
Abstract
Anillin (ANLN), a mitotic protein that regulates contractile ring assembly, has been reported as an oncoprotein. However, the function of ANLN in cancer cells, especially in the nucleus, has not been fully understood. Here, we report a role of nuclear ANLN in gene transcriptional regulation. We find that nuclear ANLN directly interacts with the RNA polymerase II (Pol II) large subunit to form transcriptional condensates. ANLN enhances initiated Pol II clustering and promotes Pol II CTD phase separation. Short-term depletion of ANLN alters the chromatin binding and enhancer-mediated transcriptional activity of Pol II. The target genes of ANLN-Pol II axis are involved in oxidoreductase activity, Wnt signaling and cell differentiation. THZ1, a super-enhancer inhibitor, specifically inhibits ANLN-Pol II clustering, target gene expression and esophageal squamous cell carcinoma (ESCC) cell proliferation. Our results reveal the function of nuclear ANLN in transcriptional regulation, providing a theoretical basis for ESCC treatment.
Similar content being viewed by others


Introduction
Anillin (ANLN) is a unique scaffold protein that interacts with cytoskeletal elements such as F-actin, myosin II, and RhoA to regulate cell division1,2. ANLN promotes mitotic progression by organizing contractile ring components, a process that lasts approximately 40–60 min3,4. In a previous study, we found that the abundance of ANLN protein mediated by the ubiquitin system is essential for esophageal squamous cell carcinoma (ESCC) cell division and malignant progression5. However, during an interphase of up to 20 h, ANLN is distributed in the nucleus and may perform unknown functions2. It is difficult to fully explain the cancer-promoting mechanism of ANLN only by regulating contractile ring assembly. Previous studies have shown that ANLN plays several non-mitotic functions in different types of cells, such as regulating cell-cell junctions, signal transduction and cell motility6,7. Whether ANLN is directly involved in these processes, especially nuclear ANLN, remains unresolved.
RNA polymerase II (Pol II) consists of 12 subunits and transcribes all protein-coding genes in eukaryotic genomes8,9. Various proteins and protein complexes interact with Pol II to regulate its activity. These complexes accumulate at specific regions within the nucleus through liquid-liquid phase separation (LLPS), previously named transcription condensates or Pol II clusters10. The central hub driving Pol II clustering is the carboxyl-terminal ___domain (CTD) of its largest subunit POLR2A, which is an intrinsically disordered region (IDR) consisting of 52 heptapeptide repeats11. Although CTD is sufficient to form phase separation droplets in vitro, the formation of Pol II clusters in the nucleus is regulated by multiple mechanisms, including RNA concentration, chromatin conformation, and interactions with transcriptional regulators containing IDRs12,13,14. Different types of Pol II clusters are involved in transcriptional activation, RNA processing and 3D genome organization15. In particular, the recently reported large Pol II clusters, which contain transcription initiation related Pol II, are enriched at super-enhancer regions16. The regulation of Pol II clusters is intimately associated with the transcriptional amplification mechanism in cancers17. Consequently, investigating the molecular mechanisms underlying Pol II clustering is essential.
Here, we identified nuclear ANLN as a transcription regulator in cancer cells, which regulates Pol II clustering and target gene expression. Mechanistically, ANLN directly interacts with the large subunit of Pol II and enhances the phase separation of Pol II CTD. Short-term depletion of ANLN alters the chromatin binding of Pol II and enhancer-mediated gene transcriptional activity. We also identified 84 direct target genes regulated by the ANLN-Pol II axis, which are associated with tumor-related signaling pathways and biological processes. The specificity of ANLN-Pol II axis in regulating gene expression may be determined based on its interaction with various transcription factors and super-enhancers. The small-molecule THZ1 specifically inhibits ANLN-Pol II clusters, target gene expression, and ESCC cell viability, providing a potential cancer treatment strategy.
Results
Nuclear ANLN directly interacts with Pol II large subunit
ANLN is present in the nucleus during interphase and on the contractile rings during mitosis, as confirmed in several types of cancer cells (Fig. 1A). Cellular fractionation further showed that ANLN in the nucleus is associated with chromatin (Fig. 1B). Previous transcriptome data from cancer cells demonstrated that knockdown of ANLN altered the expression of a series of genes; however, it remained unclear whether these changes were directly mediated by ANLN. We then asked whether the distribution of nuclear ANLN is associated with nascent RNAs. Consequently, 5-bromouridine 5’-triphosphate (BrUTP) was used to label nascent transcripts, followed by the visualization of active transcription sites using BrdU antibody18. As depicted in Fig. 1C and supplementary fig. 1A, a clear colocalization was observed between ANLN and nascent transcripts. Consistently, a variety of proteins related to gene transcription events, especially the subunits of Pol II, were identified from the ANLN interactome (Fig. 1D, Supplementary fig. 1B). Following endogenous co-immunoprecipitation, ANLN was found to exist in Pol II complexes, including phosphorylated and unphosphorylated Pol II, as well as transcription initiation factors TBP and CDK7. To identify which amino acid regions within ANLN are necessary for ANLN-Pol II interaction, a co-immunoprecipitation analysis was performed on HEK293T cells expressing different HA-ANLN deletion mutants. The N-terminal 1-300 and 454-712 aa of ANLN were sufficient to maintain interaction, but both were less effective than the wild-type (WT), while the C-terminal 712-1124 aa did not mediate the interaction (Fig. 1F). To determine whether the ANLN-Pol II interaction is direct, we generated and purified recombinant ANLN and Pol II subunits. In vitro pulldown assays showed that ANLN directly interacted with both 846-1592 aa and CTD domains in the C-terminal of Pol II large subunit under cell free conditions, but not with the N-terminal 1-845 aa or Pol II small subunit (POLR2E) (Fig. 1G, H and Supplementary fig. 1C). Consistent with this, a proximity ligation assay (PLA) confirmed that ANLN interacts with the Pol II large subunit in the nucleus (Fig. 1I, Supplementary fig. 1D).

A Representative immunofluorescence staining images of ANLN (pink) and DNA (blue) in different cell cycles. Scale bar, 5 μm. B Western blotting was performed to examine the different subcellular fractions of the indicated cells. Lamin B1 and histone H3 were used as marker proteins of the chromatin. GAPDH was used as a marker of the cytoplasm. C BrUTP was transfected into HeLa cells using liposomes for 15 min, and then cultured in fresh medium for another 15 min. Immunofluorescence staining was performed using ANLN (green) and BrdU (red) antibodies. Scale bar, 5 μm. D The enrichment analysis of ANLN interacting proteins identified by mass spectrometry was performed using Metascape database. Based on the selected terms, accumulative hypergeometric P-values and enrichment factors were calculated and used for filtering. Remaining significant terms were hierarchically clustered into a tree based on Kappa-statistical similarities among their gene memberships. The 0.3 kappa score was applied as the threshold to cast the tree into term clusters. E Immunoprecipitation was performed using ANLN antibody, and the indicated antibodies were subsequently used for the western blotting. F The indicated plasmids were transfected into HEK293T cells for 48 h, and then the cells were lysed and subjected to immunoprecipitation with HA magnetic beads. G, H The interaction between recombinant POLR2A fragments and ANLN full-length protein was examined using an in vitro pulldown assay. I Representative PLA images for the interaction between endogenous ANLN and Pol II in KYSE510 cells. Scale bar, 5 µm. The data are representative of at least three independent experiments with similar results. Source data are provided as a Source Data file.
ANLN and Pol II form condensates and colocalize at the gene TSS regions
As expected, the colocalization of ANLN and Pol II was observed in the nucleus during interphase, but not during mitosis (Supplementary fig. 2A). This observation led us to investigate the chromatin occupancy of nuclear ANLN and Pol II. A cleavage under targets and tagmentation (CUT&Tag) assay was conducted in KYSE150 cells with ANLN and Pol II antibodies. Heatmaps and profile plots showed that Pol II and ANLN colocalized in the genome, and both of them occupied the transcription start site (TSS) of many active genes (Fig. 2A, B). 62.67% of Pol II and 64.60% of ANLN signals were distributed in the gene promoter regions, with 84.38% of ANLN overlapping with Pol II signals (Fig. 2C, D). These data suggest that the ANLN-Pol II complex is more likely to function in transcription initiation.

A Heatmap shows the CUT&Tag signals of ANLN and Pol II on the genome of KYSE150 cells. B Relative enrichment of CUT&Tag signals of ANLN and Pol II on TSS-gene body region. C Pie chart of ANLN and Pol II signals distribution in different regions at the genome. D Venn diagram of ANLN and Pol II peaks in KYSE150 cells. E Representative super-resolution images of nuclear ANLN (pink) and Pol II-pS5 (green) in HeLa cells. Scale bar, 3 µm. Cells were treated with or without 5% 1,6-HD for 60 s. Scale bar, 3 μm. F, G The number (F) and average area (G) of protein clusters per cell per focal plane. Clusters smaller than 12675 nm2 were excluded based on previous studies. n = 57 (Control); n = 51 (1,6-HD). The error bars indicate the mean ± SDs. Box plots indicate the median (middle line, red), 25th and 75th percentiles (boxes), and minimum and maximum percentiles (whiskers). Two-tailed Student’s t tests were performed to determine the significance. H Relative fluorescence intensity of protein signals per cell per focal plane. n = 57 (Control); n = 51 (1,6-HD). The error bars indicate the mean ± SDs. Two-tailed Student’s t tests were performed to determine the significance. I, J Classify the identified clusters as type I (12675-25350 nm2), type II (25351-63375 nm2), and type III ( > 63375 nm2), showing the proportion of each cluster (n = 57). Scale bar, 3 µm. K Effect of 1,6-HD on the number of different types of clusters. n = 57 (Control); n = 51 (1,6-HD). The error bars indicate the mean ± SDs. Two-tailed Student’s t tests were performed to determine the significance. L Representative raw and reconstructed images of the interaction patterns between ANLN (pink) and Pol II-pS5 (green) large clusters (Type III). Scale bar, 3 µm. CUT&Tag analysis representative of two independent experiments, and other data are representative of at least three independent experiments with similar results. Source data are provided as a Source Data file.
To better understand the nuclear spatial organization of the two proteins, ANLN and initiated Pol II (Pol II-pS5) were labeled with antibodies19, and the nuclei were subsequently analyzed with super-resolution imaging. To this end, a 60 nm spatial resolution structured illumination microscopy (SIM) was used to acquire super-resolution imaging, which illuminates the sample through a highly inclined and laminated optical (HILO) method to specifically excite a narrow 0.4 µm band across the nucleus while excluding the cytoplasm above and below. It was observed in HeLa and KYSE150 cells that Pol II-pS5 generated protein clusters with different sizes, whereas ANLN only formed small-size clusters (Fig. 2E and Supplementary fig. 2B). We also found that treatment of 1,6-hexanediol (1,6-HD), commonly used to dissolve LLPS condensates by perturbing hydrophobic protein interactions20, led to a reduction in ANLN and Pol II clusters without affecting protein expression levels (Fig. 2E–H and Supplementary fig. 2D, E). To observe chromatin-bound clusters, cells were pre-extracted to release free proteins from the nucleoplasm21,22. After pre-extraction, the protein level and cluster number of nuclear ANLN decreased, while Pol II-pS5 cluster slightly decreased, indicating that these clusters partially bound to chromatin (Supplementary fig. 2F–H). We next identified the types of ANLN and Pol II-pS5 clusters in human cancer cells. Clusters smaller than 12675 nm2 were excluded based on previous studies23,24,25, Pol II-pS5 was subsequently found to form two types small-size clusters, namely type I (12675–25350 nm2) and type II (25351–63375 nm2), and a large-size type III cluster (> 63375 nm2), but ANLN rarely forms type III clusters (Fig. 2I, J and Supplementary fig. 2C). Pol II type III clusters show distinctive shapes and are particularly sensitive to 1,6-HD (Fig. 2K, L), in line with previous reports in zebrafish embryos26. Interestingly, ANLN clusters usually gather around the periphery of Pol II type III, accounting for 83.33% of the 60 fields observed (Fig. 2L). Taken together, these results suggest that ANLN and initiated Pol II form condensates and are co-located in the gene TSS regions.
