
Abstract
DOPA Decarboxylase (DDC) has been proposed as a cerebrospinal fluid (CSF) biomarker with increased concentrations in Lewy body disorders (LBDs) and highest levels in patients receiving dopaminergic treatment. Here we evaluate plasma DDC, measured by proximity extension assay, and the effect of dopaminergic treatment in three independent LBD (with a focus on dementia with Lewy bodies (DLB) and Parkinson’s disease (PD)) cohorts: an autopsy-confirmed cohort (n = 71), a large multicenter, cross-dementia cohort (n = 1498) and a longitudinal cohort with detailed treatment information (n = 66, median follow-up time[IQR] = 4[4, 4] years). Plasma DDC was not altered between different LBDs and other disease groups or controls in absence of treatment. DDC levels increased over time in PD, being significantly associated to higher dosages of dopaminergic treatment. This emphasizes the need to consider treatment effect when analyzing plasma DDC, and suggests that plasma DDC, in contrast to CSF DDC, is of limited use as a diagnostic biomarker for LBD, but could be valuable for treatment monitoring.
Similar content being viewed by others

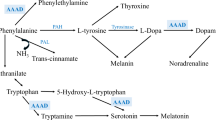

Introduction
Lewy body diseases (LBDs) are common causes for cognitive and/or physical impairment in the elderly. To date, the diagnostic process for different LBDs, such as dementia with Lewy bodies (DLB) and Parkinson’s Disease (PD), is challenging due to the large overlap in clinical and pathological characteristics with other neurodegenerative diseases such as Alzheimer’s disease (AD)1. While the seed aggregation assay has been shown valuable in detecting α-synuclein pathology in LBDs, it fails to capture other prominent disease processes such as dopaminergic impairment2. Recent studies have proposed the enzyme DOPA decarboxylase (DDC, also known as Aromatic-L-amino acid decarboxylase (AADC)) as a promising cerebrospinal fluid (CSF) biomarker for LBDs3,4,5,6,7. DDC is the last enzyme in the dopaminergic pathway, converting the substrate L-DOPA (also known as levodopa) into dopamine upon binding of the coenzyme pyridoxal phosphate. Several studies measuring DDC levels by proximity extension assay (PEA), have shown increased CSF levels of DDC in different LBDs compared with other neurodegenerative diseases and healthy controls (HC), with high diagnostic accuracy (areas under curve of up to 0.913,4,5,6). Findings on DDC levels in plasma, however, are inconsistent between studies. While one study reports increased levels of plasma DDC in LBDs compared with controls5, other studies show no difference between diagnostic groups at baseline, but report an increase in DDC levels over time already at prodromal disease stages6,7. These inconsistent findings highlight the need for further in-depth studies on peripheral DDC levels to evaluate the clinical value of plasma DDC as a potential biomarker for LBDs.
Common treatments to alleviate motor symptoms in PD target the dopaminergic pathway. Such treatments are administered orally and include e.g., L-DOPA in combination with peripheral DDC inhibitors to allow sufficient L-DOPA to cross the blood-brain barrier and reduce systemic effects, but also dopamine agonists, catechol-O-methyltransferase (COMT)-inhibitors and monoamine oxidase type-B (MAO-B) inhibitors8. Dopaminergic treatment is administered less frequently and usually at lower dosages in DLB because parkinsonism is less prominent and patients are prone to side effects9. In CSF, higher levels of DDC were detected in patients with dopaminergic medication, indicating an influence of dopaminergic treatment on CSF DDC levels3,4,5. As dopaminergic medications are administered orally, they could have an even stronger and more immediate effect on plasma than on CSF DDC. Two recent studies have shown that also plasma DDC levels are positively associated with dopaminergic treatment6,7, but further study of peripheral DDC as a biomarker for LBDs is needed. We hypothesized, that plasma DDC is not suitable as a diagnostic biomarker for LBDs due to the close association with dopaminergic treatment. We therefore investigated levels of plasma DDC across LBDs and related neurodegenerative disorders in three independent cross-sectional cohorts: one cohort with autopsy confirmation, one cross-dementia cohort, and one longitudinal cohort. Additionally, we examined the potential association of plasma DDC with dopaminergic treatment in the longitudinal cohort that had been followed for a median of 4 years.
Results
Cohort characteristics
Baseline characteristics of all cohorts are summarized in Table 1. In all cohorts, except the PPMI Cohort, participants from the disease groups were older than controls and overall participants of the Vallecas/VARS Cohort (median age = 81.0 years) were older than participants of the bPRIDE (median age = 71.0 years) and PPMI Cohorts (median age = 63.8 years). DLB and FTD groups tended towards having more men than women. Disease severity scores were worse in disease groups compared with controls and showed dose-dependent changes in the pre-disease and MCI stages.
