasive procedures (112/137; Fig. 7A). When compared with the clinical model (Fig. 7B), the clinic-RadmC model could correctly reclassified 48 benign samples as low-risk, and 202 malignant samples as high-risk, reducing the unnecessary invasive procedures for benign diseases by 35.0% (48/137), and avoiding the delayed treatment for cancer by 38.8% (202/521). When compared with single-omics model consisted of the DL-radiomics and 6bp-5mC model, the clinic-RadmC model could provide a reduction of unnecessary invasive procedures for benign cases by 13.1% ~ 16.1% (18/137; 22/137), and provide avoidance of potential delayed treatment for lung cancer by 3.1% ~ 20.3% (16/521; 106/521; Fig. 7C, D). The superior reclassification performances of the clinic-RadmC model to the clinic-Radiomics or clinic-mC model were also observed, and it could reduce the unnecessary invasive procedures for benign IPLs by 10.9% ~ 21.9%, and prevent delayed treatments for lung cancer by 3.8% ~ 13.8% (Fig. 7E, F).

A Confusion matrices illustrating the predicted outcomes generated by the multiomics model in comparison with the actual outcomes, as well as between the multiomics model and the clinical model B the DL-radiomics model C the 6bp-5mC model D the clinic-Radiomics model E and the clinic-mC model F with emphasis placed on the patients ruled in and ruled out. The dotted lines demarcate the corresponding cutoff values of the different models. The number labeled with * refer to cancer cases misclassified as low-risk samples by the x-axis model but correctly reclassified as high-risk samples by the multiomics model on y-axis, whereas the number labeled with # refer to benign cases misclassified as high-risk samples by the x-axis model but correctly reclassified as low-risk samples by the multiomics model on the y-axis. Source data are provided as a Source Data file.
Discussion
In this multi-institutional study involving a large population, we established the clinic-RadmC model by integrating clinical, fragmentomic with radiomic features and demonstrated that this multiomics model provided additional diagnostic information for lung cancer. We further revealed its superior performance to single-omics models and clinical model in predicting the malignancy risks of IPLs, with an execution time of less than one second, offering a fast, effective and noninvasive method for lung cancer detection and facilitating individual management.
Currently, chest LDCT is still a low-financial-cost and highly-sensitive method for detecting lung cancer, but 96% of IPLs are diagnosed as benign after follow-up scans; as many as 20% of IPLs are diagnosed as benign after surgical resection, and 38% after biopsy21,22. In previously published studies, radiomic analyses enable to noninvasively and longitudinally capture the radiographic phenotype and characterize the underlying pathophysiology of a tumor, providing an improved disease diagnosis accuracy23,24. A dizzying array of possibilities, such as circulating tumor DNA, DNA methylation, have also been investigated as potential diagnostic biomarkers for IPLs25,26, allowing for the optimization of early cancer diagnoses26. The fragmentation patter of cfDNA represents another class of liquid biopsy-based biomarkers in oncology, and hold promise for cancer detection11,27,28,29,30. Among the emergent class of cfDNA fragment features, end motif-based approach act as an effective method for identifying various cancers9,31,32,33, outperforming other cfDNA fragment metrics34. End motif also has the advantage that only a small number of DNA molecules required to achieve its plateau of performance comparing to other tumor markers such as copy number aberration and tumor-derived single-nucleotide variants27,31. Recent study has indicated that cfDNA motif-based method held promise in stage I lung adenocarcinoma detection9. These preliminary findings suggested the end motifs-based approach outperformed other cfDNA fragment metrics in identifying cancers, shedding light on the diagnosis of lung cancer at early stage in our study. In this context, it is conceivable that molecular biomarkers combined with radiomic might improve the cancer detection process through the noninvasive characterization of cancer biology. In 2021, Kammer et al.35 developed a biomarker model by combining clinical variables, blood-based biomarkers with radiomic features, which first demonstrated the notable strength of combined biomarkers in hastening the early diagnosis of lung cancer. In 2023, PulmonSeek Plus, established by combining clinical data, CT imaging data, and cfDNA methylation biomarkers, achieved excellent accuracy17, suggesting the superiority of the integrative model. Nevertheless, the diagnostic value of plasma cfDNA end motif profiles in 5mC- or 5hmC-enriched regions specifically for lung cancer is still underexplored, and studies evaluating the multiomics signatures for diagnosing lung cancer are mostly limited by relatively small sample sizes.
