
Abstract
Human gut Bacteroides and Parabacteroides species play crucial roles in human health and are known for their capacity to utilize diverse polysaccharides. Understanding how these bacteria utilize medicinal polysaccharides is foundational for developing polysaccharides-based prebiotics and drugs. Here, we systematically mapped the utilization profiles of 20 different medicinal polysaccharides by 28 human gut Bacteroides and Parabacteroides species. The growth profiles exhibited substantial variation across different bacterial species and medicinal polysaccharides. Ginseng polysaccharides promoted the growth of multiple Bacteroides and Parabacteroides species; in contrast, Dendrobium polysaccharides selectively promoted the growth of Bacteroides uniformis. This distinct utilization profile was associated with genomic variation in carbohydrate-active enzymes, rather than monosaccharides composition variation among medicinal polysaccharides. Through comparative transcriptomics and genetical manipulation, we validated that the polysaccharide utilization locus PUL34_Bu enabled Bacteroides uniformis to utilize Dendrobium polysaccharides (i.e. glucomannan). In addition, we found that the GH26 enzyme in PUL34_Bu allowed Bacteroides uniformis to utilize multiple plant-derived mannan. Overall, our results revealed the selective utilization of medicinal polysaccharide by Bacteroides and Parabacteroides species and provided insights into the use of polysaccharides in engineering the human gut microbiome.
Similar content being viewed by others
Introduction
Human gut Bacteroides and Parabacteroides, primary consumers of polysaccharides, play critical roles in human health and disease, such as the development1,2 and immunotherapy3,4 of cancer, colonization resistance to pathogens5, etc. They utilize endogenous (host-derived) and exogenous (e.g. plant-derived) oligo/polysaccharides, providing nutrition and vitamins to the host and other intestinal microbes5,6,7. Recent studies highlight that polysaccharides impact host health by regulating the growth and metabolic profiles of Bacteroides8,9,10. Understanding how Bacteroides and Parabacteroides utilize these polysaccharides is essential for developing novel polysaccharide-based prebiotics and drugs to promote health.
Medicinal polysaccharides from herbs and mushrooms have complex and diverse structures, comprising various glycosidic linkages (e.g., α- or β- (1 → 3)/(1 → 4)/(1 → 5)/(1 → 6)-glycosidic linkages) in backbone or branched chains and including ten or more monosaccharides, sometimes with modifications like methylation and acetylation11. Research shows that medicinal polysaccharides and other plant-derived polysaccharides (e.g. from vegetables and fruits) possess various biological activities12,13,14,15, including anti-tumor, anti-viral, anti-inflammatory, and immunomodulatory effects. Bacteroides and Parabacteroides species can be promoted by multiple medicinal polysaccharides. Ginseng polysaccharides (GPs) have been shown to enrich both Bacteroides vulgatus and Parabacteroides distasonis, thereby enhancing the response rate of PD-1/PD-L1 immunotherapy3. Ophiocordyceps sinensis polysaccharides have been found to enrich gut commensal Parabacteroides goldsteinii, leading to improvements in obesity and metabolic disorders16. Dendrobium officinale polysaccharide recall the diversity of gut microbiota, and increasing the abundance of Bacteroides, having protective effect against dextran sulfate sodium induced colitis in mice17.
Despite the advancement in polysaccharides utilization of Bacteroides, how Bacteroides utilize medicinal polysaccharides is not clear. Bacteroides and Parabacteroides are equipped with hundreds of carbohydrate-active enzymes (CAZymes) responsible for degrading polysaccharides5,18,19. The CAZymes are typically organized in clusters known as polysaccharide utilization loci (PULs), which are upregulated by specific polysaccharides6,20. Glycoside hydrolase (GH) are enzymes responsible for the hydrolysis of glycosidic bonds. The genomes of Bacteroides species usually harbor multiple PULs for utilizing different polysaccharides. Some of the mechanisms have been elucidated, for example, B. thetaiotaomicron VPI-5482 utilize starch using Sus system21; Bacteroides spp. degrade complex O-glycans found in mucins using sulfatase22; B. plebeius utilize sulphated polysaccharide porphyran utilization using porphyranases7, etc.
In this study, we characterized the growth phenotypes of 28 human gut Bacteroides and Parabacteroides species using 20 medicinal polysaccharides. We observed significant variations in growth profiles among species and polysaccharides. Notably, Dendrobium polysaccharides (DPs, a type of glucomannan) specifically induced the growth of B. uniformis DA183. Through transcriptomics and genetic manipulations, we identified the key gene cluster PUL34_Bu and the critical enzyme GH26_BuDA183 involved in DPs utilization. In vitro enzyme activity assays and molecular docking further elucidated the molecular mechanism of GH26 catalysis. These results provide valuable insights into how human gut microbes utilize medicinal polysaccharides and offer important information for developing novel polysaccharide-based drugs.
Results
Mapping the utilization profile of medicinal polysaccharides by human gut Bacteroides and Parabacteroides species
To comprehensively analyze the utilization of medicinal polysaccharides by human gut Bacteroides and Parabacteroides, we developed an in vitro gut bacterial growth profile platform showed in Fig. 1A, starting with testing three minimal media. Growth curves indicated that modified Bacteroides minimal medium (mBMM), a synthetic medium, generally supported the growth of Bacteroides and Parabacteroides species (Supplementary Fig. 1).

A The flow chart showing the experimental process of growth profile characterization for medicinal polysaccharides, including i) extraction of medicinal polysaccharides using hot water/ethanol, ii) isolation of the human gut Bacteroides species through culturomics, iii) growth profiling of Bacteroides species with minimal medium supplemented with 0.5% medicinal polysaccharides in an anaerobic chamber (85% N2, 10% H2, and 5% CO2). Growth optical density (OD) was measured and recorded every 12 h using a microplate reader. B The prevalence (bar chart) and relative abundance (point plot) of Bacteroides species based on public human gut metagenome sequence data (n = 16282). The ‘n’ refers to the number of fecal samples collected from healthy individuals in the GMrepo23 database. Bacteroides species were colored by genus, Bacteroides in blue, Parabacteroides in red, and Phocaeicola in green. Prevalence = 0.3 (red dotted line), Relative abundance = 0.1% (green dotted line). Relative abundance data are expressed as mean values ± standard deviation (SD). C The pie chart showed the coverage of core human gut Bacteroides (prevalence> 30%, mean relative abundance> 0.1%, n = 16,282). D The pie chart showed the coverage of human gut Bacteroides carbohydrate active enzymes in CAZy database. E The heat map showed the growth OD600 of 28 Bacteroides species at 48 h under 20 medicinal polysaccharides (n = 3 independent experiments). Statistical significance was determined using a two-sided unpaired t-test between each experimental group and the negative control group, with adjustments for multiple comparisons made using the Bonferroni method. The P values are indicated as follows: * < 0.05; ** < 0.01; *** < 0.001; **** < 0.0001. Each experiment was conducted in triplicate to ensure reliability. The sub-graph in the right counts the number of medicinal polysaccharides utilized by Bacteroidetes spp. The sub-graph at the bottom counts the number of Bacteroidetes spp. utilizing medicinal polysaccharides. RNPs: Radix Notoginseng Polysaccharides; GPs: Ginseng polysaccharides; PQPs: Panax Quinquefolium Polysaccharides; PSPs: Polygonatum sibiricum Polysaccharides; RAPs: Rhizoma anemarrhenae Polysaccharides; CPPs: Codonopsis Pilosula Polysaccharides; AdPs: Adenophora Polysaccharides; PLPs: Pueraria lobata Polysaccharides; APs: Astragalus polysaccharide; DPs_1 and DPs_2: Dendrobium polysaccharides; GLPs_1 and GLPs_2: Ganoderma lucidum polysaccharides; LBPs_1 and LBPs_2: Lycium barbarum polysaccharides; PPs: Poria Polysaccharides; RPPs: Radix Pseudoxtellariae Polysaccharides; AnPs: Angelica Polysaccharides; HAPs: Hovenia Acerba Polysaccharides; SPs: Scrophulariaceae Polysaccharide; PC: positive control; NC: negative control.
Bacteroides and Parabacteroides, which are highly prevalent and abundant in the human gut microbiota, encode numerous PULs. The platform includes 18 Bacteroides species, 6 Parabacteroides species, and 4 Phocaeicola species (formerly Bacteroides spp.). Some strains were chosen repetitively at the species level (e.g., B. fragilis DA486 and DA557) due to their different CAZyme profiles. To evaluate the prevalence and mean relative abundancreactivee of these Bacteroides species in the human gut microbiota, we analyzed publicly available 16S rRNA gene amplicon datasets (n = 16,282) from GMrepo23. We found that 71.4% (20/28) of the Bacteroides species had a prevalence greater than 30%, and 89.3% (25/28) had a mean relative abundance greater than 0.1% (Fig. 1B, Supplementary Tables 1, 2). The coverage of core human gut Bacteroides species (prevalence > 30%, mean relative abundance > 0.1%24) and CAZymes in Bacteroides (CAZy database25) was 100% and 70.9%, respectively (Fig. 1C, D). These selected Bacteroides ensure a systematic and comprehensive analysis of polysaccharide utilization.
Bacteroides and Parabacteroides were reported as the target of many medicinal polysaccharides in the intestine, but the mechanisms of their utilization remain underexplored. To expand our understanding, we selected 20 phylogenetic different medicinal polysaccharides with distinct structure (i.e. sugar linkage and modification) (Table 1). For instance, GPs from the Araliaceae family contained starch-like glucans, arabinogalactans, α-(1 → 4)-GalA backbone (homogalacturonan), rhamnogalacturonan-rich pectin26; Dendrobium polysaccharides (DPs_1 and DPs_2, prepared using different methods) feature backbones of →4)-β-D-Glcp-(1 → , →4)-β-D-Manp-(1 → , →4)-2-O-acetyl-β-D-Manp-(1 → , and →4)-3-O-acetyl-β-D-Manp-(1 → 27. These 20 medicinal polysaccharides exhibit multiple biological activities, including anti-tumor, antioxidant, anti-inflammatory, and immune regulation, while maintaining low toxicity and minimal side effects. More details about other medicinal polysaccharides used in this study can be found in Supplementary Table 3A and B.
