
Abstract
Salmonella utilizes type 3 secreted effector proteins to induce plasma membrane (PM) perturbations during invasion of host cells1. The effectors drive mobilization of host membranes to generate cell surface ruffles, followed by invagination and scission of the PM to generate Salmonella-containing vacuoles (SCVs)2. Here, we show that LRRK2 kinase generates membrane reservoirs exploited by Salmonella during invasion. The reservoirs are tubular compartments associated with the PM under basal conditions and are formed through the phosphorylation of RAB10 GTPase by LRRK2. Mobilization of membrane reservoirs to generate invasion ruffles mediates delivery of phosphorylated RAB10 to invasion sites. Subsequently, RAB10 dephosphorylation is required for its inactivation by a bacterial GTPase activating protein and subsequent scission of the PM. RAB10 dephosphorylation is mediated by a TLR4/PIEZO1/TMEM16F-dependent pathway and is inhibited by hyperactive variants of LRRK2. Our findings reveal how Salmonella exploits LRRK2-dependent PM dynamics during invasion and provide new insight into how LRRK2 variants can protect against bacterial infection3,4.
Similar content being viewed by others

Introduction
Salmonella enterica serovar Typhimurium (STm) is a facultative intracellular bacterial pathogen that contributes to foodborne gastroenteritis in humans5. These bacteria exhibit the ability to invade and replicate within an intracellular niche by utilizing type 3 secretion systems (T3SSs) to translocate virulence factors, known as effectors, into host cells6. STm utilizes two distinct T3SSs, encoded within Salmonella pathogenicity islands (SPI)−1 and SPI-2. The SPI-1 T3SS translocates effectors that trigger cytoskeletal rearrangements and alterations in membrane dynamics, initiating the internalization of the bacteria into SCVs. Subsequently, the SPI-2 encoded T3SS translocates effectors across the SCV membrane, facilitating intracellular growth and cell-to-cell spread7,8.
In a recent study we examined how the cooperative actions of SPI-1 T3SS effectors subvert membrane trafficking during invasion of host cells9. We observed that SopB, SopE2 and SipC promote membrane mobilization to sites of STm invasion9. Membrane was found to be mobilized from multiple cellular sources, including deep invaginations of the PM referred to as membrane reservoirs9,10. Under normal growth conditions, host RAB10 stabilizes membrane reservoirs in its GTP-bound state through interaction with its effectors EHBP1 and MICAL-L1 and through BAR ___domain proteins PACSIN2 and PACSIN39. Upon infection, SopB mediates recruitment of RAB10 from membrane reservoirs to sites of bacterial invasion9. The recruitment of host membranes to invasion sites enables exocyst-dependent exocytosis and formation of cell surface protuberances, called invasion ruffles, which enable bacterial uptake9,11 (Fig. 1a).

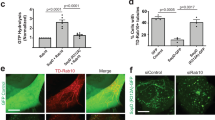
a Current model of RAB10’s roles in STm invasion. b WT Henle 407 cells were treated with 50 nM MLi-2 for 90 min and cell lysates were immunoblotted with indicated antibodies. c and d Representative images (c) and quantifications (d) of RAB10+ tubules (visualized with anti-RAB10 antibody) in WT Henle 407 cells in control condition or treated with MLi-2 as in (b). e Representative images of Henle 407 cells transfected with GFP-RAB10 and stained for phospho-RAB10 (T73), with or without MLi-2 treatment. f Line plot profile of the white arrow in the inset (control) in e. Arb. units (arbitrary units) indicate the signal densities along the chosen white arrow. g and h Representative images (g) and quantifications (h) of RAB10+ membrane reservoirs in WT Henle 407 cells transfected with GFP-RAB10 and treated with MLi-2. Arrow indicates a RAB10+ membrane reservoir structure. i and j Representative images (i) and quantifications (j) of CellMask+ membrane reservoirs in WT Henle 407 cells in control condition or treated with MLi-2 as in (b). Cells in both conditions did not have RAB10 overexpressed. k CRISPR/Cas9-mediated deletion of LRRK2. l and m, Representative images (l) and quantifications (m) of endogenous RAB10+ membrane reservoirs in indicated cells in basal growth condition. n and o, Representative images (n) and quantifications (o) of RAB10+ membrane reservoirs in RAB10 KO Henle 407 cells transfected with myc-PM-RAB10 constructs, and treated with or without MLi-2 (50 nM, 90 min). All images shown are representative images from three independent experiments. Data shown are means ± standard deviation (S.D.) for three independent experiments. At least 100 cells for each condition in each experiment were scored for the presence of RAB10-(c, g and l) or CellMask-(h) positive tubules. P values were calculated using two tailed unpaired t-test (d, h and j), one-way analysis of variance (ANOVA) (m), or two-way ANOVA (o). Scale bars, (c, g, i, l and n) 10 μm, (e, upper and lower panels) 10 μm, (e, middle panel) 3 μm. Source data are provided as a Source Data file.
The next step of invasion involves invagination and scission of the PM to generate SCVs2. Since RAB10 and its effectors can stabilize the PM in membrane reservoirs, it’s recruitment to invasion sites would be expected to adversely affect STm uptake. However, the SPI-1 T3SS effector SopD inhibits RAB10 at invasion sites through its GTPase activating protein (GAP) activity12. By converting RAB10 to its GDP-bound form, SopD promotes Dynamin−2 (DNM2) recruitment to invasion sites, thereby initiating PM scission to generate SCVs12 (Fig. 1a). SopB also contributes to PM scission at invasion sites via dephosphorylation of phosphatidylinositol-(4,5)-bisphosphate13. Altogether, the combined activities of SopB, SopE2 and SipC promote membrane mobilization and SopB and SopD promote scission, thereby allowing STm to efficiently control PM dynamics and invade host cells9.
Phosphorylation of RAB GTPases has emerged as a key regulatory mechanism14. Thus, we sought to further understand the regulation of RAB10 by examining its phosphorylation during STm infection (Fig. 1a). In other cellular contexts, LRRK2 kinase was shown to phosphorylate RAB10 on Threonine-73 (T73), thereby stabilizing RAB10 in its GTP-bound form15,16. Here, we demonstrate that LRRK2 regulates PM dynamics through a dynamic RAB10 phospho-regulation cycle that is exploited by STm during infection. Furthermore, we show that pathogenic LRRK2 variants associated with both Parkinson’s Disease (PD) and Inflammatory Bowel Disease (IBD)17,18,19 can alter PM dynamics and have the beneficial effect of inhibiting STm invasion.
Results
LRRK2 generates membrane reservoirs via RAB10 phosphorylation
By western blotting with a phospho-specific antibody, we observed significant phosphorylation of RAB10 on T73 in Henle 407 human intestinal epithelial cells under normal growth conditions (Fig. 1b). Treatment of cells with a selective LRRK2 kinase inhibitor (MLi-2)20 decreased RAB10 T73 phosphorylation (Fig. 1b). Our findings are consistent with prior studies indicating that RAB10 can be phosphorylated on T73 under normal growth conditions15,21,22.
Next, we examined the impact of RAB10 T73 phosphorylation on RAB10 localization. Prior studies revealed that RAB10 localizes to tubular structures under normal growth conditions23,24. Here, we found that treatment of cells with MLi-2 disrupted these tubular structures bearing endogenous RAB10 (i.e., detected with antibodies) (Fig. 1c, d). These data suggest that LRRK2 kinase activity is closely linked to the dynamics of the RAB10+ tubules via RAB10 phosphorylation.
With immunofluorescence microscopy we observed phospho-RAB10 T73 was enriched on GFP-RAB10+ tubules compared to elsewhere in the cell (Fig. 1e, f). Consistent with the western blot results, the immunofluorescence signal for phospho-RAB10 was decreased after MLi-2 treatment, concomitant with loss of tubular structures (Fig. 1e). These findings indicate that RAB10 is phosphorylated by LRRK2 on T73 and that the phosphorylated form of RAB10 localizes to membrane tubules under normal growth conditions.
Our recent study suggested that RAB10+ tubules represent membrane reservoirs, deep invaginations of the PM that are primed for mobilization9. Notably, more than 85% of RAB10+ tubules were found to colocalize with markers of the PM9. Based on this prior work, we hypothesized that LRRK2-mediated phosphorylation of RAB10 controls the formation of membrane reservoirs. To test this hypothesis, we labeled cells overexpressing GFP-RAB10 with the fluorescent PM probe CellMask to visualize membrane reservoirs, defined as CellMask+ tubules longer than 10 µm within the cell body9. Treatment of cells with MLi-2 disrupted these membrane reservoirs, concomitant with the loss of RAB10+ tubules (Fig. 1g, h). Membrane reservoirs in cells with endogenous RAB10 expression (i.e., without GFP-RAB10 transfection) were also inhibited by MLi-2 treatment (Fig. 1i, j). MLi-2 inhibition of LRRK2 also induced the loss of RAB10+ membrane reservoirs in SH-SY5Y human neuroblastoma cells, MCF-7 human adenocarcinoma cells and mouse embryonic fibroblasts (Supplementary Fig. 1a–f), cell types previously shown to possess these membrane reservoir structures9. To confirm that MLi-2 was not having off-target effects, we generated LRRK2 knockout (KO) cells (Fig. 1k) and compared them to a previously generated RAB10 KO cell line12. Knockout of LRRK2 disrupted the formation of membrane reservoirs visualized by immunofluorescence staining of endogenous RAB10 (Fig. 1l, m). We conclude that RAB10 phosphorylation by LRRK2 is necessary for membrane reservoir formation in a variety of cell types.
The serine/threonine protein phosphatase PPM1H is known to counteract LRRK2 signaling by dephosphorylation of RAB1025,26,27,28. Therefore, we modulated RAB10’s phosphorylation state by transfection of cells with a plasmid encoding PPM1H and monitored the dynamics of membrane reservoirs. Overexpression of WT PPM1H, but not catalytically inactive PPM1H (mutants H153D and D288A)25 suppressed RAB10 phosphorylation (Supplementary Fig. 2a) and the formation of RAB10+ membrane reservoirs (Supplementary Fig. 2b–c). Thus, increased phosphatase activity induced by PPM1H overexpression suppresses LRRK2-mediated phosphorylation of RAB10 and impacts formation of membrane reservoirs.