Hence, our study was conducted to establish a multiomics model by integrating clinical, radiomic and end motifs features from 5mC-sequencing data, aiming to provide a more accurate, stable, cost-effective and noninvasive tool for optimizing lung cancer diagnosis. Several strengths of our study might be emphasized. First, 2032 participants from 5 different clinical centers were prospectively enrolled, representing the largest sample size reported in this field to date. Second, a deep learning technique was used to extract radiomic features and establish the radiomic model. The construction of a classic radiomic model still requires the ROIs in CT images to be manually segmented in a layer-by-layer manner. Compared with the classic radiomic, deep learning techniques reduce the burden of data processing by extracting radiomic features directly from the labeled ROIs and can also achieve better and more stable performance. In addition, the existing DL-based radiomics model reported in previously published studies yielded a pooled AUC of 0.88 ~ 0.94, with a sensitivity of 63.0% ~ 84.0% and a specificity of 83% ~ 90%36,37,38,39,40,41. The DL-radiomics models in our study achieved AUCs around 0.85 and maintained comparable performance with that of previously DL-based radiomics models, indicating the appropriate application of the DL-radiomics model for further analyses in our study. Third, our study added the information to support the diagnostic value of the cfDNA 6 bp end motifs profiles in 5mC- and 5hmC-enriched regions for lung cancer. Mechanistically, the cfDNA end motifs profiles might be globally affected by the changes in chromatin accessibility and nuclease activity42, and chromatin accessibility is important for the fragmentation of cfDNA, which contributes to their preferred ends43. Accordingly, we assume that the abnormal epigenetic changes in cancer genomes might influence the chromatin accessibility and DNA endonucleases activity, leading to the changes of cfDNA end motifs9,27, and the 5mC and 5hmC modifications would exert different effects on cfDNA end motif profile, since they are 2 distinct epigenetic marks in human genomes. We investigated the performance of 4 bp and 6 bp end motifs derived from 5mC-sequencing data, and found that both of them yielded good performance (>0.80) in distinguishing malignant and benign nodules. Compared with similar assays previously reported9,10,11,30,32,33,34, both 4bp- and 6bp-5mC model kept the comparable and robust performance for lung cancer detection. Besides, the incorporation of the multidimensional epigenomics features into multiomics models might comprehensively reveal more epigenetic characteristics, such as the previously reported efficacy of multidimensional “omics” features11,44. 5hmC biomarkers have been used as notable tools for lung cancer detection in our previous studies13,14. However, in this study, after methylation information has been incorporated, adding a diagnostic score form 5hmC-sequencing data could not provide additional improvement for predicting malignancy risk of IPLs. DCA, NRI and IDI analyses further validated the similar benefits derived from the clinic-RadmC model and the clinic-Rad(h)mC model. These finding suggested that it is not the case that the more -omics information, the more accurate the lung cancer diagnosis, and the combination of information derived from clinical, radiomic, and cfDNA 5mC-sequencing data could provide sufficient performance, serving as a more cost-effective method for diagnosing lung cancer in clinical practice. Fourth, machine learning methods, such as, SVM, deep learning, and logistic analyses, were used to construct models in our study, and the interpretability analyses of the predictive models were conducted in our study. Translating AI research to the optimization of the clinical workflow is the ultimate goal of this state-of-the-art technology, which is also the benchmark indicator for measuring the clinical value of an AI model. We conducted a functional analysis for the end-motifs, exploring the importance levels of DL-based radiomic features via DeepLIFT, and ranked the importance of each feature in the clinic-RadmC model via SHAP. We further explored the biological basis of the DL-radiomics model via the published Radiogenomics dataset. These analyses might help to enhance the credibility and applicability of our predictive models in clinical practice.
We further compared the predictive performance of the clinic-RadmC model with that of a similar model, namely PulmoSeek Plus. First, despite the similar AUC, the samples size used to establish the clinic-RadmC model included many more than 543 samples used to establish the PulmoSeek Plus model, ensuring the robustness of the clinic-RadmC model. Second, the cfDNA methylation biomarkers in the PulmoSeek Plus model were identified via high throughput DNA bisulfite sequencing technology45,46, which might be complex and costly, and might limit the generalizability of this approach. Besides, the CT image features included in the PulmoSeek Plus model were evaluated manually, which might impose a greater workload on radiologists. In contrast, we applied the low-depth cfDNA sequencing technology to identify fragment patterns on 5mC- and 5hmC-enriched regions, and radiomic features were identified directly via an “end-to-end” deep learning algorithm, which improved the clinical cost-effectiveness and convenience of the clinic-RadmC model. Third, an explainability analysis of the clinic-RadmC model were performed to elucidate the mechanism underlying the prediction process from the perspective of genomic biology, and could provide insight into its diagnostic mechanism and enable broad adoption of the model. Similar explorations were rarely conducted on validated models in previously public studies. Accordingly, the clinic-RadmC model could serve as a more efficient, credible, stable, and cost-effective signature in terms of lung cancer diagnoses.
We also evaluated the clinical applicability of our established clinic-RadmC model by investigating the reclassification performance of it, since an ideal diagnostic tool for the clinical setting would be expected to facilitate individual management of IPLs. Our findings revealed that the clinic-RadmC model could not only achieve high sensitivity of 0.895 and high specificity of 0.806 in diagnosing lung cancer on the external test set, but also reduce the unnecessary invasive procedures by 10.9% ~ 35% for benign IPLs, and avoid the delayed treatment by 3.1% ~ 38.8% for lung cancer. Hence, this multiomics assay allow to use easy-accessible information from screening pipeline, including clinical, radiological and blood-derived features, to diagnose lung cancer more accurately and promote precise treatment for IPLs. Meanwhile, we also found that the clinic-RadmC model might also show potential value in confirming the suspected lung cancer cases. It showed superior performance in diagnosing lung cancer among patients aged>50 years, who were identified as high-risk population for lung cancer, as well as among those with radiological pure-solid IPLs, those with large IPLs (size≥15 mm), and pulmonary mass (size≥30 mm), indicating the great potential of it in clinical application.
