
Abstract
Human manganese superoxide dismutase (MnSOD) plays a crucial role in controlling levels of reactive oxygen species (ROS) by converting superoxide (\({{{{\rm{O}}}}}_{2}^{\bullet -}\)) to molecular oxygen (O2) and hydrogen peroxide (H2O2) with proton-coupled electron transfers (PCETs). A key catalytic residue, Tyr34, determines the activity of human MnSOD and also becomes post-translationally inactivated by nitration in various diseases associated with mitochondrial dysfunction. Tyr34 has an unusual pKa due to its proximity to the Mn metal and undergoes cyclic deprotonation and protonation events to promote the electron transfers of MnSOD. Neutron diffraction, X-ray spectroscopy, and quantum chemistry calculations in oxidized, reduced and product inhibited enzymatic states shed light on the role of Tyr34 in MnSOD catalysis. The data identify the contributions of Tyr34 in MnSOD activity that support mitochondrial function and give a thorough characterization of how a single tyrosine modulates PCET catalysis. Product inhibition occurs by an associative displacement mechanism.
Similar content being viewed by others
Introduction
About 25% of known enzymes perform redox reactions to promote cellular functions1. These enzymes, known as oxidoreductases, use proton-coupled electron transfers (PCETs)1,2,3. PCETs are required for a wide range of biological functions, including energy generation and DNA synthesis, and oxidoreductase dysfunction leads to many disease states4,5,6,7,8. While PCETs are a fundamental biochemical reaction, how they are facilitated by enzymes is not well understood due to difficulties in unraveling the precise proton transfer and electron transfer steps2,9,10. Some enzymes use multiple proton transfers per electron transfer step or vice-versa11,12,13. Defining how PCET reactions occur in oxidoreductases benefits the understanding of redox-mediated disease states and furthers therapeutic interventions14,15,16,17,18.
Some oxidoreductases use their PCETs to regulate the concentrations of reactive oxygen species (ROS) in cells. These PCETs are crucial as fluctuations of ROS levels stimulate mitophagy and programmed cell death, and excessive levels lead to the damage of DNA, proteins, and lipids19. Impairment of oxidoreductase function promotes several diseases, including cardiovascular and neurological conditions and cancer progression20,21. In particular, the oxidoreductase human manganese superoxide dismutase (MnSOD) is responsible for eliminating superoxide (\({{{{\rm{O}}}}}_{2}^{\bullet -}\)) in the mitochondrial matrix. MnSOD dysfunction contributes to a broad range of diseases including psoriasis, inflammatory bowel disease, multiple sclerosis, cardiovascular disease22, and breast and prostate cancer23,24,25,26. Overall, the PCETs of MnSOD contribute to health and longevity27,28. \({{{{\rm{O}}}}}_{2}^{\bullet -}\) is endogenously produced as a byproduct of the mitochondrial electron transport chain, and MnSOD is the only means to reduce \({{{{\rm{O}}}}}_{2}^{\bullet -}\) in the mitochondrial matrix29,30.
Within the mitochondrial matrix, MnSOD uses PCETs to decrease \({{{{\rm{O}}}}}_{2}^{\bullet -}\) concentrations rapidly and efficiently (kcat/Km > ~ 109 M-1 s-1). In the first half-reaction, \({{{{\rm{O}}}}}_{2}^{\bullet -}\) is oxidized to molecular oxygen (O2) with a Mn3+ ion (k1), and in the second half-reaction, \({{{{\rm{O}}}}}_{2}^{\bullet -}\) is reduced to hydrogen peroxide (H2O2) with a Mn2+ ion (k2). The two half-reactions regenerate the Mn3+ ion28.
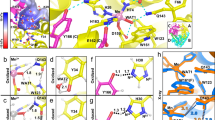
For PCET catalysis, MnSOD uses a hydrogen bond network (dashed blues lines, Fig. 1a right panel) to couple proton transfers with changes in Mn oxidation state28,31,32. The Mn ion is covalently bound to His26, His74, His163, Asp159, and a solvent molecule called WAT1. From WAT1, the hydrogen bond network extends to Asp159, Gln143, Tyr34, WAT2, His30, and Tyr166 from the adjacent subunit. To enter the active site, the substrate and solvent pass through a gateway formed by Tyr34 and His30. The distance between Tyr34(OH) and His30(Nδ1) is 5.4 Å. Hydrophobic residues Trp123, Trp161, and Phe66 hold the hydrogen bonding atoms of Asp159, WAT1, Gln143, and Tyr34 close together33. In our previous work, we investigated the protonation states of the hydrogen bond network in relation to the oxidation state of the Mn ion13.

a The active sites of tetrameric MnSOD are found in positively charged cavities made by two subunits. Solvent and substrate must pass through gateway residues His30 and Tyr34 to interact with the catalytic Mn ion. The distance between Tyr34(OH) and His30(Nδ1) is 5.4 Å. Blue dashed lines indicate hydrogen bonds. The inset indicates the chain identity derived crystallographically, where the asymmetric unit is composed of an AB dimer, and the CD dimer is generated through symmetry to form the native tetrameric assembly. b Room temperature neutron structures revealed changes in protonation among WAT1 and Gln143 and alterations in the hydrogen bond network of WAT1-Gln143-Trp123. Dotted lines indicate hydrogen bonds ≥ 1.8 Å while hashed lines indicate SSHBs that are hydrogen bonds < 1.8 Å. c A shared proton, indicated with rounded dots, was observed between His30 and Tyr166. This proton helps modulate the protonation state of Nδ1(His30) when the Mn ion changes oxidation states. d Tyr34 is deprotonated in Mn3+SOD and protonated in Mn2+SOD. There is no water within hydrogen bond distance on Tyr34 in Mn2+SOD. e Superposition of wildtype MnSOD (yellow, PDB ID 5VF9) and Tyr34Phe MnSOD (red, PDB ID 9BWR) active sites with a root-mean-square deviation of 0.07 Å among Cα atoms. Panel a was created from MnSOD X-ray structure (PDB ID 5VF9)70, and panels (b–d) were created from the neutron structures of Mn3+SOD (PDB ID 7KKS) and Mn2+SOD (PDB ID 7KKW)13. All hydrogen positions were experimentally determined except for solvent molecules in panel a that were randomly generated to accentuate the solvent in the active site funnel. All distances are in Å.
Using neutron crystallography, we previously observed how changing the Mn oxidation state shifts the pKa of active site amino acids and leads to several unusual protonation states13. Neutron diffraction is advantageous for studying PCET mechanisms because the scattering of deuterium is on par with carbon, nitrogen, and oxygen, and neutrons do not photoreduce metal ions, unlike X-rays34,35. The neutron structures of Mn3+SOD and Mn2+SOD revealed three important observations on how PCET catalysis is facilitated. First, during the Mn3+ to Mn2+ redox transition, an unconventional proton transfer occurs between Gln143 and metal-bound −OH(WAT1), leading to an unusual Gln143 amide anion and H2O (Fig. 1b). The amide anion is stabilized by two short-strong hydrogen bonds (SSHBs) with WAT1 and Trp123. SSHBs stabilize catalytic steps and enhance kinetic rates (hashed lines, Fig. 1b)36,37,38. Second, the Nδ1 of His30 is protonated only in the Mn2+ oxidation state (Fig. 1c) and a low-barrier hydrogen bond (LBHB) is formed between Tyr166 and His30 in the Mn2+ state. A LBHB is a type of SSHB where the heteroatoms transiently share a proton, and the hydrogen bond distances are nearly equivalent (1.2–1.3 Å)39. Third, Tyr34 is deprotonated in the Mn3+ oxidation state and becomes protonated in the Mn2+ state (Fig. 1d). We concluded that Tyr34 and His30 are probably the two proton donors needed during the Mn2+ to Mn3+ redox transition to form H2O2 from the protonation of \({{{{\rm{O}}}}}_{2}^{\bullet -}\) (k2)28,32,40,41. Overall, the previous MnSOD neutron structures indicate that multiple proton transfers are coupled to electron transfer events and that the active site metal leads to residues with unconventional pKas essential for PCET catalysis.
How \({{{{\rm{O}}}}}_{2}^{\bullet -}\) interacts with the active site for catalysis is unclear28,31,32. \({{{{\rm{O}}}}}_{2}^{\bullet -}\) may bind the Mn ion directly for an inner-sphere electron transfer or between His30 and Tyr34 for a long-range outer-sphere electron transfer with the Mn ion28,31,32,41. The inner sphere binding site for \({{{{\rm{O}}}}}_{2}^{\bullet -}\) is unknown and could form a six or five coordinate active site binding. The sixth site is predicted to bind opposite Asp15942. Five coordinate binding would displace Asp159 or WAT143. Quantum chemistry computational studies have postulated that the first half-reaction (k1, Mn3+ → Mn2+) proceeds through an inner-sphere electron transfer while the second half-reaction (k2, Mn2+ → Mn3+) proceeds through an outer-sphere electron transfer41. The outer-sphere mechanism for the second half-reaction is a promising hypothesis because His30 and Tyr34 have been shown to lose protons during the Mn2+ → Mn3+ redox transition (Fig. 1c, d)13. His30 and Tyr34 could protonate \({{{{\rm{O}}}}}_{2}^{\bullet -}\) in concert with long-range electron transfer to form the H2O2 product. While there is no structural evidence for how \({{{{\rm{O}}}}}_{2}^{\bullet -}\) interacts with the active site for electron transfer, modeling, and quantum chemical calculations support the second half-reaction proceeding through an outer-sphere mechanism41.
Human MnSOD is inhibited by its product, H2O2, to regulate the output of mitochondrial H2O2. Physiologically, MnSOD product inhibition is thought to be related to H2O2 acting as a second messenger31,32,44. Mitochondrially-derived H2O2 plays a role in apoptosis45,46, mitochondrial biogenesis44, and protein localization and activity47. Furthermore, mitochondrial H2O2 has been shown to modulate the activity of PTEN (phosphatase and tensin homolog), a protein tyrosine phosphatase that contributes to cancer and neurological disease48,49. Mitochondrial H2O2 may also be scavenged by peroxiredoxins (PRDXs) and glutathione peroxidases (GPXs). These enzymes are dependent on nicotinamide adenine dinucleotide phosphate (NADPH) for function50,51,52. MnSOD product inhibition regulates mitochondrial H2O2 levels independent of metabolic status. However, little is known about how MnSOD mechanistically achieves product inhibition.