Next, we generated phospho-site mutants of RAB10, a strategy that was previously used to examine the impact of LRRK2 phosphorylation on RAB10 function22,29. Since phospho-site mutants of RAB10 were reported to impair geranylgeranylation of RAB1030, we generated phospho-site mutants within a RAB10 construct artificially targeted to the PM by replacing its C-terminal cysteines with the polybasic C-terminus of K-Ras (myc-PM-RAB10)31. In prior studies we showed that myc-PM-RAB10 was sufficient to complement formation of membrane reservoirs in RAB10 KO cells9. As compared to the myc-RAB10 construct, the generated non-phosphorylatable (T73A) and phosphomimic (T73E) mutants of PM-targeted RAB10 predominantly localized on the PM instead of intracellular compartments (Supplementary Fig. 3).
In RAB10 KO cells, expression of WT and T73E, but not T73A, myc-PM-RAB10 were sufficient for membrane reservoir formation (Fig. 1n, o). Upon MLi-2 treatment, the membrane reservoirs positive for WT myc-PM-RAB10 were disassembled. In contrast, expression of the T73E phosphomimic mutant stabilized these structures in the presence of MLi-2 (Fig. 1n, o). Together, our findings indicate that phosphorylation of T73 on RAB10 by LRRK2 is both necessary and sufficient for the formation of RAB10+ membrane reservoirs under normal growth conditions.
LRRK2-dependent membrane reservoirs are exploited by STm during invasion
During STm infection, RAB10 is known to be rapidly recruited from membrane reservoirs to invasion sites9. Here, we observed phospho-RAB10 T73 enrichment at STm invasion sites at 10 min post-infection (p.i.) (Fig. 2a, upper panels). We observed no significant recruitment of either GFP-RAB10 or phospho-RAB10 to STm invasion sites in LRRK2 KO cells (Fig. 2a, lower panels), suggesting that RAB10 is recruited in the phosphorylated state. To further support this, we expressed myc-PM-RAB10 constructs in RAB10 KO cells. We observed recruitment of WT and T73E but not T73A myc-PM-RAB10 to invasion sites at 10 min p.i. (Fig. 2b, c).

a Representative images of WT Henle 407 cells transfected with GFP-RAB10 and infected with WT STm. Cells were fixed at 10 min p.i. and stained for phospho-RAB10 (T73). In this and following panels, invasion sites at 10 min p.i. were identified by positive F-actin and STm staining. The cell boundaries are depicted by the white outlines. b and c Representative images (b) and quantifications (c) of myc-PM-RAB10 recruitment to STm invasion sites. RAB10 KO Henle 407 cells were transfected with myc-PM-RAB10 WT, T73E or T73A construct, and then infected with WT STm. Cells were fixed at 10 min p.i. and stained for myc-tag. Line plot profile follows the white arrow in the ‘Merge’ channel in (b). d and e, Representative images (d) and quantifications (e) of GFP-RAB10 recruitment to STm invasion sites. RAB10 KO Henle 407 cells were transfected with GFP-RAB10 WT, Q68L or T23N construct and infected with WT STm. Cells were fixed and imaged at 10 min p.i. Line plot profile follows the white arrow in the ‘Merge’ channel in d. f Representative SEM images of invasion ruffles. WT and LRRK2 KO Henle 407 cells were infected with WT STm and fixed at 10 min p.i. g Quantifications of invasion ruffle areas identified in (f). The areas of individual invasion ruffle were measured as described in the “Methods” section. h WT and LRRK2 KO Henle 407 cells were infected with WT STm and lysed at 2 hours p.i. for CFU counting. All images shown are representative images from three independent experiments. Data shown are means ± S.D. for three independent experiments. At least 100 invasion sites (c and e) for each condition in each experiment were scored for the recruitment of RAB10. For SEM analysis of invasion ruffle area (g), 15 invasion ruffles for each condition in each experiment were scored (45 invasion ruffles in total for each condition). P values were calculated using one-way ANOVA (c and e) or two tailed unpaired t-test (g and h). Scale bars, 10 μm. Source data are provided as a Source Data file.
We also examined the role of GTP-binding for RAB10 recruitment to invasion sites using GFP-fusion constructs without PM targeting. We observed that WT and constitutively active (Q68L) GFP-RAB10 constructs displayed significant recruitment to STm invasion sites but a dominant negative (T23N) mutant of RAB10 did not (Fig. 2d, e). Together, these data suggest that RAB10 is recruited to STm invasion sites in its phosphorylated and GTP-bound form.
The mobilization of RAB10+ membrane reservoirs during STm infection was shown to be required for optimal ruffle formation and bacterial entry into host cells9. Thus, we wanted to determine if LRRK2 contributes to the formation of invasion ruffles. For this, WT and LRRK2 KO cells were infected with WT STm, and invasion ruffles were imaged with scanning electron microscopy and subsequently measured. LRRK2 KO cells displayed significantly reduced invasion ruffle size (Fig. 2f, g). Furthermore, bacterial invasion, as determined by measuring intracellular colony forming units (CFU) at 2 hours p.i., was decreased in LRRK2 KO cells compared to WT cells (Fig. 2h).
We also examined invasion ruffle sizes in RAB10 KO cells expressing WT, T73E, or T73A myc-PM-RAB10 constructs. For these experiments, we utilized fluorescence microscopy so that cells transfected with the various constructs could be identified. The expression of WT or T73E myc-PM-RAB10 construct was able to generate ruffles of normal size (Supplementary Fig. 4a, b). However, expression of the T73A mutant significantly reduced the ruffle size. Together, these data suggest that LRRK2 generates membrane reservoirs via RAB10 T73 phosphorylation that STm can exploit to facilitate its invasion.
RAB10 dephosphorylation is required for PM scission at STm invasion sites
The PM undergoes invagination at the base of invasion ruffles prior to scission of the PM and generation of SCVs12. While RAB10 is recruited to invasion sites from membrane reservoirs within the first 10 min p.i., it is normally removed from these sites within 30 min12. Removal of RAB10 from invasion sites is mediated by the STm SPI-1 T3SS effector SopD, which encodes a GAP ___domain that catalyzes GTP hydrolysis by RAB1012. During infection by SopD-deficient STm (ΔsopD mutant), RAB10 is retained at invasion sites and inhibits bacterial uptake by stabilizing invaginated regions of the PM and blocking recruitment of DNM2, a GTPase required for PM scission10,20.
The phosphorylation of RAB10 on T73 by LRRK2 has been shown to limit the accessibility of GAP proteins and thus stabilize RAB10 in its GTP-bound and active form15. Therefore, we hypothesized that RAB10 must undergo dephosphorylation at invasion sites to allow SopD-mediated inactivation of RAB10. To test this, we monitored RAB10 T73 phosphorylation upon STm invasion by western blot. We observed that RAB10 is dephosphorylated between 10 and 30 min p.i. (Fig. 3a, b), a time period consistent with the removal of RAB10 from invasion sites of WT STm12. We also observed retention of T73E, but not WT or T73A, myc-PM-RAB10 at invasion sites at 30 min p.i. (Fig. 3c, d). Similarly, constitutively active but not WT or dominant negative GFP-RAB10 displayed significant retention at STm invasion sites at 30 min p.i. (Fig. 3e, f). Together, these data suggest that RAB10 is removed from invasion sites in its dephosphorylated and GDP-bound form.

a and b Representative images (a) and quantifications (b) of phospho-RAB10 (T73) signal measured by western blot in WT Henle 407 cells infected with WT STm for indicated time. In this and following panels with the quantification of RAB10 phosphorylation by western blot, the relative phospho-RAB10 (T73) signal was calculated by comparison with the respective total RAB10 signal. c and d, Representative images (c) and quantifications (d) of PM-RAB10 retention at STm invasion sites at 30 min p.i. RAB10 KO Henle 407 cells were transfected with WT, T73E or T73A myc-PM-RAB10, and then infected with WT STm. Cells were fixed at 30 min p.i. and stained for myc-tag. In this and the following panels, invasion sites at 30 min p.i. were identified by staining of extracellular STm before permeabilization. Line plot profile follows the white arrow in the ‘Merge’ channel in c. e and f, Representative images (e) and quantifications (f) of GFP-RAB10 retention at STm invasion sites at 30 min p.i. RAB10 KO Henle 407 cells were transfected with GFP-RAB10 WT, Q68L or T23N construct, and then infected with WT STm. Cells were fixed and imaged at 30 min p.i. Data shown are means ± S.D. for three independent experiments. At least 100 invasion sites (d and f) for each condition in each experiment were scored for the retention of RAB10. P values were calculated using one-way ANOVA. Scale bars, 10 μm. Source data are provided as a Source Data file.
Next, we examined the impact of RAB10 phosphorylation on PM scission during STm invasion. For this, we used a PM scission assay developed by Terebiznik et al13. Invaginations of the PM and unsealed SCVs were visualized using a fluorescent protein targeted to the PM (PM-mCherry) via the myristoylation and palmitoylation sequences from Lyn tyrosine kinase32. CellMask was added to the medium at 30 min p.i. to label the cell surface and accessible membranes prior to fixation. The degree of scission was quantified by measuring the generation of sealed SCVs (PM-mCherry+ bacteria that are inaccessible to CellMask).
First, we examined PM scission following infection of cells with WT STm. The infection of RAB10 KO Henle cells complemented with T73E myc-PM-RAB10 displayed defective PM scission, compared to cells expressing either WT or T73A myc-PM-RAB10 (Fig. 4a, b). These findings suggest that phosphorylation of RAB10 is sufficient to impair PM scission during STm invasion.

a Representative images of WT or RAB10 KO Henle 407 cells transfected with the indicated constructs (PM-mCherry with empty vector or indicated PM-RAB10 construct), infected with WT BFP-STm for 30 min and labeled with CellMask. b Quantifications of a and Supplementary Fig. 5. Scission is represented as the percent of bacteria in SCVs. The total number of bacteria that were positive for PM-mCherry at the invasion site was quantified and the proportion of these bacteria that were negative for CellMask was determined. These bacteria were in a sealed compartment that was considered an SCV and the number was used to represent the invasion efficiency at 30 min p.i. (c and d) Representative images (c) and quantifications (d) of phospho-RAB10 (T73) signal measured by western blot in WT Henle 407 cells, or LRRK2 KO Henle 407 cells complemented with LRRK2 WT or mutant expression. The cells were infected with WT STm and cell lysates were collected and immunoblotted at 30 min p.i. for phospho-RAB10 (T73), total RAB10, or ß-actin (loading control). e WT or LRRK2 KO Henle 407 cells transfected with indicated constructs (PM-mCherry with empty vector or indicated LRRK2 construct), and then infected with WT BFP-STm SL1344 strain for 30 min and labeled with CellMask. PM scission was quantified as in c. Representative images are shown in Supplementary Fig. 6a, b. Data shown are means ± S.D. for three independent experiments. At least 25 independent cells (b and e) for each condition in each experiment were scored for PM scission. P values were calculated using two-way ANOVA. In b and e P values were calculated between the KOs (RAB10 KO or LRRK2 KO) and their respective WT controls. Scale bars, 10 μm. Source data are provided as a Source Data file.