DDC levels at baseline
Plasma DDC levels at baseline were not significantly different between diagnostic groups upon correction for confounding effects of age and sex (and center in the multicenter bPRIDE Cohort) in any of the three cohorts (Vallecas/VARS Cohort: p = 0.128; bPRIDE Cohort: p = 0.131; PPMI Cohort: p = 0.233; Fig. 1; uncorrected values are presented in Supplementary Fig. 1). We observed a positive association of plasma DDC with age in the PPMI Cohort (β (SE) = 0.021 (0.008), p = 0.007), but not in the Vallecas/VARS and bPRIDE Cohorts (Vallecas/VARS: β (SE) = −0.014 (0.014), p = 0.318; bPRIDE: β (SE) = −0.005 (0.003), p = 0.058). In the Vallecas/VARS Cohort higher levels of DDC were observed in male participants (β (SE) = 0.285 (0.139), p = 0.045) and in the bPRIDE Cohort, lower levels of DDC were observed in male participants (β (SE) = −0.116 (0.033), p = 0.0005).

Baseline plasma DDC levels across diagnostic groups are illustrated in the Vallecas/VARS Cohort (n HC (blue) = 59, n DLB (red) = 12 (a), multicenter bPRIDE Cohort (n Control (pale blue) = 374, n sMCI (light blue) = 179, n MCI-LB (pale red) = 25, n DLB (red) = 171, n pre-AD (pale yellow) = 187, n MCI-AD (light yellow) = 164, n AD (yellow) = 182, n MCI-FTD (light purple) = 46, n FTD (purple) = 170 (b), and PPMI Cohort (n HC (blue) = 33, n PD (orange) = 33 (c). DDC was measured in singlicate by PEA. Baseline plasma DDC NPX values are reported on a log2 scale and did not differ across diagnostic groups in any of the cohorts. Plasma DDC differences were assessed across groups by GLMs. Boxplots represent DDC levels across different diagnostic groups. Lines through the boxes represent the median value and lower and upper lines correspond to the first and third quartiles. Dots represent individual data points and whiskers extend to 1.5 times of the interquartile range. Source data are provided as a Source Data file. ADD Alzheimer’s disease dementia, DDC DOPA decarboxylase, DLB dementia with Lewy bodies, FTD frontotemporal dementia, HC healthy controls, LB Lewy bodies, MCI mild cognitive impairment, NPX normalized protein expression, PEA proximity extension assay, PD Parkinson’s disease, pre-AD Alzheimer’s disease at the pre-dementia stage, sMCI stable mild cognitive impairment.
In contrast, DDC levels in CSF differed significantly between groups within the subset of patients of whom plasma samples were also available (Supplementary Fig. 2). In the PRIDE Cohort, significantly lower levels of CSF DDC were observed in controls (β (SE) = −1.103 (0.180), p < 0.0001), AD (β (SE) = −0.569 (0.168), p = 0.001), and FTD (β (SE) = −0.650 (0.268), p = 0.018) compared with DLB. In the PPMI Cohort, CSF DDC levels were significantly lower in HC compared with PD (β (SE) = −0.340 (0.165), p = 0.044). Within the same subset of samples from the bPRIDE Cohort, plasma DDC levels were significantly lower in controls compared to those with DLB. However, this effect was much less pronounced than in CSF (β (SE) = −0.480 (0.217), p = 0.031). In the PPMI subcohort, plasma DDC levels were not discriminative between PD and HC (β (SE) = −0.276 (0.172), p = 0.114). In the PRIDE Cohort, CSF DDC was able to differentiate DLB from controls with an area under the curve (AUC) of 0.909 (specificity = 0.90, sensitivity = 0.85; Supplementary Fig. 3a) and from the non-DLB dementias with an AUC of 0.789 (specificity = 0.76, sensitivity = 0.77; Supplementary Fig. 3b).
Plasma DDC levels over time
Longitudinal measurements of DDC were available in the PPMI Cohort over a median follow-up time of 4 years [IQR: 4, 4]. At baseline, none of the participants in the PD group received dopaminergic treatment, but during follow-up, 31 of the patients with PD (93.9%) had started dopaminergic treatment. Plasma DDC levels increased over time in the PD group, but not in HC (PD: β (95% CI) = 0.342 (0.261–0.422), p < 0.0001; HC: β (95% CI) = 0.005 (−0.071 to 0.080), p = 0.906; Fig. 2). This effect got weaker, but remained significant upon correction for LEDD, and lost significance when only DDC measurements were included at timepoints during which no dopaminergic treatment was used, which could, however, be due to a lower number of observations (longitudinal increase corrected for use of dopaminergic treatment) (n observations = 196: PD: β (95%CI) = 0.170 (0.088–0.254), p = 0.0001; HC: β (95%CI) = 0.005 (−0.060 to 0.069), p = 0.886; no dopaminergic treatment (n observations = 146): PD: β (95%CI) = 0.152 (−0.002 to 0.316), p = 0.061; HC: β (95%CI) = 0.008 (−0.057 to 0.072), p = 0.820; Supplementary Tables 1 and 2, and Supplementary Figs. 4 and 5, respectively).