Nevertheless, several limitations of our study must be acknowledged. First, the role of all involved centers as tertiary care centers might have generated some selection bias within our sample population, since less suspicious SSNs that were likely to be stable over time were rarely referred to us. Future studies investigating the performance of the clinic-RadmC model for less suspicious pulmonary nodules are warranted. Second, the present study was only conducted in clinical centers from China. Owing to differences among disease spectra, further studies recruiting diverse non-Asian populations are needed to demonstrate the generalizability. Third, we only investigated the diagnostic value of plasma cfDNA end motif profile in our study, and future studies are of still necessity to investigate the potential value of different fragment patterns or biomarkers derived from 5mC- or 5hmC-enriched regions for cancer detection. Fourth, although the clinic-RadmC model enables the integration of clinically convenient plasma sources with LDCT data to diagnose lung cancer, its clinical benefit and cost-effectiveness in practical applications still require further study if more people are to be reached.
In conclusion, the clinic-RadmC model constructed in this study attained superior predictive performance to that of single-omics models, and could serve as a more effective, robust and noninvasive tool for optimizing lung cancer diagnoses, providing a promising avenue for improving lung cancer screening programs and reducing the global burden of this prevalent cancer.
Methods
Ethical statements
This study was conducted according to the Declaration of Helsinki, and registered in the Chinese Clinical Trial Registry (ChiCTR2100041971). The ethical committees of all involved centers, including Shanghai Pulmonary Hospital, Ningbo Hwa Mei Hospital, The First Affiliated Hospital of Nanchang University, the Affiliated Hospital of Zunyi Medical College, and The First Hospital of Lanzhou University, approved this study (L21-022, L21-022-1, L21-022-2). Written consents were obtained from participants prior to the enrollment, and they were not compensated for participation.
Participants enrollment
As shown in Fig. 1A, participants who had IPLs were prospectively recruited at the Department of Thoracic Surgery of Shanghai Pulmonary Hospital between March 2021 and December 2021 based on the following inclusion criteria: (1) aged ≥ 18 years; (2) did not have a history of malignant tumors within 5 years prior to their diagnoses; (3) did not have definite signs of tumor metastasis; (4) did not receive any anticancer treatments before their diagnoses; and (5) signed an informed consent form. Participants who had multiple resected IPLs were excluded, and those without definite pathological outcomes were also excluded to ensure the robustness of our study. Participants whose samples had inadequate DNA after extraction, whose sequencing quality control tests failed (spike-in DNA > 10 reads with capture efficiency <0.5, and sequencing reads <one million) or whose chest CT images could not be extracted were excluded. In total, 1718 participants were enrolled and randomly classified into training set, validation set, and internal test set at a ratio of 6:2:2. Further, participants from 4 clinical centers (Ningbo Hwa Mei Hospital, The First Affiliated Hospital of Nanchang University, the Affiliated Hospital of Zunyi Medical College, and The First Hospital of Lanzhou University) were recruited from September 2021 to December 2022 (Fig. 1B), and 314 participants were ultimately enrolled, serving as an independent external test set. The disease outcomes of all participants were surgery-, or biopsy-proven, and the baseline characteristics were collected via clinical records.
Plasma sample collection and processing
Approximately 10-ml peripheral venous blood samples were collected from each participant within 2 weeks prior to the initial diagnosis via BD Vacutainer EDTA tubes (Becton, Dickinson and Company, Cat# 367525), and processed for plasma isolation following the procedures: The blood was rested for 30 min at 4 °C and then centrifuged at 1000×g for 10 min at 4 °C; then, the supernatant was collected and centrifuged at 2000×g for 5 min at 4 °C. The plasma was frozen at -80 °C until DNA isolation. For cfDNA extraction, a VAHTS Serum/Plasma Circulating DNA Kit (Vazyme, Cat# N902-02) was used, and the cfDNA was quantified with a Qubit fluorometer (Life Technologies). cfDNA (10-30 ng) was applied for library construction: adapter ligation was facilitated via the VAHTS Universal DNA Library Prep Kit for Illumina (Vazyme, Cat# ND607) according to the protocol provided by the manufacturer, with spike-in controls added (0.01 pg of each amplicon per 10 ng of cfDNA). The spike-in control, including 3 distinct lambda DNA amplification products (~180 bp; one without modification and the other two with 5mC and 5hmC modifications), was prepared as follow47: lambda DNA was PCR amplified by Taq DNA Polymerase (NEB) and purified by AMPure XP beads (Beckman Counlter) in nonoverlapping ~180 bp amplicons, with a cocktail of dATP/dGTP/dTTP and one of the following: dCTP, dmCTP or 10% dhmCTP (Zymo)/90% dCTP. The employed primers sequences were as follows: dCTP FW-5′-CGTTTCCGTTCTTCTTCGTC-3′, RV-5′-TACTCGCACCGAAAATGTCA-3′; dmCTP FW-5′-GTGGCGGGTTATGATGAACT-3′, RV-5′-CATAAAATGCGGGGATTCAC-3′; 10% dhmCTP/90% dCTP FW-5′-TGAAAACGAAAGGGGATACG-3′, RV-5′-GTCCAGCTGGGAGTCGATAC-3′.