ANLN enhances initiation related Pol II clustering
Several transcription factors and coactivators have been reported to contribute to the formation of initiated Pol II clusters10,27. To investigate the role of ANLN in Pol II clustering, the expression of ANLN was silenced by siRNA in HeLa cells, and the spatial organization of Pol II was subsequently analyzed by super-resolution imaging. Results showed that depletion of ANLN specifically reduced Pol II-pS5 type III clusters, and had no effect on the number of small-size clusters and the expression level of Pol II-pS5 (Fig. 3A–C). Since ANLN also functions during mitosis, to exclude the effects of mitotic defects, we next adopted a short-term depletion method to study the role of nuclear ANLN. In line with previous reports28,29, long-term depletion (48 h) of ANLN resulted in multinucleation in approximately 80% of cells, a defective phenotype caused by mitotic failure, while short-term depletion (24 h) did not affect cell morphology and size (Supplementary fig. 3A). To precisely control the timing of ANLN depletion, a doxycycline (Dox) induced rapid knockdown of ANLN in KYSE150 cells was generated. It was observed that Dox treatment for 24 h significantly decreased the expression level of ANLN and reduced Pol II type III clusters (Supplementary fig. 3B, C, H), without affecting the small-size clusters and expression level of Pol II-pS5 (Supplementary fig. 3C, D), similar to the results observed in HeLa cells. Reintroduction of HA-tagged ANLN rescued the reduction of Pol II clusters (Supplementary fig. 3B, C). We also evaluated whether ANLN regulates additional transcriptional steps such as elongation. Following the rapid depletion of ANLN, the elongating Pol II was labeled with pS2 antibody and observed via super-resolution microscopy. Results showed that the depletion of ANLN slightly affected the generation of elongating clusters, reducing it by approximately 13.6%, without affecting the expression level of Pol II-pS2 (Supplementary fig. 3E–G). To further exclude the interference of mitotic defects, an ANLN mutant with nuclear localization defect was introduced into the ANLN rapid depletion cell line. Mutation of 5 amino acids in the previously predicted nuclear localization sequence (NLS) resulted in obvious nuclear localization defect of ANLN (Fig. 3D)30,31,32. As expected, the reduction of Pol II-pS5 clusters caused by rapid depletion of ANLN could be rescued by ANLN WT but not NLS mutant (Fig. 3D–G).

A Representative super-resolution and wide field images showing Pol II-pS5 (green) in HeLa cells after 48 h of ANLN knockdown. Scale bar, 3 μm. B The relative number of different types of Pol II-pS5 clusters after ANLN knockdown. n = 45 (si-NC); n = 45 (si-ANLN). The error bars indicate the mean ± SDs. Two-tailed Student’s t tests were performed to determine the significance. C Relative fluorescence intensity of Pol II-pS5 signals per cell per focal plane. n = 45 (si-NC); n = 45 (si-ANLN). The error bars indicate the mean ± SDs. Two-tailed Student’s t tests were performed to determine the significance. D Representative super-resolution images showing Pol II-pS5 (green) in ANLN rapid depletion KYSE150 cells introduced with ANLN WT or NLS mutant. Scale bar, 3 μm. E The relative number of different types of Pol II-pS5 clusters. n = 84 (Control); n = 66 (ANLN KD); n = 81 (ANLN KD + WT); n = 48 (ANLN KD + NLS mut). The error bars indicate the mean ± SDs. Two-tailed Student’s t tests were performed to determine the significance. F Relative fluorescence intensity of Pol II-pS5 signals per cell per focal plane. n = 84 (Control); n = 66 (ANLN KD); n = 81 (ANLN KD + WT); n = 48 (ANLN KD + NLS mut). The error bars indicate the mean ± SDs. Two-tailed Student’s t tests were performed to determine the significance. G Western blot showing the efficiency of ANLN depletion and rescue in KYSE150 cells. H Prediction of intrinsically disordered residues in ANLN and POLR2A protein by PONDR program. I, J The key amino acid regions that mediate the phase separation ability of ANLN were predicted by PSPHunter. K The indicated plasmids were transfected into HEK293T cells for 48 h, the cells were lysed and subjected to immunoprecipitation with HA magnetic beads. L Representative super-resolution images showing Pol II-pS5 (green) in ANLN rapid depletion KYSE150 cells introduced with ANLN WT or PS mutant. Scale bar, 3 μm. M The relative number of different types of Pol II-pS5 clusters. n = 42 (Control); n = 45 (ANLN KD); n = 39 (ANLN KD + WT); n = 51 (ANLN KD + PS mut). The error bars indicate the mean ± SDs. Two-tailed Student’s t tests were performed to determine the significance. N Relative fluorescence intensity of Pol II-pS5 signals per cell per focal plane. n = 42 (Control); n = 45 (ANLN KD); n = 39 (ANLN KD + WT); n = 51 (ANLN KD + PS mut). The error bars indicate the mean ± SDs. Two-tailed Student’s t tests were performed to determine the significance. O Western blot showing the efficiency of ANLN depletion and rescue in KYSE150 cells. The data are representative of at least three independent experiments with similar results. Source data are provided as a Source Data file.
ANLN was initially reported as an actin binding protein that functions during mitosis33. Therefore, it was explored whether the promotion of Pol II clustering by ANLN was dependent on actin binding. The amino acids near the actin binding ___domain of ANLN were deleted to generate different mutants, and then it was found that the deletion of 100-150 or 150-200 aa eliminated ANLN-actin interaction, but did not reduce ANLN-Pol II interaction (Supplementary fig. 3I). Further investigation found that ANLN mutants with actin interaction defects rescued the Pol II-pS5 cluster reduction caused by ANLN depletion, similar to the effect of ANLN WT (Supplementary fig. 3J–M). This indicates that the regulation of Pol II clustering by ANLN is independent of actin binding.
CTD is a hub ___domain that drives Pol II phase separation during the transcription initiation stage. Typically, transcriptional regulators bind to the CTD via its IDRs to enhance transcriptional condensate formation24,34. Indeed, we not only found that ANLN directly binds to CTD (Fig. 1H), but also identified the presence of IDR in the N-terminus of ANLN, approximately from 1 to 700 aa, using the Predictor of Natural Disordered Regions (PONDR) program (Fig. 3H)20. To determine whether ANLN enhanced Pol II clustering is related to phase separation, PSPHunter, a machine learning method, was used to predict the key amino acids driving phase separation35. PSPHunter combined with amino acid sequence analysis indicated that the IDR of ANLN protein contains three potential phase separation driving sequences, which are composed of 46 amino acids rich in stickers and spacers (Fig. 3I, J)36. Co-immunoprecipitation showed that only deletion of all 46 key amino acids attenuated the ANLN-Pol II interaction, and we defined this mutant as PS mut (ANLN phase separation mutant) (Fig. 3K). In contrast, random deletion of 50 amino acids in the IDR of ANLN did not weaken the ANLN-Pol II interaction (Supplementary fig. 3I). Furthermore, the reduction of Pol II-pS5 clusters caused by rapid depletion of ANLN could be rescued by ANLN WT but not PS mut (Fig. 3L–O). In summary, these findings suggest that nuclear ANLN promotes Pol II clustering through the key residues of its IDR, and this is a novel function related to phase separation but independent of actin binding.
ANLN promotes Pol II CTD phase separation in vitro
To determine whether ANLN promotes Pol II CTD phase separation in vitro, the proteins were expressed and purified from prokaryotic cells, and their biomolecular condensate features were examined through droplet assay. CTD or ANLN recombinant proteins formed condensates in a concentration dependent manner in dextran working buffer, while disrupting hydrophobic interactions reduced droplet formation (Fig. 4A). FRAP analysis also showed that CTD or ANLN droplets could fuse and recover rapidly after photobleaching (Fig. 4B). Importantly, ANLN increased the size and condensate fractions of CTD droplets in a concentration dependent manner (Fig. 4C, D and Supplementary fig. 4A). At near physiological concentration (0.5 μM), ANLN was distributed in the periphery of CTD droplets, similar to our observations in the nucleus, which follows the Pickering emulsion theory (Fig. 4C, D and Supplementary fig. 4B–D)37. Moreover, ANLN promoted phase separation of CTD at extremely low concentrations (Fig. 4D), and ANLN-CTD droplets were much larger than ANLN or CTD droplets alone (Fig. 4E). Taken together, these results indicate that the multivalent interactions between ANLN and Pol II CTD promote the assembly of large condensates.

A Representative images and statistical data of in vitro droplet assays with different concentrations of CTD-mEGFP (green) or ANLN-mCherry (red). Proteins were incubated in phase separation buffer with or without 10% 1,6-HD. Scale bar, 20 μm. CTD in order n = 0, 29, 53, 78, 199, 209 and 260, respectively. ANLN in order n = 0, 10, 41, 89, 107, 223 and 172, respectively. The error bars indicate the mean ± SDs. Two-tailed Student’s t tests were performed to determine the significance. B Representative images and quantification of droplet photobleaching recovery at the indicated time points. Scale bars, 5 μm. n = 3 for each condition. The error bars indicate the mean ± SDs. C, D Representative images and statistical data of in vitro droplet assays with 5 μM CTD-mEGFP (green) and different concentrations of ANLN-mCherry (red). Scale bar, 20 μm. n = 114, 100, 84, 130, 169, 76, 122, 145, 105, 162, respectively. The error bars indicate the mean ± SDs. Two-tailed Student’s t tests were performed to determine the significance. E Statistics of droplet area. The droplet size of ANLN or CTD alone and the droplet size formed by both were compared. n = 114, 10, 122, 114, 41, 145, 114, 89, 105, respectively. The error bars indicate the mean ± SDs. Two-tailed Student’s t tests were performed to determine the significance. F Droplet assays of CTD (5 μM, green) and ANLN (0.5 μM, red) in the presence of different concentrations of total RNA isolated from KYSE150 cells. Scale bar, 20 μm. n = 80, 159, 155, 155, 226, 120, 82, respectively. The error bars indicate the mean ± SDs. Two-tailed Student’s t tests were performed to determine the significance. G Droplet assays of CTD (5 μM, green) and ANLN (0.5 μM, red) in the presence of different concentrations of genomic DNA isolated from KYSE150 cells. Scale bar, 20 μm. n = 130, 212, 272, 241, 115, 146, 106, respectively. The error bars indicate the mean ± SDs. Two-tailed Student’s t tests were performed to determine the significance. H Coomassie blue staining of purified proteins. I, J Representative images and statistical data of in vitro droplet assays with 5 μM CTD-mEGFP (green) and different concentrations of ANLN-mCherry WT or PS mutant (red). Scale bar, 20 μm. n = 51, 132, 108, 128, 198, respectively. The error bars indicate the mean ± SDs. Two-tailed Student’s t tests were performed to determine the significance. K–M The distribution characteristics of CTD-mEGFP and ANLN-mCherry WT or PS mutant were analyzed using clock scan. The data are representative of at least three independent experiments with similar results. Source data are provided as a Source Data file.