We found that the bacterial growth data were consistent across three technical replicates (R = 0.96, p < 2.2e16) (Supplementary Fig. 2A–C) and between two independent experiments (R = 0.91, p < 2.2e16) (Supplementary Fig. 2D), indicating that our method is highly reproducible. Characterizing the growth phenotypes revealed diverse growth profiles among different Bacteroides and medicinal polysaccharides (Fig. 1E, Supplementary Fig. 3A–C). The statistical analysis indicated that each strain can utilize an average of 4.79 types of medicinal polysaccharides, among which some strains can utilize up to 9 medicinal polysaccharides, while others cannot utilize any of the 20 medicinal polysaccharides. Each type of medicinal polysaccharide can support the growth of an average of 6.8 strains. Some polysaccharides, such as GPs, support the growth of multiple Bacteroides and Parabacteroides species, whereas others, like DPs_2, support only a single Bacteroides species. We found that polysaccharides extracted from plants with close phylogeny resulted in similar bacterial growth profiles (Supplementary Fig. 3D). This similarity is likely due to comparable polysaccharide compositions and structures, as seen in RNPs and PQPs, which both contain Ara, Gal, GalA, Glc, GlcA, Man, and Rha, with 1 → 4 glycosidic linkages forming their linear backbones26.
Growth profiles variation among medicinal polysaccharides may be due to the differences in physicochemical properties of polysaccharides, such as monosaccharide composition, and polysaccharide structure. To determine whether polysaccharide structure or monosaccharides composition causes variations in growth profiles, we selected eight medicinal polysaccharides exhibiting different growth profile. First, we characterized the physical and chemical properties of these polysaccharides (Supplementary Fig. 4A–B, Supplementary Fig. 5A–D, and Supplementary Tables 5–7). The monosaccharide composition analysis revealed that APs, GPs, GLPs_1, and GLPs_2 were mainly composed of glucose (Glc) (92.8%, 93.1%, 72.2%, and 68.3%, respectively). DPs_1 and DPs_2 were mainly composed of mannose (Man) (81.1% and 78.7%, respectively). LBPs_1 were mainly composed of 36.9% arabinose (Ara), 22.6% galactose, and 23.4% galacturonic acid (GalA), while LBPs_2 were mainly composed of 16.0% Glc, 33.5% Gal, and 39% GalA. The selected medicinal polysaccharides showed differences in total sugar content, molecular weight and distribution, and monosaccharide composition, which indicated that the medicinal polysaccharides were diverse in physicochemical properties. Notably, we found almost no correlation between distance of monosaccharide composition and growth profile, indicating that polysaccharides with similar monosaccharide composition did not exhibit similar growth profiles (Supplementary Fig. 4C). For instance, Astragalus polysaccharide (APs) and GPs, despite having similar monosaccharide compositions, displayed markedly different human gut bacteria growth profiles. It is worth mentioning that APs consist of methylated GalAp and diverse side chains28, differing from GPs. These findings suggest that polysaccharide structure, rather than monosaccharide composition, drives growth profile variations.
The utilization profile of medicinal polysaccharides is associated with genomic variation in carbohydrate-active enzymes
The characterization of the chemical structure of polysaccharides is a recognized worldwide challenge. However, complex polysaccharides have led to the evolution of numerous diverse carbohydrate-active enzymes (CAZymes) families. In turn, the specificity of degrading enzymes can provide information on the structure of the degraded polysaccharides. The genes encoded by different gut bacteria can help us understand the utilization profile. Bacteroides encode hundreds CAZymes to utilize complex polysaccharides19. CAZymes involved in carbohydrate degradation29 include glycoside hydrolases (GHs) for hydrolyzing glycosidic linkages, polysaccharide lyases (PLs) for breaking down polysaccharides containing uronic acids, carbohydrate-binding modules (CBMs) for binding carbohydrate motifs, carbohydrate esterases (CEs) for de-esterification, and auxiliary activity enzymes (AAs) for various redox transformations. Using dbCAN (version 4.1.4)30 to analyze annotated CAZymes, we observed variations in CAZyme distribution at the genus, species, and strain levels among Bacteroides and Parabacteroides species (Fig. 2A). Clustering the 28 Bacteroides based on the Euclidean distance of CAZymes revealed that Bacteroides and Parabacteroides with similar taxonomy share similar CAZyme distribution patterns (Fig. 2A). GHs were the most widely encoded, whereas AAs were less common due to the gut’s anaerobic environment limiting oxygen-dependent AAs29.

A The heat map showed the distribution of CAZyme genes involved in carbohydrate degradation. Bacteroidetes spp. were clustered based on the Euclidean distance of CAZyme genes. CAZymes genes clustered based on the Euclidean distance of distribution in Bacteroides and Parabacteroides. Glycoside Hydrolases (GHs): hydrolysis and/or rearrangement of glycosidic bonds; Polysaccharide Lyases (PLs): non-hydrolytic cleavage of glycosidic bonds; Carbohydrate-Binding Modules (CBMs): adhesion to carbohydrates; Carbohydrate Esterases (CEs): hydrolysis of carbohydrate esters; Auxiliary Activities (AAs): redox enzymes that act in conjunction with CAZymes. B Bray curtis based principal components analysis (PCA) shows the dimension reduction analysis of carbohydrate active enzymes in Bacteroides. The dots on the graph are colored according to genus of Bacteroidetes. C Euclidean distance based principal components analysis (PCA) shows the dimension reduction analysis of polysaccharides utilization at 48 h by Bacteroides. The dots on the graph are colored according to genus of Bacteroidetes. D The point diagram displays the correlation between growth profile distance and CAZymes distribution distance. The R and P values refer to the most parsimonious model. Statistical significance was determined using a two-sided Pearson correlation test (for R value) and an ordinary least squares (OLS) regression analysis (for the linear fit). The figure also includes a 95% confidence interval around the best linear fit. Light green dots represented high dot density and dark green dots represented low dot density.
To explain the growth profile variations among different Bacteroides and Parabacteroides species, we clustered the 28 stains based on the Euclidean distance of CAZymes (Fig. 2B), and growth profile distribution (Fig. 2C). We found that Bacteroides spp. with similar CAZyme genes distribution had similar polysaccharide metabolic pattern. For example, P. dorei DA26 and P. vulgatus DA57 exhibited similar CAZyme gene distributions and polysaccharide metabolic patterns. A similar phenomenon was observed among Parabacteroides spp. Additionally, a significant correlation was observed between the distances of CAZyme distribution and growth profiles, indicating that Bacteroides and Parabacteroides with similar CAZyme distributions exhibit similar growth profiles (Fig. 2D). These results suggest that CAZyme distribution drives growth profile differences.
Transcriptomics analysis reveals that specific PULs are highly up-regulated by medicinal polysaccharides
Differential gene expression aids in discovering enzymes and pathways for utilizing specific carbon sources. First, we selected Ginseng polysaccharides (GPs) that supported the growth of multiple Bacteroides. We analyzed the transcriptome data of P. vulgatus DA57 and P. distasonis DA104 cultivated with GPs and glucose as the sole carbon sources (Fig. 3A–F, Supplementary Fig. 6A–D, and Supplementary Tables 8, and 9). Comparative transcriptome analysis revealed that the top 10 upregulated carbohydrate metabolism genes were located in PUL19 of P. vulgatus DA57 (PUL19_Pv) (Fig. 3A) and PUL29 of P. distasonis DA104 (PUL29_Pd) (Fig. 3C) as annotated by PULpy31. We refined the PUL boundaries based on the expression levels of upstream and downstream genes (Fig. 3C, E, I, and Supplementary Fig. 7C) (see “Methods” for more details). Notably, GH13_10 and GH13_20, described as α-amylases by the CAZy database25, were upregulated in both PUL19_Pv and PUL29_Pd. Given that neutral polysaccharides, including amyloid mixtures32, are the main components of GPs, these results suggest that GH13_10 and GH13_20 are crucial for GPs utilization.

A Volcano plot of P. vulgatus DA57 gene expression comparing minimal media supplemented with GPs to glucose, showing the fold-change (log2, x-axis) versus the differential significance (-log10 adjusted p-value, y-axis). The top 10 up-regulated genes are highlighted in green. Differential expression analysis was performed using DESeq2, employing a two-sided Wald test. Adjusted p-values were obtained after correcting for multiple hypothesis testing using the Benjamini and Hochberg method to control the false discovery rate (FDR). Genes with an adjusted p-value < 0.05 are considered statistically significant. The x-axis represents the log2 fold change in gene expression, while the y-axis shows the -log10 of the adjusted p-value. Each experiment was conducted in triplicate to ensure reliability. B The schematic diagram shows that PUL19 of P. vulgatus DA57 (PUL19_Pv) contains 8 genes, with a cluster size of 14,518 base pairs. susC: susC-like TonB-dependent transporter; susD: susD-like cell-surface glycan-binding protein; GH: glycoside hydrolyze; Unk: unknown functions. C Volcano plots of P. distasonis DA104 gene expression comparing minimal media supplemented with GPs to glucose. The statistical analysis is the same as in (A). D The schematic diagram shows that PUL29 of P. distasonis DA104 (PUL29_Pd) contains 8 genes, with a cluster size of 13,063 base pairs. E–F Bubble plot displays the differential expression of the E) PUL19_Pv, and F) PUL29_Pd genes and its neighboring 3 genes, utilizing GPs and glucose. G Volcano plots of B. uniformis DA183 gene expression comparing minimal media supplemented with DPs to glucose. The statistical analysis is the same as in (A). H The schematic diagram shows that PUL34_Bu contains 22 genes, with a cluster size of 39,709 base pairs. CE: carbohydrate esterase. I Bubble plot displays the differential expression of the PUL34_Bu gene and its neighboring 3 genes, utilizing DPs and glucose. The region between two vertical cyan dotted lines indicates significantly upregulated genes with consistent transcription direction.