Methylation, and hydroxymethylation profiling
For cell-free 5mC sequencing, ligated cfDNA (5–10 ng) (VAHTS Universal DNA Library Prep Kit for Illumina V3, Vazyme ND607, and QIAseq UDI Y-Adapter Kit, Qiagen) was used by the cell-free methylated DNA immunoprecipitation and high-throughput sequencing (cfMeDIP-seq)48,49. The spike-in controls were used to roughly assess the 5mC enrichment ratio instead of the methylated DNA and unmethylated DNA spike-in controls used in the cfMeDIP-seq. We added a Y-shaped long linker sequence containing index information to the cfDNA to protect the integrity of cfDNA methylome profiling process50, as described in Supplementary method 1. For cell-free 5hmC sequencing, a low-input whole-genome, cell-free 5hmC sequencing method based on selective chemical labeling (hMe-Seal) was developed51. The hMe-Seal method is a robust approach that uses β-glucosyltransferase (βGT) to selectively label 5hmC with biotin via an azide-modified glucose for the pull-down of 5hmC-containing DNA fragments for sequencing51. The standard hMe-Seal procedure requires micrograms of DNA. In our modified approach, cfDNA was first ligated with sequencing adapters, and 5hmC was selectively labeled with a biotin group. After the cfDNA containing 5hmC WAS captured via streptavidin beads, the final library was directly completed by PCR from the beads instead of eluting the captured DNA to minimize the sample loss induced during the purification steps. With our modified approach, the cell-free 5hmC could be readily sequenced from 1-10 ng of cfDNA. The detailed information of this method47 has been further described in Supplementary method 2.
cfDNA sequencing data processing and end-motif profiles identification
With respect to the processing strategy applied to the cfDNA sequencing data, the 5mC- and 5hmC-captured libraries were sequenced via 150 bp paired-end runs (300 cycles) on the illumine NovaSeq6000 platforms50. Prior to alignment, low-quality sequences were filtered using the Fastp software (version 0.21.0). BWA (version 0.7.17-r1188) was used to process sequencing reads, which were aligned to hg19 and spike-in DNA with default parameters. SAMtools (version 1.7) was used to filter the generated SAM files with parameter settings of ‘-f 3 -F 3852 -q 30’ to include high quality, primary alignment, properly paired reads, followed by converting to BAM format. Picard (version 2.25.5) was employed to sort and index filtered SAM files and to ensure the removal of duplicate reads before subsequent analyses. Bedtools (version 2.26.0) bamtobed was utilized to convert BAM alignment to BED with ‘-bedpe’ parameter. Only paired-end reads with both ends aligned to the same autosome with the correct orientation, spanning an insert size from 20 bp to 1000 bp, were used for downstream analyses. Three types of spike-in DNA sequences were included into reference; then, capture efficiency, as a quality control measurement for the 5mC and 5hmC, was calculated as counts of reads aligned to a type-specific spike-in DNA divided by counts of reads aligned to total spike-in DNA. The 4-mer (bp) end motifs profiles of the 5mC- and 5hmC-sequencing data were extracted after generating the fragment file. Then, the 4 bp end-motifs was extended to 6 bp end-motifs, and the first 6-nucleotide sequence on each 5’ end of plasma DNA fragment was identified (Fig. 2A), since the 6 bp end-motif might exhibit better diagnostic performance for cancer detection9,29. The frequency of end motifs (4 bp: 4^4 = 256; 6 bp: 4^6 = 4096) out of the total number of motifs was calculated.
Chest CT image acquisition and nodule annotation
The thin-section chest CT images that were closest to the initial diagnosis date were extracted from the picture archiving and communication (PACS) system and imported into the 3D-slicer software (version 4.10.1), with the CT scanning parameters detailed in Supplementary method 3. Each three-dimensional region of interest (ROI) was independently annotated with a bounding box by two junior thoracic surgeons (M.M. Zhao and J.J. Deng) and a junior radiologist (T.T. Wang) under the lung (level, -450 HU; width, 1500 HU) and mediastinal (level, 40 HU; width, 400 HU) window settings. The imaging subtypes, emphysema and spiculation were recorded, and IPLs were further allocated as pure ground-glass opacity (GGO), part-solid nodules or pure-solid nodules. The pure-GGOs comprised IPLs only with GGOs, and the part-solid nodules consisted of GGOs and solid components, whereas the pure-solid nodules had only solid components. The radiological solid-component size was measured, as well. The disagreement was solved via a group discussion with a senior radiologist (X.W. Sun).
