
Abstract
Therapeutic use of tiny extracellular vesicles (EVs) requires understanding cargo loading mechanisms. Here, we use a modular proximity labeling approach to identify the cargo of ciliary EVs associated with the transient receptor potential channel polycystin-2 PKD-2 of C. elegans. Polycystins are conserved ciliary proteins and cargo of EVs; dysfunction causes polycystic kidney disease in humans and mating deficits in C. elegans. We discover that polycystins localize with specific cargo on ciliary EVs: polycystin-associated channel-like protein PACL-1, dorsal and ventral polycystin-associated membrane C-type lectins PAMLs, and conserved tumor necrosis factor receptor-associated factor (TRAF) TRF-1 and TRF-2. Loading of these components to EVs relies on polycystin-1 LOV-1. Our modular EV-TurboID approach can be applied in both cell- and tissue-specific manners to define the composition of distinct EV subtypes, addressing a major challenge of the EV field.
Similar content being viewed by others

Introduction
Extracellular vesicles (EVs) are essential for cell-cell communication within an organism and across different biological kingdoms. The study of EV biology is challenging due to EVs’ small sizes, transient nature of biogenesis, diverse origins, and heterogeneity1. Various EVs coexist in biological fluids and, due to their overlapping physical properties, often co-isolate with each other, making it difficult to study individual EV subtypes. Herein, we define an EV subtype as a group of EVs that (i) are associated with a specific EV cargo, (ii) are produced in a cell-type-specific manner, and (iii) are shed from a distinct subcellular ___location. Addressing the challenge of cargo composition of individual EV subtypes in the face of EV heterogeneity is crucial for advancing EV-based diagnostics, therapeutics, and biomedical research. Of particular interest are EVs produced by the cilium, a specialized signaling compartment of the cell2,3,4,5,6,7,8. Because of the cilium’s small size (roughly 1/10,000 of the cell volume), ciliary EVs may go undetected in bulk EV preparations yet may carry important signaling components. Here, we introduce a methodology to identify the cargo of individual ciliary EV subtypes carrying polycystins via proximity labeling in the nematode worm Caenorhabditis elegans.
Human polycystin-1 (PC1 encoded by PKD1, polycystic kidney disease 1 gene) and polycystin-2 (PC2 encoded by PKD2, polycystic kidney disease 2 gene) are mutated in autosomal dominant polycystic kidney disease (ADPKD), the most common inherited cause of renal failure9,10. PKD1 and PKD2 encode a transient receptor potential (TRP) polycystin cation channel complex that functions in the primary cilia of kidney tubules and is a cargo of CD133-positive urinary EVs11,12. Urinary EV biomarkers may be useful in diagnosis and monitoring of ADPKD progression and treatment13,14. PC1- and PC2-carrying EVs are abundant in human urine11,12,15. Through a candidate screening approach for proteins that interact with the polycystins in EVs, only two validated interactors were identified in humans: (i) the autosomal recessive PKD gene product fibrocystin PKHD1 (selected via manual curation of candidate genes phenocopying the polycystin disruption)16, and (ii) EPCIP - Exosomal Polycystin 1 Interacting Protein, CU062 (selected from the mass spectrometry data of the urinary EV profiling as a candidate with correlative abundance in healthy vs. ADPKD patients)17. The precise composition and function of polycystin-carrying urinary EVs remain unknown.
In addition to their function in the kidney, polycystins also play roles in multiple organ systems, including vertebrate spinal cord morphogenesis and left-right axis patterning during embryogenesis. In the morphogenesis of the spinal cord, PKD2L1 functions in cerebral spinal fluid (CSF)-contacting neurons that are critical for precise posture control18,19,20. Whether polycystins are cargo of CSF EVs remains to be determined; however, CSF is driven in part by motile cilia and carries extracellular lipidic EV-like particles21, suggesting that EV-like particles, including ciliary EVs, might play roles in nervous system function. Another example of EV- and polycystin-involved process is the establishment of left-right asymmetry in vertebrates, where nodal flow transfers EV-like lipidic particles that were proposed to carry morphogenic signals22. Moreover, Tanaka et al. observed directional movement of polycystins (specifically, PKD1L1, polycystic kidney disease 1—like 1) on the nodal EV-like particles during gastrulation, where they were released by nodal pit cells to regulate left-right axis determination of the embryonic body23. Because polycystins are important for the proper function of many organ systems in health and disease, it is important to identify in vivo polycystin-associated partners.
Many aspects of fundamental polycystin biology were elucidated using C. elegans. In C. elegans, polycystin-1-like protein LOV-1 (Location Of Vulva gene 1) and polycystin-2-like protein PKD-2 (human Polycystic Kidney Disease 2—related) are expressed in male-specific ciliated sensory neurons that govern male mating behaviors. Both polycystins are required for males’ behavioral response to the presence of mating partners (hermaphrodites) and the ability to locate vulva. lov-1 and pkd-2 single and double mutants display similar male mating behavior defects, suggesting that both genes act in the same pathway24,25,26,27. Assessing the male mating behavior is an excellent read-out tool for the functionality of the polycystin pathway in C. elegans ciliated sensory neurons and was used to discover additional conserved components, such as homologs of human TRAFs -TRF-1 and TRF-2 (Tumor Necrosis Factor (TNF)-receptor-associated factors 1-6). Mutants of trf-1 and trf-2 are deficient in male mating behaviors to the same degree as mutants of lov-1 and pkd-2, suggesting that TRFs and polycystins act in the same pathway28. C. elegans LOV-1 and PKD-2, akin to human PC1 and PC2, colocalize and function in the cilium and extracellular vesicles (EVs)29,30. The presence of PC1 and PC2 on human urinary EVs was first discovered by Pisitkun et al.11 and followed up with an extensive characterization by Hogan et al.12 using electron microscopy. PC-1-positive EVs were observed in the vicinity of cilia, on vesicles inside the cytoplasm, and within multivesicular bodies12. The biogenesis origin of the human urinary PC1-positive EVs is unclear due to limitations of fixed preparation examination. In C. elegans, though, PKD-2 and LOV-1 are shed together on EVs from the cilium base for possible neuron-glia crosstalk and from the cilium tip into the environment for inter-animal communication29,31,32. Here, we use “the worm” to identify in vivo interactors of the polycystins in cilia and EVs, with the goal of shedding light on polycystin signaling in vertebrate systems.
We recently attempted to identify the proteome of polycystin EVs33, intending to find candidates that might shed light on the function of polycystin EVs or, better yet, conserved cargo that might help us understand the function of human polycystin EVs. We used PKD-2::GFP as a marker to track polycystin EVs in density gradients (Fig. 1a). We optimized our isolation procedure to enrich PKD-2 EVs from the bulk of other EVs, including bacterial outer membrane vesicles (OMVs) of E. coli (a food source for C. elegans), which constitute more than 99% of EVs in C. elegans cultures. Significant depletion of the bacterial OMVs enabled us to detect cargo of C. elegans EVs in fractions enriched in PKD-2::GFP EVs. We discovered that similar to mammalian systems, C. elegans EVs share similar size and density despite diverse biogenesis routes and that polycystin EVs co-isolate with many other types of EVs. Specifically, we identified almost 3000 cargo candidates that co-isolate with PKD-2::GFP EVs33. We then conducted bioinformatical filtering methods to pinpoint candidates likely of ciliated-cell origin and generated fluorescent reporters to validate their presence on EVs. None of the validated candidates were cargo of polycystin ciliary EVs. Instead, we discovered cargo of other sex-shared amphid and phasmid ciliated neurons—tetraspanin 6 (TSP-6), indicating that cilia of different cells shed EVs of different cargo compositions. We found systemic RNA interference defective gene 2 (SID-2), an RNA-binding cargo shed by polycystin-expressing neurons, but in EVs distinct from the polycystin EVs, signifying that one cilium can produce different EV subtypes. We also showed that C. elegans EVs carry RNA. We also discovered that ectonucleotide pyrophosphatase/phosphodiesterase 1, ENPP-1, is an example of cargo that might be shed in EVs via cell-specific biogenesis routes. Specifically, ENPP-1 is shed in ciliary ectosomes by inner labial type 2 (IL2) sensory neurons and in exosomes by male reproductive tract cells33. Our discovery of the enormous complexity of C. elegans EVome necessitated the development of more specific methods for identifying the composition of individual EV subtypes. Herein, we developed a system for specific and precise identification of cargo of individual EV subtypes through TurboID-mediated proximity labeling. We applied this EV-TurboID methodology and an armamentarium of C. elegans genetic tools to identify and validate the cargo of a ciliary EV subtype that carries polycystins.

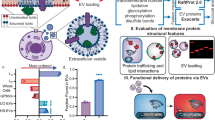
a Scheme of EV harvest and enrichment, followed by pulldown of candidate interactors biotinylated by TurboID targeted to PKD-2-carrying EVs. b Scheme of male tail sensory rays that release PKD-2 EVs. Each sensory ray of the male tail harbors a ciliated dendritic ending protruding into the environment and releasing PKD-2 EVs from the tip of its sensory cilium. c Fluorescence image of EVs released from the sensory rays carrying PKD-2::GFP and TurboID indicative of successful targeting of the biotin ligase to PKD-2-carrying EVs. See Supplementary Fig. 1 for additional images. d Identified top candidate PKD-2 interactors, validated in subsequent figures. Created in BioRender. Barr, M. (2025) https://BioRender.com/n98b409.
Results
Identification of polycystin proximity interactors within EVs
Proximity labeling by biotin ligases is a preferable method for identifying interactors of transmembrane proteins34,35. It allows for biorthogonal in vivo labeling followed by a stringent isolation procedure, thus avoiding post-lysis artefactual interactions of tag-affinity-based purifications. Additionally, proximity labeling can capture transient, low-affinity, or scarce interactions, making it suitable for the case where polycystin ciliary EVs co-isolate with many other types of EVs, even in the most optimized density gradient pipeline that eliminates more than 99% of contaminating EVs33.