Next, we examined gene expression data of Bacteroides utilizing Ganoderma lucidum polysaccharides (GLPs_1), which supported the growth of a moderate number of Bacteroides. The top 10 upregulated carbohydrate metabolism genes were located in PUL16, PUL25, and PUL39 of B. uniformis DA183 (Supplementary Fig. 6E–F, Supplementary Fig. 7A, and Supplementary Table 10). Analysis of gene cluster structures showed that these PULs encoded GH55 (i.e., exo/endo-β-1,3-glucanase) in PUL16, GH9 (i.e., endo-β-1,4-glucanase) in PUL16&25, GH3 (i.e., endo-β-1,6-glucosidase) in PUL39, and GH16_3 and GH158 (i.e., endo-β-1,3-glucanase) in PUL39 (Supplementary Fig. 7B, C). The presence of multiple PULs responsible for the same polysaccharides aligns with previous studies20. GLPs are composed of →6)-β-D-Glcp-(1 → , →6)-α-D-Galp-(1 → , and →3)-β-D-Glcp-(1 → 33. These findings indicate that PUL16, PUL25, and PUL39 are likely responsible for GLP utilization.
We also analyzed gene expression data for Bacteroides utilizing DPs_2, which selectively promoted the growth of B. uniformis. Focusing on B. uniformis DA183, we compared transcriptome data when cultivated with DPs and glucose as the sole carbon sources. The top 10 upregulated carbohydrate metabolism genes were located in PUL34 of B. uniformis DA183 (PUL34_Bu) (Fig. 3G–I, Supplementary Fig. 6E–F, Supplementary Table 11). DPs are mannose oligosaccharide with a backbone consisted of → 4)-β-D-Glcp-(1 → , → 4)-β-D-Manp-(1 → , →4)-2-O-acetyl-β-D-Manp-(1 → , and →4)-3-O-acetyl-β-D-Manp-(1 → 27. The measured monosaccharide composition of DPs confirms that mannose is the main component (Supplementary Fig. 4A, B). Two significantly upregulated glycoside hydrolases (GHs) in PUL34_Bu, GH26 and GH5_7, are described as exo-β-1,4-mannobiose and endo-β-1,4-mannanase by the CAZy database25.
PUL34_Bu gene cluster in B. uniformis is required for Dendrobium polysaccharides utilization
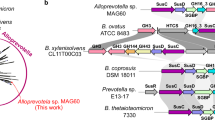
To explore the genetic basis underlying the highly specific utilization of Dendrobium polysaccharides (DPs) by B. uniformis, we utilized CRISPR/Cas-based Bacteroides genome editing tool34 to construct PUL34 knock-out mutants (Methods). To ensure successful gene knockout, we targeted two regions in the gene cluster for separate knockout (Fig. 4A). The first region (PUL34M_Bu) was a moderately sized segment with significant upregulation at the transcriptional level and genes transcribed in the same direction. The second region (PUL34S_Bu) contained three key enzymes which were isozymes of three key enzymes involved in inulin utilization35, including a SusD-like protein for polysaccharide binding in the outer membrane, a SusC-like protein for polysaccharide transport, and GH26 for glycosidic bond hydrolysis. Gel electrophoresis and DNA sequencing confirmed the successful knockout of the two regions (Fig. 4B, Supplementary Fig. 9A–D). When grown in mBMM medium with DPs as the sole carbon source, the wild-type strain grew, whereas the mutant strain did not (Fig. 4C, and D).

A The schematic diagram shows the two region of PUL34_Bu genes knock out. PUL34M_Bu indicates significantly upregulated genes with consistent transcription direction. PUL34S_Bu indicates the putative three key genes. B Nucleic acid electrophoresis diagram shows the four DNA products: products amplified with primers designed within a 1.5k region flanking the region of PUL34M_Bu (P1 ~ P4), and genomic DNA of wild-type, and mutate strain; Products amplified with primers designed within a 1.5k region flanking the region of PUL34S_Bu (P2 ~ P3), and genomic DNA of wild-type, and mutate strain. If the fragment is successfully deleted, a 3000 bp product will be amplified. In contrast, if the deletion is not successful, no product will be amplified due to the excessive length of the target band. This representative image is from a single experiment. C–D The point plot shows the growth density of C) wild-type and PUL34S knockout strains, and D) wild-type and PUL34M knockout strains in mBMM medium with DPs_2 as the sole carbon source. Data are presented as mean values ± SD. Assays were performed in technical triplicates (n = 3). E The heatmap shows the correlation between PUL34_Bu and growth density OD600. PUL34_Bu genes were identified by BLASTP (version 2.6.0)98 (coverage ≥ 50%, identify ≥ 30%). The species carrying PUL34_Bu genes were colored in cyan, otherwise, in white. The species utilizing DPs was colored gray, otherwise, in white.
To investigate the distribution of PUL34_Bu in the genomes of 27 other Bacteroides and Parabacteroides species, we performed sequence alignment with a sequence similarity threshold of 60% and a coverage threshold of 70% (Methods). We found that only B. uniformis encoded a complete PUL34_Bu gene cluster (Fig. 4E). In summary, we revealed that a complete PUL34_Bu gene cluster allowed for the highly specific utilization of DPs by B. uniformis.
GH26 enzymes is necessary for plant-derived mannan utilization in B. uniformis
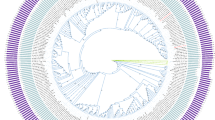
We constructed a phylogenetic tree for the 89 characterized GH26 protein sequences deposited in the CAZy database25 (Methods). B. uniformis GH26 is clustered with other enzymes (including GH26 enzymes from B. ovatus ATCC8483) annotated as Mannan endo-1,4-β-mannosidase (EC 3.2.1.78) (Fig. 5A). To explore the distribution of B. uniformis GH26 in human gut bacteria, we performed sequence alignments in UHGP database36 using DIAMOND methods37. We found that the B. uniformis GH26 orthologs were mainly present in Bacteroides spp. at the functional conserved alignments threshold (identity > 60%, coverage> 70%), including 64/66 B. uniformis, 26/32 B. cellulosilyticus, 3/17 B. ovatus, 2/2 B. stercorirosoris, and 2/2 P. dorei (Supplementary Fig. 10A, B).

A Phylogenetic tree of GH26 protein sequences, including 89 characterized GH26 and B. uniformis GH26 in this study. Organism names are indicated to the species level, next to the GenBank accession number of the GH33 protein analyzed. The tree was generated using the Maximum likelihood method. The tree label was colored by EC classification. B. uniformis GH26 in this study was marked with a red five-pointed star. The crystallized GH26 of B. ovatus was marked with a gray five-pointed star. B The point plot shows the growth density of B. ovatus ATCC8483 (blue), B. uniformis DA183 (gray), B. uniformis ATCC8492 (red), mutant strain of B. uniformis ATCC8492 deleted PUL34S (green) and another mutant strain of B. uniformis ATCC8492 deleted PUL34S (orange) utilizing 6 carbon source, such as glucose, Dendrobium officinale polysaccharide (DPs), konjac glucomannan, guar gum galactomannan, carob galactomannan, and ivory nut mannan. Data are presented as mean values ± SD. Assays were performed in technical triplicates (n = 3). C Domain structure of B. uniformis GH26. D Overall structure of B. uniformis GH26 was predicted by AlphaFold2 based on the protein sequence. The Ig-like ___domain is colored in in green, GH26 ___domain in red, galactose-binding lectin in purple, respectively. E The structure of B. uniformis GH26 (red) ___domain is superimposed on that of B. ovatus GH26 (gray) (PDB code 6HF4) (TM-score = 0.91, RMSD = 1.37 Å). F The active sites (E415 and E504), the calcium-binding sites (L198 and S201), and the substrate-binding sites (V170, A200, W207, Y342, W527, and Y528) of B. uniformis GH26 (red stick models), and the active sites (E201 and E291), the calcium-binding sites (L105 and S108), and the substrate-binding sites (V77, A107, W112, Y148, W314, and Y315) of B. ovatus GH26 (gray stick models) is conserved.
To investigate the necessity of PUL34_Bu for the utilization of plant-derived mannans by gut Bacteroides, we conducted growth profiling for B. ovatus ATCC8483, B. uniformis DA183, B. uniformis ATCC8492 (wild-type), and B. uniformis ATCC8492 ΔPUL34S_1&_2 (mutate) utilizing various plant-derived mannans, such as Dendrobium officinale polysaccharide (DPs), konjac glucomannan38, guar gum galactomannan39, carob galactomannan40, and ivory nut mannan41. We found that wild type B. uniformis and B. ovatus strains were able to utilize these multiple plant-derived mannan, while the PUL34S knock-out mutants were not able to grow (Fig. 5B). These results indicated that GH26 in B. uniformis hydrolyzes a wide range of mannan substrates.
To further understand the structural properties of B. uniformis GH26, we predicted its structure using AlphaFold242 (pLDDT=93.04). We found B. uniformis GH26 contained a signal peptide (residues 1 to 30), a bacterial Ig-like ___domain (residues 31 to 131), a GH26 ___domain (residues 135 to 564), and galactose-binding lectin (residues 239 to 354) (Fig. 5C, D). The structure of GH26 enzyme from B. ovatus ATCC8483 has been determined by crystallography (GenBank accession ALJ47537.1; PDB code 6HF4)43. Comparing with the structure of B. ovatus GH26, we found that the structure of B. uniformis GH26 contained two extra modules: 1) Ig-like ___domain which is related to the stability of the enzyme44 or serving as connector between two domains45; 2) carbohydrate binding module46,47. We found that the active sites, the calcium-binding sites, and the substrate-binding sites of B. uniformis GH26, and B. ovatus GH26 are conserved (TM-score = 0.91, RMSD = 1.37 Å) (Fig. 5E, and Supplementary Fig. 11).
The conserved residues of B. uniformis GH26 (W527 and Y528) and B. ovatus GH26 (W314 and Y315) are responsible for binding mannose by hydrogen bonds43, which indicates that the substrate of B. uniformis GH26 contains mannose. However, residues K149 responsible for binding galactose are not conserved between the two enzymes, which indicated a different substrate-binding specificity. Indeed, the substrate of B. ovatus GH26 is a galactomannan, which is structurally different from DPs (i.e. glucomannan).