Fragmentomic analyses
The principal component analysis (PCA) was performed to evaluated the potential batch effects in 5mC- and 5hmC-sequencing data between different hospital regions. The dimensions reduction of end motif profiles were conducted on the training set as follow: (1) a Wilcoxon rank-sum test was used to select differently expressed features between benign and malignant samples, and only the feature with p < 0.01 were selected; (2) a univariate logistic analysis was performed and only the features with p < 0.01 were selected for the next step; (3) the minimum redundancy and maximum relevancy method was applied to rank each selected feature depending on its relevance to malignancy, and the top 100 most significant features were selected; (4) the 100 identified features were subjected to the least absolute shrinkage and selection operator (LASSO) model. The area under the receiver operating characteristic curve (AUC) was used as the criterion, and the optimal feature set with the maximum AUC was selected as input features. Hierarchical clustering analysis was applied to evaluate the classification performance of these identified end motifs. Then, a support vector machine (SVM) was used to construct the predictive models using the end motifs selected from 5mC-sequencing data (5mC model), and 5hmC-sequencing data (5hmC model) after multiplying the coefficient identified by LASSO, whereas hyperparameter optimization was performed via a grid-search and manual fine-tuning with 5-fold cross-validation. The models based on 4 bp and 6 bp end motif features were simultaneously established, and those with the best performance were reserved for subsequent analyses. We further evaluated the diagnostic performance of the end motif profiles obtained after gradually downsampling the 5mC- and 5hmC-sequencing fragments to 25%, 50%, and 75%. In addition, the chromatin accessibility contributes to cfDNA preferred ends43, and thus, the changes in chromatin accessibility and DNA nuclease activity or the combined effect of both could have impacts on the cfDNA end motifs. Increased DNA methylation is often associated with reduced chromatin accessibility, influencing the TF binding process. Thus, we hypothesize that if the sequence pattern of the end motif resembles that of the TF binding motif, the end motif may serve as a potential binding sequence for TF. Also, epigenetic changes in the vicinity of end motif sequences may affect the TF binding52, leading to the transcription disturbance of TF regulatory genes. Accordingly, the functional exploration of end motifs was performed via 3 steps: (1) matching with the known motifs contained in the Homo sapiens Comprehensive Model Collection database to identify TFs (https://meme-suite.org/meme/tools/tomtom); (2) searching for genes regulated by the identified TFs in the TRRUST V2 database (https://www.grnpedia.org/trrust/); and (3) applying gene ontology (GO) for functional annotation and pathway enrichment analyses.
Radiomic analyses
A feature pyramid-DenseNet backbone network was applied to multiscale features from CT images, and the deep learning-based radiomic (DL-radiomics) model was then obtained via a fully connected network after fusing the CT features. The details of the image preprocessing and model construction are described in Supplementary method 4-6. The classic radiomic (C-radiomics) model was also established to evaluate the potential superiority of the deep learning technique, with the details of the procedures of constructing the C-radiomics model presented in Supplementary method 7.
To enhance the interpretability of the DL-radiomics model, the attribution scores of radiomics features were calculated by the deep learning importance features (DeepLIFT) method, which was a backward propagation method for gaining interpretability of deep neural networks53,54, to explain the contribution of input feature to the model’s output. We further explored the biological basis of the DL-radiomics model by applying the Radiogenomics dataset obtained from the Gene Expression Omnibus database. Gene set enrichment analysis (GSEA) and single-sample gene set enrichment analysis (ssGSEA) were performed to reveal the heterogeneity of the genetic pathways and the infiltration of immune cells in the tumor microenvironment among patients with different DL-radiomics scores. The processes of the biological exploration are detailed in Supplementary method 8.
Multiomics analyses
A univariate logistic regression was used to evaluate the associations between the clinical factors, 5mC model score, 5hmC model score and DL-radiomics model score with the malignancy risk of IPLs on the training set, and variables with p < 0.05 were fed into the multivariate logistic regression analyses via backward stepwise selection with the Akaike information criterion. Ultimately, all variables with p < 0.05 were selected as candidates, and the correlations among them were evaluated via the Pearson correlation coefficients analyses. Then, a series of 5 predictive models involving different feature sets were derived through (1) clinical factors only (clinical model), and adding (2) the DL-radiomics model score (clinic-Radiomics model), (3) the 5mC model score (clinic-mC model), (4) the DL-radiomics model score and 5mC model score (clinic-RadmC model), and (5) the DL-radiomics model score, 5mC model score and 5hmC model score (clinic-Rad(h)mC model) to evaluate the superiority of multiomics assays to models including peer assays. Multivariable logistic regression was used to establish the multiomics models, with a fivefold cross-validation scheme used to optimize the model hyperparameters.
Support for guiding the individual management process
The cutoff values of the models were identified based on the maximum Youden index on the training set, and the participants were classified into low-risk (the model score<cutoff values) and high-risk (the model score≥cutoff values) subgroups. Then, a hypothetical clinical management scheme was determined based on model classification results by assuming that any patients “ruled out” (identified as low-risk) would not undergo additional testing beyond follow-up chest CT scans, whereas any patients “ruled in” (identified as high-risk) would proceed directly to receive invasive procedures. The stratification efficiency was analyzed with respect to the reduction in the rate of invasive procedures unnecessary for benign diseases and potential delayed treatment for lung cancers in comparison with actual care.
Statistics and reproducibility
No statistical method was used to predetermine the sample size of this study since it was a prospective, observational study. Besides, participants were selected according to the inclusion and exclusion criterions described in Fig. 1. This study was designed as a prospective, observational, and case-control study, and all participants were classified based on their histopathological results. Thus, the experiments were not randomized, and the investigators were not blinded to group allocation during outcome assessment. However, the sample operation team was blinded to the group status during the whole sequencing process, and all CT reviewers were blinded to the clinical information during the annotation process. No sex or gender-related analysis was performed, as well.