Next, we examined PM scission following infection of cells with ΔsopD STm. In WT Henle cells, these bacteria displayed a PM scission defect compared to WT STm, consistent with previous studies12. Uptake of ΔsopD STm was enhanced in RAB10 KO cells, as expected12. Complementation of RAB10 KO cells with either WT or T73E mutant myc-PM-RAB10 restored the PM scission defect during ΔsopD STm invasion (Supplementary Fig. 5& Fig. 4b). However, expression of the T73A mutant of myc-PM-RAB10 had no effect on bacterial entry to SCVs (Supplementary Fig. 5 & Fig. 4b), consistent with its absence from invasion sites (Fig. 2b, c). Together, these data suggest that the dephosphorylation of RAB10 allows SopD-mediated removal from invasion sites, thereby allowing bacterial uptake into SCVs.
Disease-associated LRRK2 variants inhibit STm invasion by preventing RAB10 dephosphorylation
Coding variants impacting the LRRK2 kinase ___domain have been shown to cause hyper-phosphorylation of RAB10 and are associated with an increased risk for the development of Crohn’s disease (CD) and Parkinson’s disease (PD)17,18,19. In contrast, other common LRRK2 variants, including the N551K variant, are not associated with the hyperphosphorylation of RAB1033 and have been shown to be protective in multiple PD cohorts34. Based on our findings above, we hypothesized that risk-associated LRRK2 variants can alter PM dynamics during STm invasion through altered RAB10 phosphorylation.
To test this, we examined the effects of LRRK2 variant expression in LRRK2 KO Henle cells. Expression of WT LRRK2 and N551K LRRK2 restored endogenous RAB10 T73 phosphorylation to a level comparable to WT cells (Fig. 4c, d). However, the risk variants G2019S and N2081D induced RAB10 phosphorylation to a higher level under basal growth conditions (Fig. 4c, d), consistent with previous studies18. With STm invasion, RAB10 was dephosphorylated in cells expressing either WT or N551K LRRK2 at 30 min p.i. In contrast, cells expressing G2019S and N2081D LRRK2 variants maintained RAB10 phosphorylation upon STm invasion (Fig. 4c, d), suggesting that hyperactive disease-associated LRRK2 variants are able to disturb RAB10 dephosphorylation during STm invasion.
Since we determined above that the dephosphorylation of RAB10 is required for PM scission and bacterial uptake, we next examined the impact of LRRK2 variant expression on WT STm invasion. Expression of G2019S or N2081D LRRK2 in the LRRK2 KO cells was sufficient to induce a significant defect in PM scission, as indicated by the higher percentage of CellMask+ bacteria (Supplementary Fig. 6a, b & Fig. 4e). In contrast, cells expressing WT or N551K LRRK2 displayed PM scission comparable to WT Henle cells (Supplementary Fig. 6a, b & Fig. 4e). Infection of cells with ΔsopD STm led to a PM scission defect, as expected12. This scission defect was not observed in LRRK2 KO cells, consistent with our findings above showing that RAB10 is not recruited to invasion sites (Fig. 2a). However, expression of all LRRK2 alleles was sufficient to induce a PM scission defect during ΔsopD STm infection (Supplementary Fig. 6a, b & Fig. 4e). These data suggest that disease-associated LRRK2 variants induce high and stable phosphorylation of RAB10 on T73, thereby preventing SopD-mediated inactivation of RAB10 and subsequent PM scission.
TLR4 mediates RAB10 dephosphorylation during STm invasion
Next, we investigated the mechanism of RAB10 dephosphorylation at invasion sites. STm SPI-1 T3SS effectors SopB and SopD have been shown to act cooperatively in regulating RAB10+ dynamics during STm invasion12. However, mutants lacking one or both of these effectors were still capable of inducing dephosphorylation of RAB10 during infection (Fig. 5a, b). Remarkably, the supernatant of STm cultures (STm S/N), heat killed bacteria (STm dead) and bacteria lacking a functional SPI-1 T3SS and targeted artificially to the host cell surface by expression of the Yersinia pseudotuberculosis Invasin protein (ΔinvA/Inv STm)35 were sufficient to induce RAB10 dephosphorylation within 30 min (Fig. 5c, d), indicating another bacterial factor was involved.

a and b Representative images (a) and quantifications (b) of phospho-RAB10 (T73) signal measured by western blot in WT Henle 407 cells infected with the indicated STm strains. Cell lysates were collected and immunoblotted for phospho-RAB10 (T73), total RAB10, or ß-actin (loading control) at 30 min p.i. c and d, Representative images (c) and quantifications (d) of phospho-RAB10 (T73) signal measured by western blot in WT Henle 407 cells infected with WT STm, treated with WT STm supernatant (STm S/N), dead WT STm, LPS (100 ng/ml) or ΔinvA/Inv STm (invasion-deficient attenuated strain with inactivation of T3SS-1, and with invasin gene from Yersinia expressed) for 30 min. e and f Representative images (e) and quantifications of (f) phospho-RAB10 (T73) signal measured by western blot. WT, LRRK2 KO and TLR4 KO Henle 407 cells were infected with WT STm and cell lysates were collected at 10 min or 30 min p.i. and immunoblotted for TLR4, phospho-RAB10 (T73), total RAB10, or ß-actin (loading control). g and h Quantifications (g) and representative images (h) of phospho-RAB10 (T73) localization at WT STm invasion sites at the indicated times p.i. WT, LRRK2 KO or TLR4 KO Henle 407 cells were transfected with GFP-RAB10 and then infected with WT STm. Cells were fixed at 10 or 30 min p.i. and stained for phospho-RAB10 (T73) and STm. The cell boundaries in h are depicted by the white outlines. i WT Henle 407 cells and TLR4 KO Henle 407 cells were infected with WT STm and lysed at 2 hours p.i. for CFU counting. Data shown are means ± S.D. for three independent experiments. In g At least 100 invasion sites for each condition in each experiment were scored for the recruitment or the retention of RAB10. P values were calculated using one-way ANOVA (b and d), two-way ANOVA (f and g), or two tailed unpaired t-test (i). P values in d were calculated and labeled between the treated groups with the untreated control. Scale bars, 10 μm. Source data are provided as a Source Data file.
Lipopolysaccharide (LPS), a major component of the outer membrane of Gram-negative bacteria, is required for STm virulence36,37. A recent study suggested that LPS stimulation significantly decreased RAB10 T73 phosphorylation via the activation of Toll-like receptor 4 (TLR4) and downstream inhibition of LRRK2 kinase activity38. To determine if STm uses a similar strategy to promote RAB10 dephosphorylation, we treated cells with 100 ng/ml LPS for 30 min and monitored RAB10 phosphorylation via western blot. LPS treatment was sufficient to cause RAB10 dephosphorylation (Fig. 5c, d). Furthermore, LPS treatment for 30 min led to disruption of RAB10+ membrane reservoirs (Supplementary Fig. 7a, b) and reduced the size of invasion ruffles induced by WT STm (Supplementary Fig. 7c, d). However, expression of hyperactive disease-associated LRRK2 variants was sufficient to prevent RAB10 dephosphorylation induced by LPS treatment (Supplementary Fig. 8a, b).
To further examine if LPS triggers RAB10 dephosphorylation via TLR4, we generated TLR4 KO Henle 407 cells, which was confirmed by western blotting (Supplementary Fig. 9a) and loss of NF-κB activation in response to LPS treatment, as assessed by less production of mature NF-κB2 p52 (Supplementary Fig. 9a, b). Under basal growth conditions TLR4 localized to RAB10+ membrane reservoirs (Supplementary Fig. 9c). TLR4 KO cells displayed normal levels of RAB10+ membrane reservoirs (Supplementary Fig. 9d, e) and higher basal levels of RAB10 phosphorylation (Fig. 5e, f).
Phosphorylated RAB10 was recruited to STm invasion sites in TLR4 KO cells within 10 min, at levels comparable to WT cells (Supplementary Fig. 9f & Fig. 5g). Invasion ruffles of similar sizes were generated in TLR4 KO cells compared to WT cells at 10 min p.i. (Supplementary Fig. 7c, d). However, as the infection proceeded, in contrast to WT cells, TLR4 KO cells maintained high levels of RAB10 phosphorylation at 30 min (Fig. 5e, f) that was visible at STm invasion sites (Fig. 5g, h). Moreover, we also observed significant defects in PM scission at 30 min p.i. (Supplementary Fig. 9g, h) and bacterial invasion at 120 min p.i. in TLR4 KO cells (Fig. 5i). Together, these data suggest that during STm invasion, this bacterial pathogen uses LPS to stimulate TLR4 and induce RAB10 dephosphorylation at invasion sites, thereby allowing RAB10 inactivation/removal from invasion sites and the promotion of bacterial uptake.
PIEZO1 regulates plasma membrane dynamics at invasion sites via RAB10 dephosphorylation
TLR4 was found to activate macrophage activation through its interaction with PIEZO139, a mechanosensitive ion channel that regulates the influx of several cations including calcium40. Since calcium influx is required for STm invasion41,42, we hypothesized that PIEZO1 contributes to RAB10 dephosphorylation at STm invasion sites. To test this hypothesis, we treated cells with a highly selective small molecule agonist of PIEZO1, Yoda143. Upon treatment of cells with Yoda1 for 30 min, the amount of RAB10 T73 phosphorylation was significantly decreased (Fig. 6a, b). Furthermore, a loss of GFP-RAB10+ tubules was also observed upon Yoda1 treatment (Supplementary Fig. 10a, b). Treatment of cells with PIEZO1 inhibitors (GsMTx4, Gd3+, and ruthenium red)44 effectively blocked LPS- or Yoda1-induced RAB10 dephosphorylation (Fig. 6c, d & Supplementary Fig. 10c–f). These results indicate that PIEZO1 activation is both necessary and sufficient to trigger TLR4-dependent RAB10 dephosphorylation.