We opted for one of the latest generations of biotin ligases, TurboID, with rapid labeling time and improved efficiency at 20 oC (the cultivation temperature for C. elegans)36. We also opted for indirect proximity labeling (i.e., targeting TurboID via a GFP-binding nanobody ___domain) because indirect labeling shows improved performance and offers several advantages over direct fusions of the biotin ligase with a protein-of-interest37,38. Briefly, the advantages of indirect TurboID include minimizing strain generation time, using endogenous GFP-labeled strains that might already be available and characterized, and ensuring uniform expression levels of TurboID across different samples and controls37,38. The indirect TurboID shows superior results to the direct fusion approach, resulting in less noisy data and reliable identification of bona fide interactors37.
We engineered the proximity labeling enzyme TurboID to be targeted to a GFP-labeled ciliary EV cargo PKD-2::GFP via a GFP-binding nanobody ___domain. Between the GFP-binding nanobody and TurboID domains, we added a fluorescent mScarlet ___domain that would enable us to track TurboID in living animals and environmentally released EVs through super-resolution live imaging (Fig. 1a-b). Expression of anti-gfp nanobody::mScarlet::turboID in neurons resulted in a uniform distribution of the biotin ligase throughout the cell body, neurites, and cilia (Supplementary Fig. 1a-b). Co-expression of anti-gfp nanobody::mScarlet::turboID and pkd-2::gfp resulted in the biotin ligase being enriched in PKD-2-specific subcellular localizations, such as distal dendrites, cilia, and environmentally released ciliary EVs (Fig. 1c, Supplementary Fig. 1c), indicative of successful targeting of TurboID to PKD-2-carrying EVs.
To identify novel cargo of PKD-2-carrying EVs, we pulled down biotinylated proteins for mass spectrometry analysis from EV-enriched fractions of density gradients (Fig. 1a) from the strains with targeted and untargeted TurboID. The mass spectrometry results comprised a short list of peptides (Supplementary Data 1), prohibiting the application of robust statistical methods developed for large-scale proteomics projects. Thus, we ranked the cargo candidates based on their differential presence in the targeted sample compared to the untargeted control (Table 1). The identified proximity interactors of PKD-2 included two previously known cargo of PKD-2::GFP-carrying EVs: (i) LOV-124,25,30 and (ii) CIL-7 (ciliary localization gene 7, which encodes an EV biogenesis protein for polycystin EVs)31,39. No generic ciliary proteins, such as components of intraflagellar transport, Bardet-Biedl Syndrome complex BBSome, or the transition zone, were found among the identified candidates40, confirming the specificity and efficacy of our EV-TurboID approach. We selected the top six candidates for further unbiased validation: two homologs of human TNF receptor-associated factors (TRAFs) TRF-1 and TRF-2, three transmembrane lectins (PAML-1, PAML-2, CWP-5), and a novel transmembrane protein resembling an ion channel (PACL-1) (Fig. 1d, Table 1; for the complete list of identified proteins, refer to Supplementary Data 1).
The concern of many biological studies is the potential for artifacts caused by overexpression of fluorescent reporters41. The overexpression of EV cargo reporters might cause increased EV biogenesis or alter the delicate dynamics of endosomal compartments, directly or indirectly influencing EV biogenesis42. Therefore, we generated endogenous reporters for all EV cargo candidates, including PKD-2, using CRISPR/Cas9-mediated genome editing for our validation procedure. We used super-resolution microscopy to analyze candidates’ co-localization with PKD-2 within cilia and ciliary EVs (Fig. 2a).

a Scheme of fluorescent profiling along cilia and assessment of reporter colocalization. Flattened z-stacks show the ciliary presence of endogenous FP-tagged PKD-2 with TRF-1::mScarlet (b, d) and TRF-2::GFP (c, e) in the wild-type cilium (b, c) and lov-1 mutant (d, e). Representative average fluorescence profiles for each case are shown in panels b’, c’, d’, e’. Fluorescence values are normalized to the average of the minimum and maximum for each cilium. Scatter plots on panels b”, c”, d”, and e” show correlations between PKD-2 fluorescence and either TRF-1::mScarlet (b”, d”) or TRF-2::GFP (c”, e”) within shafts (blue) and tips (yellow) of cilia. Pearson correlation coefficients from a two-sided test are reported alongside unadjusted p-values. Loss of colocalization is observed in lov-1 mutants as reflected by a drop in the Pearson correlation coefficients. Correlation plots contain the combined data for all Ray B neurons of the tail. Sample sizes are indicated for each panel graph. For the full dataset that includes all rays, refer to Supplementary Figs. 2-3. Source Data are provided as SourceData_Main.xls, SourceData_SuppFig2.xls, and SourceData_SuppFig3.xls files.
Polycystin EVs carry homologs of human TRAFs
Evolutionary conservation of polycystin biology prompted us to start validation with the conserved homolog of human TRAFs. The TRAF family comprises adapter-type proteins that act downstream of receptors and mediate the assembly of cytoplasmic signaling components43. We discovered that endogenously labeled trf-1::mScarlet and trf-2::GFP are expressed at high levels only in polycystin-expressing sensory neurons that govern male mating behavior (Cephalic male neurons CEMs, tail Ray type B neurons 1-5, 7-9, and hook sensillum B neuron HOB). In these neurons, TRF-1 and TRF-2 colocalized with PKD-2 in cilia, ciliary tips, and EVs (Fig. 2b-b’-b”, c-c’-c”, Supplementary Figs. 2-3), suggesting that TRAFs might act as adapters for the polycystin complex and that polycystin EVs serve as carriers for TRAFs to engage target tissues.
Polycystins load TRAFs to ciliary EVs
To dissect functional interactions between polycystins and TRAFs, we analyzed TRAFs’ subcellular localization in the background of disrupted polycystins. We previously showed that PKD-2 is essential for LOV-1 to exit the neuronal cell body and enter the cilium and EVs, whereas LOV-1 is not required for PKD-2 ciliary and EV localization30. Therefore, we chose the lov-1 mutant background to analyze TRF-1 and TRF-2 colocalization with PKD-2 in cilia and EVs. Because the lov-1 mutation does not perturb PKD-2 ciliary and EV localization, PKD-2 served as an excellent marker of the ciliary tip and EVs in the background of disrupted polycystin complex to test whether TRAFs occupy the same ciliary regions as the polycystins. We found that in the lov-1 mutant, both TRF-1 and TRF-2 were present in the ciliary shaft but failed to become enriched at the ciliary tip and were not loaded to ciliary EVs (Fig. 2d-d’-d”, e-e’-e”, Supplementary Figs. 2-3). The absence of TRF-1 and TRF-2 ciliary tip enrichment in the lov-1 mutant indicates that LOV-1 is required for loading TRF-1 and TRF-2 to ciliary EVs, but not for their localization to the cilium shaft.
While TRF-1 and TRF-2 could still enter the ciliary shaft in the lov-1 mutant background, they exhibited a complete loss of colocalization with PKD-2 within the shaft (Pearson correlation coefficient drop from 0.53 to 0.27 for TRF-1::mScarlet Fig. 2b” vs. 2d”, and from 0.78 to 0.28 for TRF-2::GFP Fig. 2c” vs. 2e”, also see Supplementary Figs. 2a’, 3a’). The loss of colocalization suggests that LOV-1 is required for TRF-1 and TRF-2 interaction with the polycystin complex within the cilium. Consistent with the hypothesis that both TRAFs interact with the polycystin complex and thus should localize in the proximity of each other, we found a high correlation between fluorescence intensities of TRF-1::mScarlet and TRF-2::GFP within the ciliary shafts and tips in the wild-type animals (Supplementary Fig. 4a-b-b’), suggesting that both TRAFs localize together in the vicinity of the polycystin complex.
To understand whether TRAFs are required for ciliary localization of the polycystin complex, we measured the co-localization of PKD-2::GFP and LOV-1::mScarlet in cilia and EVs in the trf-1 or trf-2 mutants (Supplementary Fig. 5a-a’-b). Our data suggest that neither TRF-1 nor TRF-2 is required for PKD-2 and LOV-1 ciliary and EV localization.
TRAFs require each other for their loading to ciliary EVs
We next investigated the relationship between TRF-1 and TRF-2 in cilia and EVs. In the trf-2 mutant, TRF-1::mScarlet remained confined to the ciliary shaft, lost colocalization with PKD-2::GFP, and failed to localize to the ciliary tip and EVs (Fig. 3a-a’-a”, Supplementary Fig. 6a-a’). The loss of colocalization of TRF-1::mScarlet with PKD-2::GFP and failure to enter the ciliary tip and EVs of the trf-2 mutant occurred without significant changes in cellular TRF-1::mScarlet levels (Supplementary Fig. 6b). These data suggest that TRF-2 is required for the TRF-1 colocalization with the polycystin complex in cilia and EVs.