Continuous and categorical variables of enrolled participants were reported as means with standard deviations (SDs) and frequencies with percentages, respectively. The Pearson χ² test or Fisher’s exact test, and Student’s t test or Wilcoxon signed-rank test were used to compare the participants characteristics, when applicable. Univariate and multivariate logistic regression analyses were performed to assess the associations between different participants characteristics and the odds of malignant diseases. The AUC was used to evaluate the diagnostic performance of the models, and compared by the DeLong’s test, with confidence intervals (CIs) generated based on 10,000 bootstrap replicates. Performance metrics, including sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV), and accuracy, were calculated based the cutoff values derived from the training set. A calibration curve analysis and a decision curve analysis (DCA) were applied to determine the clinical usefulness, with net reclassification improvement (NRI) and integrated discrimination improvement (IDI) analyses applied to evaluate the clinical benefits of different approaches. The predictive performance of the models was further assessed in subgroups stratified by age (≤50 years, >50 years), radiological image type (pure-GGO, part-solid, pure-solid), and radiological solid-component size (subcentimeter nodules with size≤ 10 mm, large nodules with 15 mm≤size≤30 mm, and mass with size>30 mm). To facilitate the clinical use of the models, the Shapley additive explanations (SHAP) method was used to examine the importance levels of each predictor and provide consistent attribution values for features incorporated into the models55. The statistical analyses were two-sided, and performed in SPSS (version 23.0, IBM SPSS Statistics), R software (version 4.1.3, http://www.Rproject.org) or Python (version 3.10, https://www.python.org/), with p < 0.05 indicating statistical significance.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw 5mC- and 5hmC-sequencing data in this study have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2022), China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences, which can be accessible under the accession code of HRA003308 and HRA007190. The CT imaging dataset supporting study’s findings are not publicly shared to protect patient privacy, but are available for research use from the corresponding author. The data transmission is required to be approved by the institutional review boards of all involved centers by providing the research plan and is restricted to non-commercial academic research purposes. Request can be submitted to Y.L.S. and will receive an internal review response within 30 days. In addition, the features incorporated into the clinical, end motifs-based and multiomics models are made publicly available at Figshare with https://doi.org/10.6084/m9.figshare.26861002. The RNA-seq dataset was obtained from Gene Expression Omnibus (GEO) data under accession code GSE103584 at https://www.cancerimagingarchive.net/collection/nsclc-radiogenomics/. Source data are provided with this paper.
Code availability
Codes for extracting classic radiomic features are derived from the PyRadiomics package from Python, which are open-access. The original codes for the deep learning-based radiomics model are publicly available at GitHub with https://github.com/Bercy0616/RadmC. Codes for the clinical model, end motifs-based models and multiomics models are made publicly available at Figshare with https://doi.org/10.6084/m9.figshare.27080899.
References
Han, B. et al. Cancer incidence and mortality in China, 2022. JNCC, https://doi.org/10.1016/j.jncc.2024.01.006 (2024).
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 70, 7–30 (2020).
Koo, M. M. et al. Presenting symptoms of cancer and stage at diagnosis: evidence from a cross-sectional, population-based study. Lancet Oncol. 21, 73–79 (2020).
MacMahon, H. et al. Guidelines for management of incidental pulmonary nodules detected on ct images: From the fleischner society 2017. Radiology 284, 228–243 (2017).
Gould, M. K. et al. Evaluation of individuals with pulmonary nodules: when is it lung cancer? Diagnosis and management of lung cancer, 3rd ed: American College of. Chest Physicians Evid.-based Clin. Pract. Guidel. Chest 143, e93S–e120S (2013).
Callister, M. E. et al. British thoracic society guidelines for the investigation and management of pulmonary nodules. Thorax 70, ii1–ii54 (2015).
Lam, S. & Tammemagi, M. Contemporary issues in the implementation of lung cancer screening. Eur. Respir. Rev. 30, https://doi.org/10.1183/16000617.0288-2020 (2021).
Liu, M. C. et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 31, 745–759 (2020).
Guo, W. et al. Sensitive detection of stage I lung adenocarcinoma using plasma cell-free DNA breakpoint motif profiling. EBioMedicine 81, https://doi.org/10.1016/j.ebiom.2022.104131 (2022).
Li, Y. et al. Multi-omics integrated circulating cell-free DNA genomic signatures enhanced the diagnostic performance of early-stage lung cancer and postoperative minimal residual disease. EBioMedicine 91, https://doi.org/10.1016/j.ebiom.2023.104553 (2023).
Wang, S. et al. Multidimensional cell-free DNA fragmentomic assay for detection of early-stage lung cancer. Am. J. Respir. Crit. Care Med. 207, 1203–1213 (2023).
Liu, Q. X. et al. A noninvasive multianalytical approach for lung cancer diagnosis of patients with pulmonary nodules. Advanced Science 8, https://doi.org/10.1002/advs.202100104 (2021).
Ren, Y. et al. A highly sensitive and specific non‐invasive test through genome‐wide 5‐hydroxymethylation mapping for early detection of lung cancer. Small Methods, https://doi.org/10.1002/smtd.202300747 (2023).