DDC NPX values over time by diagnosis in the PPMI Cohort (n individuals = 66, n samples = 196). Linear mixed-effects models, corrected for the effect of age and sex, were built to analyze the association of plasma DDC NPX values with diagnosis over time. The dark lines represent the estimated marginal model of plasma DDC NPX values over up to 8 years of follow-up (HC: blue, PD: orange). The transparent areas show 95% confidence intervals around the mean estimated values. Data points show raw longitudinal DDC measurements (HC: square, PD: dot) and light lines connect measurements within individuals. Source data are provided as a Source Data file. DDC DOPA decarboxylase, HC healthy controls, NPX normalized protein expression, PD Parkinson’s disease.
Association with dopaminergic treatment and disease severity
To analyze if longitudinal increases in plasma DDC concentrations are associated with dopaminergic treatment rather than disease severity, we studied the effects of LEDD on DDC, alone and next to different disease severity measures, namely UPDRS3 total score, HY total score, and MoCA. At follow-up visits, a subset of patients had multiple disease severity scores reported for the same visit, evaluated during either a state of medication ON-, or OFF-effect. For 21 patients, different UPDRS3 scores were reported (n different UPDRS3 scores during same visit = 32) and for 3 patients different HY scores were reported for 3 patients (n different HY scores during same visit = 3), but MoCA scores did not differ between ON and OFF. We first included only those scores that were evaluated during an ON state. LEDD levels were strongly associated with plasma DDC levels, which were not affected by disease severity measures (Fig. 3, Table 2). These effects remained stable when included disease severity measures were evaluated during OFF states (Supplementary Table 3). Plasma DDC was not associated with MoCA scores (β (95%CI) = 0.207 (−0.034 to 0.427), p = 0.081), however, plasma DDC levels were significantly associated with higher HY scores during both, the ON and OFF states (ON: β (95%CI) = 0.198 (0.008–0.385), p = 0.044; OFF: β (95%CI) = 0.231 (0.014–0.434), p = 0.033; Supplementary Table 4). During OFF states, higher plasma DDC levels were associated with higher UPDRS3 total scores, but not during ON states (OFF: β (95%CI) = 0.208 (0.014–0.393), p = 0.034; ON: β (95%CI)= −0.036 (−0.179 to 0.107), p = 0.627; Supplementary Table 4).

Associations of plasma DDC NPX values with LEDD alone (pink, n individuals = 33, n samples = 96 (a) and in combination with the disease severity scores UPDRS3 (purple, n individuals = 33, n samples = 96 (b), HY (turquoise, n individuals = 33, n samples = 96 (c) and MoCA (green, n individuals = 30, n samples = 50 (d) in the patients with PD of the PPMI Cohort. Association of DDC NPX values and LEDD and disease severity measures were analyzed in the PD group by mixed-effects models, with random effect for each individual and corrected for age and sex and different disease severity scores in (b–d). Dots represent raw DDC measurements, lines represent predicted values based on mixed effects models and transparent areas represent 95% confidence intervals. Source data are provided as a Source Data file. DDC DOPA decarboxylase, HY Hoehn and Yahr scale, LEDD levodopa equivalent daily dosage, MoCA Montreal Cognitive Assessment, NPX normalized protein expression, PD Parkinson’s disease, PPMI Parkinson’s disease Progression Marker Initiative, R2c conditional R squared, R2m marginal R squared, UPDRS3 unified Parkinson’s disease rating scale part 3.
Association of plasma DDC with CSF DDC
We observed a weak association between paired plasma and CSF DDC measurements in the (b)PRIDE (β (SE) = 0.370 (0.105), R2 = 0.146, p = 0.0007) and PPMI Cohorts at baseline (β (SE) = 0.0.342 (0.123), R2 = 0.121, p = 0.007). When also follow-up measurements were included in the PPMI Cohort, i.e., also patients that received dopaminergic treatment, the association between plasma and CSF DDC became stronger (β (SE) = 0.622 (0.067), R2 = 0.336, p < 0.0001). However, upon correction for dopaminergic treatment, this effect got weaker (β (SE) = 0.451 (0.062), R2 = 0.503, p < 0.0001) Fig. 4, suggesting an influence of dopaminergic treatment on the DDC levels.

Correlations between plasma and CSF DDC measurements were analyzed by linear regression in a subset of the bPRIDE Cohort with the preceding PRIDE study (n individuals = 75 (a), in the PPMI Cohort at baseline (n individuals = 58(b), and the PPMI Cohort including follow-up visits (n individuals = 58, n samples = 174 (c). The black dots represent individual measurements, the blue lines represent predicted values based on regression analysis and transparent areas represent 95% confidence intervals. Source data are provided as a Source Data file. CSF cerebrospinal fluid, DDC DOPA decarboxylase, NPX normalized protein expression.
Discussion
In this study, we show that baseline plasma DDC levels did not differ between LBDs (including DLB and PD) and controls in three independent cohorts, in contrast to findings in CSF. Over time, DDC levels increased in PD compared with HC in the longitudinal PPMI Cohort, which appeared associated with dopaminergic medication. This indicates that plasma DDC could be a sensitive biological response marker for dopaminergic treatment.