Product inhibition occurs when the ratio of \({{{{\rm{O}}}}}_{2}^{\bullet -}\) to MnSOD is high31,32. At concentrations of \({{{{\rm{O}}}}}_{2}^{\bullet -}\) that are lower than enzyme, catalysis proceeds rapidly through the first-order reactions k1 and k2. At concentrations of \({{{{\rm{O}}}}}_{2}^{\bullet -}\) that are much greater than enzyme, catalysis proceeds through the reversible inhibition reactions k3 (first-order) and k4 (zero-order). The k3 reaction forms a product-inhibited complex (indicated by square brackets) that is unreactive to \({{{{\rm{O}}}}}_{2}^{\bullet -}\) and only disassociates through the zero-order k4 reaction that dictates the lifetime of the complex. Several kinetic models suggest that inhibition is initiated from the reaction of a Mn2+ ion with \({{{{\rm{O}}}}}_{2}^{\bullet -}\) in the absence of protonation to form [Mn3+-O22−] or [Mn3+-−OOH] (k3)31,32. The inhibited complex is then relieved by at least one protonation to yield Mn3+ and H2O2 (k4). Since k2 and k3 both use Mn2+ and \({{{{\rm{O}}}}}_{2}^{\bullet -}\) as reactants, they are competing reactions, and their ratios determine the propensity for MnSOD to become product-inhibited. Human MnSOD has equal k2 and k3 (Supplementary Table 1), which means at high \({{{{\rm{O}}}}}_{2}^{\bullet -}\) to enzyme ratios, ~50% of Mn2+ reactions with \({{{{\rm{O}}}}}_{2}^{\bullet -}\) form the inhibited complex53,54. The factors that govern k3 and k4 are unclear, although Tyr34 appears to contribute to the inhibition process55,56.
or
Tyr34 is a conserved residue in the active site of MnSOD55,57. Physiologically, the residue becomes nitrated in several human neurodegenerative diseases, leading to inactivated MnSOD58. During the PCET mechanism, Tyr34 is thought to be one of the sources of protons needed to produce H2O2 during the Mn2+ to Mn3+ redox transition28,32,57. Our previous neutron structures show that Tyr34 is poised to donate a proton in Mn2+SOD (Fig. 1d)13. Interestingly, the Tyr34Phe MnSOD variant is unable to proceed through the fast Mn2+ to Mn3+ redox transition (k2), and catalysis proceeds exclusively through the product-inhibited pathway k3 (see rate constants listed in Supplementary Table 1). When k2 <<k3 then ~99% of Mn2+ reactions with \({{{{\rm{O}}}}}_{2}^{\bullet -}\) form the inhibited complex. There is also higher retention of the inhibited complex, with half the disassociation rate compared to wildtype (see k4, Supplementary Table 1). Since the active site structure of the Tyr34Phe variant is practically identical to wildtype (Fig. 1e)57, the perturbed kinetics and enrichment of product inhibition of the Tyr34Phe variant are probably due to the loss of the hydroxyl group for proton transfer. Interestingly, the Trp161Phe variant, which does not eliminate a hydroxyl group, also primarily uses the product inhibited pathway. In fact, Trp161Phe has similar kinetics to the Tyr34Phe variant (Supplementary Table 1)33. This suggests that the contribution of Tyr34 toward catalysis is not solely proton transfer. To study the effects that govern the reactions k1-k4 of MnSOD, we performed a series of experiments with the Tyr34Phe variant.
Several studies have reported that mixing high H2O2 concentrations with Mn3+SOD leads to the formation of the product-inhibited complex that decays to Mn2+SOD33,59,60,61. Mutagenesis studies found the process to be correlated with the forward reactions k2-k4. These puzzling results may be explained by excessive concentrations of H2O2 initiating a backward reaction with Mn3+SOD to produce \({{{{\rm{O}}}}}_{2}^{\bullet -}\), and then a subsequent forward reaction of \({{{{\rm{O}}}}}_{2}^{\bullet -}\) with MnSOD to form the inhibited complex60,61. Importantly, this would provide an avenue to structurally analyze product inhibition without relying on volatile \({{{{\rm{O}}}}}_{2}^{\bullet -}\) solutions62. We combined H2O2 soaking with the Tyr34Phe variant to study the product-inhibited complex and gain new information on the MnSOD PCET mechanism and an enhanced understanding of product inhibition.
To help understand the role of the hydroxyl group of Tyr34 in MnSOD activity we used the Tyr34Phe MnSOD variant in combination with neutron crystallography, X-ray absorption spectroscopy (XAS), and quantum mechanical (QM) chemistry calculations. With neutron crystallography, we captured the product-bound, reduced, and oxidized states of Tyr34Phe MnSOD free from radiation-induced perturbations. For each state, we used XAS to determine the oxidation state and coordination number of the manganese with each treatment, and to probe the electron orbitals of the Mn ion. Solution XAS data was useful in guiding the interpretation of neutron crystal structures that sometimes have asymmetric active site structures between subunits due to the crystal lattice. Then, we used QM calculations to quantitatively interrogate the Mn ion orbitals that determine redox activity. Lastly, the Tyr34Phe data were compared with wildtype13 and Trp161Phe63 to piece together a MnSOD mechanism that describes the reactions k1-k4. With respect to oxidoreductases in general, our work presents a thorough characterization of how a single tyrosine modulates PCET catalysis. Our main finding is that Tyr34 plays a part in every MnSOD kinetic step: proton donor/acceptor, orienting nearby residues for rapid proton transfer, limiting the product inhibited complex, and keeping the lifetime of the inhibited state short. Thus, Tyr34 in MnSOD is a central regulator of H2O2 levels in the mitochondria and signaling.
Results and discussion
X-ray absorption spectroscopy (XAS) characterization of Tyr34Phe MnSOD
Tyr34Phe MnSOD was selected for this study to help understand the role of Tyr34’s hydroxyl group on the MnSOD PCET and product inhibition mechanisms. Solution XAS data is useful in guiding the interpretation of neutron crystal structures. In Mn K-edge XAS, the X-ray absorption near edge structure (XANES) contains information on the oxidation and coordination states of the Mn, while the extended X-ray absorption fine structure (EXAFS) region provides information on Mn-ligand bond distances. The Tyr34Phe MnSOD variant is advantageous in this study because it accumulates and retains the product-inhibited complex, and from that, we can infer information about the PCET mechanism (see Supplementary Table 1 for rate constants). First, we measured the XANES spectral signature to characterize four states of Tyr34Phe MnSOD: oxidized, reduced, H2O2 soaked, and \({{{{\rm{O}}}}}_{2}^{\bullet -}\) soaked (see Supplementary Fig. 2 for all Tyr34Phe, Trp161Phe and WT MnSOD HERFD-XANES spectra, and structural comparisons).
The energy of the rising edge of the XANES region increases with higher oxidation states, while the intensity of the pre-edge contains information about the coordination of the Mn ion (Fig. 2a). The pre-edge corresponds to 1 s to 3 d orbital transitions, with gains in intensity from 4p character mixing into the 3 d orbitals from symmetry distortions and/or loss of inversion symmetry64,65,66. Based on electron paramagnetic resonance (EPR) data, the Mn ion is expected to be high-spin in both Mn3+ and Mn2+ states67. For oxidized Tyr34Phe MnSOD, the rising edge is observed at higher energy than the reduced data, indicating a difference in the Mn ion valency. Given that these samples were mixed with stoichiometric excesses of redox reagents, we interpret oxidized Tyr34Phe MnSOD as Mn3+ and reduced Tyr34Phe MnSOD as Mn2+. For both oxidized and reduced Tyr34Phe MnSOD, the pre-edge intensity (i.e., the area under the curve, inset Fig. 2a) corresponds to a distorted five-coordinate trigonal bipyramidal complex that has been observed in crystal structures57,68. A strong pre-edge intensity requires electric dipole allowed transitions that can only occur due to the presence of Mn 4p character mixed into the 3 d orbitals. Such a mixing requires the absence a local inversion center around the Mn ion as is the case for a 5-coordinated complex. For a 6-coordinated complex, a sufficient amount of 4p character could only be reached by one short Mn-ligand bond distance, as it is observed for example for some Fe-O complexes66. However, neither our neutron scattering, XAS or DFT data suggest such a geometry.

a The HERFD-XANES region of Tyr34Phe MnSOD treated with potassium dichromate to isolate the Mn3+SOD resting state, sodium dithionite to isolate the Mn2+SOD resting state, and either superoxide or hydrogen peroxide to isolate the product-inhibited state. b Overlay of the Fourier transform of Mn K-edge EXAFS data [k2χ(k)] from superoxide- and peroxide-soaked Tyr34Phe MnSOD with the EXAFs spectrum in k-space shown in the inset. Due to the scattering phase shift, the distance found by the Fourier Transformation (R) is ~0.5 Å shorter than the actual distance and a 0.5 Å correction (α) was implemented. c, d Superoxide- and peroxide-soaked Tyr34Phe MnSOD with the EXAFs spectrum. Colored lines represent experimental data, while the black line is simulated EXAFS spectra fit to the experimental data.
To isolate the product-inhibited complex with XAS, we introduced \({{{{\rm{O}}}}}_{2}^{\bullet -}\) or H2O2 to Tyr34Phe MnSOD. \({{{{\rm{O}}}}}_{2}^{\bullet -}\) was used to isolate the complex from the forward reaction and H2O2 was used to attain the complex with the back reaction60,61. For both approaches, the inhibited complex has been reported to form within 44 ms of mixing with Tyr34Phe MnSOD60. Indeed, the approaches yield nearly identical XANES spectra and reveal that the same electronic structure is formed (Fig. 2a). The superoxide and peroxide-soaked spectra are like Tyr34Phe Mn2+SOD, indicating the presence of a divalent Mn ion. However, these spectra have unique features relative to Tyr34Phe Mn2+SOD, with lower intensities at ~6550-6555 eV and higher intensities at ~6565-6575 eV. Similarly, the pre-edge intensities are like Tyr34Phe Mn2+SOD to indicate that the resulting complex from either mixing \({{{{\rm{O}}}}}_{2}^{\bullet -}\) or H2O2 is five-coordinate (Fig. 2a). Altogether, by using the Tyr34Phe MnSOD variant that enriches for the product-inhibited complex, we show that an electronically distinct five-coordinate Mn2+ complex forms from either exposure to \({{{{\rm{O}}}}}_{2}^{\bullet -}\) or H2O2.
Next, we investigated the EXAFS region of the \({{{{\rm{O}}}}}_{2}^{\bullet -}\) and peroxide-soaked Tyr34Phe MnSOD K-edge spectra. The Fourier transform of the EXAFS data, χ(k), yields χ(R) that provides information on the atomic radial distribution (bond lengths) around the absorbing Mn ion (Fig. 2b–d). Overall, the k-space (inset) and R-space spectra are highly similar for \({{{{\rm{O}}}}}_{2}^{\bullet -}\) and H2O2 soaked Tyr34Phe MnSOD and yield the same Mn bond distance solutions (Table 1). For both samples, the first shell of coordination observed at ~2.1 Å is best fit by three N atoms at 2.15 Å and two O atoms at 2.11 Å to indicate a five-coordinate complex (Supplementary Table 2 and 3). These account for Nε2 atoms of His26, His74, and His163, Oδ2 atom of Asp159, and another O atom (denoted O1). As the next closest scatterer is a single atom found at 2.44 Å (denoted O2), we interpret O1 and O2 as belonging to a dioxygen species. The largest contributors to the second peak seen at ~3.0 Å are seven carbon atoms that correspond to the Cδ2 and Cε1 of the three histidines and the Cγ of the aspartate residue. Overall, the EXAFS data show that when \({{{{\rm{O}}}}}_{2}^{\bullet -}\) or H2O2 are given to Tyr34Phe MnSOD, a five-coordinate Mn complex that includes a dioxygen species is formed.