Hu, X. et al. Integrated 5-hydroxymethylcytosine and fragmentation signatures as enhanced biomarkers in lung cancer. Clinical Epigenetics 14, https://doi.org/10.1186/s13148-022-01233-7 (2022).
Sammut, S.-J. et al. Multi-omic machine learning predictor of breast cancer therapy response. Nature 601, 623–629 (2021).
Jiang, Y.-Z. et al. Integrated multiomic profiling of breast cancer in the Chinese population reveals patient stratification and therapeutic vulnerabilities. Nature Cancer, https://doi.org/10.1038/s43018-024-00725-0 (2024).
He, J. et al. Accurate classification of pulmonary nodules by a combined model of clinical, imaging, and cell-free DNA methylation biomarkers: a model development and external validation study. Lancet Digital Health 5, e647–e656 (2023).
Huang, J. et al. Distribution, risk factors, and temporal trends for lung cancer incidence and mortality. Chest 161, 1101–1111 (2022).
Zhao, W. et al. 3D Deep learning from CT scans predicts tumor invasiveness of subcentimeter pulmonary adenocarcinomas. Cancer Res. 78, 6881–6889 (2018).
Liu, Jianing et al. A self-supervised learning-based fine-grained classification model for distinguishing malignant from benign subcentimeter solid pulmonary nodules. Acad. Radio. S1076-6332, 00287–00283 (2024).
Madariaga, M. L. et al. Multidisciplinary selection of pulmonary nodules for surgical resection: Diagnostic results and long-term outcomes. J. Thorac. cardiovascular Surg. 159, 1558–1566.e1553 (2020).
Zhang, W. et al. Combination of CT and telomerase-positive circulating tumor cells improves diagnosis of small pulmonary nodules. JCI Insight, https://doi.org/10.1172/jci.insight.148182 (2021).
Liu, A. et al. Preoperative diagnosis of malignant pulmonary nodules in lung cancer screening with a radiomics nomogram. Cancer Commun. 40, 16–24 (2020).
Choi, W. et al. Radiomics analysis of pulmonary nodules in low-dose CT for early detection of lung cancer. Med. Phys. 45, 1537–1549 (2018).
Kammer, M. N. & Massion, P. P. Noninvasive biomarkers for lung cancer diagnosis, where do we stand? J. Thorac. Dis. 12, 3317–3330 (2020).
Seijo, L. M. et al. Biomarkers in lung cancer screening: achievements, promises, and challenges. J. Thorac. Oncol. 14, 343–357 (2019).
Lo, Y. M. D., Han, D. S. C., Jiang, P. & Chiu, R. W. K. Epigenetics, fragmentomics, and topology of cell-free DNA in liquid biopsies. Science 372, https://doi.org/10.1126/science.aaw3616 (2021).
Cristiano, S. et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 570, 385–389 (2019).
Ma, X. et al. Multi-dimensional fragmentomic assay for ultrasensitive early detection of colorectal advanced adenoma and adenocarcinoma. J. Hematol. Oncol. 14, https://doi.org/10.1186/s13045-021-01189-w (2021).
Nguyen, T. H. et al. Multimodal analysis of methylomics and fragmentomics in plasma cell-free DNA for multi-cancer early detection and localization. eLife 12, https://doi.org/10.7554/eLife.89083 (2023).
Jiang, P. et al. Plasma DNA end-motif profiling as a fragmentomic marker in cancer, pregnancy, and transplantation. Cancer Discov. 10, 664–673 (2020).
Liu, J., Shen, H., Chen, K. & Li, X. Large language model produces high accurate diagnosis of cancer from end-motif profiles of cell-free DNA. Briefings in Bioinformatics 25, https://doi.org/10.1093/bib/bbae430 (2024).
Shen, H., Yang, M., Liu, J., Chen, K. & Li, X. Development of a deep learning model for cancer diagnosis by inspecting cell-free DNA end-motifs. npj Precision Oncol. 8, https://doi.org/10.1038/s41698-024-00635-5 (2024).
Hou, Y., Meng, X. Y. & Zhou, X. Systematically evaluating cell‐free DNA fragmentation patterns for cancer diagnosis and enhanced cancer detection via integrating multiple fragmentation patterns. Adv. Sci. 11, https://doi.org/10.1002/advs.202308243 (2024).
Kammer, M. N. et al. Integrated biomarkers for the management of indeterminate pulmonary nodules. Am. J. Respiratory Crit. Care Med. 204, 1306–1316 (2021).
Yoo, H., Kim, K. H., Singh, R., Digumarthy, S. R. & Kalra, M. K. Validation of a deep learning algorithm for the detection of malignant pulmonary nodules in chest radiographs. JAMA Network Open 3, https://doi.org/10.1001/jamanetworkopen.2020.17135 (2020).
Venkadesh, K. V. et al. Deep learning for malignancy risk estimation of pulmonary nodules detected at low-dose screening CT. Radiology 300, 438–447 (2021).
Vaidhya Venkadesh, K. et al. Prior ct improves deep learning for malignancy risk estimation of screening-detected pulmonary nodules. Radiology 308, https://doi.org/10.1148/radiol.223308 (2023).