We and others have previously shown increased CSF levels of the enzyme DDC in different LBDs and proposed DDC as a CSF dopaminergic biomarker candidate3,4,5,6,7. Contrary to previously published findings in plasma by Pereira et al.5, but in concordance with Rutledge et al.6 and Appleton et al.7, we report no significant differences in plasma DDC between PD and controls at baseline, nor between DLB and controls or other neurodegenerative diseases even at the advanced disease stage, as represented by the autopsy-confirmed VARS Cohort. However, over time we observed that DDC increased in PD during follow-up in the PPMI Cohort, while it remained stable in the control group. Of note, no participant in the PPMI Cohort received dopaminergic treatment at baseline, but almost all participants in the PD group (n = 31 (93.9%)) started dopaminergic treatment during the time of follow-up. We found that the observed increase was likely driven by dopaminergic treatment. In line with previous findings, we found that in the CSF subset, DDC was significantly increased in DLB compared to the other groups and reached AUCs of up to 0.909. Altogether, these findings indicate that while CSF DDC has been proven to have great potential as a diagnostic biomarker, plasma DDC lacks diagnostic value for LBDs and our findings highlight the reactivity of plasma DDC to dopaminergic treatment.
The prolonged use of levodopa therapy in combination with peripheral DDC inhibitors has been shown to paradoxically increase dopamine production in serum, which could indicate an increase in DDC activity10. However, the increased supply of substrate, albeit in combination with DDC inhibitors, could also lead to an overproduction of peripheral DDC due to the excess supply of dopamine precursor and persisting shortage of dopamine. This could hence result in an increased concentration of DDC which in turn leads to a higher turnover of L-DOPA to dopamine. This hypothesis has been previously proposed by Rutledge et al.6 and aligns with the here observed longitudinal increase in plasma DDC levels within the PPMI Cohort. This increase was significantly associated with higher LEDD and was independent of disease severity outcomes. Notably, it was not observed in those cases without treatment, indicating that increasing levels of plasma DDC were primarily driven by dopaminergic treatment and not reflective of progressing disease severity. While DDC is highly expressed in the brain and involved in the dopaminergic pathway, which is impaired in LBDs, it is also peripherally expressed in tissues like the kidneys11 or leukocytes12. Changes in plasma DDC could hence reflect changes in peripheral DDC rather than brain-specific DDC changes due to neurodegenerative processes affecting the dopaminergic system. Plasma DDC could therefore be more easily affected than CSF DDC, not only by dopaminergic treatment but also by other confounders at the same time.
Previous proteomics studies have shown that, similarly to plasma DDC, also CSF DDC levels are affected by dopaminergic treatment. However, in contrast to findings in plasma, levels of CSF DDC were also significantly (albeit slightly less) increased in those cases that did not receive dopaminergic treatment6. These results are in line with our findings in subsets of the studied cohorts where paired CSF samples were available, showing significantly increased baseline (i.e., in non-medicated individuals) CSF DDC levels in DLB (PRIDE) and PD (PPMI) compared with controls, while this was not observed, or less pronounced, in plasma. The disparity between group differences of plasma and CSF DDC also becomes apparent in the association analysis between the two matrices. We report only weak associations between plasma and CSF DDC within the (b)PRIDE and PPMI Cohorts at baseline. However, when follow-up data, including medicated individuals, were included, the association within the PPMI Cohort became stronger. After adjusting for dopaminergic treatment, the predictive value of CSF DDC on plasma DDC levels decreased, indicating that dopaminergic treatment influences plasma DDC levels as well as the association of DDC within the two matrices. Previously, a strong correlation between plasma and CSF DDC has been reported. However, the influence of dopaminergic treatment on these results was not considered5. Altogether, these findings indicate that changes in plasma are more dependent on treatment than in CSF, which is also supported by previously published results3,6,7.
The strength of this study lies in the evaluation of three independent cohorts, allowing for the robust replication of our findings in treated and non-treated patients with different LBDs. However, this study is not without limitations. Studying the effect of disease severity and dopaminergic treatments on DDC levels is complicated due to the tight relation between these variables, and disease severity markers such as UPDRS3 and HY are imperfect measures. Although dopaminergic therapy is used less frequently and at lower LEDD in DLB compared with PD due to substantial side-effects13,14, and thereby likely only weakly affecting DDC levels in DLB, we did not have any information on medication use or longitudinal samples in the DLB groups in the VARS and (b)PRIDE Cohorts. Lastly, we have observed a general effect of dopaminergic treatment (converted to LEDD) on plasma DDC levels. Although the majority of patients in this study received L-DOPA in combination with a DDC inhibitor, or a MOA-B inhibitor, which have been shown to have a similar effect on DDC mRNA levels15, we cannot completely rule out that different types of dopaminergic medication could influence DDC levels differently and this should be studied further.
We found that plasma DDC was not altered in patients with LBDs compared with HC nor within patients having other neurodegenerative diseases. We showed that plasma DDC levels significantly increased over time in PD cases which was primarily driven by the use of dopaminergic treatment during follow-up. Together with previously published findings, these findings indicate that CSF DDC is a more stable marker for dopaminergic dysfunction than plasma DDC, which is however a potential marker to monitor biological treatment effects of dopaminergic medication.