The equilibrium behavior of transcriptional condensates is regulated by RNA or DNA mediated electrostatic effects12,38,39. To investigate the effect of RNA or DNA on ANLN-CTD condensates, total RNA and genomic DNA were purified from KYSE150 cells. Droplet assay showed that low RNA level promoted and high RNA level inhibited the formation of ANLN-CTD condensate (Fig. 4F), similar to previously reported mediator condensates12. Although DNA has a negative charge, its regulatory effect on ANLN-CTD condensates is smaller than that of RNA at the same concentrations (Fig. 4G). These results indicate that ANLN-Pol II CTD condensates conform to the general mechanism by which transcription initiation complexes are regulated by polyanions.
We next expressed and purified ANLN PS mut recombinant protein (lacking 46 key amino acids), and then evaluated its effect on CTD phase separation in vitro. Droplet assay showed that the ability of PS mut to promote CTD phase separation was significantly weakened compared with ANLN WT (Fig. 4H–J). The size and dynamics of condensates are regulated by protein clusters adsorbed on its interface24,37. Interestingly, ANLN WT aggregated at the edge of condensates (about 90% of the radius) and CTD was evenly distributed inside, while unstable PS mut localization led to disordered CTD distribution (Fig. 4K–M). This indicates that the absence of key phase separation driving amino acids impairs the interaction pattern of ANLN-CTD, which may reduce the steric stabilization of condensates.
ANLN regulates gene transcriptional activity and Pol II chromatin binding
To evaluate the role of ANLN in global gene expression, RNA-Seq analysis was performed under normal and ANLN knockdown conditions. The expression of thousands of genes was changed, with a total of 1227 down-regulated and 1270 up-regulated genes. Among them, 434 genes were down-regulated and 333 were up-regulated with a fold change greater than 1.5, while 168 genes were down-regulated and 75 were up-regulated with a fold change greater than 2, which highlighted the negative impact of ANLN deletion on transcription (Fig. 5A). An enrichment analysis revealed that down-regulated genes were specifically involved in several canonical cancer-related pathways, such as Wnt, KRAS, and Hippo signaling (Fig. 5B). The majority of the up-regulated genes are associated with cell stress, DNA damage response, and apoptosis, indicating the role of ANLN in cell survival (Fig. 5C). Next, we observed that the promoters of down-regulated genes usually have more ANLN binding than those of up-regulated genes, suggesting that down-regulated genes are more likely to be directly dependent on the transcriptional regulation of ANLN (Fig. 5D, E). To explore the direct effect of ANLN on gene transcriptional activity, CUT&Tag analysis of Pol II and H3K27ac was performed under the condition of short-term depletion of ANLN. Surprisingly, it was observed that the H3K27ac signal decreased near the TSS, while the deposition of Pol II on chromatin increased (Fig. 5F, G). This is similar to the previously reported abnormal pausing and accumulation of Pol II caused by topoisomerases or BRD4 deletion40,41, as the mature transcripts of down-regulated genes were typically observed to be reduced, while housekeeping or up-regulated genes were less adversely affected by ANLN depletion (Fig. 5H, I and Supplementary fig. 5A, B). Furthermore, the representative target genes regulated by ANLN were labeled by DNA fluorescence in situ hybridization (DNA FISH), and their distribution with ANLN and Pol II was observed by combined immunofluorescence (Supplementary fig. 5C). CCND1 and TXNRD1 are canonical oncogenes, which were previously found to be amplified in cancer cells42,43. We found that ANLN protein occupied most loci of target genes but less housekeeping genes, while Pol II occupied these loci indiscriminately (Supplementary fig. 5C, D). These results suggest that ANLN-mediated gene transcription is a gene-specific rather than a general transcription mechanism, which is consistent with the limited regulation of ANLN on the levels of transcription products. To further understand the effect of ANLN on Pol II activity, a previously established chromatin association biochemical assay was used to determine the affinity between Pol II and chromatin44,45. Despite the absence of ANLN results in the accumulation of Pol II CUT&Tag signals, these Pol II are readily dissociated from chromatin under high salt washing conditions, suggesting that Pol II activity may be reduced (Fig. 5J). Consistently, luciferase reporter gene assays showed that ANLN rapid depletion led to a decrease of about 20% in Pol II activity, while overexpression of ANLN WT instead of NLS mut rescued Pol II activity (Fig. 5K). Recent studies have shown that Pol II and transcriptional regulators form large-size phase-separated condensates at super-enhancers16,26, aligning with our observations of Pol II type III clusters. Therefore, the effect of ANLN depletion on super-enhancers was investigated. The super-enhancer regions of KYSE150 cell line were obtained from SEdb database46, and then the CUT&Tag signals with or without short-term knockdown of ANLN were mapped to these regions47. Compared with TSS proximal regions, short-term depletion of ANLN strongly inhibited H3K27ac signal on both typical and super enhancers (Fig. 5L–N). Taken together, the rapid depletion of ANLN expression altered Pol II chromatin binding and enhancer-mediated gene transcriptional activity, thereby regulating the expression level of a subset of specific genes.

A Volcano plots depicting gene expression changes in control and ANLN knockdown KYSE150 cells. The false discovery rates (FDRs) are extracted from DESeq2 by adjusting P-values using empirical Bayes approach. B, C Metascape database was used for enrichment analysis of differential genes in RNA-Seq data. Based on the selected terms, accumulative hypergeometric P-values and enrichment factors were calculated and used for filtering. Remaining significant terms were hierarchically clustered into a tree based on Kappa-statistical similarities among their gene memberships. The 0.3 kappa score was applied as the threshold to cast the tree into term clusters. D, E Proportion of 2-fold differentially expressed genes with or without ANLN bound within 3 kb of TSS (D), and the average signal intensity of ANLN occupancy (E). n = 168 (Down); n = 75 (Up). The error bars indicate the mean ± SDs. Two-tailed Student’s t tests were performed to determine the significance. F, G Metaplots and heatmaps showing the occupancy of H3K27ac and total Pol II measured by CUT&Tag in NC and ANLN short-term depletion (24 h) cells. H Representative track examples of down-regulated genes (TXNRD1, CCND1) and housekeeping genes (GAPDH) showing the occupancy of H3K27ac and Pol II after short-term depletion of ANLN. I FPKM of representative genes in RNA-Seq data. n = 3 for each group. The error bars indicate the mean ± SDs. Two-tailed Student’s t tests were performed to determine the significance. J Western blot showing the whole cell lysate, chromatin pellets, and chromatin supernatant in ANLN short-term depletion cells. Pellets and supernatant were obtained by eluting chromatin components with 400 mM NaCl followed by centrifugation. K pGLC or pGLB plasmids were transfected into Dox-induced ANLN knockdown KYSE150 cells or control cells. Cells were treated with Dox 24 h after transfection, and luciferase activity was measured 48 h after transfection. Renilla luciferase activity served as an internal control. n = 4 for each group. The error bars indicate the mean ± SDs. Two-tailed Student’s t tests were performed to determine the significance. L–N Metaplots comparing the average H3K27ac signal in NC and ANLN short-term depletion cells. Flow chart for identifying signals in the super-enhancer regions of KYSE150 cells (L). CUT&Tag signal is shown for typical enhancer (M) and super-enhancer regions (N). CUT&Tag analysis representative of two independent experiments, and other data are representative of at least three independent experiments with similar results. Source data are provided as a Source Data file.
Identification of direct target genes of ANLN
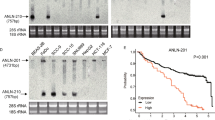
To further clarify the functional relevance of the role played by ANLN in transcriptional regulation, the direct targets of the ANLN-Pol II axis in KYSE150 cells were identified. Previous RNA-Seq or microarray studies with knockdown cells revealed ANLN regulated genes48,49. However, the genes directly targeted by ANLN are still unknown. Here, we combined RNA-Seq and CUT&Tag to screen candidate targets, especially genes with ANLN occupancy and exhibiting altered expression under short-term depletion conditions (Fig. 6A). As shown in Fig. 6B, 84 targets were identified as being directly regulated by the ANLN-Pol II axis. Consistent with this, ANLN and Pol II binding in the promoter regions of these target genes was verified by ChIP-qPCR (Fig. 6C). Functional annotations revealed that 84 target genes are associated with biological processes or signal transduction involved in cancer progression, particularly with oxidoreductase activity, cell differentiation, and Wnt signaling (Fig. 6D). The CUT&Tag signals of 12 representative targets were displayed, revealing a high degree of overlap between ANLN and Pol II peaks, especially in the promoter region (Fig. 6E). Reverse transcription-quantitative PCR (RT-qPCR) analysis further demonstrated that the expression level of 12 target genes decreased with the depletion of ANLN in a time-dependent manner (Fig. 6F). Consistently, rapid depletion of ANLN resulted in downregulation of target genes, and most of them could be rescued by overexpression of ANLN WT rather than PS mut (Supplementary fig. 6A). Given that ANLN regulates super-enhancer activity (Fig. 5N), whether the target genes are regulated by super-enhancers was investigated. A public super-enhancer database was used, which contains extensive super-enhancer information obtained from human H3K27ac ChIP-Seq data46. All 12 targets we validated were confirmed as super-enhancer regulated genes, with 9 identified in ESCC cells or esophageal epithelium samples (Fig. 6G). By mapping our CUT&Tag data to the identified super-enhancer regions in KYSE150 cell line, as expected, these regions contain abundant H3K27ac signals. ANLN and Pol II peaks were found to occupy H3K27ac enriched active sites, suggesting that the ANLN-Pol II complex regulates targets by binding to super-enhancers (Fig. 6H). Transcription factors control the aggregation and activity of Pol II by forming multiple complexes, resulting in transcriptional selectivity. To understand which transcription factors cooperate with ANLN, candidates were screened from the ANLN interactome and the Cistrome, a transcription factor database50. The results showed that there were at least 200 transcription factors directly or indirectly interacting with ANLN protein, many of which were reported as oncogenic transcription factors, such as RUVBL1/2, CSNK2A1, CEBPZ, UBTF, and EWSR1 (Fig. 6I, J). We compared the peaks of transcription factors downloaded from the Cistrome database with our ANLN and Pol II peaks and found that these transcription factors commonly co-localize with ANLN and Pol II at the promoter or super-enhancer regions of target genes (Fig. 6J and Supplementary fig. 6B–D). These transcription factors did not occupy ANLN binding sites on all target genes, indicating that ANLN co-regulates specific targets with different transcription factors in different cellular backgrounds and conditions (Fig. 6J and Supplementary fig. 6B-D). Based on these data and recent reports, we hypothesized a model: ANLN and Pol II may form large-size transcriptional condensates with multiple transcription factors at the super-enhancer regions, regulating the expression of target genes of cancer-related pathways and processes (Fig. 6K)15,16,24,26.