Our XAS data indicate that H2O2 has both a reducing effect on MnSOD and can form the inhibited complex. The reducing effect of MnSOD by H2O2 has been reported previously59,60. Thermodynamically, the process is unfavorable as the reduction potential of wildtype Mn3+SOD to Mn2+SOD is 0.4 V, and the oxidation potential of H2O2 to \({{{{\rm{O}}}}}_{2}^{\bullet -}\) is -0.85 V69. However, a 100-fold excess of H2O2 was used, probably driving a backward reaction60,61. Mutagenesis studies have indicated that forming the inhibited complex through peroxide-soaking depends on reactions k2-k4 and suggests that the \({{{{\rm{O}}}}}_{2}^{\bullet -}\) generated from the backward reaction then reacts with MnSOD in the forward direction60. In these past experiments, detection of \({{{{\rm{O}}}}}_{2}^{\bullet -}\) was lost during the dead time of the instrument (1.4 ms). As H2O2 is more stable than \({{{{\rm{O}}}}}_{2}^{\bullet -}\) in aqueous solution, we used H2O2 for the structural studies on Tyr34Phe MnSOD.
Neutron structure of the Tyr34Phe product-inhibited complex
To visualize the product-inhibited complex and the corresponding active site protonation states, we solved a cryocooled neutron structure of perdeuterated Tyr34Phe MnSOD soaked with deuterium peroxide (D2O2) at 2.30 Å resolution. For the neutron structures, proton positions can be assigned at 2.5 Å or better. We first solved the all-atom structure of the entire enzyme, excluding the active site. Then, with these phases, we carefully interrogated the active site nuclear density maps for the active site coordination and protonation states.
For chain A, the omit |Fo | - |Fc| nuclear scattering-length density next to the Mn ion was oblong at 3.0 σ (Fig. 3a). We interpreted the density as a dioxygen species with a single proton (denoted as LIG for ligand) that has taken the place of the WAT1 position observed in the resting states (Fig. 1b). The proton of LIG, D2, points toward Trp161 and appears to be interacting with the pi system (Fig. 3a). The O2(LIG) and Dε21(Gln143) atoms are close in proximity (2.2 Å apart) though LIG and Gln143 are not in optimal geometry for hydrogen bonding (Figs. 3a, b and Supplementary Fig. 1a). LIG refined well at full occupancy and led to Mn bond distances that closely resemble those measured from the EXAFS spectra (Table 1). At physiological temperatures, optical absorption spectra in the literature also suggest a displacement of WAT1 and binding of a dioxygen species, which agrees with our cryocooled diffraction data43. For chain B, the omit |Fo | - |Fc| is instead a spherical shape at 3.0 σ (Supplementary Fig. 1b), indicating a -OD molecule that has been previously observed in wildtype neutron structures13. Contrasting structures between the two subunits of the asymmetric unit are often observed for the MnSOD P6122 crystal form due to differences in solvent accessibility13,70. In chain B, the six-coordinate nuclear density near the Mn ion and −OD molecule did not correspond with the five-coordinate XAS data, making it less useful in terms of defining the PCET mechanism (Supplementary Fig. 1b). Regardless, from the nuclear density at chain A, a singly protonated dioxygen species replaces WAT1 upon D2O2 soaking and forms a complex with bond distances like those found from EXAFS spectra (Table 1).

a D2O2-soaked Tyr34Phe MnSOD for chain A with a singly-protonated dioxygen species denoted LIG. The inset highlights the elongated |Fo | - |Fc| difference density of LIG. b D2O2-soaked Tyr34Phe MnSOD for chain A rotated 90° along the horizontal axis relative to panel (a). c Tyr34Phe Mn2+SOD for chain A. Notably, this neutron structure is the lowest resolution, 2.5 Å of the three in Supplementary Table 6. d Active site overlay of wildtype Mn2+SOD (PDB 7KKW, solved at 2.3 Å resolution) and Tyr34Phe Mn2+SOD (this study, solved at 2.5 Å resolution). e Tyr34Phe Mn3+SOD for chain A. f Active site overlay of wildtype Mn3+SOD (PDB 7KKS, solved at 2.3 Å resolution) and Tyr34Phe Mn3+SOD (this study, solved at 2.3 Å resolution). Green, orange, and magenta omit |Fo | - |Fc| difference neutron scattering length density of protons displayed at 2.5σ, 3.0σ, and 3.5σ. respectively. Light blue 2|Fo | - |Fc| density is displayed at 1.0σ. Distances are in Å. Dashed lines indicate hydrogen bonds ≥ 1.8 Å, and hashed lines indicate SSHBs that are hydrogen bonds < 1.8 Å. Chain B active sites are shown in Supplementary Fig. 1.
How the product-inhibited complex is formed as well as its oxidation state and coordination, have been historically controversial31,32,61,71,72,73. The generally accepted mechanism is that inhibition is initiated from \({{{{\rm{O}}}}}_{2}^{\bullet -}\) binding Mn2+ to form a Mn3+-O22- complex (k3, Supplementary Table 1)28,31,32,74,75. Our XAS data indicates a five-coordinate Mn2+ as the identity of the complex59,60. Indeed, the neutron structure of Tyr34Phe MnSOD soaked with D2O2 yields a five-coordinated active site with a singly-protonated dioxygen species. The Mn bond distances of the solved neutron structure with Mn2+ and a hydroperoxyl anion−,O2H, resemble those from the EXAFS data. Also, the density function theory (DFT) calculations using Mn2+ and −O2H most closely match the neutron structure (Table 1). Overall, these data and calculations indicate that the inhibited complex is a five-coordinate Mn2+ complex where the WAT1 position has been replaced with −O2H.
Tyr34 orients the Gln143-WAT1 SSHB of Mn2+SOD and limits product inhibition
We next wondered whether enrichment of the product-inhibited complex in the Tyr34Phe variant was because of structural perturbations near the Mn ion or due to the absence of the Tyr34 hydroxyl group for proton transfer. To this end, we solved neutron structures of reduced and oxidized Tyr34Phe MnSOD at 2.5 and 2.3 Å resolution, respectively. For reduced Tyr34Phe MnSOD, the Mn bond distances are similar to five-coordinate wildtype Mn2+SOD (Supplementary Table 4). Furthermore, the protonation states resemble those of wildtype Mn2+SOD, where WAT1 is of the D2O form while Gln143 is deprotonated to the amide anion (Fig. 3c). Deprotonated amino acids are identified when attempts to model and refine a proton result in negative |Fo | - |Fc| difference neutron scattering length density, and all the other protons of the amino acid can be placed. Interestingly, the lack of the hydroxyl group in Tyr34Phe Mn2+SOD perturbs the orientation of Gln143 and lengthens the WAT1-Gln143 hydrogen bond (Fig. 3d). The WAT1-Gln143 SSHB of the wildtype enzyme is critical for the back-and-forth proton transfers needed for redox cycling of the Mn ion13, and the Tyr34Phe Mn2+SOD neutron structure suggests one role Tyr34 plays in catalysis is correctly positioning Gln143 for rapid PCET catalysis. Indeed, the Mn2+ to Mn3+ redox transition is greatly diminished for Tyr34Phe MnSOD (k2, Supplementary Table 1)31,32. Another consequence of the perturbation of the WAT1-Gln143 interaction is the potentially easier displacement of WAT1 by a dioxygen species to form the product-inhibited complex. Overall, the reduced Tyr34Phe MnSOD neutron structure suggests that Tyr34 orients Gln143 for a tight hydrogen bonding interaction with WAT1.
For oxidized Tyr34Phe MnSOD, the Mn ion is like wildtype with a bound −OD molecule (Fig. 3e) with similar covalent bond distances (Supplementary Table 4), and identical hydrogen bond distances for O(WAT1)-Dε21(Gln143) and Dε1(Trp123)-Oε1(Gln143) (Fig. 3f). As PCET mechanisms depend on the distribution of electrostatic vectors to propagate proton and electron transfers, the missing anionic deprotonated Tyr34 in this variant probably perturbs enzyme kinetics. Such electronic effects are indicated by the lower kinetic rates of the Mn3+ to Mn2+ redox transition for the Tyr34Phe variant (k1, Supplementary Table 1). Overall, the interactions between WAT1, Gln143, and Trp123 are the same in Tyr34Phe and wildtype Mn3+SOD making this variant. Because these active sites are practically identical the use of Tyr34Phe MnSOD and its handy kinetic data to study the product inhibited state is valid and can help interpret the wildtype mechanism.
The neutron structures of reduced and oxidized Tyr34Phe Mn2+SOD suggest the roles of Tyr34 in catalysis are (1) orient Gln143 for efficient interaction and proton transfer with WAT1 during the Mn2+ to Mn3+ PCET reaction, (2) limit formation of the product-inhibited complex by strengthening the WAT1-Gln143 hydrogen bond, and (3) contribute subtle electronic effects as a phenolate anion during the Mn3+ to Mn2+ reaction. These interpretations are supported by the kinetic rates of Tyr34Phe MnSOD31,32. For Tyr34Phe MnSOD, the fast Mn2+ to Mn3+ redox reaction is dramatically reduced (k2, Supplementary Table 1), formation of the product-inhibited complex is enriched (k3 >> k2, Supplementary Table 1), and the Mn3+ to Mn2+ redox reaction is cut in third (k1, Supplementary Table 1). A unifying theme among Tyr34Phe MnSOD and other variants studied kinetically is that the precise orientation of Gln143 is critical for catalysis. Mutating residues directly adjacent to Gln143, such as Trp161, Trp123, and Tyr34, lead to similar kinetic consequences, where the Mn2+ → Mn3+ half reaction is greatly diminished, product inhibition is enriched, and the Mn3+ → Mn2+ half reaction is slower compared to wildtype31,32. These redundant effects of the Trp161Phe, Trp123Phe, and Tyr34Phe variants suggest that a crucial role of residues neighboring Gln143 is to enforce a SSHB between WAT1 and Gln143. In the case of Tyr34, our neutron structures indicate that Tyr34 is responsible for positioning Gln143 for a SSHB with WAT1 during the Mn2+ resting state and that the strength of the bond between Gln143 and WAT1 correlates with the extent of product inhibition.