Methods
Inclusion and ethics
Ethical approval was granted by the local ethics committee of each participating center (Comite de Ética del Instituto de Salud Carlos III, Centrale Comissie Mensgebonden Onderzoek te Den Haag, Ethikkomission Uni Ulm, Regionala Etikprövningsnämnden I Lund, Etico Aziende Sanitarie Regione Umbria, Human Research Ethics Committee of St. Vincent’s Hospital Melbourne) and all participants gave their written informed consent for use of their biological material and clinical data for research purposes. Participants did not receive compensation for their participation in this study. The PPMI study is registered under the identifier NCT01141023 at ClinicalTrials.gov.
Study population
We retrospectively included a total of 1635 patients from three international cohorts: the Vallecas Alzheimer Reina Sofía (VARS) Cohort with autopsy-confirmed diagnoses, the cross-sectional multicenter blood Proteins for early Discrimination of dEmentias Joint Programme—Neurodegenerative Disease Research project (bPRIDE) Cohort and the Parkinson’s Progression Markers Initiative (PPMI) Cohort.
The VARS Cohort included 12 patients with DLB who were recruited from a clinicopathological study between 2007 and 2020 and were neuropathologically confirmed as described previously16. Additionally, we included 59 HC from the Vallecas study, a community-based prospective cohort study of aging17.
The bPRIDE Cohort consisted of a total of 1498 patients, of which 374 cognitively unimpaired individuals without underlying AD pathology were included as controls. Further, we included 179 patients with stable mild cognitive impairment (sMCI) without biomarker evidence of amyloid pathology, 25 patients with mild cognitive impairment due to Lewy bodies (MCI-LB), 171 patients with DLB, 187 cognitively unimpaired individuals with underlying AD pathology (pre-AD), 164 patients with MCI due to AD (MCI-AD), 182 patients with AD dementia (AD-dem), 46 patients with MCI due to frontotemporal dementia (MCI-FTD) and 170 patients with FTD dementia (FTD-dem). Patients from the bPRIDE Cohort were recruited from six different centers (Amsterdam Dementia Cohort18, Sant Pau Initiative on Neurodegeneration19, Ulm University Hospital20, BioFINDER Cohort21, University of Perugia22 and the Australian Imaging Biomarker and Lifestyle Flagship Study of Aging)23. Controls were individuals with subjective cognitive decline or volunteers, who had normal AD CSF biomarkers and in whom no objective cognitive impairment could be determined. Patients with MCI due to DLB or FTD presented with MCI at baseline and converted to DLB or FTD, respectively, during follow-up. All patients within the AD continuum groups (pre-AD, MCI-AD, AD) presented with abnormal CSF AD biomarkers, or amyloid PET positivity. DLB diagnoses were made according to consensus guidelines9 and primarily based on clinical presentation. Additional DaT-SPECT was performed for 57 patients and an abnormal DaT-SPECT could support the clinical diagnoses in 43 patients. Global cognitive impairment was assessed by Mini Mental State Examination.
Lastly, we included a longitudinal cohort from the PPMI repository with available PEA data (derived from Project 196 Targeted Proteomics: Olink Explore 384 Cardiometabolic panel). Detailed description of study protocols and inclusion/exclusion criteria of the PPMI cohorts are published elsewhere24. Data used in the preparation of this article was obtained on [2023-10-05] from the PPMI database (www.ppmi-info.org/access-data-specimens/download-data), RRID:SCR_006431. For up-to-date information on the study, visit www.ppmi-info.org. The PPMI Cohort included 33 HC and 33 patients with PD with longitudinal DDC measurements of up to 8 years (2 follow-up measurements per patient: median follow-up time [IQR] = 4 [4, 4] years). Information about dopaminergic medications including levodopa, dopamine agonists, COMT inhibitor, and MAO-B inhibitors, were converted into levodopa equivalent daily dosage (LEDD), according to the PPMI Data User Guide Script 7. An overview of the distribution of used medication types is presented in Supplementary Table 5. At baseline no patients in the PPMI Cohort received dopaminergic treatment. During the follow-up, 31 (93.9%) patients with PD received dopaminergic treatment (Supplementary Table 6). Motor impairment in PD was assessed by the MDS-Unified Parkinson’s disease rating scale part 3 (UPDRS3) and the Hoehn and Yahr (HY) scale. For 21 patients, two UPDRS3 scores were reported during either one or two visits, and for three patients two HY scores were reported during one visit each. These scores were evaluated during a medication ON and OFF state. The medication state reflects the response to dopaminergic treatment in regard to motor fluctuations. ON indicates a good motor response and OFF indicates a poor response, i.e., no/decreased alleviation of motor symptoms25. Cognitive impairment was assessed during follow-up visits, but not at baseline, by Montreal Cognitive Assessment (MoCA) and did not differ between ON and OFF states. All scores are expressed as total scores of all elements.
In all cohorts, DDC measurements were available at baseline, while additional longitudinal DDC measurements were available from the PPMI Cohort. All participants underwent standardized clinical evaluation at the respective center and diagnoses were made based on corresponding diagnostic consensus criteria9,26,27,28,29,30,31,32 by experts in the field.