a Flattened z-stack shows that disruption of trf-2 abrogates loading of TRF-1::mScarlet to PKD-2::GFP EVs. TRF-1::mScarlet stays in the cilium and does not reach the ciliary tip, as a representative fluorescent profiling shows (a’); fluorescence values are normalized to the average of minimum and maximum values for each cilium (n = 10 cilia). a” Scatter plot shows no correlation between PKD-2::GFP and TRF-1::mScarlet within shafts (blue) and tips (yellow) of cilia (n = 122 cilia) of the trf-2(tm5167) mutant. Pearson correlation coefficients from a two-sided test are reported alongside unadjusted p-values. For the full dataset that includes all rays, refer to Supplementary Fig. 6. Source Data are provided as SourceData_Main.xls and SourceData_SuppFig6.xls files. b Flattened z-stack shows that disruption of trf-1 abrogates ciliary localization of TRF-2::GFP, and thus, no TRF-2::GFP is loaded to PKD-2::mScarlet EVs. b’ Quantification of total fluorescence within the cilium shows that TRF-2::GFP ciliary levels in the trf-1 mutant are reduced tenfold compared to WT cilia, whereas PKD-2::mScarlet total levels remain unaffected. n = 73 cilia for WT, and 112 cilia for the trf-1(nr2014) mutant. Two-sided Wilcoxon Rank Sum test, p < 2.2e-16 for TRF-2::GFP, and p = 0.006035 for PKD-2::mScarlet. c Flattened z-stack through cell bodies of RnB neurons showing reduced cytoplasmic levels of TRF-2::GFP. c’ Quantification of cell body cytoplasmic levels of PKD-2::mScarlet and TRF-2::GFP as a ratio of average cytoplasmic fluorescence normalized by fluorescence within the nucleus on a single image plane, n = 96 cell bodies for WT, n = 147 cell bodies for the trf-1(nr2014) mutant. Two-sided Wilcoxon Rank Sum test, p < 2.2e-16 for TRF-2::GFP, and p = 0.4343 for PKD-2::mScarlet. The box-and-whiskers plots (b’ and c’) show the median values where the box covers two quartiles around the median, and the whiskers extend 1.5 quartiles from the median. Source Data are provided as SourceData_Main.xls file. d Homology of C. elegans TRFs to human TRAFs. e Working model of the molecular mechanism for loading TRFs to ciliary EVs. LOV-1 is required for loading both TRFs to ciliary EVs; in the lov-1 mutant, cilia produce EVs without TRFs. TRFs are required for loading each other to the ciliary EVs; in the trf-2 mutant, cilia produce EVs without TRF-1, whereas the trf-1 mutation abrogates TRF-2 ciliary localization. Additional supporting data for the model are presented in Supplementary Fig. 4, showing the colocalization of TRF-1::mScarlet and TRF-2::GFP, and Supplementary Fig. 5, showing that TRFs are not required for release of polycystins in ciliary EVs. Source Data are provided as SourceData_SuppFig4.xls and SourceData_SuppFig5.xls files.
Conversely, in the trf-1 mutant, TRF-2::GFP presence in cilia, including the ciliary base, was reduced by order of magnitude as compared to WT animals (Fig. 3b-b’) and undetectable in the ciliary shaft. Thus, we proceeded to examine the TRF-2::GFP cellular abundance. We found a three-fold reduction in average TRF-2::GFP abundance in cell bodies (Fig. 3c-c’, Supplementary Fig. 6c-c’). These data indicate that TRF-1 is required for high cytoplasmic levels of TRF-2, suggesting that TRF-1 might stabilize or protect TRF-2 from degradation.
Evolutionary conservation of TRAF ___domain assemblies suggests that the function of human and C. elegans TRAFs might likewise be conserved. Both human and C. elegans TRAF families comprise two types of ___domain organizations: RING ___domain-containing TRAFs (TRF-1 in C. elegans and TRAF2-6 in humans) and RING-less TRAFs (TRF-2 in C. elegans and TRAF1 in humans) (Fig. 3d). The RING ___domain confers ubiquitin ligase activity to human TRAF644,45, promoting the assembly of multimeric signaling networks presumably comprised of trimer dimers46. In Dictyostelium discoideum, the RING ___domain recruits TRAF to sites of membrane damage47. However, the specific role of RING-less TRAFs in the context of TRAF cascades remains unclear. This study shows that TRAFs act in the cilium (Fig. 3e). Specifically, our work highlights the essential role of TRF-2 in recruiting TRF-1 from the ciliary shaft to ciliary EVs.
The polycystin complex associates with and recruits a channel-like protein PACL-1 to cilia and EVs
The next polycystin EV cargo candidate was a novel four-pass transmembrane protein, T13F3.7, which we named PACL-1 (Polycystin-Associated Channel-like protein) (see Supplementary Fig. 8 for the protein name rationale). To investigate whether PACL-1 functions with the polycystins, we generated an endogenously labeled strain bearing the C-terminal fusion PACL-1::GFP. pacl-1 was expressed only in polycystin-expressing neurons. PACL-1::GFP colocalized with PKD-2::mScarlet within the ciliary shaft, ciliary tip, and polycystin EVs released into the environment (Fig. 4a-a’-a”-b-b’, Supplementary Fig. 8). We found that the ciliary localization of PACL-1::GFP was abolished by the lov-1 mutation (Fig. 4c). Specifically, in the lov-1 mutant, PACL-1::GFP remained in neuronal cell bodies and was not localized to cilia or environmentally released EVs, indicating that LOV-1 recruits the PACL-1 channel-like protein to cilia and EVs (Fig. 4d).

a Flattened z-stack showing colocalization of PKD-2::mScarlet with PACL-1::GFP in WT cilia with a representative fluorescence profile (n = 12 cilia) (a’) and a cumulative correlation plot (n = 114 cilia) (a”) showing a high level of colocalization in the ciliary tip and shaft regions. Pearson correlation coefficients from a two-sided test are reported alongside unadjusted p-values. For the full dataset that includes all rays, refer to Supplementary Fig. 8. Source Data are provided as SourceData_Main.xls and SourceData_SuppFig8.xls files. b–b’ Quantification showing colocalization of PACL-1::GFP to PKD-2::mScarlet on ciliary EVs (b) with high Pearson correlation (two-sided) coefficients for raw fluorescent intensities (b’), n = 7 animals. The box-and-whiskers plots show the median values where the box covers two quartiles around the median, and the whiskers extend 1.5 quartiles from the median. Source Data are provided as SourceData_Main.xls file. c Flattened z-stacks show that in the lov-1 mutant, PACL-1::GFP remains in the cell bodies and fails to move to dendrites and cilia. d Working model of the molecular mechanism loading PACL-1 to ciliary EVs. LOV-1 is required for PACL-1 ciliary localization. In the lov-1 mutant, cilia produce EVs without PACL-1. A rationale for the PACL-1 channel-like identity is presented in Supplementary Fig. 7.
The polycystin complex associates with distinct transmembrane C-type lectins that specify dorsal and ventral populations of cilia and EVs
Our proximity labeling also identified novel multi-pass transmembrane cargo candidates, Y70G10A.2 and F25D7.5, which possessed N-terminal extracellular EGF and C-type lectin domains (Fig. 1d). Based on the presence of these domains and candidates’ associations with polycystins, we named them Polycystin-Associated Membrane Lectins, PAML-1, and PAML-2, respectively.
We generated endogenous reporters of PAML-1 and PAML-2. paml-1::gfp expression was limited to the ventral subset of pkd-2-expressing neurons, specifically those in which cilia protrude to the ventral side of the male tail (HOB, and paired ray B neurons R2B, R4B, and R8B neurons), and paired lateral ray neurons R3B. The ventral neurons mediate vulva ___location behavior and contact-based response to hermaphrodite27. Within these ventral tail neurons, PAML-1::GFP colocalized with PKD-2::mScarlet in both cilia and EVs (Fig. 5a, Supplementary Fig. 9a-a’-b).

Flattened z-stacks showing colocalization of PKD-2 reporters with PAML-1::GFP (a) and PAML-2::mScarlet (b) in WT. PAML-1 is present in cilia of ventral HOB neuron and paired ray neurons R2B, R4B, R8B, and lateral R3B; whereas PAML-2 is present in cilia of dorsal ray neurons R1B, R5B, R7B, and lateral R9B. c Left panel shows % of EVs with colocalization of PAML-1::GFP with PKD-2::mScarlet released from ventral but not dorsal rays, and PAML-2::mScarlet with PKD-2::GFP in EVs released from dorsal but not ventral rays. The right panel shows Pearson correlation (two-sided) coefficients for each point of the left panel, n = 9 animals for PAML-1::GFP colocalization with PKD-2::mScarlet, and n = 11 animals for PAML-2::mScarlet colocalization with PKD-2::GFP. The box-and-whiskers plots show the median values where the box covers two quartiles around the median, and the whiskers extend 1.5 quartiles from the median. Source Data are provided as SourceData_Main.xls file. d Scheme of PAML-1::GFP and PAML-2::mScarlet localization in HOB and RnB neuronal cell bodies and cilia. The color of the cell body indicates the expression of the corresponding protein, and the color of the cilium indicates the presence of the corresponding protein in the cilium. Disruption of lov-1 abrogates ciliary localization of PAML-1::GFP (e) and PAML-2::mScarlet (f). g Flattened z-stack showing that disruption of the paml-1 gene results in PAML-2::mScarlet ciliary localization in ventral neurons. For the full dataset on fluorescence profiling of each ray, refer to Supplementary Figs. 9-10. Quantification of the aberrant presence of PAML-2::mScarlet in ventral cilia of paml-1(tm12527) mutants is presented in Supplementary Fig. 11. Source Data are provided as SourceData_Main.xls, SourceData_SuppFig9.xls, and SourceData_SuppFig10.xls files.
In contrast to paml-1, paml-2 was expressed in all polycystin-expressing neurons, including ventral HOB, paired ray B neurons 1-5, 7-9 of the tail (Supplementary 9a-b), and paired CEM neurons in the head. However, PAML-2::mScarlet localized to cilia and EVs solely in the dorsal rays (R1B, R5B, and R7B) and lateral ray neurons R9B (Fig. 5b, Supplementary 10c-d) and was absent from the PAML-1-carrying ventral ray cilia and EVs (Fig. 5c). These dorsal neurons mediate the contact-based response to hermaphrodite27. Overall, the spatial specialization of PAMLs’ distribution in ray cilia (Fig. 5d) suggested that the polycystin complex and PAMLs might play a role in males’ navigation of 3D space of dorsal versus ventral mating cues.
We found that PAML-1 and PAML-2 ciliary localization requires LOV-1. In the lov-1 mutant, both PAMLs remained confined to the cell body and did not localize to dendrites, cilia, or ciliary EVs (Fig. 5e-f). These data suggest that LOV-1 may recruit PAMLs to cilia and EVs, and this recruitment may occur in the neuronal cell bodies. Thus, LOV-1 acts as a pivotal scaffold protein that recruits transmembrane components of dorso–ventral specialization to cilia and polycystin-carrying EVs.