Retention of the product-inhibited complex is dependent on the Gln143 position
As the XAS and neutron crystallographic data of Tyr34Phe MnSOD demonstrated the formation of a five-coordinated Mn2+SOD with a singly-protonated dioxygen species replacing the WAT1 position, we wondered if the formation and retention of the inhibited complex could be distinguished with other variants of MnSOD. In the context of Tyr34Phe MnSOD, we also sought to define whether the enrichment of product inhibition was due to the loss of the ionizable Tyr group only or also due to the perturbation of the Gln143-WAT1 SSHB interaction. For example, the Trp161Phe MnSOD variant alters a residue directly adjacent to Gln143 and has enriched product-inhibition kinetics like those of Tyr34Phe MnSOD (Supplementary Table 1)59. For wildtype, Tyr34Phe, and Trp161Phe, we performed XANES in the HERFD mode of detection, which allows a large improvement in energy resolution and sensitivity compared to conventional XANES76,77,78. Here, we focused on comparing the reduced form of the variants to those of peroxide-soaked counterparts. As all these complexes are expected to be five-coordinated d5 with spin S = 5/2, we sought to distinguish fine details of the spectra offered by the HERFD mode of detection (detailed evaluation of these data can be found in the Supplementary Information pages 10-13 and Supplementary Fig. 3). Overall, reduced and peroxide-soaked Tyr34Phe MnSOD spectra had distinct features between each other, and the general shape trends were reproducible with conventional XANES (Fig. 2a) and HERFD-XANES (Supplementary Fig. 3a). Similar trends were observed between reduced and peroxide-soaked forms of Tyr34Phe and Trp161Phe MnSOD (Supplementary Fig. 3ab). Since the wildtype enzyme exhibits physiological product inhibition levels (k3 = k2), compared to the Tyr34Phe and Trp161Phe variants that are enriched for product inhibition (k3 >> k2 and lower k4 compared to wildtype, Supplementary Table 1), a potential explanation for the less pronounced differences for the wildtype pair is that the peroxide-soaked data may reflect a mixture of species rather than a fully isolated product-bound complex (Supplementary Fig. 3c). The XERFD-XANES data for peroxide soaked Y34F, W161F and wild-type are similar and follow these trends (Supplementary Fig. 3d). These observations indicate that retention of the inhibited complex is not exclusively dependent on a proton transfer from Tyr34 but instead on other factors of the active site. We compared our D2O2-soaked Tyr34Phe MnSOD neutron structure with that of our previous D2O2-soaked Trp161Phe MnSOD neutron structure (Supplementary Fig. 3e, PDB ID 8VHW63 and concluded that the LIG orientation in Tyr34Phe MnSOD is probably close to what would occur physiologically. When the reduced structures are compared, both variant structures have a lengthened WAT1-Gln143 interaction compared to wildtype (Supplementary Fig. 3f, PDB ID 8VHY)63. Altogether, these comparisons show the importance of the WAT1-Gln143 interaction for catalysis and hydrogen bonding correlates with longer retention of the inhibited complex.
The electronic configuration of the Mn ion
For enzymes that use metal centers to catalyze redox reactions, the arrangement of the metal 3 d orbitals determines how electrons are exchanged and how substrates orient for catalysis79. A detailed analysis of the electronic configuration of the Mn ion is presented in the Supplementary information and is summarized here. For MnSOD, the metal ion is in a distorted C3v symmetry environment with either 4 or 5 occupied electrons in the α-manifold, depending on the oxidation state of the metal67. For Mn3+ with S = 2, the eπ (xz/yz) and eσ (xy/x2-y2) α orbitals are occupied, while Mn2+ with S = 5/2 also has the z2 α orbital occupied (Supplementary Fig. 4a). The z2 α orbital exchanges electrons during redox reactions as it is the lowest unoccupied molecular orbital (LUMO) for Mn3+ and the highest occupied molecular orbital (HOMO) for Mn2+. This means that mono- or dioxygen species are most likely to bind the Mn ion along the z-axis for reactivity (Supplementary Fig. 4b). For further insight into the MnSOD metal 3 d orbitals, we compared the K-pre-edge spectra found through time-dependent DFT (TD-DFT) simulations with spectra measured from HERFD-XANES (detailed evaluation of these data can be found in the Supplementary Information pages 16-18 and Supplementary Table 5 and Fig. 4). We found that the experimental pre-edge spectra are similar between wildtype and Tyr34Phe Mn3+SOD, indicating that the Tyr34Phe variant does not significantly alter the Mn3+ ion orbital configuration (Supplementary Figs. 4c, d). Also, the H2O2-soaked Trp161Phe and Tyr34Phe complexes have similar experimental spectra though the calculated spectra suggest differences in orbital transitions (Supplementary Figs. 4g, h).

a, b Neutron structures of D2O2-soaked Tyr34Phe MnSOD (2.3 Å resolution). c, d Neutron structures of Tyr34Phe Mn2+SOD (2.5 Å resolution). e, f Neutron structures of Tyr34Phe Mn3+SOD (2.3 resolution). Blue, green, and orange omit |Fo| - |Fc| difference neutron scattering length density of protons displayed at 2.0 σ, 2.5σ, and 3.0σ, respectively. Light blue 2|Fo | - |Fc| density is displayed at 1.0σ. Distances are in Å. Dashed lines indicate hydrogen bonds ≥ 1.8 Å and hashed lines indicate SSHBs that are hydrogen bonds < 1.8 Å. Chains are colored according to the inset in Fig. 4a.
Tyr34 contributes to the pKas of Tyr166/His30 and influences the proton shuffle across the subunit interface
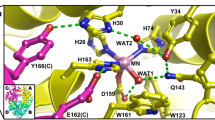
Second-sphere residues His30 and Tyr166 have unusual pKas in wildtype MnSOD and change protonation states (Fig. 1c)13. To identify whether the Tyr34 mutation to Phe affects the protonations of His30/Tyr166, we investigated the nuclear density maps of Tyr34Phe MnSOD in D2O2-soaked, reduced, and oxidized forms. For chain A of D2O2-soaked MnSOD that has a dioxygen species bound (Fig. 3a, b), the omit |Fo | - |Fc| density indicates that Tyr166 is in the neutral, protonated form while His30 is singly protonated on the Nδ1 atom (Fig. 4a). Interestingly, for chain B, Tyr166 is instead deprotonated and His30 is now protonated on Nε2 (Fig. 4b). The two residues form a 1.7 Å SSHB due to the negative charge of ionized Tyr166. Different chains of the same MnSOD neutron structure have been observed before to differ in protonation states, with the most explicable cause being differences in solvent accessibility13. However, the protonation configuration observed in chain B is unique and has not been seen before (Fig. 4b). It is not known if this proton configuration occurs in wildtype enzyme as neutron data for D2O2-soaked wildtype has yet to be collected. But we can compare reduced and oxidized Tyr34Phe MnSOD to wildtype13.
For reduced Tyr34Phe MnSOD, both chains have the same protonation states (Fig. 4c, d). Tyr166 is protonated and neutral while His30 is singly-protonated on the Nδ1 atom. The omit density for Dη(Tyr166) is elongated and spans to Nε2(His30) (Fig. 4d). Elongated density has been observed between the two residues before in reduced wildtype MnSOD13. This may be indicative of a proton transfer occurring between Tyr166 and His30, in line with the different protonation states observed in the D2O2-soaked structure (Fig. 4a, b).
The protonation states of oxidized Tyr34Phe (Fig. 4e, f) are the same as reduced Tyr34Phe (Fig. 4c, d), which is in contrast to wildtype MnSOD where the His30 protonation alters between oxidized and reduced states (Fig. 1c). This difference is probably due to the Phe mutation at position 34. In wildtype, Tyr34 is ionized when the Mn ion is oxidized (Fig. 1d). The negative charge on Tyr34 may exert electronic effects that alter the pKa of His30. Thus, Tyr34 may help modulate the pKa of nearby residues so efficient proton transfers occur for PCET catalysis.
Our three neutron structures of Tyr34Phe MnSOD highlight the importance of residues Tyr166, His30, and Tyr34 in maintaining the proton pool of the active site. Two protons are needed to protonate O22− to H2O2 during the fast Mn2+ → Mn3+ reaction (k2, Supplementary Table 1). The D2O2-soaked structure unambiguously indicates that His30 and Tyr166 shuffle protons, with proton transfers between Oη(Tyr166) and Nε2(His30) coinciding with changes in the Nδ1(His30) protonation state (Fig. 4a, d). In wildtype MnSOD, the proton between Oη(Tyr166) and Nε2(His30) was instead observed to be shared, perhaps because the wildtype structures were collected at room temperature (Fig. 1c, d). Another distinction between Tyr34Phe and wildtype MnSOD is that in the oxidized forms, the Nδ1(His30) have different protonation states. For Tyr34Phe, Nδ1(His30) is protonated (Fig. 4e, f) while in wildtype Nδ1(His30) is deprotonated (Fig. 1c). The pKas of active site residues are a result of multiple effects, including residue composition and ionization states. An ionized Tyr34 coincides with a deprotonated Nδ1(His30) in wildtype MnSOD and suggests that Tyr34 contributes to modulating the pKa of nearby residues for oxidized MnSOD. Overall, the Tyr34Phe MnSOD neutron structures help elucidate the role of Tyr166, His30, and Tyr34 in MnSOD catalysis.
Summary and comparison of Tyr34Phe active site configurations
Comparing the Tyr34Phe MnSOD neutron structures with our previous wildtype and Trp161Phe neutron structures helps our understanding of the role of Tyr34 in catalysis. For oxidized wildtype and Tyr34Phe MnSOD, the WAT1-Gln143 protonation states and hydrogen bond interaction are similar (Fig. 5a, b). However, in reduced Tyr34Phe, His30 is observed to have different protonation states compared to wildtype, and the proton between His30 and Tyr166 is not shared. For the reduced counterparts, the hydrogen bonding of Gln143 with WAT1 and Trp123 is weaker in Tyr34Phe MnSOD (Fig. 5c, d). The Gln143 hydrogen bonds in wildtype are SSHBs (<1.8 Å), while those of Tyr34Phe are typical hydrogen bonds (>1.8 Å). Reduced wildtype is observed to have a shared proton between His30 and Tyr166 while Tyr34Phe does not. However, chain B of reduced Tyr34Phe MnSOD is observed to have elongated nuclear density between His30 and Tyr166 (Fig. 4d), which indicates that a proton may be shared. For peroxide-bound Trp161Phe and Tyr34Phe, HO2- binds the Mn ion in different orientations (Fig. 5e, f). Furthermore, in Trp161Phe, a H2O2 molecule is bound with SSHBs between His30 and an anionic Tyr34. The lack of H2O2 at this site in Tyr34Phe may be due to the absence of the hydroxyl group. Overall, Tyr34 plays a role in several aspects of the active site, and several enzymatic details are revealed from this collection of neutron structures.