a and b Representative images (a) and quantifications (b) of phospho-RAB10 (T73) and total RAB10 signal measured by western blot in WT Henle 407 cells treated with the PIEZO1 agonist, Yoda1. Cell lysates were collected and immunoblotted following a 30 min treatment with either DMSO or Yoda1. Data shown are means ± S.D. for three independent experiments. c and d Representative images (c) and quantifications (d) of phospho-RAB10 (T73) signal measured by western blot in WT Henle 407 cells. The cells were treated with the PIEZO1 inhibitor GsMTx4, LPS and/or the PIEZO1 agonist Yoda1, and cell lysates were collected and immunoblotted for phospho-RAB10 (T73), total RAB10, or ß-actin (loading control) following 30 min treatment. e Representative images of TLR4 and PIEZO1 localizations in uninfected condition. WT or TLR4 KO Henle 407 cells were transfected with GFP-RAB10, or RAB10 KO Henle 407 cells were nontransfected. Then the cells were fixed and stained for TLR4 and PIEZO1. f Representative images of TLR4 and PIEZO1 localizations in at 10 min p.i. WT, TLR4 KO or RAB10 KO Henle 407 cells were infected with WT STm and then fixed at 10 min p.i. and stained for STm, TLR4 and PIEZO1. The images in the ‘Merge’ channel represent the merged accumulative fluorescence signals from the TLR4, PIEZO1 and STm channels. g and h Quantifications of TLR4 (g) and PIEZO1 (h) localizations at invasion sites in (f). i WT Henle 407 cells were untreated or pretreated with indicated PIEZO1 inhibitors, and then infected with WT STm and lysed at 2 hours p.i. for CFU counting. At least 100 invasion sites (g and h) for each condition in each experiment were scored for the recruitment of TLR4 or PIEZO1. All images shown are representative images from three independent experiments. Data shown are means ± S.D. for three independent experiments. P values were calculated using one-way ANOVA. Scale bars, 10 μm. In e and f the cell boundaries are depicted by the white outlines. Source data are provided as a Source Data file.
Given that TLR4 colocalizes with RAB10 on membrane reservoirs prior to infection (Supplementary Fig. 9c), we next explored the interaction between TLR4 and PIEZO1 both before and during STm infection. Consistent with prior findings39, we did not observe colocalization of TLR4 and PIEZO1 in uninfected cells (Fig. 6e). However, both TLR4 and PIEZO1 were recruited to STm invasion sites (Fig. 6f). TLR4 recruitment to invasion sites was dependent on RAB10 expression (Fig. 6f, g), whereas PIEZO1 recruitment occurred in a TLR4- and RAB10-independent manner (Fig. 6f, h). Together, these data suggest that the TLR4-PIEZO1 machinery is locally organized at STm invasion sites.
PIEZO1 inhibition led to a decrease in bacterial invasion at 2 hours p.i. (Fig. 6i). Ruffle formation was not affected by PIEZO1 inhibition (Supplementary Fig. 10g–h). In contrast, pretreatment with the PIEZO1 activator Yoda1 was sufficient to inhibit ruffle formation, consistent with its ability to induce RAB10 dephosphorylation (Fig. 6a, b). PIEZO1 inhibition did not affect the recruitment of phospho-RAB10 to the STm invasion sites, but did lead to retention of phospho-RAB10 at invasion sites at 30 min p.i. (Supplementary Fig. 10i–k). These findings suggest that PIEZO1 regulates PM dynamics via the phospho-regulation of RAB10 locally at invasion sites.
TMEM16F is required for local phosphatidylserine scrambling and LRRK2 removal from STm invasion sites
TMEM16F is a calcium-activated phospholipid scramblase that can translocate phosphatidylserine (PS) from the inner to the outer leaflet of the PM45. TMEM16F was previously shown to regulate mobilization of membrane reservoirs10. Furthermore, viruses are known to exploit TMEM16F during infection of host cells46,47. As such, we hypothesized that TLR4/PIEZO1-mediated signaling may promote STm infection via local activation of TMEM16F. In support of this, we observed that STm invasion was reduced in TMEM16F KO cells (Fig. 7a, b and Supplementary Fig. 11a).

a WT and TMEM16F KO Henle 407 cells were infected with WT STm and lysed at 2 hours p.i. for CFU counting. b Representative images of WT Henle 407 cells transfected with GFP-TMEM16F and myc-PM-RAB10 and infected with WT STm. Cells were fixed at 10 min p.i. and stained for myc-tag and STm. The image in the ‘Merge’ channel represents the merged accumulative fluorescence signals from the GFP-TMEM16F, myc-PM-RAB10 and STm channels. c and d Representative images (c) and quantifications (d) of GFP-TMEM16F’s localizations to STm invasion sites at 10 min p.i. WT, or RAB10 KO Henle 407 cells were transfected with GFP-TMEM16F and then infected with WT STm. Cells were fixed at 10 min p.i. and stained for STm. The images in the ‘Merge’ channel represent the merged accumulative fluorescence signals from the GFP-TMEM16F and STm channels. e A schematic model of LactC2-GFP probe labeling of local phosphatidylserine scrambling. Created in BioRender. Brumell, J. (2024) https://BioRender.com/u42p691. Representative images (middle and right panels) of WT or TMEM16F KO Henle 407 cells transfected with PM-mCherry and infected with WT or ΔsopD BFP-STm SL1344 along with the addition of LactC2-GFP probe. Cells were fixed and imaged at 30 min p.i. The cell boundaries are depicted by the white outlines. f Quantifications of LactC2-GFP localization at invasion sites in e. All images shown are representative images from three independent experiments. Data shown are means ± S.D. for three independent experiments. At least 100 invasion sites for each condition in each experiment were scored for GFP-TMEM16F (d) or LactC2-GFP (f) localization to STm invasion sites. P values were calculated using two tailed unpaired t-test (d) or two-way ANOVA (f). Scale bars, 10 μm. Source data are provided as a Source Data file.
Under normal growth conditions, we observed GFP-TMEM16F to predominantly localize to the PM, including RAB10+ membrane reservoirs (Supplementary Fig. 11b, c). Such localization was dependent on RAB10 expression since TMEM16F localized primarily to the perinuclear region in RAB10 KO cells (Supplementary Fig. 11b). However, RAB10+ membrane reservoirs were not affected in TMEM16F KO cells, indicating TMEM16F is not required for the formation of these structures (Supplementary Fig. 11c–e).
Upon infection, we observed that TMEM16F is mobilized to STm invasion sites (Fig. 7b), in a RAB10-dependent manner (Fig. 7c, d). However, TMEM16F was not required for the mobilization of RAB10+ membrane reservoirs to invasion sites (Supplementary Fig. 12a, b) or ruffle formation (Supplementary Fig. 12c, d). Therefore, we asked if the recruited TMEM16F mediates later events like local PS scrambling at invasion sites. To test this, we infected WT or TMEM16F KO cell with STm in the presence of recombinant LactC2-GFP48, a fluorescent probe that labels exofacial PS on the PM. We observed exofacial PS on host cell membranes (visualized with PM-mCherry) at the invasion sites of WT or ΔsopD STm bacteria at 30 min p.i. (Fig. 7e, f). However, these signals were not observed in TMEM16F KO cells, indicating that this scramblase is required for the transfer of PS to the exofacial leaflet of the PM locally at STm invasion sites (Fig. 7e, f).
Given our hypothesis that the scramblase activity of TMEM16F is induced by TLR4-mediated PIEZO1 activation, we next investigated whether inhibition of TLR4 or PIEZO1 reduces PS scrambling. TLR4 KO cells exhibited decreased exofacial PS signals compared to WT cells at 30 min p.i. (Supplementary Fig. 13a, b). Similarly, pretreatment of cells with the PIEZO1 inhibitor GsMTx4 significantly reduced exofacial PS exposure at STm invasion sites (Supplementary Fig. 13c, d). These data suggest that a TLR4/PIEZO1/TMEM16F pathway is activated locally at invasion sites that causes changes in PS distribution.
A recent study showed that LRRK2 localizes to curved and tubulated membranes in vitro by binding to acidic phospholipids, including PS49. Remarkably, LRRK2 was also found to have both membrane-curvature sensing and generating properties49. Since our data suggested that TMEM16F acts locally at invasion sites for PS scrambling, we hypothesized that it may act to remove LRRK2 from these sites. To test this hypothesis, we visualized LRRK2-mNeon during STm invasion. First, we observed that LRRK2 is recruited to STm invasion sites, alongside with RAB10, and both recruitments are independent of TMEM16F (Supplementary Fig. 14a, b). However, at 30 min p.i., we observed that both RAB10 and LRRK2 were absent from invasion sites in WT cells infected with WT STm (Fig. 8a, b). In contrast, TMEM16F KO cells displayed LRRK2 enrichment at invasion sites coinciding with high levels of RAB10 retention (Fig. 8a, b). Furthermore, such enrichment of LRRK2 was independent of the T3SS effector SopD, as expected (Fig. 8a, b). Together, these data suggest that TMEM16F promotes the removal of LRRK2 from invasion sites.

a and b Representative images (a) and quantifications (b) of LRRK2 localization at invasion sites at 30 min p.i. WT or TMEM16F KO Henle 407 cells were transfected with LRRK2-mNeon and myc-PM-RAB10 and infected with WT or ΔsopD STm. Cells were fixed at 30 min p.i. and stained with myc-tag. Line plot profile follows respective white arrows in the insets of the ‘Merge’ channels on the left. c and d, Representative images (c) and quantifications of (d) phospho-LRRK2 (S935) signal measured by western blot. WT, LRRK2 KO and TLR4 KO Henle 407 cells were infected with WT STm and cell lysates were collected at 30 min p.i. and immunoblotted for phospho-LRRK2 (S935), total LRRK2, or ß-actin (loading control). Data shown are means ± S.D. for three independent experiments. At least 100 invasion sites for each condition in each experiment were scored for LRRK2 localization to STm invasion sites. P values were calculated using two-way ANOVA. Scale bars, 10 μm. Source data are provided as a Source Data file.