Paired CSF and plasma PEA measurements of DDC were available for subsets of the bPRIDE Cohort from the preceding PRIDE (Proteins for early Discrimination of dEmentias) study, as well as for the PPMI Cohort (Project 196). Paired samples from the PRIDE Cohort included a total of 75 participants that were recruited from the Amsterdam Dementia Cohort. Of these participants, 29 were diagnosed with DLB, one with MCI-AD, 20 with AD, 5 with FTD and 20 participants were cognitively HC. From the PPMI Cohort, paired CSF samples were available from 30 HC and 28 participants with PD. An overview of baseline characteristics of the respective subsets is given in Supplementary Table 7 in the supplementary information.
Measurements of DDC
Plasma samples were analyzed using PEA technology (Olink Proteomics, Uppsala, Sweden) at the Olink Proteomics Analysis Service Uppsala in Sweden. Citrate plasma samples from the Vallecas/VARS Cohort were analyzed between March and April 2022, and EDTA plasma samples from the bPRIDE and PPMI Cohorts were analyzed in April 2021. In short, plasma DDC was quantified as part of the Olink Explore panel, according to the manufacturer’s protocol33. For subsets of samples from the bPRIDE Cohort (n = 75; analyzed in December 2017 and August 2018) and the PPMI Cohort (n = 250; analyzed in April 2021), CSF DDC PEA measurements were available as well. DDC is part of the cardiometabolic panel which simultaneously measures 369 proteins by employing different antibodies that are coupled to unique oligonucleotides specific for each protein in the panel. Binding of these antibodies allows for the hybridization of the coupled oligonucleotides and extension by polymerase chain reaction. Quantification takes place by next generation sequencing, and the signal is then translated back to the amount of protein of interest in the sample and reported as normalized protein expression (NPX) on a log2-scale. All analyzes were performed for each cohort by Olink Proteomics and operating staff was blinded to any clinical data. Mean intra- and inter-assay CV% were 7.9% and 12% in the Vallecas/VARS Cohort, 12.4% and 13.5% in the bPRIDE Cohort, and 3.8% and 5.6% in the PPMI Cohort, respectively.
Statistics and reproducibility
Continuous variables for cohort descriptives at baseline were described as means with standard deviation or median with interquartile range, and compared using t-tests or ANOVA, while categorical variables were compared by Pearson’s Χ2-test. DDC levels in plasma or CSF at baseline were compared between groups per cohort by generalized linear models (GLM, stats R package34(version 4.3.2)) corrected for age, sex, and if applicable different centers, with DLB or PD as the reference category. Differences in plasma DDC levels over time between diagnostic groups were analyzed by linear mixed effects models with random intercept for each patient and corrected for age and sex as well as additional correction for LEDD (lmerTest R package35 (version 3.1-3)). Associations of DDC levels with LEDD and disease severity measures were analyzed by linear mixed effects models with random intercept for each patient and corrected for age and sex. For all regression analyses, standardized effect sizes are reported. Associations between plasma DDC and CSF DDC were analyzed by linear regression. Discriminative potential of CSF DDC for the differentiation of DLB from other groups was assessed by receiver operating characteristics (ROC, pROC R package36 (version 1.18.5)) analysis. No statistical method was used to predetermine sample size. PEA DDC measurements were excluded from the analysis when quality control did not pass due to measured levels below the limit of detection. Statistical analyses were performed with R version 4.0.334. A significance level of α = 0.05 was accepted.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data, methods, and manuals regarding PEA proteome analysis from the PPMI Cohort are available online from the PPMI database (www.ppmi-info.org/access-data-specimens/download-data), RRID:SCR_006431. Anonymized patient-level data from the VARS/Vallecas and bPRIDE Cohorts will be made available in public repositories in the future and can be requested from the corresponding author in the meantime. Source data are provided with this paper.
References
Foguem, C. & Manckoundia, P. Lewy body disease: clinical and pathological “overlap syndrome” between synucleinopathies (Parkinson Disease) and tauopathies (Alzheimer Disease). Curr. Neurol. Neurosci. Rep. 18, 24 (2018).
Russo, M. J. et al. High diagnostic performance of independent alpha-synuclein seed amplification assays for detection of early Parkinson’s disease. Acta Neuropathol. Commun. 9, 179 (2021).
Del Campo, M. et al. CSF proteome profiling reveals biomarkers to discriminate dementia with Lewy bodies from Alzheimer's disease. Nat. Commun. 14, 5635 (2023).
Paslawski, W. et al. Large-scale proximity extension assay reveals CSF midkine and DOPA decarboxylase as supportive diagnostic biomarkers for Parkinson’s disease. Transl. Neurodegener. 12, 42 (2023).
Pereira, J. B. et al. DOPA decarboxylase is an emerging biomarker for Parkinsonian disorders including preclinical Lewy body disease. Nat. Aging 3, 1201–1209 (2023).
Rutledge, J. et al. Comprehensive proteomics of CSF, plasma, and urine identify DDC and other biomarkers of early Parkinson’s disease. Acta Neuropathol. 147, 52 (2024).