a Wildtype oxidized resting state from chain B of PDB ID 7KKS13. b Tyr34Phe oxidized resting state from chain A (present study, Figs. 3e, 4e). c Wildtype reduced resting state from chain B of PDB ID 7KKW13. d Tyr34Phe reduced resting state from chain A (present study, Figs. 3c, 4c). e The product-inhibited complex of Trp161Phe, consisting of a HO2- molecule bound to a reduced active site. From chain B of PDB ID 8VHW63. f The product-inhibited complex of Tyr34Phe, consisting of a HO2- molecule bound to a reduced active site. From chain A of the present study (Figs. 3a, b, 4a). Dashed lines represent normal hydrogen bonds (> 1.8 Å), wide-dashed represent SSHBs (hydrogen bonds < 1.8 Å), and round-dotted lines represent a shared proton. The portrayal of hydrogen bond lengths in 2D are not representative of those seen experimentally in 3D.
First, Tyr34 helps control the charge at the active site and provides an environment conducive to charge-dependent proton and electron transfers. For example, Tyr34 and His30 both lose protons when the active site is oxidized and gain protons when the active site is reduced (Figs. 5a, 5c, 1c, d). As a result, we previously hypothesized that Tyr34 and His30 protonate O22- during the fast Mn2+ → Mn3+ reaction (k2, Supplementary Table 1)13. In the Tyr34Phe variant, the His30 protonation state remains consistent between resting states, which implicates Tyr34 in influencing the pKa of His30 through electronic effects (Figs. 5b, d, 4c-f). These charge effects of Tyr34 may be a partial contributor to the deficient catalysis observed for the Tyr34Phe variant (Supplementary Table 1).
Second, Tyr34 plays a significant role in the fast Mn2+ → Mn3+ reaction as suggested by the very low k2 for Tyr34Phe (Supplementary Table 1). Comparison of the resting Mn2+SOD neutron structures of wildtype and Tyr34Phe indicates that Tyr34 enforces a strong WAT1-Gln143 interaction (Figs. 5c, d, 3d). This interaction is important for PCET, where the WAT1 → Gln143 proton transfer coincides with the Mn2+ → Mn3+ redox reaction (Fig. 1b)13. Another potential reason for diminished k2 catalysis for the Tyr34Phe variant is the loss of an ionizable group that could protonate O22- or HO2-. However, the Trp161Phe variant also has a nearly extinguished k2 and maintains the number of ionizable groups (Supplementary Table 1). The shared feature for these two variants is the weakened WAT1-Gln143 interaction (Fig. 4f). Since our previous structures indicate that Tyr34 loses a proton during the Mn2+ → Mn3+ reaction (Fig. 1d), it is possible that the WAT1 → Gln143 proton transfer precludes a Tyr34 → O22− proton transfer. Overall, our structures indicate Tyr34 contributes to orienting WAT1 and Gln143 for a proton transfer event during the Mn2+ → Mn3+ redox reaction.
Third, the formation of the product-inhibited complex is dependent on Tyr34. The complex is characterized by an HO2− molecule replacing the WAT1 position in the Mn2+ oxidation state (Fig. 5e, f, Supplementary Fig. 3e). As such, the capacity to form the inhibited complex may correlate with the ease an HO2- molecule can displace WAT1. The Tyr34Phe and Trp161Phe variants have a higher propensity to accumulate the inhibited complex, and both have a weakened WAT1-Gln143 interaction in the Mn2+ oxidation state that would permit easier displacement of WAT1 (Fig. 4f). This suggests that the Tyr34-Gln143-WAT1 hydrogen bond network suppresses product inhibition.
Lastly, retention of the product-inhibited complex correlates with the strength of hydrogen bonding between HO2− and Gln143. The inhibited complexes captured in the Tyr34Phe and Trp161Phe variants reveal two different HO2− binding orientations and hydrogen bond interactions (Fig. 5e, f, Supplementary Fig. 3e). The hydrogen bonding between HO2− and Gln143 is stronger in Trp161Phe compared to Tyr34Phe, and this stronger interaction correlates with a slower disassociation of the inhibited complex for Trp161Phe (k4, Supplementary Table 1). As protonation of HO2− to H2O2 is required for relief of the inhibited complex, the hydrogen bonding of Gln143 with HO2− may compete with a H2O → HO2− proton transfer to form H2O2. Comparison of the product-inhibited complex of Tyr34Phe and Trp161Phe provides clues into the relief of the inhibited complex.
Suggested mechanism
From our collection of neutron structures and XAS data, we have constructed a mechanism for the fast reaction pathways k1 and k2 and the reversible product inhibition reaction pathways k3 and k4 (Fig. 6). Note that our data describe the inhibited complex as Mn2+-containing (Figs. 2a, Supplementary Fig.3a–c), in contrast with several mechanistic models that assign a Mn3+ oxidation state to the complex31,32,33,59,60. Furthermore, in the absence of data that describe how \({{{{\rm{O}}}}}_{2}^{\bullet -}\) interacts with an oxidized active site, \({{{{\rm{O}}}}}_{2}^{\bullet -}\) reacting with the Mn3+ ion is represented only by Mn3+ gaining an electron (Fig. 6). For a reduced active site, \({{{{\rm{O}}}}}_{2}^{\bullet -}\) likely binds between His30 and Tyr34 for PCET to form H2O213,41. Overall, the mechanism delineates how MnSOD’s reversible inhibition pathway may branch from the fast reaction pathway.

The associating and dissociating molecules are indicated by the reaction on the left side of the figure for each panel. a, b The fast Mn3+ → Mn2+ half-reaction corresponding to k1. Note that the electrons are contributed to by superoxide binding. The ___location of this binding site is still unknown, so this is indicated with a thick arrow. c, d The fast Mn2+ → Mn3+ half-reaction corresponding to k2. e, f Formation of the inhibited complex that is dependent on the presence of H2O2 during the reduction of the Mn3+ ion. In the absence of H2O2, the enzyme proceeds through the fast Mn3+ → Mn2+ half-reaction (k1). g, h Relief of the inhibited complex. Our data describe the inhibited complex as Mn2+-containing in contrast to other published mechanistic models31,32,33,59,60. In the absence of data that indicate how \({{{{\rm{O}}}}}_{2}^{\bullet -}\) interacts with an oxidized active site, \({{{{\rm{O}}}}}_{2}^{\bullet -}\) reacting with the Mn3+ ion is represented only by Mn3+ gaining an electron. For a reduced active site, \({{{{\rm{O}}}}}_{2}^{\bullet -}\) likely binds between His30 and Tyr34 for PCET to form H2O213,41. Dashed lines represent normal hydrogen bonds (>1.8 Å), wide-dashed represent SSHBs (hydrogen bonds < 1.8 Å), and round-dotted lines represent a shared proton. The portrayal of hydrogen bond lengths in 2D are not representative of those seen experimentally in 3D.
For the k1 reaction that represents the fast Mn3+→ Mn2+ half-reaction, reduction of the Mn ion occurs alongside three proton transfers, Gln143 → WAT1 and protonation of His30 and Tyr34 by solvent (Fig. 6a). In the reduced state, anionic Gln143 is stabilized by SSHBs with Trp123, WAT1, and Tyr34 (Fig. 6b). For the k2 reaction that represents the fast Mn2+ → Mn3+ half-reaction, a WAT1 → Gln143 proton transfer occurs alongside protonation and reduction of \({{{{\rm{O}}}}}_{2}^{\bullet -}\) (Fig. 6c). Here, \({{{{\rm{O}}}}}_{2}^{\bullet -}\) participates in a long-range electron transfer and is protonated by His30 and Tyr34 to form H2O2 (Fig. 6d). If Mn3+ gains an electron while the resultant H2O2 is still present, H2O2 donates a proton to His30 and subsequently replaces WAT1 to form the product inhibited complex (k3, Fig. 6e, f). The inhibited complex is characterized by the inability to perform the WAT1 → Gln143 proton transfer and prohibits fast catalysis (Fig. 6f). Relief of the inhibited complex is achieved by protonation of HO2− by a solvent molecule (k4, Fig. 6g). H2O2 departs the active site and is replaced by WAT1 to form the Mn2+ resting state (Fig. 6h). From relief of inhibition, fast catalysis may proceed again through k2 (Fig. 6c, d).
The key determinant of whether product inhibition is engaged is the His30 proton donor during Mn3+ reduction. If H2O2 is the proton donor, the inhibited complex is formed (k3, Fig. 6e). If a solvent molecule is the proton donor, the enzyme proceeds through fast catalysis (k1, Fig. 6a). This explains why high concentrations of \({{{{\rm{O}}}}}_{2}^{\bullet -}\) lead to product inhibition, as \({{{{\rm{O}}}}}_{2}^{\bullet -}\) may enter the active site before H2O2 departs. Tyr34 may also serve as a potential proton acceptor for H2O2 since Tyr34 also gains a proton during reduction to Mn2+. However, Tyr34 is not necessary to form the inhibited complex, as indicated by our Tyr34Phe neutron structure (Fig. 3a, b). Our work provides insights into the PCET mechanism of MnSOD, especially in the context of product inhibition.
Altogether, our investigation reveals how a single tyrosine residue has a profound effect on PCET catalysis. Tyr34 plays a part in every MnSOD kinetic step from its roles of (1) acting as a proton donor/acceptor, (2) orienting nearby molecules Gln143 and WAT1 for proton transfer, (3) limiting the formation of the inhibited complex, and (4) shortening the lifetime of the inhibited complex. These roles of Tyr34 place the residue as a central regulator of H2O2 output in the mitochondria. H2O2 produced from MnSOD has several cellular effects, including stimulating apoptotic signaling pathways45,46, coordinating protein localization and activity47, and mitochondrial biogenesis44. Inactivation of Tyr34 by nitration is observed in neurological disease and further highlights the physiological role of Tyr3458. In total, the work provides insight into how a PCET enzyme facilitates catalysis and how an oxidoreductase molecularly facilitates mitochondrial function.
Methods
Perdeuterated expression and purification
For deuterated protein expression of MnSOD, the pCOLADuet-1 expression vector harboring full-length cDNA of MnSOD was transformed into Escherichia coli BL21(DE3) cells. Transformed cells were grown in D2O minimal media within a bioreactor vessel using D8-glycerol as the carbon source80. Induction was performed with 1 mM isopropyl β-D-thiogalactopyranoside, 8 mM MnCl2, and fed D8-glycerol until an OD600 of 15.0. Expression was performed at 37 °C for optimal Mn metal incorporation81. Harvested cell pastes were stored at -80 °C until purification. For protein purification (with hydrogenated reagents), cells were resuspended in a solution of 5 mM MnCl2 and 5 mM 3-(N-morpholino)propanesulfonic acid (MOPS), pH 7.8. Clarified lysate was incubated at 55 °C to precipitate contaminant proteins that were subsequently removed by centrifugation. Next, soluble protein was diluted with an equal volume of 50 mM 2-(N-morpholino)ethanesulfonic acid (MES) pH 5.5, yielding a final concentration of 25 mM. Measurement of pH verified a value of 5.5 after dilution. Protein was applied onto a carboxymethyl sepharose fast flow column (GE Healthcare) and eluted with a sodium chloride gradient that contained 50 mM MES pH 6.5.