TLR4 stimulation by LPS treatment in macrophages has been found to increase LRRK2 phosphorylation at Serine 935 (S935), a hallmark of 14-3-3 protein binding and RAB10 dephosphorylation38,50. 14-3-3 promotes the cytoplasmic localization of its binding partners and serves as a regulator of LRRK2-mediated cellular functions51. We observed an increase in LRRK2 S935 phosphorylation at 30 min p.i. (Fig. 8c, d), concomitant with the removal of LRRK2 from STm invasion sites (Fig. 8a, b) and dephosphorylation of RAB10 (Figs. 3a, b and 4c, d). In contrast, TLR4 KO cells displayed significantly lower S935 phosphorylation at 30 min p.i. (Fig. 8c, d). Together, these findings suggest that the TLR4-mediated signaling pathway promotes LRRK2 dissociation from STm invasion sites to the cytosol.
TMEM16F reduces RAB10 phosphorylation and promotes DNM2 recruitment at STm invasion sites
Since TMEM16F was required for the removal of LRRK2 from STm invasion sites, we hypothesized that this scramblase also contributes to the reduction of RAB10 phosphorylation normally observed during infection (Figs. 3a, b and 4c, d). To test this, we examined the phosphorylation of RAB10 in WT and TMEM16F KO cells upon STm infection. TMEM16F KO cells displayed stable RAB10 phosphorylation at 30 min p.i. in contrast to rapid RAB10 dephosphorylation observed in WT cells (Fig. 9a, b). By immunofluorescence staining we also observed retention of robust phospho-RAB10 signal at invasion sites in TMEM16F KO cells but not in WT cells (Supplementary Fig. 15a, b). TMEM16F expression was also required for RAB10 dephosphorylation following LPS treatment (Supplementary Fig. 15c, d). Together, these data suggest that TMEM16F contributes to RAB10 dephosphorylation, in part, by initiating the removal of LRRK2 from STm invasion sites.

a and b Representative images (a) and quantifications of (b) phospho-RAB10 (T73) signal measured by western blot. Henle 407 cells were infected with WT STm for 30 min and immunoblotted for indicated antibodies. c Representative images of Henle 407 cells transfected with GFP-RAB10 and DNM2-mCherry and infected with WT or ΔsopD STm. Cells were fixed at 30 min p.i. and stained for STm. Line plot profile follows respective white arrows in the insets of the ‘Merge’ channels on the left. d and e Quantifications of RAB10 (d) or DNM2 (e) localization at invasion sites at 30 min p.i. Data shown are means ± S.D. for three independent experiments. At least 100 invasion sites (d and e) for each condition in each experiment were scored for RAB10 or DNM2 localizations to invasion sites. P values were calculated using two-way ANOVA. Scale bars, 10 μm. Source data are provided as a Source Data file. f Model depicting the results of this study. Prior to infection, RAB10 is recruited to the plasma membrane (PM) where it stabilizes tubular invaginations (membrane reservoirs)9. LRRK2 may also contribute to membrane tubulation through binding to acidic phospholipids, including phosphatidylserine (PS), on the cytoplasmic leaflet of the PM49. RAB10’s T73 phosphorylation by LRRK2 maintains RAB10 in a GTP-bound state by preventing its interaction with GTPase activating proteins (GAPs)16. In turn, phosphorylated RAB10 promotes LRRK2 activity at the membrane in a feed-forward activation loop71. With STm infection, membrane reservoirs can be redistributed to STm invasion sites by carrier vesicle delivery and/or tubule resorption, helping to generate invasion ruffles9. Phosphorylated and GTP-bound RAB10 is recruited to STm invasion sites and generates invaginated portions of the PM containing bacteria. Bacterial lipopolysaccharide (LPS) stimulates TLR4 to induce PIEZO1-mediated calcium influx39, a signal that activates the PS scramblase TMEM16F72. PS scrambling promotes LRRK2 release from the membrane, enabling RAB10 dephosphorylation by PPM1H and/or other phosphatases25. Dephosphorylated RAB10 is targeted by SopD, a Salmonella T3SS effector with GAP activity12. GDP-bound RAB10 interacts with DNM255, promoting invaginated PM scission to generate SCVs12.
TMEM16F was previously shown to regulate DNM2 activation10. However, whether such TMEM16F activation impacts DNM2 activity directly or indirectly remains unclear. Prior studies showed that RAB10 inactivation at STm invasion sites is required for DNM2-mediated PM scission and bacterial uptake12. Therefore, we asked whether TMEM16F mediates DNM2 recruitment to STm invasion sites. We observed that TMEM16F KO cells displayed significantly higher RAB10 retention (Fig. 9c, d and Supplementary Fig. 15a, b) and lower DNM2 recruitment (Fig. 9c, e) at invasion sites. As expected, cells infected with bacteria lacking SopD, the T3SS effector with GAP activity towards RAB1012, displayed a similar phenotype (Fig. 9c–e). Furthermore, we observed defective PM scission in TMEM16F KO cells at 30 min p.i. (Supplementary Fig. 15e, f). These data support the notion that during STm invasion, TMEM16F-mediated LRRK2 removal and RAB10 dephosphorylation are required for subsequent SopD-mediated RAB10 removal and DNM2 recruitment.
Discussion
Our findings demonstrate that LRRK2 regulates PM dynamics and provide new insight into the mechanisms of STm invasion (see Model in Fig. 9f). First, LRRK2 helps to establish membrane reservoirs coordinated by RAB10. Phosphorylation on T73 maintains RAB10 in its GTP-bound state, allowing its effectors to initiate/stabilize tubular membrane reservoirs. A recent study indicates that LRRK2 itself can tubulate membranes in a cell-free system through binding to acidic phospholipid bilayers, including PS, on the cytoplasmic leaflet of the PM49. Thus, LRRK2 may form membrane reservoirs through both direct effects on the membrane and through the phosphorylation of RAB10.
The membrane reservoirs established by LRRK2 can be mobilized to promote the generation of invasion ruffles that mediate STm uptake. We postulate that the mobilization of these pre-existing membrane reservoirs can occur via carrier vesicles. This model is supported by several findings. First, we observed that RAB10 is recruited to invasion sites in its phosphorylated and active (GTP-bound) state. Since GDP dissociation inhibitors (GDIs) are known to bind RAB GTPases in their GDP-bound state52, this indicates that RAB10 is not recruited to invasion sites via the cytosol. Second, components of the exocyst tethering complex are known to be recruited to STm invasion sites11,53, consistent with membrane delivery to these sites via vesicle fusion. Importantly, we recently showed that one exocyst compoment, EXOC2, is localized to RAB10+ membrane reservoirs prior to infection and is recruited to invasion sites in a RAB10-dependent manner9. Third, we observed TMEM16F was not required for ruffle formation and exofacial PS was only visualized at STm invasion sites. This argues that TMEM16F does not regulate membrane reservoir mobilization and that PS scrambling is a highly localized event. However, beyond membrane delivery via carrier vesicles, cell membranes employ a variety of strategies to respond and adapt to different extracellular signals. For example, the opening or resorption of PM reservoirs is thought to provide the additional membrane required at the cell surface for various biological processes10,54. Therefore, these mechanisms may also contribute to membrane reservoir mobilization and ruffle formation during STm invasion.
Following RAB10-mediated membrane delivery and ruffle formation, PM scission and bacterial entry into SCVs requires rapid inhibition and removal of RAB10 from invasion sites12. A key step in PM scission involves rapid dephosphorylation of RAB10 at invasion sites. Our findings indicate that bacterial LPS stimulates RAB10 dephosphorylation via a TLR4/PIEZO1/TMEM16F pathway that culminates in removal of LRRK2 from invasion sites. Dephosphorylated RAB10 is then targeted by SopD, a Salmonella T3SS effector with GAP activity12. Subsequently, GDP-bound RAB10 interacts with DNM255, promoting scission of the invaginated PM to generate SCVs12.
Investigation of LRRK2’s activities at the cellular and molecular levels reveals its involvement in numerous cellular processes, primarily through the phospho-regulation of its substrates, many of which are RAB GTPases, including RAB1056. Accumulating evidence suggests that LRRK2 plays a role in intracellular vesicle trafficking and organelle maintenance, including the Golgi, endosomes, and lysosomes57. The targeting and recruitment of LRRK2 to these endomembranes results in the local accumulation of phosphorylated RAB10, thereby activating downstream pathways. For example, following lysosomal membrane damage, LRRK2 is recruited to lysosomes where it phosphorylates RAB10 and facilitates the recruitment of motor adapter protein JIP4 in a kinase-dependent manner to promote lysosomal tubulation58. In this study, we revealed that LRRK2 also regulates plasma membrane dynamics (including tubulation) through RAB10 phospho-regulation, both prior to and during STm invasion.
We observed a disruption of PM dynamics through expression of hyperactive LRRK2 variants. This may provide an evolutionary advantage in the face of STm (a prevalent foodborne pathogen) and other invasive microbes. Indeed, recent studies suggest an essential role for LRRK2’s kinase activity in promoting host cellular defenses against intracellular STm, including inflammasome activation3 and an itaconate-dependent defense pathway4. Furthermore, the disease-associated hyperactive LRRK2 variants may amplify these mechanisms, displaying additional advantages in the defense against STm infection3,4. Our findings are in line with these recent studies, underscoring LRRK2’s central role in host defense mechanisms59.
In addition to pathogen defense, hyperactive LRRK2 variants are closely associated with disease development in the central nervous system (e.g., PD) and gastrointestinal tract (e.g., CD)17,18,19. For example, pathogenic variants of LRRK2 have been found to hyper-phosphorylate RAB10, promoting its binding with the autophagy receptor OPTN (optineurin) and disruption of mitophagy, an event which potentially contributes to PD development22. Thus, LRRK2 is emerging as an important regulator of both PM and organelle homeostasis. It will be important to investigate how LRRK2 coordinates intracellular and extracellular signals. Unraveling the pathogenic mechanisms underlying LRRK2 hyperactive variants and their aberrant regulation of PM and intracellular membrane homeostasis, potentially through RAB10+ membrane reservoirs, is crucial for future studies to obtain a comprehensive understanding of PD and CD disease pathogenesis.