A Flowchart of the process used to identify the genes directly targeted by ANLN. B Heatmap showing the RNA-Seq expression and CUT&Tag signals of 84 direct target genes. C ChIP-qPCR experiments measuring ANLN and Pol II binding enrichment on promoter regions of target genes. n = 3 for each group. The error bars indicate the mean ± SDs. D Metascape database was used for enrichment analysis of 84 target genes. Based on the selected terms, accumulative hypergeometric P-values and enrichment factors were calculated and used for filtering. Remaining significant terms were hierarchically clustered into a tree based on Kappa-statistical similarities among their gene memberships. The 0.3 kappa score was applied as the threshold to cast the tree into term clusters. E Representative tracks of target genes showing the occupancy of ANLN and Pol II. F Relative mRNA levels of target genes after ANLN knockdown in KYSE150 cells were measured by RT-qPCR. n = 3 for each group. The error bars indicate the mean ± SDs. Two-tailed Student’s t tests were performed to determine the significance. G H3K27ac ChIP-Seq data from SEdb database were used to identify the super-enhancer regions of representative target genes, and the graph shows the information of each super-enhancer regions. H Representative tracks of super-enhancer regions occupancy of ANLN, Pol II and H3K72ac. I Screen potential transcription factors from the Cistrome database and ANLN interactome. J Occupancy of ANLN, Pol II and different transcription factors in the promoter and super-enhancer regions of representative target gene. K A hypothetical model: ANLN, Pol II and different transcription factors form an active transcriptional cluster at super-enhancer regions. CUT&Tag analysis representative of two independent experiments, and other data are representative of at least three independent experiments with similar results. Source data are provided as a Source Data file.
THZ1 inhibits ANLN-Pol II clusters and ESCC cell proliferation
We previously reported the small-molecule FW-04-806 (F806), which inhibits ANLN expression in the cytoplasm and suppresses mitosis and proliferation of ESCC cells5. However, there is a lack of effective inhibitors targeting the nuclear ANLN function. Inspired by recent evidence51,52, we asked whether super-enhancer inhibitors target the ANLN-Pol II axis in ESCC cells. Small-molecule THZ1 has been shown to suppress the progression of ESCC by inhibiting transcripts regulated by super-enhancers53. To reveal the direct role of THZ1, a treatment condition was determined that did not cause cell morphology changes and cell death (Supplementary fig. 7, 0.1 μM for 3 h). Super-resolution imaging showed that a 3 h THZ1 treatment almost completely dissolved the type III clusters of ANLN-Pol II, with less impact on small-size clusters (Fig. 7A–D). Based on this observation, we wondered whether the target genes of the ANLN-Pol II axis are sensitive to THZ1. To examine this, a previously published transcriptome data of THZ1-treated ESCC cells were downloaded and analyzed53. As expected, THZ1 treatment resulted in global downregulation of steady-state mRNA levels and super-enhancer-regulated genes, and 84 target genes of the ANLN-Pol II axis were found to be particularly sensitive to THZ1 treatment (Fig. 7E, F). RT-qPCR analysis validated several representative target genes and found that THZ1 treatment led to a time-dependent decrease in the expression of these genes (Fig. 7G). Given the previous findings that F806 reduces cytoplasmic ANLN expression and specifically suppresses ESCC cell growth5,54, we subsequently assessed the effect of THZ1 on ESCC cell viability. The MTS cell proliferation assay showed that THZ1 inhibited the viability of seven ESCC cell lines in a concentration-dependent manner, with an IC50 range of approximately 0.009–0.167 μM (Fig. 7H). Notably, compared to ESCC cell lines, THZ1 exhibits a significantly lower cytotoxic effect on normal cell lines such as human embryonic kidney cells (HEK293T) and human umbilical vein endothelial cells (HUVEC) (Fig. 7H). To enhance the treatment efficacy and reduce drug dosage, a dual drug combination strategy was established to simultaneously inhibit the functions of ANLN in both cytoplasm and nucleus. Results showed that the combined effect of F806 and THZ1 was 30-60% greater than the sum of their individual effects, which was validated in three ESCC cell lines (Fig. 7I). These data demonstrate that THZ1 specifically inhibits the type III clusters and target genes of ANLN-Pol II, as well as the proliferation of ESCC cell lines.

A KYSE150 cells were treated with 0.1 μM THZ1 for 3 h, ANLN (pink) and Pol II (green) clusters were observed with super-resolution imaging. Scale bar, 3 μm. B, C Effect of THZ1 on the number of different types of clusters. n = 42 (Control); n = 45 (THZ1). The error bars indicate the mean ± SDs. Two-tailed Student’s t tests were performed to determine the significance. D Relative fluorescence intensity of protein signals per cell per focal plane. n = 42 (Control); n = 45 (THZ1). The error bars indicate the mean ± SDs. Two-tailed Student’s t tests were performed to determine the significance. E, F Heatmap (E) and box plots (F) showing expression changes of all protein-coding genes (Total, n = 11376), super-enhancer-associated genes (SE, n = 652), and target genes (TG, n = 84) with or without THZ1 treatment. The error bars indicate the mean ± SDs. Two-tailed Student’s t tests were performed to determine the significance. G KYSE150 cells were treated with 0.1 μM THZ1, the mRNA expression of the indicated target genes was measured. The error bars indicate the mean ± SDs. Two-tailed Student’s t tests were performed to determine the significance. H The indicated cell lines were treated with different concentrations of THZ1 for 48 h, and cell viability was subsequently analyzed by MTS assay. The error bars indicate the mean ± SDs. I ESCC cell lines were treated with low toxicity THZ1 (0.04 μM) or/and F806 (20 μM) for 48 h, and cell viability was measured by MTS assay. n = 3 for each group. The error bars indicate the mean ± SDs. The data are representative of at least three independent experiments with similar results. Source data are provided as a Source Data file.
Discussion
ANLN is a potential therapeutic target, which is overexpressed in various types of cancer and promotes cancer cell proliferation and progression2. Besides regulating cell division, several studies have shown that ANLN contributes to signal transduction within cancer cells6,48, but it remains uncertain whether this is mediated by nuclear ANLN. The function of nuclear ANLN has been under fierce debate, and it is difficult to define the cancer-promoting mechanism of ANLN. In this study, we identified a direct interacting protein of nuclear ANLN by combining mass spectrometry and biochemical studies, observed their distribution characteristics by super-resolution imaging, and then employed short-term depletion and NLS mutation strategies to study the function of nuclear ANLN, effectively eliminating the interference of mitotic defects. Briefly, we identified nuclear ANLN as a transcriptional regulator that promotes initiated Pol II clustering and target gene expression based on direct interaction with the Pol II large subunit. ANLN was found to regulate the expression of specific gene subsets by mediating the activity of typical and super enhancers. ANLN-Pol II may cooperate with different transcription factors to bind and form active transcriptional clusters in super-enhancer regions. THZ1, a super-enhancer inhibitor, preferentially suppresses the ANLN-Pol II clusters, target gene expression, and ESCC cell proliferation. Our work established a direct relationship between nuclear ANLN and Pol II transcription factory, unveiling the authentic role of ANLN in transcriptional regulation, and provided a crucial insight for cancer research targeting ANLN.
ANLN promotes the formation of Pol II-pS5 clusters, but has a lesser effect on Pol II-pS2 clusters, indicating that ANLN mainly plays a role in the transcription initiation stage (Supplementary fig. 3B–H). However, increasing evidence suggests that transcriptional regulators are frequently involved in multiple transcription steps. For example, the elongation factor SPT5 was reported to regulate Pol II stabilization, enhancer activation, elongation, and termination55. In addition, PHF3 was found to facilitate Pol II phase separation and transcription elongation, while also negatively regulating the mRNA stability of certain genes34. Similarly, RUVBL2 is a positive regulator of Pol II clusters. Although depleting RUVBL2 leads to a global reduction in nascent transcripts, it also disrupts the nonsense-mediated decay (NMD) pathway, resulting in increased mRNA stability24. Therefore, both upregulation and downregulation of static gene expression was observed after RUVBL2 depletion, similar to ANLN knockdown (Fig. 5A). Consistently, a series of proteins involved in RNA metabolism were identified by the ANLN interactome. Therefore, future research should focus on elucidating the functions of ANLN in different transcription steps.
The interactions between intrinsically disordered proteins (IDPs) typically exhibit dynamic and multivalent features. Unlike structured proteins, IDPs lack a fixed fold and their binding partners exhibit multiple interaction modes, particularly dynamic multivalent interactions (DMI)56,57. DMI of IDPs and their partners leads to a dynamic ensemble of protein complexes with heterogeneous conformations, promiscuous binding, stoichiometry, and kinetics, allowing for fine-tuning of the interaction profile serving in promiscuous functional roles56,57. Our data showed that the N-terminal 1-700 aa of ANLN as well as Pol II CTD contained IDR (Fig. 3H). The full-length ANLN interacts with the C-terminal and CTD of Pol II large subunit (Fig. 1G, H), while Pol II large subunit interacts with three short sequences on the IDR of ANLN (Fig. 3K). This complex multiple-site interactions may contribute to the rapid formation and breakage of interactions at the binding interface, which is prevalent in nuclear pore complexes as well as transcription activator-coactivator complexes56. Flory-Huggins theory describes LLSP polymers composed of IDPs through a stickers-and-spacers model36. Glycine and proline tend to form spacers, while stickers are typically composed of aromatic amino acids such as tyrosine, phenylalanine, and tryptophan. The parts involved in attractive interactions are considered as stickers, while the parts scattered between stickers but not significantly driving attractive interactions are regarded as spacers. Non-covalent interactions between stickers within or from different chains induce reversible physical crosslink formation36. We found that deletion of phase separation driving sequences on ANLN IDR weakened the ANLN-Pol II interaction, and these sequences were rich in glycine, proline, and aromatic amino acids (Fig. 3I–K). Therefore, these key amino acids may follow the stickers-and-spacers model to promote multivalent interactions between ANLN and Pol II, thereby generating large condensates.
The regulation of Pol II clusters by nuclear actin, especially under serum stimulation, was previously reported to depend on actin polymerization58. This process requires the participation of N-WASP, an actin nucleation-promoting factor58. However, the precise molecular mechanisms of how actin scaffolds Pol II clusters on specific genes in response to stimuli remain to be elucidated. On the other hand, the actin binding protein Myosin VI has been demonstrated to possess similar functions59, highlighting the extensive role of actin and its binding proteins in Pol II clusters and gene transcription regulation. ANLN was initially identified as an actin binding protein that promotes F-actin recruitment at the contractile rings during mitosis30. However, our data indicate that the promoting effect of ANLN on Pol II clustering is independent of actin binding (Supplementary fig. 3I–M). The promotion of Pol II clustering and phase separation by ANLN is determined by 46 key phase separation driving residues on its IDR (Figs. 3I–O and 4H–J), providing an insight into the regulatory role of actin binding proteins in Pol II clustering.