To uncover mechanisms behind the selective presence of PAML-2 solely in dorsal cilia (Fig. 5d), we examined the localization of PAML-2::mScarlet in paml-1 mutant animals. In the absence of ventral paml-1 expression, PAML-2::mScarlet ectopically localized to the cilia of ventral ray neurons (Fig. 5g). The PAML-2 total abundance in ventral cilia of the paml-1 mutant reached 25-60% of the levels observed in dorsal neurons (Supplementary Fig. 11a). The ectopic ciliary presence of dorsal lectin PAML-2::mScarlet in ventral neurons of paml-1 mutants was accompanied by a 2-4-fold increase in PAML-2::mScarlet levels in the ventral cell bodies (Supplementary Fig. 11a’). These data suggest that in the absence of the PAML-1 ventral lectin, a compensatory mechanism boosts the production or stabilization of PAML-2 in ventral neurons.
The polycystin complex is not essential for the shedding of ciliary EVs as evidenced by tracking cargo CWP-5
The final validated candidate interactor of PKD-2 was CWP-5 (Coexpressed With Polycystins 548), a single-pass transmembrane protein. We generated the endogenous reporter CWP-5::GFP and found cwp-5::gfp expression in all pkd-2-expressing neuronal cell bodies except R2B (Supplementary Fig. 12). Abundant ciliary localization of CWP-5::GFP was observed in all but R2B and R8B neurons (Fig. 6a, Supplementary 12, 13a-a’).

a Flattened z-stacks showing colocalization of PKD-2::mScarlet with CWP-5::GFP. CWP-5 is enriched in the ciliary transition zone with low levels of PKD-2::mScarlet. a’ Representative average fluorescence profile of CWP-5::GFP and PKD-2::mScarlet along the cilium (n = 20 cilia). Note the prominent presence of CWP-5::GFP in the areas with low levels of PKD-2::mScarlet – the transition zone (TZ) and the neck of the cilium. For the full dataset on fluorescence profiling of each ray, refer to Supplementary Figs. 12-13. Flattened z-stacks showing that disruption of lov-1 (b) and pkd-2 (c) does not alter CWP-5::GFP ciliary and EV localization. d Working model of the molecular mechanism for loading CWP-5::GFP to cilia and ciliary EVs. CWP-5 ciliary localization and release in EVs is independent of polycystins LOV-1 and PKD-2. Source data are provided as SourceData_Main.xls and SuppFig13.xls files.
Unlike PACL-1 and PAMLs, CWP-5 did not exhibit complete colocalization with PKD-2 within the cilium. Specifically, we found CWP-5::GFP enriched in the transition zone and the compartment proximal to the ciliary tip (“neck”), two exclusion areas for PKD-2 reporters (Fig. 6a-a’). The absence of complete colocalization was also evident through the analysis of the fluorescence correlation between PKD-2::mScarlet and CWP-5::GFP (Supplementary Fig. 13a-a’) with much lower correlation coefficients between PKD-2 and CWP-5 than between PKD-2 and PACL-1 or PAMLs (Supplementary Fig. 8a’, 9a’, 10c’). The ciliary and EV localization of CWP-5::GFP was independent of LOV-1 and PKD-2 (Fig. 6b-c, Supplementary Fig. 13a-a’), indicating that CWP-5::GFP is transported to the cilium and loaded to polycystin-carrying EVs independently from the polycystin complex (Fig. 6d).
The polycystin complex and its proximity interactors act in the same genetic pathway
Since the polycystin pathway functions in male sensory neurons to regulate mating behavior, we next tested whether the new interactor PACL-1 was involved in the same process by assessing polycystin-related male mating behavior—response to hermaphrodite contact and vulva ___location26 (Fig. 7a). Male response behavior is initiated when ray sensory neurons of the male tail come in contact with a potential mate. Next, the male backs along the hermaphrodite’s body until encountering the vulva24,25,26. The male then stops at the vulva, coordinates his movements to the hermaphrodite’s, and proceeds to the next step. This vulva ___location behavior requires the HOB neuron and other neurons in the male tail27,49. Mutations in the polycystins pkd-2 and lov-1 result in defects in response and vulva ___location steps indicative of the major role of polycystin-expressing neurons in these behaviors in C. elegans.

a A diagram showing steps in the male mating behavior controlled by the polycystin pathway—response to hermaphrodite and vulva ___location. b Assessment of male mating behavior shows that the pacl-1 mutant is deficient in responding to hermaphrodite contact. Number of mating trails conducted were: n = 6 WT, 3 pacl-1(tm12580), 5 pkd-2(sy606), and 3 pacl-1(tm12580)rescue. Vulva ___location behavior of the pacl-1 mutants could not be assessed due to their severe response defect. Assessment of male mating behavior shows that the paml-1; paml-2 double mutant is deficient in responding to hermaphrodite contact (c), and ___location of vulva (d, e). Number of mating trails conducted were: n = 8 WT, 3 paml-1(tm12527), 3 paml-2(my155), and 8 paml-1(tm12527); paml-2(my155) double mutant. 20 adult males were tested in each mating trial. Bar graphs represent mean values +/- SD. One-way ANOVA analyzed data with the Tukey post hoc adjustment. Source data are provided as SourceData_Main.xls file.
Response and vulva ___location behavioral deficiencies are also exhibited by the trf-1 and trf-2 mutants, indicating that TRAFs and polycystins act in the same genetic pathway28. Thus, we tested whether the newly discovered interactors PACL-1 and PAMLs also act in the polycystin pathway.
The pacl-1 mutant males were tested alongside the wild-type and pkd-2-null males, where wild-type and the pkd-2 mutant served as positive and negative controls, respectively. We found that 90% of wild-type males and only 10% of the pacl-1 mutant males responded to the presence of mates within 5 min. The pacl-1 null males were deficient in the response behavior to the same severe degree as the pkd-2 null males (Fig. 7b, one-way ANOVA with the Tukey post hoc adjusted p-value < 0.001). To confirm that the deficiency of the pacl-1 mutant was due to the disrupted PACL-1 function, we generated a rescue strain carrying an extrachromosomal array with the wild-type pacl-1 genomic sequence in a pacl-1 mutant background. The rescued strain exhibited normal response behavior indistinguishable from the wild-type (one-way ANOVA with the Tukey post hoc test) (Fig. 7b). These data indicate that the auxiliary ion channel-like protein PACL-1 functions in the male-specific polycystin-expressing sensory neurons where pacl-1 is required for polycystin-mediated response behavior.
To investigate whether PAMLs are involved in the polycystin pathway within the sensory neurons, we tested the male mating behavior of the paml-1 and paml-2 null mutants. The response to hermaphrodite of the wild-type (92%) was not significantly different from the single paml-1 (90%) and paml-2 (87%) mutants. However, the paml-1; paml-2 double null mutant exhibited significantly reduced response to hermaphrodite contact (70%) (one-way ANOVA with Tukey post hoc test adjusted p < 0.01) (Fig. 7c).
The paml-1; paml-2 double mutant response defect was not as severe as that of the pkd-2 mutant (6%), which allowed us to score the vulva ___location step of the male mating behavior. We measured the efficiency of the vulva ___location by dividing the number of positive vulva locations by the total number of vulva encounters. We found that, on average, 91% of wild-type, 81% of the paml-1, and 89% of the paml-2 single mutants could find a vulva in the experimental time frame. In the double paml-1; paml-2 mutants, only 50% of males found the vulva, which was significantly different from the wild-type and single mutant strains (one-way ANOVA with Tukey post hoc test adjusted p < 0.05) (Fig. 7d). Vulva ___location efficiencies of wild-type males (91%) and the single paml-2 mutants (88%) were not statistically different with the majority of males stopping at the vulva only on the first encounter (Fig. 7e). The single paml-1 mutant was slightly deficient as compared to wild-type (81%, one-way ANOVA with Tukey post hoc test adjusted p < 0.01). The efficiency of the double mutant was reduced to 47% (one-way ANOVA with Tukey post hoc test adjusted p < 0.001), with the majority of males stopping at the vulva only on the second encounter (Fig. 7e). Taken together, our data are consistent with ventral PAML-1 and dorsal PAML-2 acting redundantly to fine-tune the polycystin-mediated mating behaviors.
Previous studies showed that the CWP-5 transmembrane lectin is involved in the polycystin-mediated response to the hermaphrodite50, but not vulva ___location behavior.
In summary, our data show that lov-1, pkd-2, trf-1, trf-2, pacl-1, paml-1, paml-2 genes are required for response and vulva ___location behavior, with paml-1 and paml-2 acting redundantly. These gene products may act cell autonomously in ciliated male-specific sensory neurons and/or cell non-autonomously in ciliary EVs. Entangling the site(s) of action of the polycystin signaling complex in neuronal cilia versus ciliary EVs is the challenging future direction.
Discussion
This study used TurboID-mediated proximity labeling to discover cargo associated with polycystin-2 ciliary EVs of C. elegans. We took a modular indirect approach generating TurboID with an anti-GFP nanobody ___domain that tethered the proximity labeling enzyme to a GFP-labeled cargo of interest. This EV-TurboID approach has many implications for the future of the EV field as opposed to direct fusions of the biotin ligase with a cargo of interest. Expression of the anti-gfp nanobody::mScarlet::turboID under cell- or tissue-specific promoters in strains with endogenously GFP-tagged proteins enables one to address EV cargo composition in a cell-specific manner. Our previous work showed that EVs with shared cargo can be generated via distinct routes and thus might differ drastically in content33. The application of modular proximity labeling in cell- and tissue-specific manner is a robust strategy for dissecting the composition of EV subtypes and individual EVs with unprecedented resolution.
We discovered a regulated hierarchical assembly of signaling components on ciliary EVs at a single-cell, single-EV level in C. elegans. Specifically, our data show that LOV-1 is responsible for recruiting polycystin-associated cargo to polycystin ciliary EVs (Fig. 8). An association with LOV-1 was essential and a prerequisite for EV cargo loading. Our study suggests that PKD-2 first assembles with LOV-1, prompting PACL-1 and PAMLs to join the complex. In our model, this complex then acquires TRF-1 and TRF-2, likely within the ciliary shaft, before entering the ciliary tip and shedding in ciliary EVs. Conversely, CWP-5 ciliary and EV localization is polycystin-independent and achieved via an unidentified mechanism. This level of precision in analyzing individual EV components and EV cargo loading in vivo remains unattainable in vertebrate models.