We observed that DPs_1 also induced the growth of B. finegoldii DA347, which encodes a GH26 gene in its genome (Fig. 4E). This observation prompted us to perform sequence and structural alignments of the GH26 from B. finegoldii DA347. Sequence alignment showed low similarity among GH26 from B. uniformis DA183, B. ovatus ATCC8483, and B. finegoldii DA347. The sequence identity is 50.5% (56.0% coverage) between B. uniformis DA183 and B. ovatus ATCC8483, 49.1% (52.0% coverage) between B. uniformis DA183 and B. finegoldii DA347, and 48.3% (93.0% coverage) between B. ovatus ATCC8483 and B. finegoldii DA347 (Supplementary Fig. 12A). Structural alignment showed that B. finegoldii DA347 encodes an enzyme structurally similar to B. ovatus ATCC8483 GH26 (TM-score = 0.98, RMSD = 1.03 Å) (Supplementary Fig. 12B-C). Moreover, the active sites (E217 and E321), the calcium-binding sites (L109 and S112), and the substrate-binding sites (W116, W353, and Y354) of GH26 from B. finegoldii DA347 (cyan stick models), and the active sites (E201 and E291), the calcium-binding sites (L105 and S108), and the substrate-binding sites (W112, W314, and Y315) of B. ovatus GH26 (gray stick models) are conserved (Supplementary Fig. 12D). These results may explain its ability to grow using DPs_1.
The enzymatic activity and specificity of Bacteroides GH26 enzyme
To further characterize the enzyme activity and substrate specificity of Bacteroides GH26, we expressed and purified the GH26 enzyme from B. uniformis DA183 and B. ovatus ATCC8483 (Supplementary Fig. 13A, B) and tested them with 13 substrates, including 6 mannans, 3 potential substrates from structurally similar proteins or the same protein family, and 4 well-defined substrates as potential negative controls (Supplementary Table 3C). Reducing-end production was measured using the standard 3,5-dinitrosalicylic acid (DNS) assay with mannose as the standard (Methods). We found that both enzymes hydrolyzed all six mannans, but not the other seven substrates (Fig. 6A, and Supplementary Table 16A, B).

A The bar chart shows the reducing-end mannose produced by GH26 BuDA183 (cyan) and GH26 BoATCC8483 (gray) using 13 mannan substrates. The enzyme concentration was 1.25 mg/mL, substrate concentration was 5 mg/mL, reaction temperature was 37 °C, and reaction time was 30 min. The reducing end is the terminus of a carbohydrate chain that has a free aldehyde or ketone group capable of reducing copper ions in the DNS assay. Data are presented as mean values ± SD. Assays were performed in technical triplicates (n = 3). B–D The dot plot shows the reducing-end production over time for six substrates hydrolyzed by GH26 BuDA183 (Wild type), GH26 BuDA183 E415A, and E504A (Catalytic site mutation). The substrates are: konjac glucomannan (yellow), Dendrobium officinale polysaccharides (red), galactosyl mannabiose (green), carob galactomannan (blue), guar gum galactomannan (pink), and ivory nut mannan (purple). The enzyme concentration was 125 nM, substrate concentration was 5 mg/mL, reaction temperature was 37 °C, and reaction time was 10, 20, 30, 60, 120 min. Data are presented as mean values ± SD. Assays were performed in technical triplicates (n = 3). E–F Interactions of GH26 BuDA183 (Wild type) with galactosyl mannabiose, and 1-kestose are visualized in 3D (left) and 2D (right). Substrates are shown as green sticks or circle, interacting residues as gray sticks or circle, and catalytic sites as cyan sticks or circle. Interaction forces are displayed as dotted lines. G The reaction system is the same as in Panel (B–D), except the enzyme is GH26 BuDA183 A363K (Different residues interchanges).
The putative catalytic residues are two Glu residues (E201 and E291 in B. ovatus ATCC8483 and E415 and E504 in B. uniformis DA183), which hydrolyze mannans via a double-displacement general acid−base mechanism48. We performed point mutations on the two catalytic sites of B. uniformis GH26 (E415A and E504A) and found that mutation of either Glu residue leads to loss of enzyme activity, indicating that both catalytic sites are necessary for enzyme activity (Fig. 6B, and Supplementary Table 17).
To investigate the structural basis of different substrate specificity, we performed molecular docking simulations for 5 structurally defined substrates (1 reactive substrate and 4 non-reactive substrates) using AutoDock Vina (v1.2.0)49. We predicted their binding affinities and ranked the docking poses of the enzyme-substrate complexes using CSM-carbohydrate50. The predicted binding free energy of the non-reactive substrates was only slightly higher than that of the reactive galactosyl mannobiose, and the non-catalyzed trehalose even had a lower binding free energy. This indicates that substrate specificity cannot be solely explained by comparing the binding free energies of the substrates. Notably, the catalytic residues were found in the interactive residues of enzyme-reactive substrate complexes, but not in those of enzyme-non-reactive substrate complexes, suggesting that substrate reactivity depends on their ability to interact with catalytic residues (Fig. 6C, D and Supplementary Fig. 14A–C).
Among the six reactive mannans, GH26 from B. uniformis DA183 showed the highest activity toward glucomannan (e.g. konjac mannan) (Fig. 6B), while GH26 from B. ovatus ATCC8483 had the highest activity toward galactomannan (Supplementary Fig. 15A). To investigate the role of differential substrate-binding sites in the activity of GH26 from B. uniformis DA183 and B. ovatus ATCC8483, we identified the unique differential site (A363 in B. uniformis DA183 and K149 in B. ovatus ATCC8483) through structural alignment and interchanged the residues. Interestingly, the GH26 BoATCC8483 K149A mutant showed significantly reduced activity toward guar gum galactomannan and became most active toward konjac glucomannan (Supplementary Fig. 15C). Unexpectedly, the enzymatic activity of the GH26 BuDA183 A363K mutant showed a slight increase and did not result in a significant change in substrate specificity, unlike the GH26 BoATCC8483 K149A mutant, suggesting that other unpredicted differences in substrate-binding sites contribute to the distinct substrate preferences of the two enzymes (Fig. 6G, and Supplementary Table 17).
To evaluate the role of CBM ___domain in GH26 BuDA183, we firstly compared the enzymatic activity of full-length wild type and CBM truncated GH26. Interestingly, no significant difference in enzyme activity was observed between wild type GH26 (Km=3.75 ± 1.44 mg/mL, Kcat = 678.23 ± 101.54 s-1) and CBM truncated GH26 (Km=3.81 ± 1.63 mg/mL, Kcat = 656.68 ± 110.27 s-1) (Supplementary Fig. 13A–B, Supplementary Fig. 16A–B, and Supplementary Table 18). Notably, some studies have shown that CBM ___domain provided thermo-protection for certain carbohydrate-hydrolyzing enzymes51,52. By comparing the enzyme activities of wild-type GH26 and CBM-truncated GH26 at different temperatures, we found that the CBM-truncated GH26 became less thermostable. Specifically, the Tm values were ~55°C for wild-type GH26 and 50°C for the CBM-truncated variant (Supplementary Fig. 16C, and Supplementary Table 19). This indicates that the possible function of the CBM ___domain of GH26 is related to thermostability.
Discussion
Manipulating the gut microbiome with polysaccharides is a promising therapeutic strategy for various human diseases53,54,55. Understanding how gut bacteria utilize medicinal polysaccharides will guide the selection of polysaccharides for microbiome manipulation. However, knowledge in this area is still limited. To address this gap, we systematically mapped the utilization profiles of 20 different medicinal polysaccharides by 28 human gut Bacteroides and Parabacteroides species. We found that different Bacteroides and Parabacteroides species showed distinct utilization profiles of medical polysaccharides, which was associated with genomic variation in carbohydrate-active enzymes. Through comparative transcriptomics and genetical manipulation, we validated that the polysaccharide utilization locus PUL34_Bu enabled B. uniformis to utilize Dendrobium polysaccharides. We discovered that the GH26 enzyme in B. uniformis and B. ovatus, annotated as a mannosidase, was essential for the utilization of multiple plant-derived mannans (e.g. Dendrobium polysaccharides). Overall, our study provides a general framework to profile the utilization of plant-derived polysaccharides by human gut bacteria and to understand the underlying molecular mechanisms. The highly specific utilization of some medicinal polysaccharide (e.g. Dendrobium polysaccharides) also highlight the potential of glycan-based prebiotics and drugs for targeted modulation of the human gut microbiome.
B. uniformis is a prominent member of the human gut microbiota and plays a crucial role in maintaining gut health. Specifically, strain CECT 7771 ameliorates metabolic and immunological dysfunction in mice with high-fat-diet-induced obesity56 and enhances metabolic and immune benefits when combined with fiber in obese mice57. Additionally, B. uniformis and its preferred substrate, α-cyclodextrin, improve endurance exercise performance in both mice and human males58. Strain JCM5828 regulates colon NF-κB and MAPK signaling pathways by inhibiting IL-17 signaling, thereby ameliorating colitis development59. Strain F18-22 alleviates intestinal inflammation by improving gut dysbiosis60. Moreover, B. uniformis produces 3-succinylated cholic acid (3-sucCA), which alleviates metabolic dysfunction-associated steatohepatitis (MASH)61. Engineering B. uniformis to enhance its probiotic properties holds significant promise for therapeutic applications. By modifying B. uniformis to more efficiently degrade specific polysaccharides or produce beneficial metabolites, such as 3-sucCA, we can potentially improve its ability to colonize the gut and exert positive health effects. Engineered strains could also be designed to deliver therapeutic molecules directly to the gut, offering a targeted approach to treating gastrointestinal disorders.
Growth profile variations reflect differences in polysaccharide structure, suggesting the potential for developing quality control markers for medicinal polysaccharides by comparing growth profiles. Quality control in polysaccharide drug manufacturing is challenging due to their complexity, chemical diversity, and batch-to-batch variations62. Current methods have been employed for characterizing medicinal polysaccharides, such as 1) quantifying the sugar content based on colorimetry, 2) determining the molecular weight and distribution based on high-performance liquid chromatography (HPLC), and 3) measuring monosaccharide composition based on HPLC. However, such methods do not capture the high-dimensional structure of polysaccharides, which is closely related to their bioactivities. Growth profiles can provide valuable insights into the structural characteristics of polysaccharides.
In this study, it was observed that a polysaccharide can simultaneously up-regulate multiple PULs, and partial gene upregulation in other PULs was also noted, similar to B. dorei’s response to human milk oligosaccharides8. These phenomena are potentially due to the functional redundancy of PULs. For example, both PUL72 and PUL16 in B. thetaiotaomicron are involved in processing high-mannose N-glycans63,64. Knockout of six PULs up-regulated by pectin intervention revealed that none were absolutely necessary for complete degradation of the specific pectin preparation20. Moreover, B. thetaiotaomicron expresses multiple PULs to respond to host mucin O-glycans, ensuring its survival and metabolism even when some PULs are inactive; for instance, deletion of ECF-σ gene BT1053 or insertion in susC-like gene BT1042 did not significantly affect growth on neutral O-glycans65.