We attempted to overexpress ANLN in several human normal cells and to observe the expression of 12 target genes. Interestingly, the expression levels of only a few target genes were changed (Supplementary Fig. 8A–D). Therefore, the regulation of most identified target genes by ANLN may be specific in cancer cells. The mechanism of ANLN regulating gene expression may change in different cellular backgrounds, as our data show that ANLN regulates gene expression not only with Pol II, but also with cancer-related transcription factors (Fig. 6I–K and Supplementary Fig. 6B–D). Therefore, exploring the transcriptional regulation function of nuclear ANLN in different normal cells requires a lot of additional work. On the other hand, ANLN has unique non-mitotic functions in different normal cells and tissues, such as regulating neuronal cell migration and growth cone extension by stabilizing the F-actin network at the leading edge7. It was also found that ANLN stabilizes adherens junctions and tight junctions by recruiting RhoA in embryonic cells60. Overall, our future work will further focus on these unique functions and mechanisms of ANLN in normal and cancer cells.
Methods
Cell culture, cell lines generation and cell treatments
KYSE150, KYSE510, KYSE140, KYSE180, KYSE70, TE1, TE3 and NCM460 cells were maintained in RPMI-1640 medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (GIBCO), 100 U/mL penicillin G, and streptomycin. DMEM medium (GIBCO) supplemented with 10% fetal bovine serum was used for HEK293T, HeLa and FHC cell culture. HUVEC cells were grown in a fully supplemented Endothelial Cell Medium (ScienCell). ARPE-19 cells were cultured with DMEM-F12 containing 10% fetal bovine serum. ESCC cell lines were established and provided by Dr. Shimada Yutaka (Faculty of Medicine, Kyoto University, Japan) and Dr. Nishihira (Tohoku University School of Medicine, Japan)61,62,63. NCM460 cells were established and provided by Dr. M.P. Moyer (University of Texas, San Antonio, TX)64. ARPE-19 (GNHu45) and HeLa (SCSP-504) cells were obtained from National Collection of Authenticated Cell Cultures (Shanghai, China). HEK293T (CRL-3216) and FHC (CRL-1831) cells were obtained from American Type Culture Collection (ATCC). Cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2. Cell lines were authenticated by short tandem repeat profiling and were routinely tested for mycoplasma contamination5. For small interfering RNA (siRNA) knockdown, siRNAs targeting human ANLN or negative control siRNA were diluted in Opti-MEM reduced serum medium (Life Technologies, 31985070) and mixed with siRNA Transfection Reagent (Life Technologies, 13778150), according to the manufacturer’s instructions. Cells were harvested 24 or 48 h after transfection5. Dox-induced ANLN-depleted and control cells were generated in KYSE150 cells as previously described65. Short hairpin RNA (shRNA) targeting the 3’ UTR region of ANLN was constructed into a Dox-induced expression plasmid and stably expressed in KYSE150 cells by lentiviral infection. For rapid depletion of ANLN, cells were treated with 2 μg/mL Dox for 24 h. The sequences of siRNA and shRNA are listed in Supplementary Data 1. For 1,6-HD (Sigma, 240117) treatment, HeLa cells were cultured in 24 well plates containing glass coverslips, treated with 5% 1,6-HD for 60 s, immediately fixed with 4% paraformaldehyde and then used for immunofluorescence assay. For THZ1 (MedChemExpress, HY-80013) treatment, KYSE150 cells were cultured in 24 well plates, treated with different concentrations of THZ1 for 3 h, and observed with an inverted microscope to determine a low toxicity concentration, which was selected for immunofluorescence assay. For pre-extraction, cells were treated with cold cytoskeleton buffer (0.5% Triton X-100, 10 mM PIPES pH 6.8, 3 mM MgCl2, 200 mM NaCl, 300 mM sucrose) for 5 min at 4 °C, followed by fixation with 4% paraformaldehyde, and the samples were used for immunofluorescence assay21,22.
Plasmids
CTD-mEGFP and mEGFP were cloned into pET28a-sumo expression vector (gift from Professor Xiong Ji, Peking University, China)24,66. POLR2A-1-845 and 846-1592 were cloned into pGEX-6P-1 vector. Full-length ANLN and its mutants were cloned into pBOBI-C-3×HA, pET32a and pET28a-sumo vectors. The C-terminal of ANLN was fused with mCherry-tag and then cloned into pET28a-sumo vector. pRL-TK, pGLC (pGL3-Control) and pGLB (pGL3-Basic) plasmids were purchased from Promega. All plasmids were sequenced at the Beijing Genomics Institute (BGI). Primers used in the study are listed in Supplementary Data 1.
Western blotting
Cells were lysed in SDS sample buffer and proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a 0.45 μm polyvinylidene fluoride membrane5,67. Subsequently, the membrane was blocked with 5% non-fat milk in TBST for 1 h, and then incubated with antibodies against ANLN (1:3000, Abcam, ab211872), Pol II-total (1:5000, Abcam, ab26721), Pol II-pS5 (1:5000, Abcam, ab5131), Pol II-pS2 (1:5000, Abcam, ab238146), TBP (1:2000, Abcam, ab220788), CDK7 (1:2000, Abcam, ab256787), Lamin B1 (1:10000, Proteintech, 66095-1-Ig), Histone H3 (1:3000, Cell Signaling Technology, 4499S), α-Tubulin (1:5000, Proteintech, HRP-66031), GAPDH (1:5000, Proteintech, 60004-1-Ig), GFP-tag (1:10000, Proteintech, HRP-66002), His-tag (1:5000, TransGen Biotech, HT501-01) and HA-tag (1:3000, Cell Signaling Technology, C29F4). Proteins were detected by incubation with horseradish peroxidase-conjugated secondary antibodies and visualized using ECL substrate (Biosharp). The raw data of western blotting have been submitted to the Source Data file.
Cellular fractionation
Cells were washed with PBS and resuspended in 400 μL of Buffer A (10 mM HEPES pH 7.4, 5 mM MgCl2, 250 mM sucrose, 1% protease inhibitor cocktail) and incubated on ice for 5 min. NP-40 (1%) was added to a final concentration of 0.1%68. Subsequently, nuclei were pelleted by centrifugation at 940 g for 10 min at 4°C. The cytoplasmic fraction was removed and the pellet was washed with 1 mL buffer A. After centrifugation at 940 g for 10 min, the supernatant was removed and the pellet was resuspended in 200 μL buffer B (10 mM HEPES pH 7.9, 150 mM NaCl, 0.1 mM EDTA, 1 mM dithiothreitol, 1% NP40 and 1% protease inhibitor cocktail) and incubated on ice for 10 min. Soluble nuclear proteins were isolated from chromatin by centrifugation at 2600 g for 5 min44. To determine the affinity between Pol II and chromatin, chromatin components were further washed using a high salt buffer (buffer C). Specifically, the pellet was solubilized using sonication in buffer C (10 mM HEPES pH 7.9, 400 mM NaCl, 0.1 mM EDTA, 1% NP40, 1 mM dithiothreitol and 1% protease inhibitor cocktail) and the association of Pol II to chromatin was analyzed by western blotting44,45.
Immunofluorescence assay
Cells on glass coverslips were washed twice with PBS and then fixed with 4% paraformaldehyde for 15 min. After three times washing with PBS, cells were permeabilized with 0.1% Triton X-100 for 8 min. After washing three times with PBS, cells were blocked with 5% BSA for 1 h at 25 °C, and then incubated with antibodies against ANLN (1:2000, Abcam, ab211872), Pol II (1:500, Abcam, ab26721), Pol II-pS5 (1:4000, Abcam, ab5131) or Pol II-pS2 (1:4000, Abcam, ab238146) at 4 °C overnight. Cells were washed three times with PBS and incubated with Alexa fluor 647 donkey anti-rabbit IgG (1:200, Jackson, 711-605-152), Alexa fluor 488 donkey anti-mouse IgG (1:200, Jackson, 715-545-150) and DAPI (1:2000, Beyotime, C1005) for 1 h in the dark. Coverslips were subsequently mounted onto slides containing anti-fading reagent (Beyotime, P0126)5.
PLA assay
For PLA duolink experiment, cells were fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.1% Triton X-100 for 10 min, and blocked with 5% BSA for 1 h. Cells were incubated with ANLN and/or Pol II antibodies overnight at 4 °C. Cells were subsequently treated with a Proximity Ligation Assay reagent (Sigma) according to the manufacturer’s instructions. Briefly, cells were incubated with diluted secondary probes at 37 °C for 1 h, and then ligation solution was added at 37 °C for 30 min. Then, amplification solution was added and incubated at 37 °C for 100 min69. Cells were incubated with DAPI (1:1000) for 10 min and observed using LSM800 confocal laser scanning microscope (Carl Zeiss, Oberkochen, Germany).
Super-resolution imaging and data analysis
Super-resolution imaging within the nucleus was performed using commercialized Hessian-SIM, termed HIS-SIM (High Intelligent and Sensitive SIM) provided by CSR Biotech (Guangzhou) Co., Ltd70,71. Images were acquired using a 100×/1.5 NA oil immersion objective (Olympus). For image reconstruction, SIM Pattern Denoise (notch filter) was used to remove periodic artifacts based on the previous description (equation 17 in supplementary information of Huang et al.70). The parameters of notch filter were as follows: v = 2, a0 = 1/(2*sigma*sigma), sigma=5. In addition, a 3D Pattern Denoise (equation 11 in Gustafsson et al., parameter w = 0.3) was used to suppress reconstruction artifacts72. Sparse deconvolution was performed according to the method and parameters reported by Zhao et al.73. All raw images were super-resolution reconstructed using the same parameters, and those reconstructed images were further analyzed by Fiji software. For cluster identification, the average threshold of at least 20 images was calculated by using the “Auto Threshold” in Fiji software, and then a “12-Infinity (pixel units)” in “Analyze Particles” was employed to measure the number of clusters24. Clusters smaller than 12675 nm2 (12 pixel units) were excluded, and the same threshold was applied to the images in each condition23,24,25. The number of clusters per cell per focal plane from independent fields was counted and normalized, and then statistical analysis was done using a t-test.
DNA FISH combined with immunofluorescence
KYSE150 cells were fixed with 4% paraformaldehyde, and then covered with a reaction chamber to perform the following reactions. The samples were dehydrated and denatured with methanol, and the hybridization buffer (10% formamide in 2×SSC) containing specific targeting probes was added to the chamber and incubated at 37 °C overnight. The samples were washed three times with PBST, followed by ligation of targeting probes in ligation mix (0.2 U/µL T4 DNA ligase in 1×ligation buffer with 0.2 µg/µL BSA) at 25 °C for 3 h. Next, samples were washed three times with PBST and subjected to rolling-circle amplification (1 U/µL Phi29 DNA polymerase, 1×Phi29 buffer, 0.25 µM dNTP, 0.2 µg/µL BSA, 5% glyceryl and 2 µM dUTP) at 30 °C overnight. After washing twice with PBST, samples were incubated with fluorescent detection probes in 2×SSC with 10% formamide at 37 °C for 1 h. For DNA FISH combined with immunofluorescence, the prepared FISH samples were treated with 0.1% Triton X-100 for 15 min at 25 °C, and then washed three times with PBS. Subsequently, samples were incubated with PBS containing 5% BSA for 1 h at 25 °C, and then incubated with the primary antibody at 4 °C overnight. After washing three times with PBS, Alexa fluor secondary antibodies were mixed into PBS and incubated with the samples for 1 h at 25 °C. Samples were washed three times with PBS and then immersed in the anti-fading reagent for super-resolution imaging74. The sequences of DNA FISH probes used in this study are listed in Supplementary Data 1.