Our work presents evidence that a single genetic change (a polycystin-1 mutation) can significantly alter the content of EVs and their potential signaling properties (Fig. 8). In our study, mutations in the polycystin-1-like protein LOV-1 resulted in the release of EVs lacking many signaling components. Transferring this knowledge to a human setting might mean that patients with PC1 mutation experience different EV signaling within pathological tissues where altered EVs might serve as a pathology-exacerbating factor. This hypothesis aligns with studies in which EVs isolated from the cystic fluid of ADPKD patients promote cystogenesis in 3D cell cultures51.
Moreover, human polycystin-1 mutations cause more severe polycystic kidney disease than polycystin-2 mutations52, aligning with our hypothesis that different ADPKD patient mutations might have distinct content of cilia and/or their EVs, potentially impacting disease progression. The idea of signaling between cells via polycystin EVs is supported by studies where soluble fragments of the PC1 C-type lectin ectodomain act as a ligand and activate the polycystin complex in cultured mammalian cells53, suggesting that polycystin EVs might signal between cells via their extracellular domains. Our work highlights the importance of understanding polycystin function in EVs and the potential application of single EV analysis for non-invasive early diagnosis of ADPKD.
The connection of the polycystin pathway with the ubiquitin-conjugating machinery has been reported for other species. For example, in Drosophila, the enrichment of the PKD2 homolog AMO at the sperm flagellar tip requires the function of two proteins related to ubiquitination: (i) E3 ubiquitin-protein ligase dKPC1 (homolog of human RNF123) and (ii) adapter protein dKPC2 (homolog of human UBAC1, ubiquitin-associated ___domain-containing 1). Disruption of the PKD2 homolog AMO or either dKPC1 or dKPC2 elicits similar deficiencies in male sperm fertility54, suggesting that the Drosophila polycystin pathway uses components of ubiquitination machinery.
In this work, we found a connection between the polycystin pathway and the TRAF-related ubiquitin-conjugating system. Specifically, TRF-1 (an adapter with a ubiquitin-ligase-like RING ___domain) is required for cytoplasmic abundance of TRF-2 (a TRAF without RING) and its presence in the ciliary shaft. In contrast, TRF-1 enters the cilium independently of TRF-2 but requires TRF-2 for its colocalization with the polycystin complex and loading to polycystin EVs. Neither TRF-1 nor TRF-2 is required for the polycystin complex ciliary localization and release in the form of ciliary EVs.
Given the high conservation of the TRAF family and the polycystins between humans and C. elegans, their functional interactions might likewise be conserved. This hypothesis is supported by the fact that Traf6 knock-out mice phenocopy skeletal ciliopathies, including exencephaly, problems with neural tube closure, shortening of long bones and tooth agenesis in neonates, focal alopecia, delayed pigmentation, and underdevelopment of skin glands, including sweat and sebaceous glands55,56,57,58. These phenotypes mimic ciliopathies of humans, such as NEK1, DYNC2H1, and IHH mutations (shortening of long bones), tooth agenesis in patients with BBSome variants, sparse hair and focal alopecia of EDA patients59,60,61,62,63,64. Mutations in TRAF7 (unusual TRAF possessing the RING ___domain but lacking the MATH ___domain) cause skull-base meningiomas and congenital heart disease in humans and are important for intraflagellar transport and ciliogenesis in Xenopus65. Our work and the literature suggest that human TRAFs might act in the cilium, possibly in tandem with polycystins.
Our data from C. elegans show that disrupted polycystins affect the subcellular localization of polycystin-associated channel-like protein PACL-1. This finding also parallels an aspect of human polycystin biology. Specifically, polycystin dysfunction in ADPKD patients is associated with deregulated monovalent ion transport that directly contributes to cyst enlargement66,67, suggesting that human polycystins might regulate the function, localization, or expression of monovalent channels, resulting in deregulated ion homeostasis and abnormal fluid accumulation. Whether PACL-1 functions as a channel and how it interacts with the polycystins warrants further investigation.
Polycystins’ connection with transmembrane C-type lectins of dorso–ventral neuronal function hints at conserved involvement of polycystins in the sensation of spatial orientation as observed in cerebrospinal fluid-contacting neurons (CSF-cNs) of zebrafish18,19,20 and in the left-right organizer of the early mouse embryo23. In C. elegans, polycystin complex PKD-2LOV-1 associates with either ventral-specific or dorsal-specific transmembrane lectins, resulting in the production of two distinct subtypes of polycystin-carrying EVs marked by the presence of either PAML-1 or PAML-2. We hypothesize that the mutually exclusive presence of PAMLs on the polycystin EVs stems from the different affinities of PAML-1 and PAML-2 for the polycystin complex. Specifically, in the ventral ray B neurons where PAML-2 is co-expressed with PAML-1, the polycystin complex may preferentially bind PAML-1, resulting in the release of ciliary EVs with the PAML-1 transmembrane lectin. In contrast, in the dorsal ray B neurons, PAML-1 is not expressed and thus does not compete with PAML-2 for binding to the polycystin complex, resulting in the production of EVs carrying the PAML-2 marker. Elucidation of the functional significance of these two populations of polycystin-carrying EVs is a subject for future investigation. Besides PAMLs, the CWP-5 transmembrane lectin also demonstrated cell-specific presence in cilia and EVs, prompting us to put forward several hypotheses about EV loading mechanisms and the fate of specific EVs, detailed in the legend of Supplementary Fig. 12. A provoking hypothesis suggests the possibility of the ray 2B cilium acting as a receiver of EVs released from other ray B neurons. We also propose that C-lectin domains present at the ciliary tips of sensory neurons might have differential affinities to different cuticle glycans along the hermaphrodite’s body68 and thus provide positional information to the male during the mating process.
In summary, our study presents a new application of proximity labeling for EV subtyping that we used to identify and validate bona fide cargo of polycystin EVs of C. elegans. We established a connection between polycystins and three classes of proteins (channel-like, transmembrane C-type lectins, and TRAFs). Our study offers a blueprint for further exploration of EV biology.
Limitations of study
A technical limitation of the study concerns colocalization analysis. Colocalization of fluorescent reporters does not imply direct binding. Thus, we cannot rule out an indirect association between components of the polycystin complex, nor can we establish exact binding sites. We can only conclude that validated proteins exist in close proximity to each other, within 10 nm of the TurboID-mediated biotin-labeling radius. However, the newly discovered functional interactions between these polycystin EV-associated cargo imply that they act in the same molecular cascade. We can conclude that the TRAFs, PAMLs, and PACL-1 act cell-autonomously in male sensory neurons to control polycystin-mediated mating behaviors. However, the way in which EVs influence male mating behaviors and the range of ciliary EV bioactivities remain subjects of intense studies.
Conceptually, our study uses C. elegans to discover the fundamental biology of polycystins. Thus, not all the findings might directly apply to human health, but some aspects of polycystin functioning might be either evolutionarily conserved or functionally convergent.
Methods
Strains and cultural conditions
Animals were cultured at 20 oC using the OP50 E. coli strain as a food source. For culturing animals for EV isolation, see sections below.
Mutant strains carrying alleles paml-1(tm12527), pacl-1(tm12580), trf-1(nr2014), and trf-2(tm5167) were backcrossed at least six times to eliminate most background mutations. The paml-2(my155) allele was generated using CRISPR/Cas9-mediated genome editing and outcrossed three times.
Refer to Supplementary Fig. 14 for diagrams of genomic structures of trf-1, trf-2, paml-1, paml-2, pacl-1, lov-1, and pkd-2 and corresponding mutant alleles used in the study.
All strains used in the study are reported in Supplementary Table 1.
Generation of transgenic strains
To generate the plasmid carrying Pklp-6::anti-GFP nanobody::mScarlet::TurboID, we used a splice PCR for seamless fusion. The nanobody-encoding sequence was amplified from the pGEX6P1-GFP-Nanobody plasmid (a gift from Kazuhisa Nakayama, Addgene plasmid #61838). The mScarlet-encoding sequence was amplified from pJR022 (a gift from Paul Sternberg, Addgene plasmid #198820). The TurboID-encoding sequence was amplified from the 3xHA-TurboID_pAS31 plasmid (a gift from Jessica Feldman, Addgene plasmid #118220). The klp-6 promoter was amplified from genomic DNA (2000 bp upstream of the start codon).
To generate a rescue plasmid for the pacl-1(tm12580) mutant, we fused a wild-type genomic sequence of the pacl-1 locus spanning the 2 kb promoter region and the entire pacl-1-coding sequence to a GFP-encoding sequence from a plasmid of Andrew Fire (pPD95_75 Addgene plasmid # 1494).
To generate strains with stable transgene expression, the plasmid carrying transgene-of-interest was mixed with a rescue plasmid for the wild-type pha-1 gene69 and injected into the gonads of young adult hermaphrodites bearing the pacl-1(tm12580) and pha-1(e2123 temperature-sensitive mutation, lethal at 22 oC and permissive at 15 oC). Injected P0 hermaphrodites, the resulting F1 generation, and F2 stable lines were grown at the restrictive 22 oC temperature to ensure the expression and maintenance of the transgene70.
Generation of endogenously labeled strains
To validate mass spectrometry findings, we generated endogenous reporters using CRISPR/Cas9-mediated genome editing as described in Dokshin et al.71. All generated strains were verified by Sanger sequencing and are reported in Supplementary Table 1. Also, refer to Supplementary Fig. 12d for the summary of expression patterns within the polycystin-expressing neurons for all newly generated endogenous reporters used in the study. Sequences of all newly generated alleles and transgenes used in the study are included in the Supplementary Information alongside their genotyping conditions (Supplementary Tables 2–4).