Notably, several B. uniformis strains, including B. uniformis DA183, contain two GH26 enzymes in their genomes, located in different PULs. These enzymes may have evolved to target different substrates or function under different conditions. For example, in B. ovatus ATCC8483, the PUL encodes two GH26 β-mannanases, BoMan26A and BoMan26B, with distinct activities: BoMan26A primarily forms mannobiose, while BoMan26B, which is surface-exposed, degrades galactomannan into a diverse mixture of oligosaccharides66. This functional redundancy and specialization ensure efficient degradation and utilization of complex galactomannans.
Future research should explore strain-levels variations in glycan utilization, as seen with B. ovatus using mucin67,68. With decreasing sequencing cost, synthetic communities (SynComs) are being considered for growth profile characterization due to their well-defined compositions and reproducibility69,70. As demonstrated in this study, differences in growth profiles and CAZyme distribution can reflect variations in polysaccharide structures. Therefore, novel growth profiles may aid in identifying polysaccharides with potential novel structures. Additionally, analyzing the proteins that degrade these polysaccharides provides valuable insights into their complex sugar chain architectures19. Advances in CAZyme functional prediction may improve our understanding of medicinal polysaccharide structures through PUL transcriptome analysis. Finally, identified PULs can serve as markers for the genetic engineering of gut bacteria (e.g. PULs for inulin utilization35), and tools for enhancing probiotic colonization (e.g. PULs for porphyrin polysaccharide utilization7).
Methods
Collection and preservation of human stool samples
In this study, all 119 human participants provided informed consent, with approval from the Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences (SIAT-IRB-200315-H0438). Freshly collected stool samples from healthy donors were promptly transferred to an anaerobic chamber (Vinyl Anaerobic Chambers, Coylab, USA). Each sample (3 g) was resuspended in 15 mL of 20% glycerol (v/v, in sterile phosphate-buffered saline, with 0.1% L-cysteine hydrochloride), vortexed for homogenization, and filtered through sterile nylon mesh to remove large particles. Aliquots of the processed suspensions were stored in sterile cryovials at −80°C for long-term preservation, ready for subsequent isolation of gut Bacteroides and Parabacteroides.
Isolation and cultivation of gut Bacteroides and Parabacteroides
Bacteroides uniformis ATCC 8492 and Bacteroides ovatus ATCC 8483 were purchased from ATCC. The 28 human gut Bacteroides and Parabacteroides used in this study were isolated from the feces samples of SIAT cohort. Fecal samples thawed from -80°C were diluted and spread onto modified Yeast Casitone Fatty Acids (mYCFA) agar plates (Supplementary Table 12). The plates were incubated anaerobically at 37°C for 2–3 days in an atmosphere of 85% N2, 5% CO2, and 10% H2. Single colonies were picked up, streaked onto fresh plates, and further incubated anaerobically at 37°C for another 2–3 days. This purification process was repeated once to obtain pure strains. Isolated strains were then transferred into liquid medium and incubated at 37°C for 2 days. We then amplified and sequenced the full-length 16S rRNA genes using the PCR primer pair (27 F 5’-AGAGTTTGATCMTGGCTCAG-3’; 1492 R 5’-GGTTACCTTGTTACGACTT-3’). Strains that produced double peaks in their sequences were either discarded or subjected to another round of purification. All purified strains were stored at −80°C in a glycerol suspension (20%, v/v) containing 0.1% cysteine. Taxonomic classification of isolates was determined by comparing their full-length 16S rRNA gene sequences to those present in the Genome Taxonomy Database (GTDB release 207)71 (Supplementary Table S1A). For culture purposes, the Bacteroides species were cultivated in brain heart infusion medium (BHIS) supplemented with 5 mg/L hemin, 0.5 mg/L vitamin K3, and 1 g/L L-cysteine (Supplementary Table 12). The cultivation was carried out in an anaerobic chamber with a gas composition of 85% N2, 10% H2, and 5% CO2, maintained at a temperature of 37 °C.
Minimal medium test
To determine a minimal medium suitable for the growth of Bacteroides species, we tested three minimal media: modified Bacteroides minimal medium (mBMM)72, modified Yeast Casitone Fatty Acids (mYCFA) medium73, and modified M9 minimal (mM9) medium74,75, all supplemented with 0.5% glucose (Supplementary Table 12). Using an anaerobic workstation with a gas mixture of 85% N2, 10% H2, and 5% CO2, the modified basic medium was dispensed into a 96-well plate. The bacterial cultures were washed twice with anaerobic PBS, and the optical density at 600 nm (OD600) of the bacterial solution was adjusted to 0.1. Growth curves indicated that both mBMM and mYCFA generally supported the growth of Bacteroides species (Supplementary Fig. 1). Compared to mYCFA, mBMM, being a synthetic medium with a clear composition, provided more stable measurements of polysaccharide utilization.
Prevalence and abundance of Bacteroides species
The abundance and prevalence of the strains chosen in our study was counted. We screened 16282 healthy human metagenome from publicly available metagenomic data by search with defined filter conditions (experiment_type = ‘Metagenomics’ AND QCStatus = ‘Good runs’ AND host age > 5 AND Country=is not null AND Recent Antibiotics Use = ‘No’ AND Phenotype = ‘Health’) in GMrepo to represented the global health human gut microbiota. Species with equally weighted average frequency of occurrence prevalence> 30% and mean relative abundance > 0.1% (n = 16282) was defined as core species.
Measurement of bacterial growth profiles
The mBMM medium was added to a 96-well plate in an anaerobic workstation (85% N2, 10% H2, and 5% CO2). The medium was supplemented with 0.5% ddH2O (negative control), 0.5% glucose (positive control), or 0.5% medicinal polysaccharides (treatment). Next, the bacteria were washed twice with anaerobic PBS, and the OD600 of the bacteria solution was adjusted to 0.1. The bacteria were then inoculated at a 1:10 ratio into the 96-well plate and cultured at 37 °C. The growth OD was measured and recorded every 12 h using a microplate reader.
Extraction of medicinal polysaccharides
Seventeen herbs were purchased from Bozhou city (Anhui, China) and authenticated by and identified by one of authors, Dr. Ji Yang. The voucher specimens were deposited at Faculty of Health Sciences (FHS), University of Macau. The more detailed information about herbs see Supplementary Table 3A. Among various extraction methods (e.g., hot water/ethanol, alkaline, enzymatic), hot water extraction is the most widely used due to its ease of operation, high solubility in hot water, and minimal damage to the polysaccharides15,26. Extraction of polysaccharides was performed by soaking the medicinal pieces in 10 times their volume of deionized water for 30 minutes, followed by boiling at high heat (500 W) and simmering at low heat (250 W) for 30 minutes. The hot liquid was then filtered through a 200-mesh cloth, discarding the residue. This process was repeated with another 10 times the volume of deionized water, boiling at high heat (500 W) and simmering at low heat (250 W) for 25 minutes, and filtering again while hot. Both filtrates were combined and reduced to a low temperature (60°C) using vacuum pressure. After cooling, three times the volume of anhydrous ethanol was added to precipitate the polysaccharides while constantly stirring, and the mixture was left to stand overnight at 4°C. The precipitate was separated from the liquid using a vacuum pump filter, washed three times with anhydrous ethanol, and vacuum-dried at 60°C to obtain the final product. For medicinal polysaccharides_2 (e.g., DPs_2), the extraction process was similar to that of medicinal polysaccharides_1 (e.g., DPs_1), with the additional step of dialysis to remove molecules smaller than 3.5 kDa.
Determination of total sugar content
Total sugar content was measured using the protocol76,77,78 as follows. Specifically, glucose (Mw 180 Da) obtained from Sigma (St. Louis, MO, USA) were used to establish the calibration curve. Glucose standard solution with a concentration of 0.1 g/L was prepared. For each polysaccharide sample, medicinal polysaccharides solution with a concentration of 0.6 g/L were prepared. The mixture was heated to facilitate dissolution and then was filtered using a 0.22 μm filter element until the solution became clear. 6% phenol solution was added to glucose standard and medicinal polysaccharides solution. The mixture was thoroughly shaken to ensure proper mixing and stand for 30 minutes. The spectrophotometer at a wavelength of 490 nm (using an LED light source and CMOS photosensitive element) was preheated. The preheating process is typically completed by the end of the reaction, and the meter reading becomes stable. The zero-point calibration was performed with phenol solution without glucose added, and the one-point calibration with a black baffle (Supplementary Table S3). Then, the absorption rate of each sample was measured three times. The standard curve was calculated using linear regression with the data of glucose standard sample measured above. The total sugar content was calculated by substituting the absorption rate of medicinal polysaccharides. In this experiment, with a sample concentration of 0.06 g/L, the sugar content of a sample can be calculated using the formula:
When the sugar concentration is x, the absorption rate is y, the standard curve is y=ax+b, and the sample concentration is c (0.06 g/L in this experiment).
Determination of molecular weight and distribution
Molecular weight and distribution were measured according to the method79,80,81,82 as follows. Specifically, dextran standards with different molecular weights (Mw 1 kDa, 5 kDa,25 kDa, 50 kDa, 80 kDa,670 kDa) obtained from Sigma (St. Louis, MO, USA) were used to establish the calibration curve. Based on the gel column chromatography principle, there is an exponential relationship between the molecular weight (Mw) and the retention time (t) of a compound. A mathematical model with the expression is established to represent this relationship:
In this study, the molecular weight distribution of polysaccharide samples was analyzed using an HPLC system with the following parameters: Mobile phase: NaCl solution (0.2 mol/L), Flow rate: 0.6 mL/min, Single sampling amount: 20 µL, Sample solution concentration: 5 g/L, Chromatographic column: TSK-gel G3000PWXL (7.8 × 300 mm) column, Column temperature: 40°C, Detector: Refractive index (RI) detector. The standard sugar samples with known molecular weights and medicinal polysaccharides samples were dissolved water with final concentration of 5 mg/mL, and filtered using a 0.22 µm filter element until they are clear. The retention time of each sample was recorded. The standard curve was calculated using exponential regression with the data from the standard samples. Substituting the peak retention time of medicinal polysaccharides samples, molecular weight was calculated.