Immunoprecipitation assay
Cells were lysed in EBC buffer (50 mM Tris pH 7.5, 120 mM NaCl, 0.5% NP-40, 1% protease inhibitor cocktail), followed by brief sonication75. After centrifugation at 17,600 g for 10 min at 4 °C, the supernatant was incubated with 2 μg ANLN antibody (Santa Cruz, sc-271814) or normal mouse IgG (Santa Cruz, sc-2025) and 20 μL of protein A/G magnetic beads (MedChemExpress, HY-K0202) at 4 °C overnight. The immunoprecipitated protein complexes were washed four times with EBC buffer; the supernatant was removed; the complexes were resuspended in 60 μL SDS sample buffer, boiled for 10 min, and assayed using western blotting. For the interaction between ANLN mutants and Pol II, HEK293T cells were transfected with full-length or mutated HA-ANLN for 48 h, followed by immunoprecipitation using HA magnetic beads (Thermo Fisher Scientific, 88837).
Protein expression and purification
Transetta (DE3) chemically competent cells (TransGen Biotech, CD801-03) were transformed with plasmids overnight, and the cells were grown in LB medium (10 g/L tryptone, 5 g/L yeast extract, 10 g/L NaCl) at 37 °C to an OD600 nm value of 1.0, induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (Amresco, 0487) and incubated at 16 °C for 24 h. Bacteria were collected by centrifugation at 2600 g for 5 min and lysed with GST lysis buffer (4.3 mM Na2HPO4, 1.47 mM KH2PO4, 137 mM NaCl pH 7.3, 0.1% Triton X-100) and 1 mg/mL lysozyme (Sigma, 62971). The lysates were centrifuged at 10,400 g for 10 min, and the supernatant was incubated with GST Bind Resin (Millipore, 70541-5) or His Bind Resin (Millipore, 70666-3) for 3 h at 4 °C. Then, washed six times with GST lysis buffer or His wash buffer (50 mM sodium phosphate, 300 mM NaCl, 20 mM imidazole). Subsequently, the purified protein was obtained by eluting the resin with GST elution buffer (50 mM Tris-HCl pH 8.0 and 50 mM reduced form of glutathione) or His elution buffer (50 mM sodium phosphate, 300 mM NaCl, and 200 mM imidazole) for 1 h. Purified proteins were quantified using a pierce BCA protein assay kit (Thermo Fisher Scientific, 23225). The purity and concentration of the proteins were further confirmed by coomassie brilliant blue staining5,24,76.
In vitro pulldown assay
For GST-pulldown, GST-tag protein was incubated with GST Bind Resin in GST lysis buffer at 4 °C for 2 h. Resins were washed twice with GST lysis buffer and then incubated with His-tag protein for 4 h at 4 °C. Resins were washed six times with PBST buffer and the supernatant was removed. Resins were boiled in SDS sample buffer for 10 min and analyzed by western blotting5,77. For GFP-pulldown, 50 nM mEGFP or CTD-mEGFP was incubated with GFP magnetic beads in 50 μL of binding buffer (25 mM Tris-HCl pH 8.0, 50 mM NaCl, 1 mM DTT, 5% glycerol, and 0.03% Triton X-100) for 1 h at 4 °C. The beads were washed three times with binding buffer, different concentrations of His-ANLN proteins were added, and then incubated on ice for 30 min. The beads were washed three times with binding buffer, the supernatant was removed, then the beads were resuspended in SDS sample buffer and boiled for 10 min. Samples were subsequently detected by western blotting24.
Droplet assay
In vitro droplet assays were used to investigate the physicochemical properties of ANLN and Pol II CTD proteins24,66,78. Different concentrations of ANLN-mCherry or/and CTD-mEGFP proteins were diluted in phase separation buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl and 16% dextran). The mixture was incubated at 25 °C in the dark for 30 min, and then transferred to 96 well glass bottom plate. The droplet formation was immediately examined using an LSM880 confocal laser scanning microscope (Carl Zeiss, Oberkochen, Germany) objective based on the same parameters. Genomic DNA was isolated from KYSE150 cells using a gDNA miniprep kit (Biomiga, GD2211-01), and DNA extracts were treated with RNase and then used for droplet assay. Total RNA was extracted from KYSE150 cells using Trizol (Thermo Fisher Scientific, 15596018), and RNase inhibitor was included for assays involving RNA. The concentration of total RNA or DNA was measured using a Nanodrop 2000 (Thermo Fisher Scientific). For image analysis, Fiji software was used to process the images to measure the area of droplets24. The images were converted to 8-bit and set with an “Auto Threshold”, using “Watershed” to make sure the boundary of the droplet could be identified, and then calculate the area of droplets larger than 5 μm2. To analyze the condensed fraction, the integrated intensities in all droplets of the acquired field (I-in) and the total intensity outside the droplets (I-out) were measured using Fiji. The condensed fraction is calculated as (I-in)/((I-in)+(I-out))24,79. FRAP analysis was performed on a CSIM 110 confocal microscope (Sunny Technology) and imaged immediately after droplet formation. The droplets were imaged once before bleaching. The selected rectangular area on CTD-mEGFP droplets was bleached with 488 nm laser for 1 s, or ANLN-mCherry droplets were bleached with 561 nm laser for 1 s. After bleaching, recovered droplets were imaged every 20 s. Fiji software was used to quantify the fluorescence intensity of the bleached area, and then GraphPad Prism 8 software was used to analyze the molecular diffusion38.
Mass spectrometry analysis
KYSE150 cells were lysed using EBC buffer, and endogenous ANLN was enriched with protein A/G magnetic beads and ANLN antibody. Proteins were separated by SDS-PAGE and visualized using coomassie brilliant blue. The gel slices were cut to cubes and subjected to in-gel digestion. Peptides were redissolved in HPLC buffer A (0.1% formic acid) following desalting and injected into the Nano-LC system (Thermo Fisher Scientific, EASY-nLC 1200). Peptides were separated by a C18 column (75 μm inner-diameter × 25 cm, 3 μm C18) with a 75 min HPLC gradient at a flow rate of 300 nL/min. The HPLC gradient was as follows: 5% to 7% solvent B (0.1% formic acid in 80% acetonitrile) in 2 min, 7% to 22% solvent B in 50 min, 22% to 38% solvent B in 14 min, 38% to 100% solvent B in 3 min and hold for 6 min at 100% solvent B. The HPLC elute was electrosprayed into an Orbitrap Ecilpse Tribrid mass spectrometer, and Spray voltage was set to 2.4 KV. Mass spectrometry analysis was performed in data-dependent mode, with an automatic switch between a full MS scan and an MS/MS scan in the orbitrap. For MS1 survey scan, the resolution was 60,000 and the MS2 spectra were acquired with 15,000 resolution. For database search, MS raw files were processed with Proteome Discoverer 3.0. Trypsin specificity was applied for the peptide sequence search and a maximum of two missed cleavages was allowed. Carbamidomethylation on cysteine was specified as a fixed modification. Oxidation of methionine and acetylation on the peptide N terminus were set as variable modifications. Mass tolerances for precursor ions were set at ± 10 ppm and ± 0.02 Da for MS/MS80.
RNA-Seq and data analysis
KYSE150 cells with ANLN knockdown for 24 and 48 h were harvested for total RNA extraction. Cells were lysed with Trizol reagent and total RNA was extracted according to the manufacturer’s instructions, and then the concentration of RNA was measured by Nanodrop 2000. Samples were delivered to Novogene (Beijing) for poly(A) RNA library generation, and the final libraries were sequenced using Illumina HiSeq Xten PE15024,58. For RNA-Seq data analysis, the paired-end sequencing fastq files were removed adaptors and low-quality reads with Cutadapt V1.18. Whole-transcriptome reads were aligned using the STAR V2.7.3a with the Ensembl Homo_sapiens_Ensembl_GRCh38. And the bam files after removing duplicates were used to count the genes with the featureCounts method in Subred V2.0.058. Differential analysis was performed with DESeq2 V1.44.081. Biological processes and pathway enrichment analyses were performed using the Metascape database82.
CUT & Tag and data analysis
We performed CUT&Tag experiments according to the manufacturer’s instructions (Vazyme, TD904). Briefly, 1 × 105 cells were treated with wash buffer and incubated with Concanavalin A beads for 10 min at 25 °C. Samples were incubated with ANLN, Pol II or H3K27ac antibodies at 4 °C overnight. Then, the diluted secondary antibody was incubated with the samples at 25 °C for 1 h. After washing, the mixture was treated with Dig-300 buffer containing Hyperactive pA/G-Transposon Pro for 1 h at 25 °C. After washing, Trueprep Tagment Buffer L was added to the mixture and incubated at 37 °C for 1 h. Subsequently, 2 μL of 10% SDS and 1 pg spike-in DNA were added to the samples and incubated at 55 °C for 10 min after mixing. Concanavalin A beads were removed, and the supernatant was mixed with DNA extract beads for 20 min at 25 °C. The beads were washed three times, after removing the supernatant, ddH2O was added to resuspend the beads, and then PCR amplification was performed using the primers provided by the kit. VAHTS DNA Clean Beads were added to the sample, and incubated at 25 °C for 5 min after mixing. Supernatant was removed, the beads were washed twice with 80% ethanol, and the DNA on the beads was eluted with ddH2O. DNA libraries were subjected to paired-end sequencing on a single lane of the Illumina NovaSeq system. For the CUT&Tag data analysis, the paired-end reads were aligned to the human genome build GRCh38/hg38 using Bowtie. The MACS2 software was used for peak identification. Alignment files in the bam format were transformed into read coverage files (bigwig format) using DeepTools for visualization in Integrative Genome Viewer. Genomic binding profiles were generated using the bamCompare method in DeepTools. Heatmaps of CUT&Tag signals were generated using computeMatrix and plotHeatmap functions in DeepTools83. To visualize the CUT&Tag signals of Pol II and ANLN on target genes, bigwig files were imported into IGV software to display the binding peaks on target genes84. To investigate super-enhancers, our H3K27ac CUT&Tag data were mapped to the super-enhancer region of KYSE150 cells. The super-enhancer region bed file of KYSE150 cell line was downloaded from SEdb database. Bigwig files were previously generated, and then imported into DeepTools along with super-enhancer bed files to generate genomic binding profiles using computeMatrix46,47.
RT-qPCR
Cells were lysed with Trizol and total RNA was extracted, followed by reverse transcription with HiScript III 1st Strand cDNA Synthesis Kit (Vazyme, R312) to generate cDNA. ChamQ Universal SYBR-qPCR Master Mix (Vazyme, Q711), cDNA and specific primers were mixed, and then RT-qPCR was performed with ABI 7500 (Applied Biosystems, USA). A 2-∆∆Ct method was used to analyze the data and quantify the expression level of genes85. Primers used for RT-qPCR can be found in Supplementary Data 1.