Crystallization
Perdeuterated Tyr34Phe MnSOD crystals were grown in a microgravity environment aboard the International Space Station (ISS)82. Crystals growth was achieved in Granada Crystallization Boxes (GCBs, Triana) through capillary counterdiffusion using fused quartz capillary tubes (VitroCom) that had inner diameters of 2.0 mm and outer diameters of 2.4 mm83. 25 mg ml-1 protein-filled capillaries were plugged with 40 mm of 2% agarose (w/w) and inserted into GCBs filled with precipitating agent composed of 4 M potassium phosphate, pH 7.8. The pH of the phosphate buffer was achieved through 91:9 ratios of K2HPO4:KH2PO4. The GCBs were delivered to the ISS by SpX-17 as part of the Perfect Crystals NASA payload and returned to earth 1 month later on SpX-18. The crystals within GCBs were observed to be resilient against travel damage and were placed within carry-on baggage during further aircraft travels to the UNMC Structural Biology Core Facility and ORNL. Perdeuterated Tyr34Phe MnSOD crystals were 0.3-0.6 mm3 in size, and further details of microgravity crystallization have been published82. For X-ray diffraction, crystals were grown in 1.8 M potassium phosphate, pH 7.8 by hanging-drop vapor diffusion. Protein (23 mg ml-1) and reservoir solution were mixed at a 1:1 ratio to give a 4.0 µL drop. Crystals for X-ray diffraction were less than 0.1 mm3 in size and were fully grown after 14 d.
Crystal manipulations
For deuterium exchange, microgravity-grown crystals were first placed in 1 mL of hydrogenated 4 M potassium phosphate pH 7.8. Deuterium was introduced with 0.1 mL incremental additions every 2 min of 4 M deuterated potassium phosphate (K2DPO4:KD2PO4) pD 7.8 (calculated by adding 0.4 to the measured pH reading) for a total of five times and a net volume addition of 0.5 mL. After 10 min, 0.5 mL of the solution was removed leading to a 1 mL solution consisting of 33% deuterium. The process was repeated enough times to gradually increase the deuterium content to ~100%. The 4 M deuterated potassium phosphate also served as the cryoprotectant for the cryocooling process. Further details of the process were published84.
For redox manipulation, the deuterated potassium phosphate solutions were supplemented with either 6.4 mM potassium permanganate (KMnO4) to achieve the Mn3+ oxidation state or 300 mM sodium dithionite (Na2S2O4) to achieve the Mn2+ state. Crystals were either sealed in capillaries or 9-well glass plates to maintain the desired oxidation state. Redox reagents were not used for the Tyr34Phe MnSOD structure soaked with D2O2. The crystal was in the as is resting state, which is >90% oxidized. The dioxygen-bound complex was achieved by supplementing the cryoprotectant that the crystal was immersed in with D2O2 at a final concentration of 1% w/v (~0.28 M) and soaking for 1 min before cryocooling. Flash-cooling was performed with an Oxford diffraction cryostream85. Further details of ligand cryotrapping for neutron crystallography were published84.
Crystallographic data collection
Time-of-flight, wavelength-resolved neutron Laue diffraction data were collected from perdeuterated crystals using the MaNDi instrument86,87 at the Oak Ridge National Laboratory Spallation Neutron Source with wavelengths between 2 to 4 Å. Sample sizes ranged from 0.3 to 0.6 mm3 and data were collected to 2.30 Å resolution for oxidized and D2O2-soaked structures, while the reduced structure was collected to 2.5 Å resolution (Supplementary Table 6). Crystals were held in stationary positions during diffraction, and successive diffraction frames were collected along rotations of the Φ axis. X-ray diffraction data were collected using a Rigaku FR-E SuperBright home source or the Stanford Synchrotron Radiation Lightsource (SSRL) beamline 14–1 (Supplementary Table 6).
Crystallographic data processing and refinement
Neutron data were integrated using the MANTID software package88,89,90 and wavelength-normalized and scaled with LAUENORM from the Daresbury Laue Software Suite91. X-ray diffraction data were processed using HKL-300092. Refinements of both neutron and X-ray models were completed separately with PHENIX.REFINE from the PHENIX suite93. The refinements were intentionally performed separately due to the known perturbations that X-rays have on the solvent structure, metal redox state, and metal coordination34,94. The X-ray model was first refined against its corresponding data set and subsequently used as the starting model for neutron refinement. Torsional backbone angle restraints were derived from the X-ray model and applied to neutron refinement using a geometric target function with PHENIX.REFINE93. Mn-ligand restraints for neutron refinement were derived from DFT calculations rather than the X-ray model to remove any influence of photoreduction. The neutron refinement was performed by modeling the D atoms of the active site last to limit phase bias. Initial rounds of refinement to fit protein structure included only non-exchangeable D atoms, which have stereochemical predictable positions. Afterward, H/D atoms were modeled onto the position of each amide proton, and occupancy was refined. In general, the asymmetric units of the neutron crystal structures had a deuterium content of ~85% for the amide backbone, and areas with low deuterium exchange (<50 %) coincided with the presence of hydrogen bonds forming a secondary structure. Next, exchangeable proton positions of residues outside the active site (e.g., hydroxyl group of serine/tyrosine) were manually inspected for obvious positive omit |Fo | - |Fc| neutron scattering length density at a contour of 2.5σ or greater and modeled as a fully occupied deuterium. If the density was not obvious, and there was no chemically sensible reason for the residue to be deprotonated (which is the case for residues outside the active site), the proton position was H/D occupancy refined. D2O molecules outside the active site were then modeled and adjusted according to the nuclear density. Last, D atoms of the active site were modeled manually. At the active site, a residue is considered deprotonated when (1) attempts to model and refine a proton result in negative |Fo| - |Fc| difference neutron scattering length density, (2) all the other protons of the residue can be placed, and (3) the heavy atom that is deprotonated acts as a hydrogen-bond acceptor. As chemically ideal covalent bond distances of D atoms were ensured during model building and refinement, small deviations from D atom positions and omit |Fo | - |Fc| neutron scattering length density centers were expected from the data resolution (2.3–2.5 Å). To test if the active site ligands for chain A and chain B were truly different we performed a refinement forcing chain B to be identical to chain A. This resulted in difference density that supported our original structural interpretation of the D2O2-soaked structure (Supplementary Figs. 1a and 1b). Chain B is different than chain A as shown.
X-ray absorption spectroscopy measurements
A solution of 3 mM MnSOD (~70 mg ml-1) in 25 mM potassium phosphate pH 7.8 was treated with 8 mM potassium dichromate to isolate the Mn3+SOD resting state, 200 mM sodium dithionite to isolate the Mn2+SOD resting state, and either 20 mM superoxide or 280 mM (1% w/v) hydrogen peroxide for the product-inhibited state. For EXAFS spectra the sample included 30% ethylene glycol to avoid water crystals. Samples were mounted in 150 µl mylar cells. Superoxide stock solutions were generated by mixing 1.3 M potassium superoxide (KO2, Sigma Aldridge) in a mixture of dry DMSO (Thermo Scientific) and 0.30 M di-benzo-18-crown-6-ether (Thermo Scientific) following published protocols59,60,62,95,96. The concentration of superoxide in the DMSO/18-crown solution was measured by a UV-Vis spectrophotometer at a wavelength of 250 nm with a molar extinction coefficient of 2686 M-1 cm-1 62,97. Superoxide solutions were stored at 4 °C under a layer of argon in glass vials sealed with rubber septum.
Mn K-edge HERFD-XANES spectra were recorded at beamline 15-2 of the Stanford Synchrotron Radiation Lightsource (SSRL), while Mn K-edge EXAFS spectra were collected at beamline 7-3. At both beamlines, data were collected at 10 K using a liquid He cryostat, and the incident energy was tuned to the first derivative of an internal Mn foil at 6539 eV. X-ray irradiation was carefully monitored so that two subsequent scans of the same spot did not have photoreduction differences, and different spots along samples were scanned. When appropriate, aluminum foil was inserted into the beam path to attenuate the incident flux. For HERFD-XANES measurements, a Johann-type hard X-ray spectrometer with six Ge(333) analyzer crystals was used with a liquid-nitrogen cooled Si(311) double crystal monochromator, and energy was calibrated to a glitch with measurement of Mn Foil. For EXAFS, measurements were recorded with a 30-element Ge solid-state detector, and a Si(220) monochromator at Φ = 90° was used.
X-ray absorption spectroscopy data analysis
EXAFS data reduction, averaging, and refinement were carried out with the LARCH software package98. Refinement of the k2χ(k) EXAFS data used phases and amplitudes obtained from, FEFF99. For each fit, optimization of the radial distribution around the absorbing Mn ion (r) and the Debye-Waller factor (σ2) was performed. The goodness-of-it was evaluated by χ2 values, reduced χ2 values and R-factors.
Computational methods
All DFT calculations were performed with the ORCA quantum chemistry package version 5.0 using the B3LYP functional, the def2-TZVP basis set for all atoms, and the CPCM solvation model100,101,102,103. For geometry optimizations, the full active site (i.e., residues shown in the right panel of Fig. 1a) was included from the neutron structure where the peptide backbone was truncated and Cα was fixed. All atoms for residues Trp161, Phe66, and Tyr166 were fixed with the exception of hydroxyl Tyr166 proton to mimic the packing found in the native enzyme. The Mn ion used the high-spin quintet and sextet states for trivalent and divalent systems, respectively, per experimental observations67. A dense integration grid and tight convergence were enforced. For CPCM calculations, the dielectric constant was 80.4. The atomic radii were 2.04 Å for carbon, 1.86 Å for nitrogen, 1.32 Å for hydrogen, 1.82 Å for oxygen, and 2.40 Å for manganese. The solvent probe radius was 1.3 Å.
For TD-DFT calculations, the Mn ion was instead assigned the core property basis set, CP(PPP)104,105. The geometry-optimized model was truncated to only the Mn ion and its immediate ligands. Including all active site residues for TD-DFT did not significantly alter the simulated spectra. Computed Mn K pre-edge data were plotted using a Gaussian broadening of 1 eV, and a 32.3 eV energy correction was applied in line with previous studies64,106.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Coordinates and structure factors for neutron and X-ray crystallographic data presented in this study have been deposited in the Protein Data Bank. (PDB 9BVY, PDB 9BW2, PDB 9BWM, PDB 9BWQ, and PDB 9BWR. All relevant data supporting the key findings of this study are available within the article, its Supplementary Information files and in the Source Data file or from the corresponding author upon request. Source data are provided with this paper.
References
McDonald, A. G., Boyce, S. & Tipton, K. F. ExplorEnz: the primary source of the IUBMB enzyme list. Nucleic Acids Res 37, D593–D597 (2009).
Costentin, C., Robert, M. & Saveant, J. M. Concerted proton-electron transfers: electrochemical and related approaches. Acc. Chem. Res. 43, 1019–1029 (2010).