Methods
Plasmids
WT, CA (Q68L) and DN (T23N) peGFP-RAB10 were obtained from Dr. Marci Scidmore60. The myc-PM-RAB10 construct was obtained from Dr. Suzanne Pfeffer31. For pCMV-PM-mCherry, myristoylation and palmitoylation sequences from Lyn tyrosine kinase32 were fused to mCherry. The restriction sites BamHI and NotI in the peGFP-N1 vector were used for cloning (with the eGFP removed) using the primer pair, 5’-TGCAGGATCCGCCACCATGGGCTGCATTAAAAGCAAACGCAAAGATATGGTGAGCAAGGGCGAGGAGGATAACATG-3’ and 5’- TGCAGCGGCCGCTTACTTGTACAGCTCGTCCATGCCGCCGGT-3’. The GFP-LRRK2 WT and G2019S plasmids were gifts from Mark Cookson (Addgene plasmid # 25044 and 25045, respectively)61. The N551K GFP-LRRK2, N2081D GFP-LRRK2, T73A myc-RAB10-tk, and T73E myc-RAB10-tk mutants were generated using the Q5 Site-Directed Mutagenesis Kit (New England Biolabs), according to the manufacturer’s instructions. The N551K GFP-LRRK2 mutant used the primer pair, 5’-CAGCTTTGAAgAGGTTCATTG-3’ and 5’-TAGGACCAGTTTGTGAATATC-3’. The N2081D GFP-LRRK2 mutant used the primer pair, 5’-GAAGTTTCCAgATGAGTTTGATG-3’ and 5’-AAACCCTCTACTATTCTAC-3’. The T73A myc-RAB10-tk mutant used the primer pair, 5’-GCGATTTCACgccATCACAACCTC-3’ and 5’-TCCTGGCCTGCTGTATCC-3’. T73E myc-RAB10-tk used the primer pair, 5’-GCGATTTCACgagATCACAACCTCCTACTACAG-3’ and 5’-TCCTGGCCTGCTGTATCC-3’. The GFP-TMEM16F construct was obtained from Dr. Tsung-Yu Chen62. The DNM2-mCherry construct was a gift from Christien Merrifield (Addgene plasmid #27689) and was described previously12. The LRRK2-mNeon construct was obtained from Dr. Mark Cookson. All constructs were verified by DNA sequencing (TCAG, Toronto, Canada).
Cell culture and transfections
Henle 407 cells were obtained from the American Type Culture Collection (ATCC) (ATCC CCL-6). Although Henle 407 cell cultures have been shown to contain HeLa cell chromosomes, our Henle 407 cells were used between passages 5-25 and maintained a distinct morphology relative to HeLa cells. STR profiling of Henle 407 cells were done with GenePrint 10 System (Promega), by TCAG (Toronto, Canada). RAB10 KO Henle 407 cells were previously described12. LRRK2 KO Henle 407 cells, TLR4 KO Henle 407 cells and TMEM16F KO Henle 407 cells were constructed using CRISPR/Cas9 tools and described in the ‘CRISPR Knockout’ section below. SH-SY5Y cells (ATCC CRL-2266), MEF (ATCC, SCRC-1008) cells and MCF-7 (ATCC, HTB-22) cells were obtained from ATCC. All cells used were authenticated and tested negative for mycoplasma by ATCC and The Hospital for Sick Children Biobank. Cell cultures were maintained in growth medium (DMEM with 4.5 g/L glucose (Wisent) supplemented with 10% v/v FBS (Wisent)) at 37 °C with 5% CO2. For microscopy experiments, Henle 407 cells were seeded in 24-well tissue culture plates containing 12 mm coverslips at a density of 6 × 104 cells/well or 3 × 104 cells/well at either 24 h or 48 h before use, respectively. For live cell imaging, cells were seeded in μ-Slide 8-well chambers (ibidi) 24 h before use at a density of 4.0 × 104 cells/well. For immunoblotting, cells were seeded in 6-well tissue culture plates at a density of 4 × 105 cells/well 24 h before any treatment and cell lysate collection. Transfections were performed using X-treme GENE9 (Roche) or GeneJuice (Millipore) according to the manufacturer’s instructions. For cells seeded in each well of a 24-well plate, a total of 1 μg of plasmid DNA was used for transfection. For cells in each well of a 8-well chamber, a total of 0.5 μg of plasmid DNA was used for transfection.
CRISPR Knockout
To disrupt specific gene expression in Henle 407 cells, specific single-guide RNA (sgRNA) was designed using the online tool http://guides.sanjanalab.org/. Custom sgRNA oligonucleotides were synthesized by Sigma Aldrich. The sgRNA sequences used for LRRK2 KO are: #1, 5’-CACCGTGATTCCAACATCATAACAG-3’ and 5’-AAACCTGTTATGATGTTGGAATCAC-3’; #2, 5’-CACCGGCTCTTTGGATAGGAACTGG-3’ and 5’-AAACCCAGTTCCTATCCAAAGAGCC-3’; #3, 5’-CACCGGAAAGAAATGTAATGTGGGG-3’ and 5’-AAACCCCCACATTACATTTCTTTCC-3’.The sgRNA sequences used for TLR4 KO are: #1, 5’-ACCTGAGCTTTAATCCCCTG-3’ and 5’-CAGGGGATTAAAGCTCAGGT-3’; #2, 5’-CAGAGTCCCAGCCAGGCGCG-3’ and 5’-CGCGCCTGGCTGGGACTCTG-3’; #3, 5’-TTTCTGGTCTCACGCAGGAG-3’ and 5’-CTCCTGCGTGAGACCAGAAA-3’. The sgRNA sequences used for TMEM16F KO are: #1, 5’-CACCGATATGGGGGTGATAGCTGAG-3’ and 5’-AAACCTCAGCTATCACCCCCATATC-3’; #2, 5’-CACCGAGGACCAGTAGTACACTAGG-3’ and 5’-AAACCCTAGTGTACTACTGGTCCTC-3’; #3, 5’-CACCGTACCAGGCGTCTAAACTGGG-3’ and 5’-AAACCCCAGTTTAGACGCCTGGTAC-3’. The CRISPR/Cas9 vector pSpCas9 (BB)−2A-Puro (pX459) was obtained from Dr. Chi-Chung Hui63. For ligation into the BbsI site of pX459, a CACCG sequence was added to the 5’ flanking sequences of the sense oligonucleotides and an AAAC sequence was added to the 5’ flanking sequences of the anti-sense oligonucleotides. WT Henle 407 cells were transfected with the ligated vector and 48 h later the transfected cells were selected by puromycin (2 µg/ml) for another 48 h. Single cells were then transferred into a 96-well plate and allowed to grow until confluent. Knockout efficiency was determined by western blot analysis.
Bacterial Strains and Infections
Unless indicated, infections were performed with WT STm SL134464. Mutants in the STm SL1344 background lacking SopB (ΔsopB)65, SopD (ΔsopD)66 or both SopB and SopD (ΔsopBΔsopD)67 were described previously. A previously established approach was used for infection of epithelial cells, using late-log STm cultures as inocula68. ΔinvA/Inv STm 14028S was described previously35. WT and ΔsopD BFP-STm SL1344 were described previsouly12. Bacteria were pelleted at 10,000 × g for 2 min and resuspended in D-PBS, pH 7.2. For immunofluorescence staining, the bacteria were diluted 1:50 in D-PBS and added to cells at 37 °C for 10 min. This dilution of bacteria results in an infection ratio of ~325 bacteria per host cell, which is suitable for immunofluorescence staining at 10 min p.i. following washing and fixation, as described previously35. For cells fixed at 10 min p.i., cells were fixed with 2.5 % v/v paraformaldehyde in PBS at 37 °C for 10 min followed by immunofluorescence staining protocols. For cells fixed at 30 min p.i., the cells were washed three times with PBS and fresh growth media was added. Cells were put back to 37 °C incubator until 30 min p.i. Cells were then fixed with 2.5% v/v paraformaldehyde in PBS at 37 °C for 10 min followed by immunofluorescence staining (described below).
For the treatment of cells with STm culture supernatant (STm S/N), the bacterial culture was centrifuged (6000 × g, 20 min) and the supernatant collected and filtered using a 0.2 µm Millipore filter. The cells were treated with 500 μL STm S/N and subsequently harvested at 30 min. For the treatment of heat killed bacteria (STm dead), bacteria were pelleted at 10,000 × g for 2 min and resuspended in PBS, pH 7.2. Bacteria were then heat killed at 75 °C for 10 min. The dead bacteria were diluted 1:20 in PBS and added to cells at 37 °C for 30 min before cell were lysed for western blotting. To confirm the absence of live bacteria in ‘STm S/N’ or ‘STm dead’, 100 μL STm S/N or heat killed bacteria solution were plated on LB and incubated overnight at 37 °C.
For the CFU replication assay, the bacteria, subcultured as described above, were diluted 1:100 and added to cells at 37 °C for 10 min. Cells were then washed three times with PBS and growth medium was added to each well. The sample was incubated at 37 °C for an additional 20 min, at which point the media was changed to growth medium containing 100 μg/ml Gentamicin until 2 hours post-infection. To solubilize bacteria for the replication assay, the cells were washed twice with PBS, followed the resuspension of the cells in 1 ml 1% v/v Triton X-100, 0.1% w/v SDS in PBS. Serial dilutions were performed, and the dilutions plated on LB agar plates and cultured at 37 °C for overnight. CFUs were manually counted the following day.
Immunofluorescence
For all fixed microscopy-based experiments, cells were fixed with 2.5% v/v paraformaldehyde (PFA) (EM Sciences) in PBS for 10 min at 37 °C, unless indicated otherwise. Immunostaining was performed as previously described69 using the following primary antibodies: rabbit monoclonal anti-phospho-RAB10 T73 (Abcam, ab230261) at 1:100, mouse monoclonal anti-C-myc 9E10 (Thermo, MA1-980) at 1:500, rabbit polyclonal anti-Salmonella (BD Transduction, 229481) at 1:100, rabbit polyclonal anti-TLR4 (Novus Biologicals, NB100-56581) at 1:100, mouse monoclonal anti-PIEZO1 (Novus Biologicals, NBP2-75617) at 1:100 and chicken polyclonal anti-GFP (Rockland, 600-901-215S) at 1:500. The following secondary antibodies were used in this study: Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen, A-11029), Alexa Fluor 405-conjugated goat anti-rabbit IgG (Invitrogen, A-31556), Alexa Fluor 488-conjugated goat anti-rabbit IgG (Invitrogen, A-11034), Alexa Fluor 488-conjugated goat anti-chicken IgG (Invitrogen, A-32931) and Alexa Fluor 568-conjugated goat anti-rabbit IgG (Invitrogen, A-11011). Alexa Fluor 647 Phalloidin (Thermo) was used as a selective stain for F-actin. CellMask Deep Red Plasma membrane stain (Thermo) was used as a membrane impermeant dye to identify compartments open to the extracellular space and was used without cell permeabilization. Recombinant LactC2-GFP probe was used to label local phosphatidylserine scrambling and was added with fluorescent-tagged STm before washing and fixation at 30 min p.i.