Technical details of using modular TurboID
The overall scheme of the experiment included the isolation of EVs from two strains. The control strain carried the anti-GFP nanobody::mScarlet::TurboID-encoding sequence under the klp-6 (kinesin-like protein 6) promoter driving expression in IL2, CEMs, RnBs, and HOB neurons. Thus, in the control strain, the anti-GFP nanobody::mScarlet::TurboID protein was not targeted to any specific cell ___location, its localization appeared uniform throughout the neurons, including cell bodies, nuclei, axons, dendrites, and cilia (Supplementary Fig. 1a-c). The sample strain carried TurboID under the klp-6 promoter and the Ppkd-2::pkd-2::gfp transgene expressed in CEMs, RnBs, and HOB neurons. In this sample strain, TurboID followed the subcellular localization of PKD-2::GFP in cell bodies and cilia and was specifically loaded and enriched in PKD-2::GFP carrying EVs (Fig. 1b, Supplementary Fig. 1b-c).
TurboID-based enzymatic labeling of neuronal ciliary EVs in C. elegans
To initiate the culture, 240 hermaphrodites at the larval stage 4 were transferred to 40 6-cm normal growth medium (NGM) plates (6 animals per plate) with E. coli OP50 lawns and grown for two generations. On day 6, each plate was chunked into quarters. The chunks were transferred into 15 cm plates with high growth medium (HGM) (3 g of NaCl, 2.5 g of peptone, and 20 g of agar per 1 L supplemented with 4 mL of cholesterol stock (5 mg/mL of ethanol), 1 mL 1 M CaCl2, 1 mL 1 M MgSO4, and 25 mL 1 M potassium phosphate buffer pH 6.0) seeded with a full lawn of E. coli OP50 (1.8 mL/plate). Animals were allowed to propagate for two more generations at 22 oC until they consumed most of the bacteria and were not starved. In total, about 4 million adult worms were used to isolate EVs for each replicate.
We chose to grow animals on standard E. coli OP50 lawn, and not on a biotin auxotrophic strain MG1655bioB:kan, without administration of a bolus of biotin before EV harvest because previous studies of indirect labeling showed that these conditions are unnecessary and do not result in better outcomes37.
Harvesting EVs of C. elegans
On the day of EV harvest, we washed C. elegans off the HGM plates with the M9 buffer (3 g KH2PO4, 6 g Na2HPO4, 5 g NaCl, 1 mL of 1 M MgSO4 per 1 L) into 15 mL conical tubes. We centrifuged the collected animal suspension at 3000 g for 15 min to pellet the worms and collect the EV-containing supernatant. To increase EV yields, we resuspended animals in fresh M9 buffer and repeated the centrifugation step (15 min at 3000 × g) twice, collecting the supernatant with released EVs each time.
To clear the EV-containing supernatant of residual bacteria and large debris, we centrifuged it at 10,000 × g (8000 rpm in SW28 swinging bucket rotor, Beckman Coulter) for 30 min at 4 oC. We repeated this step 3 times, collecting the more clarified supernatant. After the final clarification step, we pelleted EVs from the cleared supernatant onto a 36% iodixanol cushion at 100,000 g (28,000 rpm in SW28 swinging bucket rotor, Beckman Coulter) for 2 h at 4 oC. The cushion was prepared by mixing three parts of 60% iodixanol (OptiPrep, Sigma #D1556) and two parts of 8% sucrose to achieve a final density of 1.2 g/L.
To concentrate pelleted EVs into a smaller volume for further loading onto density gradients, we resuspended the collected EVs in the M9 buffer. We pelleted the EV suspension again in a smaller volume tube at 100,000 × g (30,000 rpm SW41 swinging bucket rotor, Beckman Coulter) onto a 36% iodixanol cushion.
Before loading the concentrated EV suspension onto density gradients, we ensured the density of the suspension was less than 1.1 g/mL by diluting it with M9 buffer (typically, that meant adding 2 mL of fresh M9 buffer to 1 mL of concentrated EV suspension). Using a thin glass Pasteur pipette, we loaded the diluted EV suspension onto the top of density gradients (1.1–1.2 g/mL), prepared by the freeze-thaw method described in Nikonorova et al.16. To drive TurboID-labeled EVs to their respected density within the tube, we conducted isopycnic centrifugation at 100,000 × g (30,000 rpm, 16 h, at 4 oC).
To collect TurboID-containing EVs, we fractionated the equilibrated gradients on the BioComp Piston Gradient Fractionator into 30 fractions of 375 mL each. To examine fractions for PKD-2::GFP and anti-GFP nanobody::mScarlet::TurboID EVs we used a super-resolution microscope - Zeiss LSM880 with Airyscan, equipped with the Plan-Apochromat 63x/1.4 Oil DIC M27 (FWD = 0.19 mm), CG = 0.17 mm objective (Zeiss item no.: 420782-9900-000). We layered an array of representative droplets from each fraction onto a glass slide. We used double-adhesive stickers to form chambers for the droplets (SecureSeal spacers by Electron Microscopy Sciences, Cat# 70327-9S). Finding the focal plane with most EVs was done in four steps: (i) manually focusing on the very edge of the water droplet closest to the objective, (ii) shifting the center of the field of view away from the edge toward the center of the droplet by about 100–200 μm, (iii) automatic focusing to find the plane with EVs, and (iv) acquisition of a Z-stack of 3 planes–one at the automatic focal plane, and two are above and below, 0.5 μm apart. Image acquisition was performed in the R-S mode (Resolution vs Sensitivity), designed to capture very dim and/or fast dynamic processes72. Once the most enriched fractions were identified, we proceeded to the extraction of biotinylated proteins.
Extraction of biotinylated proteins from C. elegans EVs
In the extraction of biotinylated proteins, we followed the protocol of Branon et al.36, which was the first successful proximity labeling applied to C. elegans. Specifically, EVs were lysed with RIPA buffer and then incubated with streptavidin-conjugated beads (Pierce Streptavidin Magnetic beads, ThermoFisher #88816; 25 μl of the bead suspension per 300 μg of total protein). Afterward, beads were washed twice with the RIPA buffer, once with 1 M KCl, once with 0.1 M Na2CO3, once with 2 M urea in 10 mM Tris-HCl (pH 8.0), and five times with 50 mM NH4HCO3. We transferred the beads to a fresh tube during the final wash. The beads were then submitted to the Rutgers Proteomics facility for on-bead trypsin digestion and protein identification.
On-beads digestion
Trypsin digestion was performed at 37 oC for 4 h in 20 μl volume of 50 mM NH3HCO4 with the bead pellet and 0.2 μg of trypsin. After four hours of the initial incubation, another 0.2 μg of trypsin was added and incubated at 37 oC overnight. Following overnight digestion, the solution was separated from the magnetic bead pellet, and its pH was adjusted to 3.0 with 10% formic acid. The sample was desalted with a Stage tip before the LC-MSMS.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS)
Samples were analyzed by LC-MS using Nano LC-MS/MS (Dionex Ultimate 3000 RLSCnano System) interfaced with Orbitrap Eclipse Tribrid (ThermoFisher) at the Rutgers Proteomics facility. Samples were loaded onto a fused silica trap column Acclaim PepMap 100, 75 μm × 2 cm (ThermoFisher). After washing for 5 min at 5 µl/min with 0.1% TFA, the trap column was brought in-line with an analytical column (Nanoease MZ peptide BEH C18, 130 A, 1.7 μm, 75 μm × 250 mm, Waters) for LC-MS/MS. Peptides were fractionated at 300 nL/min using a segmented linear gradient 4–15% B in 30 min (where A: 0.2% formic acid, and B: 0.16% formic acid, 80% acetonitrile), 15–25% B in 40 min, 25–50% B in 44 min, and 50–90% B in 11 min. Solution B returns at 4% for 5 min for the next run.
The scan sequence began with an MS1 spectrum (Orbitrap analysis, resolution 120,000, scan range from M/Z 350–1600, automatic gain control (AGC) target 1E6, maximum injection time 100 ms). The top S (3 s) and dynamic exclusion of 60 s were used to select Parent ions for MSMS. Parent masses were isolated in the quadrupole with an isolation window of 1.4 m/z, automatic gain control (AGC) target 1E5, and fragmented with higher-energy collisional dissociation with a normalized collision energy of 30%. The fragments were scanned in Orbitrap with a resolution of 30,000. The MSMS scan range was determined by the charge state of the parent ion, but the lower limit was set at 100 am.
Database search
The peak list of the LC-MSMS was generated by Thermo Proteome Discoverer (v. 2.4) into MASCOT Generic Format (MGF) and searched against UniProt proteome database for C. elegans, E. coli, custom sequences of GFP, the anti-GFP nanobody, mScarlet, TurboID, and a database composed of common lab contaminants using the Rutgers Proteomics facility in-house version of X!Tandem (GPM Furry, Craig and Beavis, 2004). Search parameters are as follows: fragment mass error: 20 ppm, parent mass error: +/− 7 ppm; fixed modification: none; flexible modifications: oxidation on methionine; protease specificity: trypsin (C-terminal of R/K unless followed by P), with 1 miss-cut at preliminary search and 5 miss-cut during refinement. Only spectra with log(e) < −2 were included in the final report.
Enrichment analysis
The top six candidates for verification were chosen based on their respective spectral count enrichments in the samples representing TurboID targeted to PKD-2::GFP vs. the sample with no targeting (Table 1).
Super-resolution fluorescence imaging and analysis
For in vivo imaging, we anesthetized one-day-old adult C. elegans males with 10 mM levamisole solution in the M9 buffer and mounted animals onto thin 10% agarose pads. Animals were imaged within 30 min of mounting. Super-resolution imaging was performed on a Zeiss LSM880 confocal system equipped with Airyscan Super-Resolution Detector, Single Photon Lasers (488 nm for GFP and 561 nm for mScarlet), motorized XY Stage with Z-Piezo and T-PMT. All images were taken using the Plan-Apochromat 63x/1.4 Oil DIC M27 (FWD = 0.19 mm), CG = 0.17 mm objective (Zeiss item no.: 420782-9900-000). Image acquisition was performed in the R-S mode (Resolution vs Sensitivity) to capture very dim and/or fast dynamic processes. For analysis of fluorescence profiles through cilia of RnB neurons, fluorescence values were normalized to the average of minimum and maximum values for each cilium.