Appleton, E. et al. DOPA-decarboxylase is elevated in CSF, but not plasma, in prodromal and de novo Parkinson’s disease. Transl. Neurodegener. 13, 31 (2024).
Pringsheim, T. et al. Dopaminergic therapy for motor symptoms in early Parkinson disease practice guideline summary: a report of the AAN guideline subcommittee. Neurology 97, 942–957 (2021).
McKeith, I. G. et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology 89, 88–100 (2017).
van Rumund, A. et al. Peripheral decarboxylase inhibitors paradoxically induce aromatic L-amino acid decarboxylase. NPJ Parkinsons Dis. 7, 29 (2021).
Mappouras, D. G., Stiakakis, J. & Fragoulis, E. G. Purification and characterization of L-dopa decarboxylase from human kidney. Mol. Cell Biochem. 94, 147–156 (1990).
Kokkinou, I., Nikolouzou, E., Hatzimanolis, A., Fragoulis, E. G. & Vassilacopoulou, D. Expression of enzymatically active L-DOPA decarboxylase in human peripheral leukocytes. Blood Cells Mol. Dis. 42, 92–98 (2009).
Lucetti, C. et al. Levodopa response in dementia with lewy bodies: a 1-year follow-up study. Parkinsonism Relat. Disord. 16, 522–526 (2010).
Goldman, J. G., Goetz, C. G., Brandabur, M., Sanfilippo, M. & Stebbins, G. T. Effects of dopaminergic medications on psychosis and motor function in dementia with Lewy bodies. Mov. Disord. 23, 2248–2250 (2008).
Berry, M. D., Juorio, A. V., Li, X. M. & Boulton, A. A. Aromatic L-amino acid decarboxylase: a neglected and misunderstood enzyme. Neurochem. Res. 21, 1075–1087 (1996).
Ortega-Cruz, D. et al. A novel histological staging of hippocampal sclerosis that is evident in gray matter loss in vivo. Alzheimers Dement. 19, 3028–3040 (2023).
Garo-Pascual, M. et al. Brain structure and phenotypic profile of superagers compared with age-matched older adults: a longitudinal analysis from the Vallecas Project. Lancet Healthy Longev. 4, e374–e385 (2023).
van der Flier, W. M. & Scheltens, P. Amsterdam dementia Cohort: performing research to optimize care. J. Alzheimers Dis. 62, 1091–1111 (2018).
Alcolea, D. et al. The Sant Pau Initiative on Neurodegeneration (SPIN) Cohort: a data set for biomarker discovery and validation in neurodegenerative disorders. Alzheimers Dement.5, 597–609 (2019).
Oeckl, P. et al. Targeted mass spectrometry suggests beta-synuclein as synaptic blood marker in Alzheimer’s disease. J. Proteome Res. 19, 1310–1318 (2020).
Mattsson, N. et al. Plasma tau in Alzheimer's disease. Neurology 87, 1827–1835 (2016).
Wojdala, A. L. et al. Trajectories of CSF and plasma biomarkers across Alzheimer’s disease continuum: disease staging by NF-L, p-tau181, and GFAP. Neurobiol. Dis. 189, 106356 (2023).
Fowler, C. et al. Fifteen years of the Australian imaging, biomarkers and lifestyle (AIBL) study: progress and observations from 2,359 older adults spanning the spectrum from cognitive normality to Alzheimer’s disease. J. Alzheimers Dis. Rep. 5, 443–468 (2021).
Parkinson Progression Marker, I. The Parkinson Progression Marker Initiative (PPMI). Prog. Neurobiol. 95, 629–635 (2011).
Lees, A. J. The on-off phenomenon. J. Neurol. Neurosurg. Psychiatry 52, 29–37 (1989).
Albert, M. S. et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 270–279 (2011).
McKeith, I. G. et al. Research criteria for the diagnosis of prodromal dementia with Lewy bodies. Neurology 94, 743–755 (2020).
Barker, M. S. et al. Proposed research criteria for prodromal behavioural variant frontotemporal dementia. Brain 145, 1079–1097 (2022).
Berg, D. et al. MDS research criteria for prodromal Parkinson’s disease. Mov. Disord. 30, 1600–1611 (2015).
McKhann, G. M. et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 263–269 (2011).
Rascovsky, K. et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134, 2456–2477 (2011).
Postuma, R. B. et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 30, 1591–1601 (2015).
Assarsson, E. et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS ONE 9, e95192 (2014).
Team, R. C. R: A language and environment for statistical computing. (R Foundation for Statistical Computing, Vienna, Austria, 2020).
Kuznetsova, A., Brockhoff, P. B. & Christensen, R. H. B. lmerTest package: tests in linear mixed effects models. J. Stat. Softw. 82, 1–26 (2017).
Robin, X. et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 12, 77 (2011).