Chang, C. J., Chang, M. C., Damrauer, N. H. & Nocera, D. G. Proton-coupled electron transfer: a unifying mechanism for biological charge transport, amino acid radical initiation and propagation, and bond making/breaking reactions of water and oxygen. Biochim Biophys. Acta 1655, 13–28 (2004).
Albers, D. S. & Beal, M. F. Mitochondrial dysfunction and oxidative stress in aging and neurodegenerative disease. J. Neural Transm. Suppl. 59, 133–154 (2000).
Gao, L., Laude, K. & Cai, H. Mitochondrial pathophysiology, reactive oxygen species, and cardiovascular diseases. Vet. Clin. North Am. Small Anim. Pr. 38, 137–155 (2008).
Lipinski, B. Hydroxyl radical and its scavengers in health and disease. Oxid. Med Cell Longev. 2011, 809696 (2011).
Reece, S. Y. & Seyedsayamdost, M. R. Long-range proton-coupled electron transfer in the Escherichia coli class Ia ribonucleotide reductase. Essays Biochem 61, 281–292 (2017).
Bhat, A. H. et al. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 74, 101–110 (2015).
Nielsen, J. E. & McCammon, J. A. Calculating pKa values in enzyme active sites. Protein Sci. 12, 1894–1901 (2003).
Maliekal, J. et al. Comparison and contrasts between the active site PKs of Mn-superoxide dismutase and those of Fe-superoxide dismutase. J. Am. Chem. Soc. 124, 15064–15075 (2002).
Pannwitz, A. & Wenger, O. S. Proton-coupled multi-electron transfer and its relevance for artificial photosynthesis and photoredox catalysis. Chem. Commun. (Camb.) 55, 4004–4014 (2019).
Bhowmick, A. et al. Structural evidence for intermediates during O(2) formation in photosystem II. Nature 617, 629–636 (2023).
Azadmanesh, J., Lutz, W. E., Coates, L., Weiss, K. L. & Borgstahl, G. E. O. Direct detection of coupled proton and electron transfers in human manganese superoxide dismutase. Nat. Commun. 12, 2079 (2021).
Shrishrimal, S., Kosmacek, E. A., Chatterjee, A., Tyson, M. J. & Oberley-Deegan, R. E. The SOD Mimic, MnTE-2-PyP, Protects from Chronic Fibrosis and Inflammation in Irradiated Normal Pelvic Tissues. Antioxid. (Basel) 6, 87 (2017).
Mapuskar, K. A. et al. Utilizing superoxide dismutase mimetics to enhance radiation therapy response while protecting normal tissues. Semin Radiat. Oncol. 29, 72–80 (2019).
Batinic-Haberle, I. & Spasojevic, I. Complex chemistry and biology of redox-active compounds, commonly known as SOD mimics, affect their therapeutic effects. Antioxid. Redox Signal 20, 2323–2325 (2014).
Stover, K. et al. Topically applied manganese-porphyrins BMX-001 and BMX-010 display a significant anti-inflammatory response in a mouse model of allergic dermatitis. Arch. Dermatol Res. 308, 711–721 (2016).
You, C., Huang, R., Wei, X., Zhu, Z. & Zhang, Y. P. Protein engineering of oxidoreductases utilizing nicotinamide-based coenzymes, with applications in synthetic biology. Synth. Syst. Biotechnol. 2, 208–218 (2017).
Shields, H. J., Traa, A. & Van Raamsdonk, J. M. Beneficial and detrimental effects of reactive oxygen species on lifespan: a comprehensive review of comparative and experimental studies front. Cell Dev. Biol. 9, 628157 (2021).
Wallace, D. C. Mitochondria and cancer. Nat. Rev. Cancer 12, 685–698 (2012).
De Gaetano, A. et al. Mitophagy and Oxidative Stress: The Role of Aging Antioxidants (Basel), 10 (2021).
Fukai, T. & Ushio-Fukai, M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal 15, 1583–1606 (2011).
Pinero, J. et al. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res 48, D845–D855 (2020).
Li, C. & Zhou, H. M. The role of manganese superoxide dismutase in inflammation defense. Enzym. Res. 2011, 387176 (2011).
Nowak-Kiczmer, M., Niedziela, N., Zalejska-Fiolka, J. & Adamczyk-Sowa, M. Evaluation of antioxidant parameters of multiple sclerosis patients’ serum according to the disease course. Mult. Scler. Relat. Disord. 77, 104875 (2023).
Souiden, Y. et al. MnSOD and GPx1 polymorphism relationship with coronary heart disease risk and severity. Biol. Res 49, 22 (2016).
Perry, J. J., Shin, D. S., Getzoff, E. D. & Tainer, J. A. The structural biochemistry of the superoxide dismutases. Biochim Biophys. Acta 1804, 245–262 (2010).
Azadmanesh, J. & Borgstahl, G. E. O. A Review of the Catalytic Mechanism of Human Manganese Superoxide Dismutase. Antioxid. (Basel) 7, 25 (2018).
Cadenas, E. & Davies, K. J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med 29, 222–230 (2000).
Drose, S. & Brandt, U. The mechanism of mitochondrial superoxide production by the cytochrome bc1 complex. J. Biol. Chem. 283, 21649–21654 (2008).
Abreu, I. A. & Cabelli, D. E. Superoxide dismutases-a review of the metal-associated mechanistic variations. Biochim Biophys. Acta 1804, 263–274 (2010).
Sheng, Y. et al. Superoxide dismutases and superoxide reductases. Chem. Rev. 114, 3854–3918 (2014).
Hearn, A. S. et al. Kinetic analysis of product inhibition in human manganese superoxide dismutase. Biochemistry 40, 12051–12058 (2001).
O’Dell, W. B., Bodenheimer, A. M. & Meilleur, F. Neutron protein crystallography: A complementary tool for locating hydrogens in proteins. Arch. Biochem Biophys. 602, 48–60 (2016).
Carugo, O. & Djinovic Carugo, K. When X-rays modify the protein structure: radiation damage at work. Trends Biochem Sci. 30, 213–219 (2005).
Remer, L. C. & Jensen, J. H. Toward a general theory of hydrogen bonding: the short, strong hydrogen bond. J. Phys. Chem. A 104, 9266–9275 (2000).
Gerlt, J. A., Kreevoy, M. M., Cleland, W. & Frey, P. A. Understanding enzymic catalysis: the importance of short, strong hydrogen bonds. Chem. Biol. 4, 259–267 (1997).
Kumar, P., Agarwal, P. K. & Cuneo, M. J. On the case of the misplaced hydrogens. Chembiochem. 22, 288–297 (2020).
Cleland, W. W., Frey, P. A. & Gerlt, J. A. The low barrier hydrogen bond in enzymatic catalysis. J. Biol. Chem. 273, 25529–25532 (1998).
MacMillan-Crow, L. A. & Thompson, J. A. Tyrosine modifications and inactivation of active site manganese superoxide dismutase mutant (Y34F) by peroxynitrite. Arch. Biochem Biophys. 366, 82–88 (1999).
Srnec, M., Aquilante, F., Ryde, U. & Rulisek, L. Reaction mechanism of manganese superoxide dismutase studied by combined quantum and molecular mechanical calculations and multiconfigurational methods J. Phys. Chem. B,113, 6074–6086 (2009).
Lah, M. S. et al. Structure-function in Escherichia coli iron superoxide dismutase: comparisons with the manganese enzyme from Thermus thermophilus. Biochemistry 34, 1646–1660 (1995).
Whittaker, M. M. & Whittaker, J. W. Low-temperature thermochromism marks a change in coordination for the metal ion in manganese superoxide dismutase. Biochemistry 35, 6762–6770 (1996).
Palma, F. R. et al. Mitochondrial superoxide dismutase: what the established, the intriguing, and the novel reveal about a key cellular redox switch. Antioxid. Redox Signal 32, 701–714 (2020).
Dasgupta, J. et al. Manganese superoxide dismutase protects from TNF-alpha-induced apoptosis by increasing the steady-state production of H2O2. Antioxid. Redox Signal 8, 1295–1305 (2006).
Cadenas, E. Mitochondrial free radical production and cell signaling. Mol. Asp. Med. 25, 17–26 (2004).
Riemer, J., Schwarzlander, M., Conrad, M. & Herrmann, J. M. Thiol switches in mitochondria: operation and physiological relevance. Biol. Chem. 396, 465–482 (2015).
Connor, K. M. et al. Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. J. Biol. Chem. 280, 16916–16924 (2005).
Yu, Q. et al. Mitochondrial hydrogen peroxide activates PTEN and inactivates Akt, leading to autophagy inhibition-dependent cell death in neuronal models of Parkinson’s. Dis. Mol. Neurobiol. 60, 3345–3364 (2023).
Cox, A. G., Winterbourn, C. C. & Hampton, M. B. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem J. 425, 313–325 (2009).
Ekoue, D. N., He, C., Diamond, A. M. & Bonini, M. G. Manganese superoxide dismutase and glutathione peroxidase−1 contribute to the rise and fall of mitochondrial reactive oxygen species which drive oncogenesis. Biochim Biophys. Acta Bioenerg. 1858, 628–632 (2017).
Munro, D., Banh, S., Sotiri, E., Tamanna, N. & Treberg, J. R. The thioredoxin and glutathione-dependent H2O2 consumption pathways in muscle mitochondria: Involvement in H2O2 metabolism and consequence to H2O2 efflux assays. Free Radic. Biol. Med. 96, 334–346 (2016).
Sheng, Y. et al. Comparison of two yeast MnSODs: mitochondrial Saccharomyces cerevisiae versus cytosolic Candida albicans. J. Am. Chem. Soc. 133, 20878–20889 (2011).
Abreu, I. A. et al. The kinetic mechanism of manganese-containing superoxide dismutase from Deinococcus radiodurans: a specialized enzyme for the elimination of high superoxide concentrations. Biochemistry 47, 2350–2356 (2008).
Greenleaf, W. B. et al. Role of hydrogen bonding in the active site of human manganese superoxide dismutase. Biochemistry 43, 7038–7045 (2004).
Hearn, A. S. et al. Amino acid substitution at the dimeric interface of human manganese superoxide dismutase. J. Biol. Chem. 279, 5861–5866 (2004).
Guan, Y. et al. Crystal structure of Y34F mutant human mitochondrial manganese superoxide dismutase and the functional role of tyrosine 34. Biochemistry 37, 4722–4730 (1998).
Demicheli, V., Moreno, D. M. & Radi, R. Human Mn-superoxide dismutase inactivation by peroxynitrite: a paradigm of metal-catalyzed tyrosine nitration in vitro and in vivo. Metallomics 10, 679–695 (2018).
Cabelli, D. E. et al. Role of tryptophan 161 in catalysis by human manganese superoxide dismutase. Biochemistry 38, 11686–11692 (1999).
Hearn, A. S., Tu, C., Nick, H. S. & Silverman, D. N. Characterization of the product-inhibited complex in catalysis by human manganese superoxide dismutase. J. Biol. Chem. 274, 24457–24460 (1999).