Determination of monosaccharide composition
Monosaccharide composition was measured according to the method80,82,83,84, as detailed below. Specifically, the monosaccharide composition of polysaccharides samples was analyzed using HPLC with the following parameters: mobile phase: acetonitrile supplemented with 0.1 mol/L phosphate buffer solution, flow rate: 0.6 mL/min, single sampling amount: 20 µL, chromatographic column: Cosmosil 5C18-PAQ (4.6 × 250 mm), column temperature: 40 °C, detector: Ultraviolet (UV) detector. Each monosaccharide (Man, Glc, Gal, Rha, Fuc, Xyl, Ara, GalA, GlcA) obtained from Sigma (St. Louis, MO, USA) was mixed at a concentration of 10 mM. We prepared a series of monosaccharide standard solutions and calibrated the liquid chromatography system using these standards. The medicinal polysaccharides samples were completely hydrolyzed and performed pre-column derivatization with 1-phenyl-3-methyl-5-pyrazolinone. The retention time was compared with the standard monosaccharide samples to determine the types of monosaccharides present in the polysaccharide sample. Through measuring peak area ratio of each monosaccharide, the proportion of each monosaccharide in the polysaccharide sample was calculated.
Transcriptome sequencing and analysis
Bacteroides species were cultured in BHIS medium supplemented with 5 mg/L hemin, 0.5 mg/L vitamin K3, and 1 g/L cysteine in an anaerobic chamber (Coy; 85% N2, 10% H2, and 5% CO2) at 37 °C. The bacteria were washed three times with anaerobic PBS, and the optical density at 600 nm (OD600) of the bacterial solution was adjusted to 0.1. The bacteria were then inoculated at a 1:10 ratio into a 96-well plate and cultured at 37 °C. The OD600 was measured and recorded every 15 minutes using a microplate reader. After reaching the logarithmic growth phase (Supplementary Fig. 6A–C), the bacteria were collected by centrifugation at 5000 x g for 2 minutes. The collected bacteria in the logarithmic growth period were stored at -80°C before transcript sequencing.
Total RNA was extracted from the sample using the Trizol method, including DNase treatment to remove genomic DNA, and assessed for quality using the NanoDrop One (Thermo, A30221) and Agilent 4200 TapeStation (Agilent, G2991AA). Ribosomal RNA was removed using the Ribo-Zero rRNA Removal Kit (Epicenter, 15066012) designed for bacteria. For library construction, the NEBNext Ultra II Directional RNA Library Prep Kit (NEB, e7760) for Illumina was employed: mRNA was fragmented into short segments, single-stranded cDNA was synthesized using random hexamers, double-stranded cDNA was generated with dUTP substitution, purified using AMPure XP beads (Beckman, A63880), and the U-containing second strand was digested using USER enzyme (NEB, M5508). End repair, A-tailing, and adapter ligation were performed on the purified cDNA, followed by fragment size selection using AMPure XP beads. PCR amplification was conducted, and PCR products were purified with AMPure XP beads to obtain the final library. Libraries were inspected using the Agilent 4200 TapeStation, and only qualified libraries were sequenced on the Illumina platform (Illumina, HiSeq 2000). Key improvements included ensuring DNase treatment during RNA extraction, using a bacterial-specific rRNA removal kit, generating strand-specific libraries, verifying adapter compatibility with prokaryotic RNA, and adjusting fragment size selection for prokaryotic transcript lengths.
After obtaining the sequencing data for each sample, it is important to assess the quality of the data and remove any low-quality reads to ensure the reliability of subsequent analyzes. The software used for quality control is fastp85, and the following quality control criteria are applied: 1) Minimum base quality score of 15; 2) Sliding window size of 4 with an average base quality score greater than 20; 3) Minimum read length of 75 bp; 4) Default values for other parameters. The sequencing data, after quality control, is aligned to the ribosomal RNA sequences in the Rfam database using Bowtie286 to remove rRNA reads. The remaining reads are then aligned to the reference genome using Bowtie2, resulting in a BAM file containing the alignment results. The alignment results are further processed to generate read counts using RSEM87. For gene expression analysis, we utilize the read counts88 to identify differentially expressed genes among multiple sample groups.
Genomic annotation of Polysaccharide Utilization Locus (PUL)
PULs of Bacteroides were annotated by PULpy31. It is worth noting that different species of B. uniformis can have varying numbers and architectures of PULs, and these differences can also be observed between different annotated methods, such as PULpy31 and PUL prediction algorithm89 used by PULDB. The boundaries of the PUL were further recorrected based on gene expression differences. Specifically, by using a comparative transcriptomic analysis of Bacteroides spp. cultured in minimal medium supplemented with polysaccharides or glucose, the PULs with the highest upregulation was identified. Then, the transcriptomes of the upstream and downstream genes of this PULs were compared. Genes that were consistently upregulated within and outside the predicted PULs were defined as recorrected PULs.
Construction of B. uniformis knockout mutants
Due to the difficulty of genetic manipulation in wild Bacteroides strains, we performed PUL34_Bu knockout in the type strain B. uniformis ATCC8492 (Supplementary Fig. 8). Recipient B. uniformis ATCC8492 for conjugation were cultured in BHIS medium in an anaerobic chamber (Coy; 85% N2, 10% H2, and 5% CO2) at 37 °C. Donor Escherichia coli S17-1 (λpir) were cultured in Luria–Bertani (LB) medium at 37 °C. The DNA fragments (Supplementary Table 13A) of sgRNA or sgRNA-homologous arms (1 kb repair templates) were cloned into the plasmid backbone by NEBuilder HiFi DNA assembly master mix (NEB, E2621S). The primers (Supplementary Table 13B) used to amplify plasmid skeleton, sgRNA, repair template and mutant detection were synthetized by Sangon Biotech. The knock out plasmids conjugated into B. uniformis ATCC8492. And then the Bacteroides strains were diluted 1:100 in BHI medium with aTc induced for 24 h (aTc final concentration 100 ng/ml). Cultures were diluted by 100,101,102 and 103-fold and spread onto BHI-aTc agar plate and incubated at 37 °C for 40 h. Colonies were identified by PCR and sequencing.
Sequence and structure alignment
The phylogenetic tree of enzymes from the GH26 family was constructed with MUSLE90 and constructed a phylogenetic tree using the Maximum likelihood method91 for the 89 characterized GH26 protein sequences deposited in the CAZy database25. The GH26 protein 3D structure of our B. uniformis DA183 were predicted using AlphaFold242. The predicted local distance difference test value (pLDDT) was calculated using AlphaFold2. The catalytic ___domain of B. uniformis DA183 GH26 was aligned to its phylogenetic neighboring GH26 in B. ovatus ATCC848343 (GenBank accession ALJ47537.1; PDB code 6HF4) using US-align92 to compute the TM-score and RMSD.
Bacteroides GH26 enzyme expression and purification
Sequences coding for full-length wild-type, point mutant, and CBM-truncated Bacteroides GH26 proteins (Supplementary Table 14) were optimized for expression in E. coli, chemically synthesized (Tsingke Biotechnology Co., Ltd.), and cloned into the pET28a plasmid with a C-terminal 6× His-tag between the NdeI and XhoI sites. The plasmids were transformed into E. coli BL21(DE3). The signal peptide boundary was predicted using SignalP (version 6.0)93. The signal peptide was removed to ensure soluble expression. The CBM ___domain boundary was predicted using Chainsaw94. E. coli BL21(DE3) cells containing the pET28a plasmid were grown in 500 mL of LB medium with 50 μg/mL kanamycin at 37 °C, 200 rpm until OD600 ≈ 0.6. Expression was induced by adding 0.2 mM IPTG, and the culture was continued for 20 h at 16°C, 150 rpm. Cells were lysed in 50 mL of lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 7.4) using a high-pressure cell crusher. The lysate was centrifuged at 4°C, 10,000 × g for 1 h, and the supernatant was incubated with 5 mL of Ni-NTA agarose resin (Yeasen, 20502ES50) for 1 h at 4°C with head-over-tail rotation. The resin was poured into a gravity flow column and washed three times with 50 mL of wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 7.4). The protein was eluted with elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, pH 7.4).
Bacteroides GH26 enzyme activity assay
Konjac glucomannan, guar gum galactomannan, carob galactomannan, ivory nut mannan, barley glucan, tamarind xyloglucan was from Megazyme (Bray, Ireland). Corn cob xylan, 1,1-Kestotetraose, and 1-Kestose was from TCI. Galactosyl mannobiose was from ACMEC; Raffinose was from HUICH. Trehalose was from Huhui (Shanghai) Biotechnology Co., Ltd. The specific activity was measured using the standard DNS-reducing sugar assay43,95 using 1.25 mg/ml GH26 BuDA183 and GH26 BoATCC8483 and 5 mg/mL 13 substrates (Supplementary Table 3C) in 50 mM potassium phosphate buffer, pH 6.5. The incubation time was 30 min at 37 °C. The enzyme activity for point mutant and CBM truncated GH26 was measured in the same way as the standard DNS assay using 125 nM GH26 enzyme and 5 mg/mL six reactive substrates (Fig. 6A) in 50 mM potassium phosphate buffer, pH 6.5, at 10, 20, 30, 60, 120 min. The incubation time was 30 min at 37 °C. Mannose was used to obtain a concentration standard curve (Supplementary Table 15).
Enzyme Kinetics
Michaelis-Menten kinetics was measured for 125 nM full-length wild type, CBM truncated, and the variants A363K in triplicate using the DNS activity assay43,95 but varying time and substrate concentration. All reactions had a total volume of 50 μL and contained 1 mM CaCl2 and konjac glucomannan (1.25, 2.5, 3.75, 5, 6.25, 7.5, 8.75, and 10 mg/L). The reaction was stopped by heat to 100°C for 1 min. The obtained initial rates were used to generate Michaelis-Menten curves via nls96 function in R from which Km and Kcat values were estimated.