Acknowledgements
The authors wish to thank the participants of the Vallecas Alzheimer’s Reina Sofía (VARS) Cohort and The Vallecas study and their family members. We also want to acknowledge the personnel at the Reina Sofía Center. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 860197 (K.B., C.E.T., E.A.J.W.). The VARS cohort and the Vallecas Study have been supported by the Queen Sofia Foundatio (P.S.J., A.R., M.M.). The SPIN cohort received funding from the Fondo de Investigaciones Sanitarias (FIS), Instituto de Salud Carlos III (PI21/00791, PI14/01126, PI17/01019, PI20/01473, PI13/01532, PI16/01825, PI18/00335, PI19/00882, PI18/00435, PI22/00611, INT19/00016, INT23/00048, PI17/01896 and AC19/00103) and the CIBERNED program (Program 1, Alzheimer Disease to AL), jointly funded by Fondo Europeo de Desarrollo Regional, Unión Europea, “Una manera de hacer Europa”. The SPIN cohort was also supported by the National Institutes of Health (NIA grants 1R01AG056850-01A1; R21AG056974; and R01AG061566), by Generalitat de Catalunya (2017-SGR-547, SLT006/17/125, SLT006/17/119, SLT002/16/408), “Marató TV3” foundation grants 20141210, 044412 and 20142610, a grant from the Fundació Bancaria La Caixa to RB (DABNI project), Fundació Catalana Síndrome de Down and Fundació Víctor Grífols i Lucas (A.L., D.A.). PPMI—a public-private partnership—is funded by the Michael J. Fox Foundation for Parkinson’s Research and funding partners, including 4D Pharma, Abbvie, AcureX, Allergan, Amathus Therapeutics, Aligning Science Across Parkinson’s, AskBio, Avid Radiopharmaceuticals, BIAL, BioArctic, Biogen, Biohaven, BioLegend, BlueRock Therapeutics, Bristol-Myers Squibb, Calico Labs, Capsida Biotherapeutics, Celgene, Cerevel Therapeutics, Coave Therapeutics, DaCapo Brainscience, Denali, Edmond J. Safra Foundation, Eli Lilly, Gain Therapeutics, GE HealthCare, Genentech, GSK, Golub Capital, Handl Therapeutics, Insitro, Jazz Pharmaceuticals, Johnson & Johnson Innovative Medicine, Lundbeck, Merck, Meso Scale Discovery, Mission Therapeutics, Neurocrine Biosciences, Neuron23, Neuropore, Pfizer, Piramal, Prevail Therapeutics, Roche, Sanofi, Servier, Sun Pharma Advanced Research Company, Takeda, Teva, UCB, Vanqua Bio, Verily, Voyager v. 25MAR2024 Therapeutics, the Weston Family Foundation and Yumanity Therapeutics.
Author information
Authors and Affiliations
Contributions
K.B. performed the analysis, interpreted the results, and drafted the manuscript. E.A.J.W. supervised the analysis and drafting of the manuscript. M.D.C.M. and C.E.T. designed the study. P.S.J., A.R., M.M. contributed to data collection and assembly from the VARS/Vallecas Cohort. J.D.D., G.B., L.V., D.A., S.H., S.V., N.M.C., K.V., C.J.F., L.B., M.K.S., Z.H., D.N.R., L.G., A.T., J.F., Y.P., A.L., W.F., J.H., M.O., O.H., L.P., C.L.M., A.L., C.E.T., and M.D.C.M. contributed to data collection and assembly from the bPRIDE Cohort. All authors reviewed the manuscript and approved the final version for submission.
Corresponding author
Ethics declarations
Competing interests
D.A. participated in advisory boards from Fujirebio-Europe, Roche Diagnostics, Grifols S.A. and Lilly, and received speaker honoraria from Fujirebio-Europe, Roche Diagnostics, Nutricia, Krka Farmacéutica S.L., Zambon S.A.U. and Esteve Pharmaceuticals S.A. L.G, participated on advisory boards for, and received writing honoraria and travel grants from Almirall, Biogen, Euroimmun, Fujirebio, Lilly, Merck, Mylan, Novartis, Roche, Sanofi, Siemens Healthineers and Teva. A.L. participated in advisory boards from Almirall, Biogen, Eisai, Fujirebio-Europe, Grifols, Novartis, Roche, Otsuka Pharmaceutical, Nutricia, Zambón, y NovoNordisk. C.E.T. has a collaboration contract with ADx Neurosciences, Quanterix, and Eli Lilly, performed contract research or received grants from AC-Immune, Axon Neurosciences, BioConnect, Bioorchestra, Brainstorm Therapeutics, Celgene, EIP Pharma, Eisai, Fujirebio, Grifols, Instant Nano Biosensors, Merck, Novo Nordisk, PeopleBio, Roche, Siemens, Toyama, Vivoryon. She is editor of Alzheimer Research and Therapy and serves on editorial boards of Medidact Neurologie/Springer, and Neurology: Neuroimmunology & Neuroinflammation. She had speaker contracts for Roche, Grifols, and Novo Nordisk. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Mohamed Salama, who co-reviewed with Nourhan Shebl; John O’Brien, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bolsewig, K., Willemse, E.A.J., Sánchez-Juan, P. et al. Increased plasma DOPA decarboxylase levels in Lewy body disorders are driven by dopaminergic treatment. Nat Commun 16, 1139 (2025). https://doi.org/10.1038/s41467-025-56293-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56293-z