Dual-luciferase reporter assay
Dox-induced ANLN knockdown cells were seeded into a 96 well plate overnight, and then the cells were co-transfected with HA-ANLN, pRL-TK, and pGLC or pGLB plasmids. After 24 h, the medium was replaced with fresh culture containing Dox, and then the cells were cultured for another 24 h. According to the manufacturer’s protocol, cells were lysed using a Dual-Luciferase Reporter Assay System Kit (Promega) and mixed with luciferase assay substrate, and luciferase activity was subsequently measured on a GloMax™ 96 Microplate Luminometer (Promega)86.
ChIP-qPCR
Cells were fixed in 1% paraformaldehyde for 10 min at 25 °C, and then quenched with 125 mM glycine for 5 min, followed by two washes with PBS. Cells were lysed with lysis/wash buffer containing 1% protease inhibitor cocktail and transferred to nuclease-free tubes. Cells were lysed by mechanically pipetting up and down several times. Samples were centrifuged at 17,600 g for 10 min at 4 °C, and the supernatant was removed. Pellets were resuspended in a shearing buffer containing 1% protease inhibitor cocktail and sonicated using a Covaris sonicator. Then, supernatant was transferred to a new nuclease-free tube by centrifugation at 20,400 g for 10 min at 4 °C. The supernatant was incubated with dilution buffer containing 1% protease inhibitor cocktail and ANLN antibody or normal mouse IgG at 4 °C overnight. Subsequently, protein G beads were added to the samples and incubated for 6 h at 4 °C. The beads were washed four times with cold lysis/wash buffer and twice with TE buffer. DNA samples were obtained by reverse crosslinking using 5 M NaCl at 65 °C overnight. Purified DNA was quantified using Qubit dsDNA Assay Kit (Thermo Fisher Scientific, Q33231), then measured by qPCR using specific primers at the promoter region of target genes, and data were analyzed using the 2-∆∆Ct method87. The primers used are shown in Supplementary Data 1.
Cell viability assay (MTS assay)
Cell viability assays were performed using the CellTiter 96® Aqueous One Solution Cell Proliferation Assay kit (Promega, G3581) according to previous protocols. 5000 cells were seeded into 96 well plates overnight, and the culture medium was removed, and the cells were treated with culture medium containing different concentrations of THZ1. Fresh culture medium was replaced after 48 h of incubation. 20 μL MTS was added to each well and cells were incubated at 37 °C for 2 h. Subsequently, the absorbance was measured at 492 nm using an enzyme-labeled instrument (Thermo Fisher Scientific, Multiskan MK3)88.
Statistical analysis
All experiments were performed at least three times independently, unless otherwise indicated in the figure legends. Data are expressed as mean ± standard deviation. Two-tailed Student’s t-tests were performed using GraphPad Prism 8 software, and differences in means were considered statistically significant at P < 0.05. The exact P-value is provided in the corresponding figure or legend.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available within the paper and their Supplementary Information. Source data are provided with this paper. The CUT&Tag and RNA-Seq data generated in this study have been deposited in NCBI’s Gene Expression Omnibus (GEO) repository under GEO series accession code GSE285915 and GSE285916. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the iProX partner repository89,90 with the dataset identifier PXD059453. PSPHunter was used to predict the key amino acids driving phase separation (http://psphunter.stemcellding.org/). The information of super-enhancer regions was obtained from the SEdb database (http://www.licpathway.net/sedb). Transcriptome data of THZ1-treated ESCC cells were obtained from the GEO database (accession code: GSE76860. ChIP-Seq data of transcription factors were downloaded from the Cistrome database (http://cistrome.org/db). Accession code for CSNK2A1 ChIP-Seq data: GSM1415123. Accession code for CEBPZ ChIP-Seq data: GSM935274. Accession code for RUVBL2 ChIP-Seq data: GSM2877266. Accession code for RUVBL1 ChIP-Seq data: GSM2574761. Accession code for UBTF ChIP-Seq data: GSE91935. Accession code for EWSR1 ChIP-Seq data: GSM2472082. Source data are provided with this paper.
References
Piekny, A. J. & Glotzer, M. Anillin is a scaffold protein that links RhoA, actin, and myosin during cytokinesis. Curr. Biol. 18, 30–36 (2008).
Tuan, N. M. & Lee, C. H. Role of Anillin in Tumour: From a Prognostic Biomarker to a Novel Target. Cancers (Basel) 12, 1600 (2020).
Straight, A. F., Field, C. M. & Mitchison, T. J. Anillin binds nonmuscle myosin II and regulates the contractile ring. Mol. Biol. Cell 16, 193–201 (2005).
Ma, H. T. & Poon, R. Y. C. Synchronization of HeLa Cells. Methods Mol. Biol. 1524, 189–201 (2017).
Cao, Y. F. et al. Targeting USP10 induces degradation of oncogenic ANLN in esophageal squamous cell carcinoma. Cell Death Differ. 30, 527–543 (2023).
Wang, D., Chadha, G. K., Feygin, A. & Ivanov, A. I. F-actin binding protein, anillin, regulates integrity of intercellular junctions in human epithelial cells. Cell Mol. Life Sci. 72, 3185–3200 (2015).
Tian, D. et al. Anillin Regulates Neuronal Migration and Neurite Growth by Linking RhoG to the Actin Cytoskeleton. Curr. Biol. 25, 1135–1145 (2015).
Sainsbury, S., Bernecky, C. & Cramer, P. Structural basis of transcription initiation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 16, 129–143 (2015).
Huang, J. & Ji, X. Never a dull enzyme, RNA polymerase II. Transcription 14, 49–67 (2023).
Cho, W.-K. et al. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415 (2018).
Lu, H. et al. Phase-separation mechanism for C-terminal hyperphosphorylation of RNA polymerase II. Nature 558, 318–323 (2018).
Henninger, J. E. et al. RNA-Mediated Feedback Control of Transcriptional Condensates. Cell 184, 207–225.e224 (2021).
Lee, R. et al. CTCF-mediated chromatin looping provides a topological framework for the formation of phase-separated transcriptional condensates. Nucleic Acids Res. 50, 207–226 (2022).
Jaeger, M. G. et al. Selective Mediator dependence of cell-type-specifying transcription. Nat. Genet 52, 719–727 (2020).
Li, C., Li, Z., Wu, Z. & Lu, H. Phase separation in gene transcription control. Acta. Biochim .Biophys. Sin. (Shanghai) 55, 1052–1063 (2023).
Du, M. et al. Direct observation of a condensate effect on super-enhancer controlled gene bursting. Cell 187, 331–344.e317 (2024).
Song, L. et al. Hotspot mutations in the structured ENL YEATS ___domain link aberrant transcriptional condensates and cancer. Mol. Cell 82, 4080–4098.e4012 (2022).
Vreugde, S. et al. Nuclear Myosin VI Enhances RNA Polymerase II-Dependent Transcription. Mol. Cell 23, 749–755 (2006).
Harlen, K. M. & Churchman, L. S. The code and beyond: transcription regulation by the RNA polymerase II carboxy-terminal ___domain. Nat. Rev. Mol. Cell Biol. 18, 263–273 (2017).
Sun, M. et al. NuMA regulates mitotic spindle assembly, structural dynamics and function via phase separation. Nat. Commun. 12, 7157 (2021).
Ratnayeke, N., Baris, Y., Chung, M., Yeeles, J. T. P. & Meyer, T. CDT1 inhibits CMG helicase in early S phase to separate origin licensing from DNA synthesis. Mol. Cell 83, 26–42.e13 (2023).
Petropoulos, M. et al. Transcription–replication conflicts underlie sensitivity to PARP inhibitors. Nature 628, 433–441 (2024).
Chen, X. et al. Study of RNA Polymerase II Clustering inside Live-Cell Nuclei Using Bayesian Nanoscopy. ACS Nano 10, 2447–2454 (2016).
Wang, H. et al. The transcriptional coactivator RUVBL2 regulates Pol II clustering with diverse transcription factors. Nat. Commun. 13, 5703 (2022).
Zhang, H., Shao, S. & Sun, Y. Characterization of liquid–liquid phase separation using super-resolution and single-molecule imaging. Biophys. Rep. 8, 2–13 (2022).
Pancholi, A. et al. RNA polymerase II clusters form in line with surface condensation on regulatory chromatin. Mol. Syst. Biol. 17, e10272 (2021).
Boija, A. et al. Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 175, 1842–1855.e1816 (2018).
Kim, H., Johnson, J. M., Lera, R. F., Brahma, S. & Burkard, M. E. Anillin Phosphorylation Controls Timely Membrane Association and Successful Cytokinesis. PLoS Genet. 13, e1006511 (2017).
Pedersen, R. S. et al. Profiling DNA damage response following mitotic perturbations. Nat. Commun. 7, 13887 (2016).
Oegema, K., Savoian, M. S., Mitchison, T. J. & Field, C. M. Functional analysis of a human homologue of the Drosophila actin binding protein anillin suggests a role in cytokinesis. J. Cell Biol. 150, 539–552 (2000).
Chen, A., Akhshi, T. K., Lavoie, B. D. & Wilde, A. Importin beta2 Mediates the Spatio-temporal Regulation of Anillin through a Noncanonical Nuclear Localization Signal. J. Biol. Chem. 290, 13500–13509 (2015).
Beaudet, D., Akhshi, T., Phillipp, J., Law, C. & Piekny, A. Active Ran regulates anillin function during cytokinesis. Mol. Biol. Cell 28, 3517–3531 (2017).
Zhang, L. & Maddox, A. S. Anillin Curr. Biol. 20, R135–R136 (2010).
Appel, L.-M. et al. PHF3 regulates neuronal gene expression through the Pol II CTD reader ___domain SPOC. Nat. Commun. 12, 6078 (2021).
Sun, J. et al. Precise prediction of phase-separation key residues by machine learning. Nat. Commun. 15, 2662 (2024).
Choi, J.-M., Holehouse, A. S. & Pappu, R. V. Physical Principles Underlying the Complex Biology of Intracellular Phase Transitions. Annu. Rev. Biophys. 49, 107–133 (2020).
Folkmann, A. W., Putnam, A., Lee, C. F. & Seydoux, G. Regulation of biomolecular condensates by interfacial protein clusters. Science 373, 1218–1224 (2021).
Zhou, R. et al. CTCF DNA-binding ___domain undergoes dynamic and selective protein–protein interactions. iScience 25, 105011 (2022).
Shao, W. et al. Phase separation of RNA-binding protein promotes polymerase binding and transcription. Nat. Chem. Biol. 18, 70–80 (2022).
Das, S. K. et al. MYC assembles and stimulates topoisomerases 1 and 2 in a “topoisome. Mol. Cell 82, 140–158.e112 (2022).
Muhar, M. et al. SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. Science 360, 800–805 (2018).
Liu, W. et al. Large-scale and high-resolution mass spectrometry-based proteomics profiling defines molecular subtypes of esophageal cancer for therapeutic targeting. Nat. Commun. 12, 4961 (2021).