Molecular docking and visualizations
The oligosaccharide substrates were modeled using GLYCAM-Web website50 (Complex Carbohydrate Research Center, University of Georgia, Athens, GA (http://www.glycam.com)) or derived from the RCSB Protein Data Bank (PDB). Ligand docking was performed using AutoDock Vina (v1.2.0)49 employing standard parameters. The active-site of GH26 was derived from the Bo GH26 structure (PDB, 6HF4)43 by structural superposition with US-align92. The produced nine different ligand conformations for each docking complex were analyzed with CSM-carbohydrate, out of which the conformation with the lowest delta-G (binding energy) value was selected for further studying the ligand-receptor interactions by Discovery studio visualizer97.
Statistics & reproducibility
No statistical method was used to predetermine sample size. No data were excluded from the analyzes. Statistical significance for growth profile assays in Fig. 1E and Supplementary Fig. 3A was determined using a two-sided unpaired t-test between each experimental group and the negative control, with multiple comparisons adjusted using the Bonferroni method; each experiment was conducted in triplicate to ensure reliability. For comparative transcriptome analysis, differential expression was assessed using DESeq2 with a two-sided Wald test, and adjusted p-values were calculated using the Benjamini and Hochberg method to control the false discovery rate (FDR); genes with an adjusted p-value < 0.05 were considered statistically significant. Representative images from single experiments are shown for nucleic acid gel electrophoresis and protein SDS-PAGE, while data for in vitro enzyme activity characterization are presented as mean values ± SD, with assays performed in technical triplicates (n = 3) to ensure reliability.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All sequencing data generated in this study have been submitted to the ENA BioProject database (https://www.ebi.ac.uk/ena/browser/search) under accession number PRJEB75723. Other data are available in the Supplementary information and Supplementary Data. Source data are provided with this paper.
Change history
27 January 2025
In the version of this article initially published, Supplementary Tables 1–19 were not present and are now available via the HTML version of the article.
References
Garrett, W. S. The gut microbiota and colon cancer. Science 364, 1133–1135 (2019).
Sun, L. et al. Bile salt hydrolase in non-enterotoxigenic Bacteroides potentiates colorectal cancer. Nat. Commun. 14, 755 (2023).
Huang, J. et al. Ginseng polysaccharides alter the gut microbiota and kynurenine/tryptophan ratio, potentiating the antitumour effect of antiprogrammed cell death 1/programmed cell death ligand 1 (anti-PD-1/PD-L1) immunotherapy. Gut 71, 734–745 (2022).
Tanoue, T. et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 565, 600–605 (2019).
Zafar, H. & Saier, M. H. Jr Gut Bacteroides species in health and disease. Gut Microbes 13, 1848158 (2021).
Beller, Z. W. et al. Inducible CRISPR-targeted “knockdown” of human gut Bacteroides in gnotobiotic mice discloses glycan utilization strategies. Proc. Natl Acad. Sci. 120, e2311422120 (2023).
Hehemann, J.-H. et al. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature 464, 908–912 (2010).
Kijner, S., Ennis, D., Shmorak, S., Florentin, A. & Yassour, M. CRISPR-Cas-based identification of a sialylated human milk oligosaccharides utilization cluster in the infant gut commensal Bacteroides dorei. Nat. Commun. 15, 105 (2024).
Panwar, D., Shubhashini, A. & Kapoor, M. Complex alpha and beta mannan foraging by the human gut bacteria. Biotechnology Advances, 108166 (2023).
Buzun, E. et al. A bacterial sialidase mediates early-life colonization by a pioneering gut commensal. Cell Host Microbe 32, 181–190. e189 (2024).
Kou, F. et al. A review of Ganoderma lucidum polysaccharides: Health benefit, structure–activity relationship, modification, and nanoparticle encapsulation. International journal of biological macromolecules, 125199 (2023).
Lin, T.-L. et al. Role of gut microbiota in identification of novel TCM-derived active metabolites. Protein Cell 12, 394–410 (2021).
Yu, Y., Shen, M., Song, Q. & Xie, J. Biological activities and pharmaceutical applications of polysaccharide from natural resources: A review. Carbohydr. Polym. 183, 91–101 (2018).
Shan-Shan, S., Kai, W., Ke, M., Li, B. & Hong-Wei, L. An insoluble polysaccharide from the sclerotium of Poria cocos improves hyperglycemia, hyperlipidemia and hepatic steatosis in ob/ob mice via modulation of gut microbiota. Chin. J. Nat. Med. 17, 3–14 (2019).
Shi, L. Bioactivities, isolation and purification methods of polysaccharides from natural products: A review. Int. J. Biol. Macromolecules 92, 37–48 (2016).
Wu, T.-R. et al. Gut commensal Parabacteroides goldsteinii plays a predominant role in the anti-obesity effects of polysaccharides isolated from Hirsutella sinensis. Gut 68, 248–262 (2019).
Zhang, Y. et al. Identification of the core active structure of a Dendrobium officinale polysaccharide and its protective effect against dextran sulfate sodium-induced colitis via alleviating gut microbiota dysbiosis. Food Res. Int. 137, 109641 (2020).
Martens, E. C., Chiang, H. C. & Gordon, J. I. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell host microbe 4, 447–457 (2008).
Lapébie, P., Lombard, V., Drula, E., Terrapon, N. & Henrissat, B. Bacteroidetes use thousands of enzyme combinations to break down glycans. Nat. Commun. 10, 2043 (2019).
Martens, E. C. et al. Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLOS Biol. 9, e1001221 (2011).
Foley, M. H., Cockburn, D. W. & Koropatkin, N. M. The Sus operon: a model system for starch uptake by the human gut Bacteroidetes. Cell. Mol. Life Sci. 73, 2603–2617 (2016).
Luis, A. S. et al. A single sulfatase is required to access colonic mucin by a gut bacterium. Nature 598, 332–337 (2021).
Wu, S. et al. GMrepo: a database of curated and consistently annotated human gut metagenomes. Nucleic acids Res. 48, D545–D553 (2020).
Liu, C. et al. Enlightening the taxonomy darkness of human gut microbiomes with a cultured biobank. Microbiome 9, 119 (2021).
Drula, E. et al. The carbohydrate-active enzyme database: functions and literature. Nucleic acids Res. 50, D571–D577 (2022).
Qi, H. et al. Comparisons of isolation methods, structural features, and bioactivities of the polysaccharides from three common Panax species: A review of recent progress. Molecules 26, 4997 (2021).
Kuang, M.-T. et al. Structural characterization and hypoglycemic effect via stimulating glucagon-like peptide-1 secretion of two polysaccharides from Dendrobium officinale. Carbohydr. Polym. 241, 116326 (2020).
Yin, J.-Y. et al. Separation, structure characterization, conformation and immunomodulating effect of a hyperbranched heteroglycan from Radix Astragali. Carbohydr. Polym. 87, 667–675 (2012).
Wardman, J. F., Bains, R. K., Rahfeld, P. & Withers, S. G. Carbohydrate-active enzymes (CAZymes) in the gut microbiome. Nat. Rev. Microbiol. 20, 542–556 (2022).
Yin, Y. et al. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic acids Res. 40, W445–W451 (2012).
Stewart, R. D., Auffret, M. D., Roehe, R. & Watson, M. Open prediction of polysaccharide utilisation loci (PUL) in 5414 public Bacteroidetes genomes using PULpy. bioRxiv, 421024, https://doi.org/10.1101/421024 (2018).
Guo, M., Shao, S., Wang, D., Zhao, D. & Wang, M. Recent progress in polysaccharides from Panax ginseng CA Meyer. Food Funct. 12, 494–518 (2021).
Li, J. et al. Purification, structural characterization, and immunomodulatory activity of the polysaccharides from Ganoderma lucidum. Int. J. Biol. macromolecules 143, 806–813 (2020).
Zheng, L. et al. CRISPR/Cas-based genome editing for human gut commensal bacteroides species. ACS Synth. Biol. 11, 464–472 (2022).
García-Bayona, L. & Comstock, L. E. Streamlined genetic manipulation of diverse bacteroides and parabacteroides isolates from the human gut microbiota. MBio 10, 01762–01719 (2019).
Almeida, A. et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol. 39, 105–114 (2021).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. methods 18, 366–368 (2021).
Zhou, N. et al. Konjac glucomannan: a review of structure, physicochemical properties, and wound dressing applications. J. Appl. Polym. Sci. 139, 51780 (2022).
Mudgil, D., Barak, S. & Khatkar, B. S. Guar gum: processing, properties and food applications—a review. J. food Sci. Technol. 51, 409–418 (2014).
Lazaridou, A., Biliaderis, C. G. & Izydorczyk, M. S. Structural characteristics and rheological properties of locust bean galactomannans: a comparison of samples from different carob tree populations. J. Sci. Food Agriculture 81, 68–75 (2001).
Ghysels, S. et al. Fast pyrolysis of mannan-rich ivory nut (Phytelephas aequatorialis) to valuable biorefinery products. Chem. Eng. J. 373, 446–457 (2019).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Bågenholm, V. et al. A surface-exposed GH26 β-mannanase from Bacteroides ovatus: Structure, role, and phylogenetic analysis of BoMan26B. J. Biol. Chem. 294, 9100–9117 (2019).
Okano, H., Kanaya, E., Ozaki, M., Angkawidjaja, C. & Kanaya, S. Structure, activity, and stability of metagenome‐derived glycoside hydrolase family 9 endoglucanase with an N‐terminal Ig‐like ___domain. Protein Sci. 24, 408–419 (2015).
Liberato, M. V. et al. Molecular characterization of a family 5 glycoside hydrolase suggests an induced-fit enzymatic mechanism. Sci. Rep. 6, 23473 (2016).
Murray, P. A., Kern, D. G. & Winkler, J. R. Identification of a galactose-binding lectin on Fusobacterium nucleatum FN−2. Infect. Immun. 56, 1314–1319 (1988).
Kuhaudomlarp, S., Gillon, E., Varrot, A. & Imberty, A. LecA (PA-IL): A galactose-binding lectin from Pseudomonas aeruginosa. Lectin Purification and Analysis: Methods and Protocols, 257-266 (2020).
Bolam, D. N. et al. Mannanase A from Pseudomonas fluorescens ssp. cellulosa is a retaining glycosyl hydrolase in which E212 and E320 are the putative catalytic residues. Biochemistry 35, 16195–16204 (1996).