Image analysis
Imaging was performed on adult males 2–5 h past the L4 molting stage. At the minimum 10 and up to 30 age-matched animals were imaged and analyzed for each condition, including images in Figs. 1c, 4c, 5e-f, 6b-c, and Supplementary Figs. 1b-c, 10a-b, 12a.
Quantification of fluorescent protein abundance analysis was performed in Fiji (as schematically shown in Fig. 2a). For profiling the fluorescence intensities along the cilium, acquired Z-stacks spanning all rays were flattened to maximum intensity projections. A line of 10-pixel width was drawn through the tip of the cilium through the shaft and the base toward the dendrite to extract corresponding numerical fluorescence values for reporters of interest. For each cilium, these extracted values were normalized to the average of the maximum and minimum values of the fluorescence range. Normalization allowed us to represent the relative distribution of the fluorescent reporters along the cilium on a scale from 0 to 2 (Supplementary Fig. 15a). Then, normalized profiles for all measured cilia of each ray were aligned by the maximal fluorescence of ciliary tips and averaged across different animals.
To analyze the relative distribution of a fluorescent protein along the cilium we used fluorescent profile graphs. We plotted the average value of normalized fluorescence intensity as a solid line over the distance along the cilium; an upper half of the interquartile (a spread between the 50th and 75th percentiles of the data) is indicated as a semi-transparent ribbon. These diagrams were useful in analyzing cargo ciliary tip enrichment and relative cargo distribution along the ciliary compartments (as for comparison of CWP-5::GFP and PKD-2::mScarlet).
To assess the colocalization of two fluorescent reporters along the cilium, we used correlation plots where normalized fluorescence intensities for the two reporters in question were plotted on x- and y-axes, respectively (each normalized value ranging from 0 to 2). Pearson correlation coefficient (R) was calculated to measure the strength and direction of a linear relationship between two normalized intensity values (Supplementary Fig. 15b).
Quantification of total levels of a fluorescent reporter in the cilium (as for Fig. 3b’) was performed as follows. A line was drawn through the ciliary tip (marked by the ciliary tip marker PKD-2::mScarlet) to the ciliary base. The numerical values of fluorescence intensities along the line were extracted. The mean/average value along the entire line spanning the tip, the shaft, and the base was calculated. Then, the mean values of the mutant were normalized to the WT values and analyzed using the Wilcoxon Rank Sum test.
Quantification of fluorescent protein abundance in the cell bodies for Fig. 3c’ was performed in Fiji. A line was drawn through a whole cell on a single plane. Rendered fluorescence values across the cytoplasm and the nucleus were compared using the Wilcoxon Rank Sum test, which showed that the nuclear level remained unchanged, whereas the cytoplasmic level varied (Supplementary Fig. 6c’). Thus, we used nuclear levels as a normalization factor to compare cytoplasmic levels of PKD-2::mScarlet and TRF-2::GFP in WT and the trf-1(nr2014) mutant using the Wilcoxon Rank Sum test (Fig. 3c’).
Quantification of colocalization of PACL-1 and PAMLs with PKD-2 on EVs was performed in Fiji as described in Walsh et al.30 using ComDet V.0.5.5 plugin that is freely available.
Assessment of male mating behavior
The male mating behavior assay was performed in a blind-to-genotype manner in three to five biological replicates (20 animals per genotype in each replicate) as described previously24,27 with minor modifications. Briefly, we used fresh E. coli OP50 lawns of 10 μl each, seeded 2 h before the assay, dried in a 37 oC incubator, then cooled to room temperature. We then populated the mating spots with ten 1-day-old virgin adult unc-31(e169) hermaphrodites. The uncoordinated unc-31 mutation reduces movement, making hermaphrodites easy mating targets for males. After a 30 min acclimation period, we placed an adult virgin male on the mating spot with the hermaphrodites and observed male behavior for five minutes. Adult virgin males were age-matched by selecting larval stage 4 (L4) 24 h and culturing with other L4 males of the same genotype before the assay. Response efficiency was calculated as the percentage of males transitioning from forward roaming to backward scanning after contacting a hermaphrodite. The efficiency of vulva ___location was calculated for each male who successfully responded to hermaphrodite contact as the number of positive vulva locations divided by the total number of vulva encounters. Vulva ___location efficiencies of individual males were then averaged across the genotypic populations within each biological replicate. Statistical analysis was conducted to analyze average values for each biological replicate using one-way ANOVA with the Tukey post hoc test.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The proteomics data generated in this study have been deposited in the MassIVE database under accession code MSV000094641. The z-stacks that were used to produce images for all the figures have been deposited to FigShare (https://doi.org/10.6084/m9.figshare.27638175)73. Source data are provided as Source Data files for fluorescent profiling and mating efficiency measurements. Source data are provided with this paper.
References
Welsh, J. A. et al. Minimal information for studies of extracellular vesicles (MISEV2023): from basic to advanced approaches. J. Extracell. Vesicles 13, e12404 (2024).
Luxmi, R. & King, S. M. Cilia-derived vesicles: an ancient route for intercellular communication. Semin Cell Dev. Biol. 129, 82–92 (2022).
Wang, J. & Barr, M. M. Ciliary extracellular vesicles: Txt msg organelles. Cell Mol. Neurobiol. 36, 449–457 (2016).
Wang, J., Barr, M. M. & Wehman, A. M. Extracellular vesicles. Genetics 227, iyae088 (2024).
Volz, A.-K. et al. Bardet-Biedl syndrome proteins modulate the release of bioactive extracellular vesicles. Nat. Commun. 12, 5671 (2021).
Ojeda Naharros, I. & Nachury, M. V. Shedding of ciliary vesicles at a glance. J. Cell Sci. 135, jcs246553 (2022).
Hilgendorf, K. I., Myers, B. R. & Reiter, J. F. Emerging mechanistic understanding of cilia function in cellular signalling. Nat. Rev. Mol. Cell Biol. 25, 1–19 (2024).
Anvarian, Z., Mykytyn, K., Mukhopadhyay, S., Pedersen, L. B. & Christensen, S. T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 15, 199–219 (2019).
Bergmann, C. et al. Polycystic kidney disease. Nat. Rev. Dis. Prim. 4, 50 (2018).
Ward, C. J. et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat. Genet. 30, 259–269 (2002).
Pisitkun, T., Shen, R.-F. & Knepper, M. A. Identification and proteomic profiling of exosomes in human urine. Proc. Natl. Acad. Sci. USA 101, 13368–13373 (2004).
Hogan, M. C. et al. Characterization of PKD protein-positive exosome-like vesicles. J. Am. Soc. Nephrol. 20, 278–288 (2009).
Raby, K. L., Horsely, H., McCarthy-Boxer, A., Norman, J. T. & Wilson, P. D. Urinary exosome proteomic profiling defines stage-specific rapid progression of autosomal dominant polycystic kidney disease and tolvaptan efficacy. BBA Adv. 1, 100013 (2021).
Ali, H. et al. Global analysis of urinary extracellular vesicle small RNAs in autosomal dominant polycystic kidney disease. J. Gene Med. 26, e3674 (2024).
Hogan, M. C. et al. Identification of biomarkers for PKD1 using urinary exosomes. J. Am. Soc. Nephrol. 26, 1661–1670 (2015).
Wang, S. et al. Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Mol. Cell Biol. 27, 3241–3252 (2007).
Lea, W. A. et al. Polycystin-1 interacting protein-1 (CU062) interacts with the ectodomain of polycystin-1 (PC1). Cells 12, 2166 (2023).
Wyart, C., Carbo-Tano, M., Cantaut-Belarif, Y., Orts-Del’Immagine, A. & Böhm, U. L. Cerebrospinal fluid-contacting neurons: multimodal cells with diverse roles in the CNS. Nat. Rev. Neurosci. 24, 540–556 (2023).
Orts-Del’Immagine, A. et al. Sensory neurons contacting the cerebrospinal fluid require the Reissner fiber to detect spinal curvature in vivo. Curr. Biol. 30, 827–839.e4 (2020).
Djenoune, L. et al. The dual developmental origin of spinal cerebrospinal fluid-contacting neurons gives rise to distinct functional subtypes. Sci. Rep. 7, 719 (2017).
Thouvenin, O. et al. Origin and role of the cerebrospinal fluid bidirectional flow in the central canal. eLife 9, e47699 (2020).
Tanaka, Y., Okada, Y. & Hirokawa, N. FGF-induced vesicular release of Sonic hedgehog and retinoic acid in leftward nodal flow is critical for left-right determination. Nature 435, 172–177 (2005).
Tanaka, Y., Morozumi, A. & Hirokawa, N. Nodal flow transfers polycystin to determine mouse left-right asymmetry. Dev. Cell 58, 1447–1461.e6 (2023).
Barr, M. M. & Sternberg, P. W. A polycystic kidney-disease gene homologue required for male mating behaviour in C. elegans. Nature 401, 386–389 (1999).
Barr, M. M. et al. The Caenorhabditis elegans autosomal dominant polycystic kidney disease gene homologs lov-1 and pkd-2 act in the same pathway. Curr. Biol. 11, 1341–1346 (2001).
Barr, M. M. & Garcia, L. R. Male mating behavior. in WormBook: The Online Review of C. elegans Biology [Internet] (WormBook, 2006).
Liu, K. S. & Sternberg, P. W. Sensory regulation of male mating behavior in Caenorhabditis elegans. Neuron 14, 79–89 (1995).
Wang, J. et al. Cell-specific transcriptional profiling of ciliated sensory neurons reveals regulators of behavior and extracellular vesicle biogenesis. Curr. Biol. 25, 3232–3238 (2015).
Wang, J. et al. C. elegans ciliated sensory neurons release extracellular vesicles that function in animal communication. Curr. Biol. 24, 519–525 (2014).
Walsh, J. D. et al. Tracking N- and C-termini of C. elegans polycystin-1 reveals their distinct targeting requirements and functions in cilia and extracellular vesicles. PLoS Genet. 18, e1010560 (2022).