Bull, C., Niederhoffer, E. C., Yoshida, T. & Fee, J. A. Kinetic studies of superoxide dismutases: properties of the manganese-containing protein from Thermus thermophilus. J. Am. Chem. Soc. 113, 4069–4076 (1991).
Valentine, J. S. & Curtis, A. B. A convenient preparation of solutions of superoxide aniom and the reaction of superoxide anion with a copper (II) complex. J. Am. Chem. Soc. 97, 224–226 (1975).
Azadmanesh, J. et al. Revealing the atomic and electronic mechanism of human manganese superoxide dismutase product inhibition. Nat. Commun. 15, 5973 (2024).
Leto, D. F. & Jackson, T. A. Mn K-edge X-ray absorption studies of oxo- and hydroxo-manganese(IV) complexes: experimental and theoretical insights into pre-edge properties. Inorg. Chem. 53, 6179–6194 (2014).
Westre, T. E. et al. A multiplet analysis of Fe K-edge 1s → 3d pre-edge features of iron complexes. J. Am. Chem. Soc. 119, 6293–6314 (1997).
Kroll, T. et al. Effect of 3d/4p mixing on 1s2p resonant inelastic x-ray scattering: electronic structure of oxo-bridged iron dimers. J. Am. Chem. Soc. 143, 4569–4584 (2021).
Vance, C. K. & Miller, A. F. Novel insights into the basis for Escherichia coli superoxide dismutase’s metal ion specificity from Mn-substituted FeSOD and its very high E(m). Biochemistry 40, 13079–13087 (2001).
Braun, A. et al. X-ray spectroscopic study of the electronic structure of a trigonal high-spin Fe(IV) horizontal lineO complex modeling non-heme enzyme intermediates and their reactivity. J. Am. Chem. Soc. 145, 18977–18991 (2023).
Leveque, V. J., Vance, C. K., Nick, H. S. & Silverman, D. N. Redox properties of human manganese superoxide dismutase and active-site mutants. Biochemistry 40, 10586–10591 (2001).
Azadmanesh, J., Trickel, S. R. & Borgstahl, G. E. O. Substrate-analog binding and electrostatic surfaces of human manganese superoxide dismutase. J. Struct. Biol. 199, 68–75 (2017).
Carrasco, R., Morgenstern-Badarau, I. & Cano, J. Two proton-one electron coupled transfer in iron and manganese superoxide dismutases: A density functional study. Inorg. Chim. Acta 360, 91–101 (2007).
Abreu, I. A., Rodriguez, J. A. & Cabelli, D. E. Theoretical studies of manganese and iron superoxide dismutases: superoxide binding and superoxide oxidation. J. Phys. Chem. B 109, 24502–24509 (2005).
Sheng, Y. et al. Six-coordinate manganese(3+) in catalysis by yeast manganese superoxide dismutase. Proc. Natl Acad. Sci. USA 109, 14314–14319 (2012).
Bonetta Valentino, R. et al. The structure-function relationships and physiological roles of MnSOD mutants. Biosci. Rep. 42, BSR20220202 (2022).
Andres, C. M. C., Perez de la Lastra, J. M., Andres Juan, C., Plou, F. J. & Perez-Lebena, E. Superoxide anion chemistry-its role at the core of the innate immunity. Int. J. Mol. Sci., 24, 1841 (2023).
Hamalainen, K., Siddons, D. P., Hastings, J. B. & Berman, L. E. Elimination of the inner-shell lifetime broadening in x-ray-absorption spectroscopy. Phys. Rev. Lett. 67, 2850–2853 (1991).
de Groot, F. M. F., Krisch, M. H. & Vogel, J. Spectral sharpening of the Pt L edges by high-resolution x-ray emission. Phys. Rev. B, 66, 195112 (2002).
Kroll, T. et al. Resonant inelastic X-ray scattering on ferrous and ferric bis-imidazole porphyrin and cytochrome c: nature and role of the axial methionine-Fe bond. J. Am. Chem. Soc. 136, 18087–18099 (2014).
Solomon, E. I. Preface forum: functional insight from physical methods on metalloenzymes. Inorg. Chem. 44, 723–726 (2005).
Azadmanesh, J., Trickel, S. R., Weiss, K. L., Coates, L. & Borgstahl, G. E. O. Preliminary neutron diffraction analysis of challenging human manganese superoxide dismutase crystals. Acta Crystallogr F. Struct. Biol. Commun. 73, 235–240 (2017).
Whittaker, M. M. & Whittaker, J. W. Metallation state of human manganese superoxide dismutase expressed in Saccharomyces cerevisiae. Arch. Biochem Biophys. 523, 191–197 (2012).
Lutz, W. E. et al. Perfect crystals: microgravity capillary counterdiffusion crystallization of human manganese superoxide dismutase for neutron crystallography. NPJ Microgravity 9, 39 (2023).
Ng, J. D., Gavira, J. A. & Garcia-Ruiz, J. M. Protein crystallization by capillary counterdiffusion for applied crystallographic structure determination. J. Struct. Biol. 142, 218–231 (2003).
Azadmanesh, J., Lutz, W. E., Coates, L., Weiss, K. L. & Borgstahl, G. E. O. Cryotrapping peroxide in the active site of human mitochondrial manganese superoxide dismutase crystals for neutron diffraction. Acta Crystallogr F. Struct. Biol. Commun. 78, 8–16 (2022).
Kwon, H., Langan, P. S., Coates, L., Raven, E. L. & Moody, P. C. E. The rise of neutron cryo-crystallography. Acta Crystallogr D. Struct. Biol. 74, 792–799 (2018).
Coates, L. et al. The macromolecular neutron diffractometer MaNDi at the spallation neutron source. J. Appl Crystallogr 48, 1302–1306 (2015).
Coates, L., Stoica, A. D., Hoffmann, C., Richards, J. & Cooper, R. The macromolecular neutron diffractometer (MaNDi) at the spallation neutron source, oak ridge: enhanced optics design, high-resolution neutron detectors and simulated diffraction. J. Appl Crystallogr 43, 570–577 (2010).
Arnold, O. et al. Mantid—Data analysis and visualization package for neutron scattering and μ SR experiments. Nucl. Instrum. Meth A 764, 156–166 (2014).
Sullivan, B. et al. BraggNet: integrating Bragg peaks using neural networks. J. Appl Crystallogr 52, 854–863 (2019).
Sullivan, B. et al. Improving the accuracy and resolution of neutron crystallographic data by three-dimensional profile fitting of Bragg peaks in reciprocal space. Acta Crystallogr D. Struct. Biol. 74, 1085–1095 (2018).
Campbell, J. W. LAUEGEN, an X-windows-based program for the processing of Laue diffraction data. J. Appl. Crystallogr. 28, 228–236 (1995).
Minor, W., Cymborowski, M., Otwinowski, Z. & Chruszcz, M. HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr D. Biol. Crystallogr 62, 859–866 (2006).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D. Biol. Crystallogr 68, 352–367 (2012).
Garman, E. F. & Weik, M. Radiation damage in macromolecular crystallography. Methods Mol. Biol. 1607, 467–489 (2017).
O’Young, C. & Lippard, S. J. Reactions of superoxide anion with copper(II) salicylate complexes. J. Am. Chem. Soc. 102, 4920–4924 (1980).
McClune, G. J. & Fee, J. A. Stopped flow spectrophotometric observation of superoxide dismutation in aqueous solution. FEBS Lett. 67, 294–298 (1976).
Kim, S., DiCosimo, R. & Filippo, J. S. Spectrometric and chemical characterization of superoxide. Anal. Chem. 51, 679–681 (1979).
Newville, M. Larch: an analysis package for XAFS and related spectroscopies. J. Phys.: Conf. Ser. 430, 012007 (2013).
Rehr, J. J., Kas, J. J., Vila, F. D., Prange, M. P. & Jorissen, K. Parameter-free calculations of X-ray spectra with FEFF9. Phys. Chem. Chem. Phys. 12, 5503–5513 (2010).
Neese, F., Wennmohs, F., Becker, U. & Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 152, 224108 (2020).
Takano, Y. & Houk, K. N. Benchmarking the conductor-like polarizable continuum model (CPCM) for aqueous solvation free energies of neutral and ionic organic molecules. J. Chem. Theory Comput 1, 70–77 (2005).
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 (2005).
Becke, A. D. Becke’s three parameter hybrid method using the LYP correlation functional. J. Chem. Phys. 98, 5648 (1993).
Neese, F. Prediction and interpretation of the 57Fe isomer shift in Mössbauer spectra by density functional theory. Inorg. Chim. Acta 337, 181–192 (2002).
DeBeer George, S., Petrenko, T. & Neese, F. Prediction of iron K-edge absorption spectra using time-dependent density functional theory. J. Phys. Chem. A 112, 12936–12943 (2008).
Roemelt, M. et al. Manganese K-edge X-ray absorption spectroscopy as a probe of the metal-ligand interactions in coordination compounds. Inorg. Chem. 51, 680–687 (2012).
Acknowledgements
This research was supported by the NIH (R01-GM145647) and NASA EPSCoR (NE−80NSSC17M0030 and NE-NNX15AM82A) to G.E.O.B. The UNMC Structural Biology Core Facility was funded by the Fred and Pamela Buffett NCI Cancer Center Support Grant (P30CA036727). The research at Oak Ridge National Laboratory (ORNL) Spallation Neutron Source was sponsored by the Scientific User Facilities Division, Office of Basic Energy Sciences, US Department of Energy. The Office of Biological and Environmental Research supported research at ORNL Center for Structural Molecular Biology (CSMB) using facilities supported by the Scientific User Facilities Division, Office of Basic Energy Sciences, US Department of Energy. Use of the Stanford Synchrotron Radiation Lightsource (SSRL), SLAC National Accelerator Laboratory, is supported by the US Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences under Contract DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (P30GM133894). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH. Quantum chemical computations were completed using the Holland Computing Center of the University of Nebraska, which receives support from the Nebraska Research Initiative.
Author information
Authors and Affiliations
Contributions
J.A. purified protein, performed X-ray/neutron crystallography and X-ray spectroscopy experiments, processed and analyzed data, performed quantum chemistry calculations, and wrote the manuscript. K.S., L.R.S., W.E.L., J.J.L., E.C., and M.D. purified protein, participated in data collection/refinement, helped make figures and performed instrument maintenance. S.K. and A.N. prepared superoxide solution and performed experiments. L.C., D.A.A.M., and K.W. performed neutron crystallography experiments and processed data. T.K. performed X-ray spectroscopy experiments, processed data, and performed quantum chemistry calculations. G.E.O.B. acquired funding, designed experiments, and participated in writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Azadmanesh, J., Slobodnik, K., Struble, L.R. et al. The role of Tyr34 in proton coupled electron transfer and product inhibition of manganese superoxide dismutase. Nat Commun 16, 1887 (2025). https://doi.org/10.1038/s41467-025-57180-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57180-3