Chengdi Wang et al. Data-driven risk stratification and precision management of pulmonary nodules detected on chest computed tomography. Nat. Med. https://doi.org/10.1038/s41591-024-03211-3 (2024).
Wulaningsih, W. et al. Deep learning models for predicting malignancy risk in ct-detected pulmonary nodules: a systematic review and meta-analysis. Lung, https://doi.org/10.1007/s00408-024-00706-1 (2024).
Massion, P. P. et al. Assessing the accuracy of a deep learning method to risk stratify indeterminate pulmonary nodules. Am. J. Respiratory Crit. Care Med. 202, 241–249 (2020).
Han, D. S. C. et al. The Biology of Cell-free DNA Fragmentation and the Roles of DNASE1, DNASE1L3, and DFFB. Am. J. Hum. Genet. 106, 202–214 (2020).
Jiang, P. et al. Preferred end coordinates and somatic variants as signatures of circulating tumor DNA associated with hepatocellular carcinoma. PNAS 115, https://doi.org/10.1073/pnas.1814616115 (2018).
Rompala, G. et al. Profiling neuronal methylome and hydroxymethylome of opioid use disorder in the human orbitofrontal cortex. Nat. Commun. 14, https://doi.org/10.1038/s41467-023-40285-y (2023).
Liang, W. et al. Accurate diagnosis of pulmonary nodules using a noninvasive DNA methylation test. J. Clinic. Invest. 131, https://doi.org/10.1172/jci145973 (2021).
Liang, W. et al. Non-invasive diagnosis of early-stage lung cancer using high-throughput targeted DNA methylation sequencing of circulating tumor DNA (ctDNA). Theranostics 9, 2056–2070 (2019).
Song, C.-X. et al. 5-Hydroxymethylcytosine signatures in cell-free DNA provide information about tumor types and stages. Cell Res. 27, 1231–1242 (2017).
Shen, S. Y. et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 563, 579–583 (2018).
Shen, S. Y., Burgener, J. M., Bratman, S. V. & De Carvalho, D. D. Preparation of cfMeDIP-seq libraries for methylome profiling of plasma cell-free DNA. Nat. Protoc. 14, 2749–2780 (2019).
Cao, F. et al. Integrated epigenetic biomarkers in circulating cell-free DNA as a robust classifier for pancreatic cancer. Clinic. Epigenet. 12, https://doi.org/10.1186/s13148-020-00898-2 (2020).
Song, C.-X. et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 29, 68–72 (2011).
Chang, W. et al. Dynamic changes in whole genome DNA methylation, chromatin and gene expression during mouse lens differentiation. Epigenetics & Chromatin 16, https://doi.org/10.1186/s13072-023-00478-7 (2023).
Cho, H. J. et al. Interpretable meta-learning of multi-omics data for survival analysis and pathway enrichment. Bioinformatics 39, https://doi.org/10.1093/bioinformatics/btad113 (2023).
Li, J. et al. Deep-LIFT: deep label-specific feature learning for image annotation. IEEE Trans. Cybern. 52, 7732–7741 (2022).
Lundberg S. M., E. G., Lee S.-I. Consistent individualized feature attribution for tree ensembles. arXiv preprint arXiv:180203888 (2018).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (82241238 to D.X.; 82241237 to L.X.L.; 92259205, 91959126 to C.C.; 92474114 to Y.L.S.; 82302317 to B.X.H), Ningbo Top Medical and Health Research Program (No.2022030208 to G.F.Z.), Clinical Research Foundation of Shanghai Pulmonary Hospital (FKLY20016 to M.M.Z.; 2021LY0311 to C.C.), The 1·3·5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYYC23024 to D.X.), Research Fund of West China Hospital (2020HXFH006 to D.X.) and the China Postdoctoral Science Foundation (2021M700341 to B.X.H). We would like to thank the MultiomIcs claSSIfier for pulmOnary Nodules (MISSION) Collaborative Group for their supports and efforts. We are also grateful to the clinical research coordinators, Xiaomin Zhu and Chong Zhu, who helped to collect the plasma samples. Then, we thank all the participants, without whom this research would not have been possible.
Author information
Authors and Affiliations
Contributions
M.Z., G.X., B.H., Y.L.S., D.Z., and C.C. designed the study. M.Z., G.X., and B.H. performed the data analyses, built the models and wrote this paper. J.D., T.W., Y.Z., S.L., Y.W., Y.H., and T.C. helped to collected and annotated the CT images. J.Z., Z.Y., X.H., D.X., and L.L. developed the sequencing methodology. L.G., W.Q., Y.X.S., M.Y., G.Z., B.Y., and M.M. helped to collect the blood samples. X.S. helped to annotate the CT images. Y.L.S., D.X., D.Z., and C.C. conceived the study and edited the manuscript. All authors had access to all the raw datasets. All authors reviewed, revised, discussed the results and contributed the final version of the manuscript, and were responsible for the final decision to submit the manuscript for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Idoia Ochoa, Rajat Thawani and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, M., Xue, G., He, B. et al. Integrated multiomics signatures to optimize the accurate diagnosis of lung cancer. Nat Commun 16, 84 (2025). https://doi.org/10.1038/s41467-024-55594-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55594-z