Wang, J. et al. Sensory cilia act as a specialized venue for regulated extracellular vesicle biogenesis and signaling. Curr. Biol. 31, 3943–3951.e3 (2021).
Wang, J., Nikonorova, I. A., Gu, A., Sternberg, P. W. & Barr, M. M. Release and targeting of polycystin-2-carrying ciliary extracellular vesicles. Curr. Biol. 30, R755–R756 (2020).
Nikonorova, I. A. et al. Isolation, profiling, and tracking of extracellular vesicle cargo in Caenorhabditis elegans. Curr. Biol. 32, 1924–1936.e6 (2022).
Moreira, C. M. et al. Impact of inherent biases built into proteomic techniques: Proximity labeling and affinity capture compared. J. Biol. Chem. 299, 102726 (2023).
Gingras, A.-C., Abe, K. T. & Raught, B. Getting to know the neighborhood: using proximity-dependent biotinylation to characterize protein complexes and map organelles. Curr. Opin. Chem. Biol. 48, 44–54 (2019).
Branon, T. C. et al. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 36, 880–887 (2018).
Holzer, E., Rumpf-Kienzl, C., Falk, S. & Dammermann, A. A modified TurboID approach identifies tissue-specific centriolar components in C. elegans. PLoS Genet. 18, e1010150 (2022).
Xiong, Z. et al. In vivo proteomic mapping through GFP-directed proximity-dependent biotin labelling in zebrafish. eLife 10, e64631 (2021).
Maguire, J. E. et al. Myristoylated CIL-7 regulates ciliary extracellular vesicle biogenesis. Mol. Biol. Cell 26, 2823–2832 (2015).
Yi, P., Li, W.-J., Dong, M.-Q. & Ou, G. Dynein-driven retrograde intraflagellar transport is triphasic in C. elegans sensory cilia. Curr. Biol. 27, 1448–1461.e7 (2017).
Prelich, G. Gene overexpression: uses, mechanisms, and interpretation. Genetics 190, 841–854 (2012).
Dixson, A., Dawson, T. R., Di Vizio, D. & Weaver, A. M. Context-specific regulation of extracellular vesicle biogenesis and cargo selection. Nat. Rev. Mol. Cell Biol. 24, 454–476 (2023).
Park, H. H. Structure of TRAF family: current understanding of receptor recognition. Front. Immunol. 9, 1999 (2018).
Das, A. et al. The structure and ubiquitin binding properties of TRAF RING heterodimers. J. Mol. Biol. 433, 166844 (2021).
Yin, Q. et al. E2 interaction and dimerization in the crystal structure of TRAF6. Nat. Struct. Mol. Biol. 16, 658–666 (2009).
Das, A., Foglizzo, M., Padala, P., Zhu, J. & Day, C. L. TRAF trimers form immune signalling networks via RING ___domain dimerization. FEBS Lett. 597, 1213–1224 (2023).
Raykov, L., Mottet, M., Nitschke, J. & Soldati, T. A TRAF-like E3 ubiquitin ligase TrafE coordinates ESCRT and autophagy in endolysosomal damage response and cell-autonomous immunity to Mycobacterium marinum. Elife 12, e85727 (2023).
Portman, D. S. & Emmons, S. W. Identification of C. elegans sensory ray genes using whole-genome expression profiling. Dev. Biol. 270, 499–512 (2004).
Susoy, V. et al. Natural sensory context drives diverse brain-wide activity during C. elegans mating. Cell 184, 5122–5137.e17 (2021).
Miller, R. M. & Portman, D. S. A latent capacity of the C. elegans polycystins to disrupt sensory transduction is repressed by the single-pass ciliary membrane protein CWP-5. Dis. Model Mech. 3, 441–450 (2010).
Ding, H., Li, L. X., Harris, P. C., Yang, J. & Li, X. Extracellular vesicles and exosomes generated from cystic renal epithelial cells promote cyst growth in autosomal dominant polycystic kidney disease. Nat. Commun. 12, 4548 (2021).
Fencl, F. et al. Genotype-phenotype correlation in children with autosomal dominant polycystic kidney disease. Pediatr. Nephrol. 24, 983–989 (2009).
Ha, K. et al. The heteromeric PC-1/PC-2 polycystin complex is activated by the PC-1 N-terminus. Elife 9, e60684 (2020).
Li, W. et al. A genetic screen in Drosophila reveals an unexpected role for the KIP1 ubiquitination-promoting complex in male fertility. PLoS Genet. 16, e1009217 (2020).
Naito, A. et al. TRAF6-deficient mice display hypohidrotic ectodermal dysplasia. Proc. Natl. Acad. Sci. 99, 8766–8771 (2002).
Naito, A. et al. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells 4, 353–362 (1999).
Lomaga, M. A. et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 13, 1015–1024 (1999).
Lomaga, M. A. et al. Tumor necrosis factor receptor-associated factor 6 (TRAF6) deficiency results in exencephaly and is required for apoptosis within the developing CNS. J. Neurosci. 20, 7384–7393 (2000).
Peschel, N. et al. Molecular pathway-based classification of ectodermal dysplasias: first five-yearly update. Genes 13, 2327 (2022).
Kantaputra, P. et al. Dental anomalies in ciliopathies: lessons from patients with BBS2, BBS7, and EVC2 mutations. Genes 14, 84 (2022).
Handa, A., Voss, U., Hammarsjö, A., Grigelioniene, G. & Nishimura, G. Skeletal ciliopathies: a pattern recognition approach. Jpn J. Radio. 38, 193–206 (2020).
Reimer, A., He, Y. & Has, C. Update on genetic conditions affecting the skin and the kidneys. Front Pediatr. 6, 43 (2018).
David, A. et al. Isolated and syndromic brachydactylies: Diagnostic value of hand X-rays. Diagn. Interv. Imaging 96, 443–448 (2015).
Katsanis, N. Ciliary proteins and exencephaly. Nat. Genet. 38, 135–136 (2006).
Mishra-Gorur, K. et al. Pleiotropic role of TRAF7 in skull-base meningiomas and congenital heart disease. Proc. Natl. Acad. Sci. USA 120, e2214997120 (2023).
Jouret, F. & Devuyst, O. Targeting chloride transport in autosomal dominant polycystic kidney disease. Cell. Signal. 73, 109703 (2020).
Sudarikova, A. V., Vasileva, V. Y., Sultanova, R. F. & Ilatovskaya, D. V. Recent advances in understanding ion transport mechanisms in polycystic kidney disease. Clin. Sci. 135, 2521–2540 (2021).
Link, C. D., Silverman, M. A., Breen, M., Watt, K. E. & Dames, S. A. Characterization of Caenorhabditis elegans lectin-binding mutants. Genetics 131, 867–881 (1992).
Granato, M., Schnabel, H. & Schnabel, R. pha-1, a selectable marker for gene transfer in C. elegans. Nucleic Acids Res. 22, 1762–1763 (1994).
Evans, T. Transformation and microinjection. WormBook https://doi.org/10.1895/wormbook.1.108.1 (2006).
Dokshin, G. A., Ghanta, K. S., Piscopo, K. M. & Mello, C. C. Robust genome editing with short single-stranded and long, partially single-stranded DNA donors in Caenorhabditis elegans. Genetics 210, 781–787 (2018).
Wu, X. & Hammer, J. A. ZEISS Airyscan: optimizing usage for fast, gentle, super-resolution imaging. Methods Mol. Biol. 2304, 111–130 (2021).
Nikonorova, I. & Barr, M. M. Polycystins recruit cargo to distinct ciliary extracellular vesicle subtypes. https://doi.org/10.6084/m9.figshare.27638175.v1 (2025).
Davis, P. et al. WormBase in 2022—data, processes, and tools for analyzing Caenorhabditis elegans. Genetics 220, iyac003 (2022).
Acknowledgements
We are grateful to Haiyan Zheng and David Sleat for assistance with the mass spectrometry analysis; Gloria Androwski and Helen Ushakov for outstanding technical assistance; Premal Shah and John Favate for sharing and maintenance of the BioComp fractionator that was instrumental in this work, Laura Bianchi, Paul DeCaen, Monica Driscoll, Joel Rosenbaum, and Erich Schwarz for discussions; members of the Rutgers C. elegans community for thought-provoking questions. We also thank WormBase74 and WormBook which were used daily during this project. The work was funded by grants from the National Institutes of Health (NIH) DK116606, DK059418, and NS120745 to M.M.B.; NIH K12 GM093854 INSPIRE (IRACDA NJ/NY) postdoctoral fellowships to K.C.J, PKD Foundation fellowship to J.W., and intramural pilot Rutgers Core Facility grant to I.A.N. Protein identification was performed in the Biological Mass Spectrometry Facility of Robert Wood Johnson Medical School at Rutgers University supported by the NIH instrumentation grants S10OD025140 and S10OD016400 with intramural grants to I.A.N. Some strains were provided by the Caenorhabditis Genetics Center (CGC), which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). Some mutants were obtained at the National Bioresource Project of the Tokyo Women’s Medical University School of Medicine, Japan. The Center for C. elegans Anatomy and WormAtlas provided valuable anatomical and ultrastructural resources (R24OD010943).
Author information
Authors and Affiliations
Contributions
Conceptualization, I.A.N. and M.M.B.; methodology, I.A.N., J.W., J.D.W, M.M.B.; investigation, I.A.N., E.D., K.C.J, J.S.; visualization, I.A.N.; funding acquisition, M.M.B., I.A.N.; project administration, I.A.N., M.M.B.; supervision, I.A.N., J.W., M.M.B.; writing—original draft, I.A.N.; writing—review & editing, I.A.N., K.C.J., J.W., and M.M.B.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Frederik Verweij who co-reviewed with Bárbara Adem and Jelle van den Bor; Guangshuo Ou, Michał Turek, Christopher Ward and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nikonorova, I.A., desRanleau, E., Jacobs, K.C. et al. Polycystins recruit cargo to distinct ciliary extracellular vesicle subtypes in C. elegans. Nat Commun 16, 2899 (2025). https://doi.org/10.1038/s41467-025-57512-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57512-3