Eberhardt, J., Santos-Martins, D., Tillack, A. F. & Forli, S. AutoDock Vina 1.2. 0: New docking methods, expanded force field, and python bindings. J. Chem. Inf. modeling 61, 3891–3898 (2021).
Nguyen, T. B., Pires, D. E. & Ascher, D. B. CSM-carbohydrate: protein-carbohydrate binding affinity prediction and docking scoring function. Brief. Bioinforma. 23, bbab512 (2022).
dos Santos, C. R. et al. Molecular insights into substrate specificity and thermal stability of a bacterial GH5-CBM27 endo-1, 4-β-D-mannanase. J. Struct. Biol. 177, 469–476 (2012).
Roske, Y., Sunna, A., Pfeil, W. & Heinemann, U. High-resolution crystal structures of Caldicellulosiruptor strain Rt8B. 4 carbohydrate-binding module CBM27-1 and its complex with mannohexaose. J. Mol. Biol. 340, 543–554 (2004).
Zhao, L. et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 359, 1151–1156 (2018).
O’Keefe, S. J. et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nat. Commun. 6, 1–14 (2015).
Sorbara, M. T. & Pamer, E. G. Microbiome-based therapeutics. Nat. Rev. Microbiol. 20, 365–380 (2022).
Gauffin Cano, P., Santacruz, A., Moya, Á. & Sanz, Y. Bacteroides uniformis CECT 7771 ameliorates metabolic and immunological dysfunction in mice with high-fat-diet induced obesity. (2012).
López-Almela, I. et al. Bacteroides uniformis combined with fiber amplifies metabolic and immune benefits in obese mice. Gut Microbes 13, 1–20 (2021).
Morita, H. et al. Bacteroides uniformis and its preferred substrate, α-cyclodextrin, enhance endurance exercise performance in mice and human males. Sci. Adv. 9, eadd2120 (2023).
Yan, Y. et al. Bacteroides uniformis-induced perturbations in colonic microbiota and bile acid levels inhibit TH17 differentiation and ameliorate colitis developments. npj Biofilms Microbiomes 9, 56 (2023).
Dai, W. et al. Discovery of Bacteroides uniformis F18-22 as a Safe and Novel Probiotic Bacterium for the Treatment of Ulcerative Colitis from the Healthy Human Colon. Int. J. Mol. Sci. 24, 14669 (2023).
Nie, Q. et al. Gut symbionts alleviate MASH through a secondary bile acid biosynthetic pathway. Cell 187, 2717–2734. e2733 (2024).
Zeng, P., Li, J., Chen, Y. & Zhang, L. The structures and biological functions of polysaccharides from traditional Chinese herbs. Prog. Mol. Biol. Transl. Sci. 163, 423–444 (2019).
Cuskin, F. et al. Human gut Bacteroidetes can utilize yeast mannan through a selfish mechanism. Nature 517, 165–169 (2015).
Sastre, D. E. et al. Human gut microbes express functionally distinct endoglycosidases to metabolize the same N-glycan substrate. Nat. Commun. 15, 5123 (2024).
Martens, E. C., Roth, R., Heuser, J. E. & Gordon, J. I. Coordinate regulation of glycan degradation and polysaccharide capsule biosynthesis by a prominent human gut symbiont. J. Biol. Chem. 284, 18445–18457 (2009).
Bågenholm, V. et al. Galactomannan catabolism conferred by a polysaccharide utilization locus of Bacteroides ovatus: enzyme synergy and crystal structure of a β-mannanase. J. Biol. Chem. 292, 229–243 (2017).
Patnode, M. L. et al. Strain-level functional variation in the human gut microbiota based on bacterial binding to artificial food particles. Cell host microbe 29, 664–673. e665 (2021).
Odamaki, T. et al. Comparative genomics revealed genetic diversity and species/strain-level differences in carbohydrate metabolism of three probiotic bifidobacterial species. Int. J. Genomics 2015, 567809 (2015).
van Leeuwen, P. T., Brul, S., Zhang, J. & Wortel, M. T. Synthetic microbial communities (SynComs) of the human gut: design, assembly, and applications. FEMS Microbiol Rev 47, https://doi.org/10.1093/femsre/fuad012 (2023).
Cheng, A. G. et al. Design, construction, and in vivo augmentation of a complex gut microbiome. Cell 185, 3617–3636.e3619 (2022).
Parks, D. H. et al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic acids Res. 50, D785–D794 (2022).
Bacic, M. K. & Smith, C. J. Laboratory maintenance and cultivation of bacteroides species. Curr. Protoc. Microbiol. https://doi.org/10.1002/9780471729259.mc13c01s9 (2008).
Liu, W., Zhu, W. Y., Yao, W. & Mao, S. Y. Isolation and identification of a lactate-utilizing, butyrate-producing bacterium and its primary metabolic characteristics. Wei Sheng Wu Xue Bao 47, 435–440 (2007).
Fischer, E. & Sauer, U. Metabolic flux profiling of Escherichia coli mutants in central carbon metabolism using GC-MS. Eur. J. Biochem 270, 880–891 (2003).
Portnoy, V. A., Herrgård, M. J. & Palsson, B. Aerobic fermentation of D-glucose by an evolved cytochrome oxidase-deficient Escherichia coli strain. Appl Environ. Microbiol 74, 7561–7569 (2008).
DuBois, M., Gilles, K. A., Hamilton, J. K. & Rebers, P. t. & Smith, F. Colorimetric method for determination of sugars and related substances. Anal. Chem. 28, 350–356 (1956).
Dubois, M., Gilles, K., Hamilton, J., Rebers, P. & Smith, F. A colorimetric method for the determination of sugars. Nature 168, 167–167 (1951).
Sun, L. et al. Structural characterization of rhamnogalacturonan domains from Panax ginseng CA Meyer. Carbohydr. Polym. 203, 119–127 (2019).
Vidal, S., Williams, P., O’neill, M. & Pellerin, P. Polysaccharides from grape berry cell walls. Part I: tissue distribution and structural characterization of the pectic polysaccharides. Carbohydr. Polym. 45, 315–323 (2001).
Zhang, X. et al. Total fractionation and characterization of the water-soluble polysaccharides isolated from Panax ginseng CA Meyer. Carbohydr. Polym. 77, 544–552 (2009).
Kazemi, M., Khodaiyan, F., Labbafi, M., Hosseini, S. S. & Hojjati, M. Pistachio green hull pectin: Optimization of microwave-assisted extraction and evaluation of its physicochemical, structural and functional properties. Food Chem. 271, 663–672 (2019).
Feng, Z. et al. The model polysaccharide potato galactan is actually a mixture of different polysaccharides. Carbohydr. Polym. 313, 120889 (2023).
Yang, X., Zhao, Y., Wang, Q., Wang, H. & Mei, Q. Analysis of the monosaccharide components in Angelica polysaccharides by high performance liquid chromatography. Anal. Sci. 21, 1177–1180 (2005).
Liu, J. et al. Multi-level fingerprinting and cardiomyocyte protection evaluation for comparing polysaccharides from six Panax herbal medicines. Carbohydr. Polym. 277, 118867 (2022).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. methods 9, 357–359 (2012).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinforma. 12, 1–16 (2011).
Dillies, M.-A. et al. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief. Bioinforma. 14, 671–683 (2013).
Terrapon, N., Lombard, V., Gilbert, H. J. & Henrissat, B. Automatic prediction of polysaccharide utilization loci in Bacteroidetes species. Bioinformatics 31, 647–655 (2015).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids Res. 32, 1792–1797 (2004).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evolution 24, 1586–1591 (2007).
Zhang, C., Shine, M., Pyle, A. M. & Zhang, Y. US-align: universal structure alignments of proteins, nucleic acids, and macromolecular complexes. Nat. methods 19, 1109–1115 (2022).
Teufel, F. et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 40, 1023–1025 (2022).
Wells, J. et al. Chainsaw: protein ___domain segmentation with fully convolutional neural networks. Bioinformatics 40, btae296 (2024).
Miller, G. L. Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal. Chem. 31, 426–428 (1959).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing (Vienna, Austria, 2018). https://www.R-project.org/.
Visualizer, D. Discovery studio visualizer. 2. Accelrys Software Inc. (2005).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinforma. 10, 1–9 (2009).
Acknowledgements
We would like to thank Thanh Binh Nguyen from The University of Queensland for his assistance with the calculation of delta-G (binding energy) value and scoring for the dominant docking complex using the CSM-carbohydrate tool. This work was funded by regular grants (File no. 0063/2022/A2, 0018/2024/RIB1 and 0137/2024/AMJ) supported by the Macau Science and Technology Development Fund and the Ministry of Science and Technology of the People’s Republic of China. This work was also supported by the 2020 Young Qihuang Scholar funded by the National Administration of Traditional Chinese Medicine and the FDCT Funding Scheme for Postdoctoral Researchers of Higher Education Institutions (0017/2021/APD). This work is also financially supported by the Start-up Research Grant of University of Macau (SRG2022-00020-FHS), the 2023 State Key Laboratory of Quality Research in Chinese Medicine (UM) Internal Research Grant (IRG) (SKL-QRCM-IRG2023-001 & 005/2023/SKL), and the FSCPO project (type 1 and type 2) from Faculty of Health Sciences, University of Macau. This work received support from Shenzhen Key Laboratory for the Intelligent Microbial Manufacturing of Medicines (ZDSYS20210623091810032). This work is also funded by the National Natural Science Foundation of China under grant number 32201313.
Author information
Authors and Affiliations
Contributions
Z.Q., H.L., and J.Y. performed experiments and analyzed data with assistance from L.Z., J.H., Z.W., C.X., W.Z., and X.X.Z.Q. wrote the paper. E.L.L., L.D., and H.Z. conceived and guided the projects. L.S., Y.Z., Y.X., and J.L. provided herb resources and discussed results and approved the paper. Y.-Z.Z., L.Y., and L.L. gave comment to the data analysis and manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Diego Sastre, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Qu, Z., Liu, H., Yang, J. et al. Selective utilization of medicinal polysaccharides by human gut Bacteroides and Parabacteroides species. Nat Commun 16, 638 (2025). https://doi.org/10.1038/s41467-025-55845-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-55845-7
This article is cited by
-
Metagenomic analysis revealing links between age, gut microbiota and bone loss in Chinese adults
npj Metabolic Health and Disease (2025)