Confocal microscopy
Unless otherwise indicated, cells were imaged using a Quorum spinning disk microscope (Leica DMI6000B inverted fluorescence microscope with a Yokogawa spinning disk head and Hamamatsu ORCA Flash 4 sCMOS camera) with a 10x or 20x, 1.0 NA objective, or a 63x, 1.4 NA oil immersion objective and Volocity 6.3 acquisition software (Quorum). Confocal z-stacks of 0.3 μm were acquired. Images were analyzed with the Volocity software and then imported and assembled in Adobe Illustrator for labeling. For live cell imaging, cells were seeded in μ-Slide 8-well glass bottom chambers (Ibidi). 24 h after seeding, growth media was replaced with live cell imaging media (RPMI with L-Glutamine & 25 mM HEPES (Wisent) supplemented with 10% v/v FBS (Wisent)) containing the respective treatment condition. Cells were imaged at 37 °C using Leica DMI 6000B inverted fluorescence microscope with a Yokogawa spinning disk head and Hamamatsu ImagEM X2 camera. Images were acquired with a z-spacing of 0.5 μm.
Scanning electron microscopy
Henle 407 cells were pretreated with indicated conditions and infected with WT STm and fixed with 2.5% v/v glutaraldehyde in PBS, pH 7.4, for 2 h. Samples were dehydrated using an ethanol gradient (50%, 70%, and 90% v/v in water) for 15 min each, followed by three exchanges with 100% ethanol for 15 min each. Dehydrated samples were dried in a Bal-Tec CPD030 critical point dryer (32 °C, 75 bar) and sputter coated with 10 nm gold in a Leica EM ACE200 high vacuum sputter coater. Imaging was done with a HITACHI FlexSEM 1000 II scanning electron microscope. The images were then imported into Fiji (NIH)70 and the individual invasion ruffle ROIs (containing one or more bacteria) were manually drawn for area measurements. At least 10 invasion ruffles were counted in each condition and repeated for three independent experiments (At least 30 invasion ruffles in total for each condition).
Images analysis
The images acquired from confocal microscopy were imported into Fiji (NIH)70. For invasion site recruitment measurements, F-actin and STm staining were used to denote the invasion sites at 10 min p.i. An enrichment of the indicated protein’s signal at the STm invasion site, relative to the signal in the cytosol, was considered a positive recruitment and manually quantified12. For invasion site retention measurements, immunostaining of bacteria before permeabilization and after permeabilization was used. Invasion sites (30 min p.i.) were identified by positive staining of extracellular STm before permeabilization. An enrichment of the indicated protein’s signal at invasion sites, relative to the signal in the cytosol, was considered a positive retention signal at invasion sites at 30 min p.i. and manually quantified. For PM scission assays, the degree of PM scission is represented as the percent of bacteria in SCVs, as described previously12. The total number of bacteria that were positive for PM-mCherry underlying the invasion site was quantified and the proportion of these bacteria that were negative for CellMask was determined. CellMask-negative bacteria are in a sealed compartment that was considered an SCV and the number was used to represent the invasion efficiency, at 30 min p.i. For colocalization analysis, line plot profiles were obtained by using the plot profile function in Fiji (NIH), as described previously24. To determine the number of cells with GFP-RAB10+ tubules, green-fluorescing (transfected) cells were manually counted for the presence or absence of the GFP-RAB10+ tubules.
Western blots
Cell lysates were resolved by 8% or 10% SDS-PAGE, transferred to PVDF membrane (Bio-Rad), and probed with antigen-specific primary antibodies. The following primary antibodies were used for western blot detection: rabbit monoclonal anti-LRRK2 (Abcam, ab133474) at a dilution of 1:1000, rabbit monoclonal anti-phospho-LRRK2 S935 (Abcam, ab133450) at 1:1000, rabbit monoclonal anti-phospho-RAB10 T73 (Abcam, ab230261) at 1:1000, mouse monoclonal anti-RAB10 (Sigma, SAB5300028) at 1:1000, mouse monoclonal anti-TLR4 (Abcam, ab22048) at 1:1000, rabbit polyclonal anti-TLR4 (Novus Biologicals, NB100-56581) at 1:1000, rabbit polyclonal anti-NF-κB2 p100/p52 (Cell Signaling, 4882S) at 1:1000, rabbit polyclonal anti-TMEM16F (Sigma, HPA038958) at 1:1000, mouse monoclonal (6C5) anti-GAPDH (Millipore, MAB374) at 1:10000, mouse monoclonal anti-ß-actin (Sigma, A5441) at 1:10000. Blocking was performed with 5% skim milk, except for the rabbit monoclonal anti-phospho-RAB10 T73 antibody, which were instead performed with 5% bovine serum albumin. For the secondary antibodies, peroxidase-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch, 11-035-144) or peroxidase-conjugated goat anti-mouse IgG (Jackson ImmunoResearch, 035-146) were used and detection was performed using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo). The results were analyzed using Image Lab v6.1 (BioRad).
Drug treatment
For LRRK2 inhibition, Henle 407 cells were treated with MLi-2 (Abcam) at a final concentration of 50 nM in DMEM for 90 min. For PIEZO1 activation, Henle 407 cells were treated with Yoda1 (Sigma Aldrich), resuspended in DMSO, at a final concentration of 10 or 30 µM in DMEM for 30 min. For PIEZO1 inhibition, Henle 407 cells were treated with 10 µM GsMTx4 (MedChemExpress), 30 µM Gd3+ (Sigma), or 30 µM ruthenium red (Electron Microscopy Sciences) in DMEM for 30 min, as described previously44. For LPS treatment, Henle 407 cells were treated with LPS at a final concentration of 100 ng/ml in DMEM for 30 min.
Statistics
Statistical analyzes were conducted using GraphPad Prism v9.0. The mean +/− standard deviation (S.D.) is shown in the figures, and P values were calculated using either an independent sample t-test, one-way ANOVA, two-way ANOVA or two-tailed Mann–Whitney test, as indicated in the figure legends. Tukey’s HSD test was used as post-hoc test for two-way ANOVA. P values were also included in corresponding graphs to denote the statistical significance.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data needed to evaluate the conclusions in the manuscript are present in the paper or the supplementary materials. Source data are provided with this paper. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. Source data are provided with this paper.
References
Pillay, T. D. et al. Speaking the host language: how Salmonella effector proteins manipulate the host. Microbiol. (N. Y) 169, 1342 (2023).
Finlay, B. B., Ruschkowski, S. & Dedhar, S. Cytoskeletal rearrangements accompanying salmonella entry into epithelial cells. J. Cell Sci. 99, 283–296 (1991).
Liu, W. et al. LRRK2 promotes the activation of NLRC4 inflammasome during Salmonella Typhimurium infection. J. Exp. Med. 214, 3051 (2017).
Lian, H. et al. Parkinson’s disease kinase LRRK2 coordinates a cell-intrinsic itaconate-dependent defence pathway against intracellular Salmonella. Nat. Microbiol. 2023 8:10 8, 1880–1895 (2023).
Mead, P. S. et al. Food-related illness and death in the United States. Emerg. Infect. Dis. 5, 607–625 (1999).
Kubori, T. et al. Supramolecular structure of the Salmonella typhimurium type III protein secretion system. Science 280, 602–605 (1998).
Hume, P. J., Singh, V., Davidson, A. C. & Koronakis, V. Swiss army pathogen: the salmonella entry toolkit. Front Cell Infect Microbiol 7, 348 (2017).
Szeto, J., Namolovan, A., Osborne, S. E., Coombes, B. K. & Brumell, J. H. Salmonella-containing vacuoles display centrifugal movement associated with cell-to-cell transfer in epithelial cells. Infect. Immun. 77, 996–1007 (2009).
Zhu, H. et al. Salmonella exploits membrane reservoirs for invasion of host cells. Nat. Commun. 15, 3120 (2024).
Deisl, C., Hilgemann, D. W., Syeda, R. & Fine, M. TMEM16F and dynamins control expansive plasma membrane reservoirs. Nat Commun 12, 4990 (2021).
Nichols, C. D. & Casanova, J. E. Salmonella-directed recruitment of new membrane to invasion foci via the host exocyst complex. Curr. Biol. 20, 1316–1320 (2010).
Boddy, K. C. et al. Salmonella effector SopD promotes plasma membrane scission by inhibiting Rab10. Nat Commun 12, 4707 (2021).
Terebiznik, M. R. et al. Elimination of host cell Ptdlns(4, 5)P2 by bacterial SigD promotes membrane fission during invasion by Salmonella. Nat. Cell Biol. 4, 766–773 (2002).
Xu, L., Nagai, Y., Kajihara, Y., Ito, G. & Tomita, T. The regulation of Rab GTPases by phosphorylation. Biomolecules 11, 1340 (2021).
Liu, Z., Xu, E., Zhao, H. T., Cole, T. & West, A. B. LRRK2 and Rab10 coordinate macropinocytosis to mediate immunological responses in phagocytes. EMBO J. 39, 1–21 (2020).
Liu, Z. et al. LRRK2 phosphorylates membrane-bound Rabs and is activated by GTP-bound Rab7L1 to promote recruitment to the trans-Golgi network. Hum. Mol. Genet 27, 385–395 (2018).
Ahmadi Rastegar, D. & Dzamko, N. Leucine rich repeat kinase 2 and innate immunity. Front Neurosci. 14, 193 (2020).
Hui, K. Y. et al. Functional variants in LRRK2 confer pleiotropic effects on risk for Crohn’s disease and Parkinson’s disease. Sci Transl Med 10, eaai7795 (2018).
Kars, M. E. et al. The landscape of rare genetic variation associated with inflammatory bowel disease and Parkinson’s disease comorbidity. Genome Med 16, 66 (2024).
Fell, M. J. et al. MLi-2, a potent, selective, and centrally active compound for exploring the therapeutic potential and safety of LRRK2 kinase inhibition. J. Pharm. Exp. Ther. 355, 397–409 (2015).
Atashrazm, F. et al. LRRK2-mediated Rab10 phosphorylation in immune cells from Parkinson’s disease patients. Mov. Disord. 34, 406–415 (2019).
Wauters, F. et al. LRRK2 mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy 16, 203–222 (2020).
Kawai, K., Nishigaki, A., Moriya, S., Egami, Y. & Araki, N. Rab10-positive tubular structures represent a novel endocytic pathway that diverges from canonical macropinocytosis in RAW264 macrophages. Front Immunol 12, 649600 (2021).