Lee, D. et al. Induction of Oxidative Stress Through Inhibition of Thioredoxin Reductase 1 Is an Effective Therapeutic Approach for Hepatocellular Carcinoma. Hepatology 69, 1768–1786 (2019).
Amatullah, H. et al. Epigenetic reader SP140 loss of function drives Crohn’s disease due to uncontrolled macrophage topoisomerases. Cell 185, 3232–3247.e3218 (2022).
Dykhuizen, E. C. et al. BAF complexes facilitate decatenation of DNA by topoisomerase IIα. Nature 497, 624–627 (2013).
Wang, Y. et al. SEdb 2.0: a comprehensive super-enhancer database of human and mouse. Nucleic Acids Res. 51, D280–D290 (2023).
Narita, T. et al. Enhancers are activated by p300/CBP activity-dependent PIC assembly, RNAPII recruitment, and pause release. Mol. Cell 81, 2166–2182.e2166 (2021).
Wang, A. et al. ANLN-induced EZH2 upregulation promotes pancreatic cancer progression by mediating miR-218-5p/LASP1 signaling axis. J. Exp. Clin. Cancer Res. 38, 347 (2019).
Zeng, S. et al. Transcriptome sequencing identifies ANLN as a promising prognostic biomarker in bladder urothelial carcinoma. Sci. Rep. 7, 3151 (2017).
Mei, S. et al. Cistrome Data Browser: a data portal for ChIP-Seq and chromatin accessibility data in human and mouse. Nucleic Acids Res. 45, D658–D662 (2017).
Thandapani, P. Super-enhancers in cancer. Pharm. Ther. 199, 129–138 (2019).
Sengupta, S. & George, R. E. Super-Enhancer-Driven Transcriptional Dependencies in Cancer. Trends Cancer 3, 269–281 (2017).
Jiang, Y. Y. et al. Targeting super-enhancer-associated oncogenes in oesophageal squamous cell carcinoma. Gut 66, 1358–1368 (2017).
Li, L. Y. et al. Macrolide analog F806 suppresses esophageal squamous cell carcinoma (ESCC) by blocking β1 integrin activation. Oncotarget 6, 15940–15952 (2015).
Song, A. & Chen, F. X. The pleiotropic roles of SPT5 in transcription. Transcription 13, 53–69 (2022).
Weng, J. & Wang, W. Dynamic multivalent interactions of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 62, 9–13 (2020).
Arai, M., Suetaka, S. & Ooka, K. Dynamics and interactions of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 84, 102734 (2024).
Wei, M. et al. Nuclear actin regulates inducible transcription by enhancing RNA polymerase II clustering. Sci. Adv. 6, eaay6515 (2020).
Hari-Gupta, Y. et al. Myosin VI regulates the spatial organisation of mammalian transcription initiation. Nat. Commun. 13, 1346 (2022).
Reyes, CiaraC. et al. Anillin Regulates Cell-Cell Junction Integrity by Organizing Junctional Accumulation of Rho-GTP and Actomyosin. Curr. Biol. 24, 1263–1270 (2014).
Shimada, Y., Imamura, M., Wagata, T., Yamaguchi, N. & Tobe, T. Characterization of 21 newly established esophageal cancer cell lines. Cancer 69, 277–284 (1992).
Nishihira, T. et al. Characteristics of two cell lines (TE-1 and TE-2) derived from human squamous cell carcinoma of the esophagus. Gan 70, 575–584 (1979).
Kuriya, Y. et al. A new cell line (TE-3) derived from human squamous cell carcinoma of the esophagus. Tohoku J. Exp. Med. 139, 377–387 (1983).
Moyer, M. P., Manzano, L. A., Merriman, R. L., Stauffer, J. S. & Tanzer, L. R. NCM460, a normal human colon mucosal epithelial cell line. Vitr. Cell Dev. Biol. Anim. 32, 315–317 (1996).
Kappel, S., Matthess, Y., Zimmer, B., Kaufmann, M. & Strebhardt, K. Tumor inhibition by genomically integrated inducible RNAi-cassettes. Nucleic Acids Res. 34, 4527–4536 (2006).
Wang, H., Zhou, R. & Ji, X. Droplet formation assay for investigating phase-separation mechanisms of RNA Pol II transcription and CTCF functioning. STAR Protoc. 4, 102202 (2023).
Zhan, X. H. et al. LOXL2 Upregulates Phosphorylation of Ezrin to Promote Cytoskeletal Reorganization and Tumor Cell Invasion. Cancer Res. 79, 4951–4964 (2019).
Wang, X. et al. RPAP2 regulates a transcription initiation checkpoint by inhibiting assembly of pre-initiation complex. Cell Rep. 39, 110732 (2022).
Zhu, C. et al. Profilin-1 regulates DNA replication forks in a context-dependent fashion by interacting with SNF2H and BOD1L. Nat. Commun. 13, 6531 (2022).
Huang, X. et al. Fast, long-term, super-resolution imaging with Hessian structured illumination microscopy. Nat. Biotechnol. 36, 451–459 (2018).
Fu, Y. et al. Real-time imaging of RNA polymerase I activity in living human cells. J. Cell Biol. 222, e202202110 (2023).
Gustafsson, M. G. L. et al. Three-Dimensional Resolution Doubling in Wide-Field Fluorescence Microscopy by Structured Illumination. Biophys. J. 94, 4957–4970 (2008).
Zhao, W. et al. Sparse deconvolution improves the resolution of live-cell super-resolution fluorescence microscopy. Nat. Biotechnol. 40, 606–617 (2021).
Wu, X. et al. Spatial multi-omics at subcellular resolution via high-throughput in situ pairwise sequencing. Nat. Biomed. Eng. 8, 872–889 (2024).
Inuzuka, H. et al. Acetylation-Dependent Regulation of Skp2 Function. Cell 150, 179–193 (2012).
Cheng, Y. W. et al. P300/CBP-associated factor (PCAF)-mediated acetylation of Fascin at lysine 471 inhibits its actin-bundling activity and tumor metastasis in esophageal cancer. Cancer Commun. 41, 1398–1416 (2021).
Li, D. J. et al. KAT8/SIRT7-mediated Fascin-K41 acetylation/deacetylation regulates tumor metastasis in esophageal squamous cell carcinoma. J. Pathol. 263, 74–88 (2024).
Li, Y. et al. RNA Pol II preferentially regulates ribosomal protein expression by trapping disassociated subunits. Mol. Cell 83, 1280–1297.e1211 (2023).
Li, C. H. et al. MeCP2 links heterochromatin condensates and neurodevelopmental disease. Nature 586, 440–444 (2020).
Zhao, J. et al. A PARylation-phosphorylation cascade promotes TOPBP1 loading and RPA-RAD51 exchange in homologous recombination. Mol. Cell 82, 2571–2587.e2579 (2022).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Zheng, Y.-Q. et al. Discovery and validation of combined biomarkers for the diagnosis of esophageal intraepithelial neoplasia and esophageal squamous cell carcinoma. J. Proteomics 304, 105233 (2024).
Zhang, H. et al. Reversible phase separation of HSF1 is required for an acute transcriptional response during heat shock. Nat. Cell Biol. 24, 340–352 (2022).
Janssens, D. H. et al. Automated CUT&Tag profiling of chromatin heterogeneity in mixed-lineage leukemia. Nat. Genet. 53, 1586–1596 (2021).
Pan, D. et al. Splicing factor hnRNPA1 regulates alternative splicing of LOXL2 to enhance the production of LOXL2Δ13. J. Biol. Chem. 300, 107414 (2024).
Zhang, X. D. et al. The interaction of lncRNA EZR-AS1 with SMYD3 maintains overexpression of EZR in ESCC cells. Nucleic Acids Res. 46, 1793–1809 (2018).
Li, L. Y. et al. Interplay and cooperation between SREBF1 and master transcription factors regulate lipid metabolism and tumor-promoting pathways in squamous cancer. Nat. Commun. 12, 4362 (2021).
Li, R. Y. et al. Cisplatin-induced pyroptosis is mediated via the CAPN1/CAPN2-BAK/BAX-caspase-9-caspase-3-GSDME axis in esophageal cancer. Chem. Biol. Interact. 361, 109967 (2022).
Ma, J. et al. iProX: an integrated proteome resource. Nucleic Acids Res. 47, D1211–d1217 (2019).
Chen, T. et al. iProX in 2021: connecting proteomics data sharing with big data. Nucleic Acids Res. 50, D1522–d1527 (2022).
Acknowledgements
We thank the members of our laboratory for insightful discussions. We thank Prof. Xiong Ji, School of Life Sciences, Peking-Tsinghua Center for Life Sciences, Peking University, for providing the CTD-mEGFP plasmid and valuable suggestions. We thank Dr. Hao-nan Zheng from the Xiong Ji laboratory for engaging in helpful discussions. We thank Dr. Meng-Jie Sun from the College of Life Sciences, Peking University for providing valuable advice. We thank Dr. Shi Yu from the Department of Chemistry, Fudan University for providing valuable suggestions. We thank Lian-Di Liao and Wan Lin from Shantou University Medical College for their assistance in cell culture. We thank Guangzhou CSR Biotech Co., Ltd and their technicians for providing the super-resolution image reconstruction method and helpful discussions. We thank Sunny Technology and their technicians for providing technical support for FRAP. We thank Spatial FISH Biotechnology, Co., Ltd and their technicians for providing technical support for DNA FISH. This work was supported by grants from the National Natural Science Foundation of China (82403130 to Y.F.C., 82273108 to E.M.L., 82173034 to L.Y.X.), the Innovative Team Grant of Guangdong Department of Education (2021KCXTD005 to E.M.L.), 2020 Li Ka Shing Foundation Cross-Disciplinary Research Grant (2020LKSFG07B to E.M.L.), the Open Project of Anhui Provincial Key Laboratory of Tumor Evolution and Intelligent Diagnosis and Treatment (KFKT202406 to E.M.L.), the Natural Science Foundation of Guangdong Province (2022A1515010058 to L.P.), the Postdoctoral Fellowship Program of China Postdoctoral Science Foundation (GZC20231516 to Y.F.C.), and the fellowship from the China Postdoctoral Science Foundation (2024M751924 to Y.F.C.).
Author information
Authors and Affiliations
Contributions
E.M.L., L.Y.X., and Y.F.C. conceived and designed the project. K.Z. provided useful suggestions for the project and helped with mass spectrometry analysis. Y.F.C. wrote the manuscript and performed most of the experiments. H.W. provided technical support and useful suggestions for droplet and pulldown assay. Y.S. and W.Q.S. performed most of the bioinformatic analysis of CUT&Tag and RNA-Seq. B.B.T. helped with the PLA assay. L.P. and Y.M.Z. provided technical support for CUT&Tag and ChIP-qPCR. B.B.T., Y.Q.W., T.F. and H.Y.Z. helped with plasmid construction. L.Y.X. and E.M.L. revised the manuscript, and all authors read and approved the final manuscript. E.M.L., L.Y.X., L.P. and Y.F.C. obtained fundings to support the project and supervised this work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cao, YF., Wang, H., Sun, Y. et al. Nuclear ANLN regulates transcription initiation related Pol II clustering and target gene expression. Nat Commun 16, 1271 (2025). https://doi.org/10.1038/s41467-025-56645-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56645-9