Etoh, K. & Fukuda, M. Rab10 regulates tubular endosome formation through KIF13A and KIF13B motors. J. Cell Sci. 132, jcs226977 (2019).
Berndsen, K. et al. PPM1H phosphatase counteracts LRRK2 signaling by selectively dephosphorylating rab proteins. Elife 8, e50416 (2019).
Waschbüsch, D. et al. Structural basis for the specificity of PPM1H phosphatase for Rab GTPases. EMBO Rep 22, e52675 (2021).
Yeshaw, W. M. et al. Localization of PPM1H phosphatase tunes Parkinson’s disease-linked LRRK2 kinase-mediated Rab GTPase phosphorylation and ciliogenesis. Proc. Natl Acad. Sci. USA 120, e2315171120 (2023).
Kluss, J. H. et al. Lysosomal positioning regulates Rab10 phosphorylation at LRRK2+ lysosomes. Proc. Natl Acad. Sci. USA 119, e2205492119 (2022).
Ysselstein, D. et al. LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson’s disease patients. Nat Commun 10, 5570 (2019).
Dhekne, H. S. et al. A pathway for parkinson’s disease LRRK2 kinase to block primary cilia and sonic hedgehog signaling in the brain. Elife 7, 1–26 (2018).
Gomez, R. C., Wawro, P., Lis, P., Alessi, D. R. & Pfeffer, S. R. Membrane association but not identity is required for LRRK2 activation and phosphorylation of Rab GTPases. J. Cell Biol. 218, 4157–4170 (2019).
Teruel, M. N., Blanpied, T. A., Shen, K., Augustine, G. J. & Meyer, T. A versatile microporation technique for the transfection of cultured CNS neurons. J. Neurosci. Methods 93, 37–48 (1999).
Ng, A. S. L. et al. Case-control analysis of leucine-rich repeat kinase 2 protective variants in Alzheimer’s disease. Neurobiol. Aging 64, 157.e7–157.e9 (2018).
Wang, X. et al. Understanding LRRK2 kinase activity in preclinical models and human subjects through quantitative analysis of LRRK2 and pT73 Rab10. Sci. Rep. 11, 12900 (2021).
Steele-Mortimer, O. et al. The invasion-associated type III secretion system of Salmonella enterica serovar Typhimurium is necessary for intracellular proliferation and vacuole biogenesis in epithelial cells. Cell Microbiol 4, 43–54 (2002).
Nevola, J. J., Stocker, B. A. D., Laux, D. C. & Cohen, P. S. Colonization of the mouse intestine by an avirulent Salmonella typhimurium strain and its lipopolysaccharide-defective mutants. Infect. Immun. 50, 152–159 (1985).
Murray, G. L., Attridge, S. R. & Morona, R. Regulation of Salmonella typhimurium lipopolysaccharide O antigen chain length is required for virulence; identification of FepE as a second Wzz. Mol. Microbiol 47, 1395–1406 (2003).
Nazish, I. et al. Abrogation of LRRK2 dependent Rab10 phosphorylation with TLR4 activation and alterations in evoked cytokine release in immune cells. Neurochem Int 147, 1–28 (2021).
Geng, J. et al. TLR4 signalling via Piezo1 engages and enhances the macrophage mediated host response during bacterial infection. Nat Commun 12, 3519 (2021).
Ridone, P., Vassalli, M. & Martinac, B. Piezo1 mechanosensitive channels: what are they and why are they important. Biophys. Rev. 11, 795–805 (2019).
Ruschkowski, S., Rosenshine, I. & Brett Finlay, B. Salmonella typhimurium induces an inositol phosphate flux in infected epithelial cells. FEMS Microbiol Lett. 95, 121–126 (1992).
Pace, J., Hayman, M. J. & Galán, J. E. Signal transduction and invasion of epithelial cells by S. typhimurium. Cell 72, 505–514 (1993).
Syeda, R. et al. Chemical activation of the mechanotransduction channel Piezo1. Elife 4, e07369 (2015).
Cinar, E. et al. Piezo1 regulates mechanotransductive release of ATP from human RBCs. Proc. Natl Acad. Sci. USA 112, 11783–11788 (2015).
Suzuki, J. et al. Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. J. Biol. Chem. 288, 13305–13316 (2013).
Tang, D. et al. Scramblases and virus infection. Bioessays 44, e2100261 (2022).
Zaitseva, E. et al. Fusion stage of HIV-1 entry depends on virus-induced cell surface exposure of phosphatidylserine. Cell Host Microbe 22, 99–110.e7 (2017).
Doktorova, M. et al. Cell membranes sustain phospholipid imbalance via cholesterol asymmetry. Preprint bioRxiv https://doi.org/10.1101/2023.07.30.551157 (2023).
Wang, X. et al. Membrane remodeling properties of the Parkinson’s disease protein LRRK2. Proc. Natl Acad. Sci. 120, e2309698120 (2023).
Li, X. et al. Phosphorylation-dependent 14-3-3 binding to LRRK2 is impaired by common mutations of familial Parkinson’s disease. PLoS One 6, e17153 (2011).
Muslin, A. J. & Xing, H. 14-3-3 proteins: regulation of subcellular localization by molecular interference. Cell Signal 12, 703–709 (2000).
Müller, M. P. & Goody, R. S. Molecular control of Rab activity by GEFs, GAPs and GDI. Small GTPases 9, 5–21 (2018).
Braun, V. & Brumell, J. H. Bacterial invasion: entry through the exocyst door. Curr Biol 20, R677-9 (2010).
Kosmalska, A. J. et al. Physical principles of membrane remodelling during cell mechanoadaptation. Nat. Commun. 2015 6:1 6, 1–11 (2015).
Li, Z. et al. Maturation of lipophagic organelles in hepatocytes is dependent upon a Rab10/Dynamin-2 complex. Hepatology 72, 486–502 (2020).
Kuwahara, T. & Iwatsubo, T. The Emerging Functions of LRRK2 and Rab GTPases in the Endolysosomal System. Front Neurosci. 14, 528270 (2020).
Bonet-Ponce, L. & Cookson, M. R. LRRK2 recruitment, activity, and function in organelles. FEBS J. 289, 6871–6890 (2022).
Bonet-Ponce, L. et al. LRRK2 mediates tubulation and vesicle sorting from lysosomes. Sci Adv 6, eabb2454 (2020).
Herbst, S. & Gutierrez, M. G. LRRK2 in Infection: Friend or Foe? ACS Infect. Dis. 5, 809 (2019).
Rzomp, K. A., Scholtes, L. D., Briggs, B. J., Whittaker, G. R. & Scidmore, M. A. Rab GTPases are recruited to chlamydial inclusions in both a species-dependent and species-independent manner. Infect. Immun. 71, 5855 (2003).
Greggio, E. et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 23, 329–341 (2006).
Nguyen, D. M., Chen, L. S., Yu, W. P. & Chen, T. Y. Comparison of ion transport determinants between a TMEM16 chloride channel and phospholipid scramblase. J. Gen. Physiol. 151, 518–531 (2019).
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013).
Hoiseth, S. K. & Stocker, B. A. D. Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature 291, 238–239 (1981).
Steele-Mortimer, O. et al. Activation of Akt/protein kinase B in epithelial cells by the Salmonella typhimurium effector sigD. J. Biol. Chem. 275, 37718–37724 (2000).
Jiang, X. et al. The related effector proteins SopD and SopD2 from Salmonella enterica serovar Typhimurium contribute to virulence during systemic infection of mice. Mol. Microbiol 54, 1186–1198 (2004).
Bakowski, M. A., Cirulis, J. T., Brown, N. F., Finlay, B. B. & Brumell, J. H. SopD acts cooperatively with SopB during Salmonella enterica serovar Typhimurium invasion. Cell Microbiol 9, 2839–2855 (2007).
Steele-Mortimer, O., Méresse, S., Gorvel, J. P., Toh, B. H. & Finlay, B. B. Biogenesis of Salmonella typhimurium-containing vacuoles in epithelial cells involves interactions with the early endocytic pathway. Cell Microbiol 1, 33–49 (1999).
Brumell, J. H., Rosenberger, C. M., Gotto, G. T., Marcus, S. L. & Finlay, B. B. SifA permits survival and replication of Salmonella typhimurium in murine macrophages. Cell Microbiol 3, 75–84 (2001).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 2012 9:7 9, 676–682 (2012).
Vides, E. G. et al. A feed-forward pathway drives LRRK2 kinase membrane recruitment and activation. Elife 11, e79771 (2022).
Suzuki, J., Umeda, M., Sims, P. J. & Nagata, S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468, 834–840 (2010).
Acknowledgements
JHB holds the Pitblado Chair in Cell Biology. AMM holds a Canada Research Chair (Tier 1). Infrastructure for the Brumell Laboratory was provided by a John Evans Leadership Fund grant from the Canadian Foundation for Innovation and the Ontario Innovation Trust. We thank Paul Paroutis for help with confocal microscopy, Ali Darbandi for help with electron microscopy and Spencer Freeman for providing recombinant LactC2-GFP. This work was supported by the Canadian Institutes of Health Research (FDN#154329 to JHB, FDN#143202 to SG and FDN#408445 to AMM), the Natural Sciences and Engineering Research Council (RGPIN#04330 to JHB), the Leona M. and Harry B. Helmsley Charitable Trust to AMM, and the National Institutes of Health Research (NIDDK RC2DK122532 and NIDDK RC2DK118640 to AMM).
Author information
Authors and Affiliations
Contributions
Conceptualization: H.Z., J.H.B. Methodology: H.Z., A.M.S. Investigation: H.Z., A.M.S., B.R.Y., R.L., M.T.B., C.L., M.E.M. Visualization: H.Z., A.M.S., R.L. Funding acquisition: J.H.B. Project administration: J.H.B., A.M.S. Supervision: J.H.B., A.M.M., S.G., M.C., B.R. Writing–original draft: H.Z., J.H.B. Writing–review & editing: H.Z., A.M.S., B.R.Y., R.L., S.G., J.H.B.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks James Casanova, Huan Lian and Virginie Stévenin for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, H., Sydor, A.M., Yan, BR. et al. Salmonella exploits LRRK2-dependent plasma membrane dynamics to invade host cells. Nat Commun 16, 2329 (2025). https://doi.org/10.1038/s41467-025-57453-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57453-x
