
Abstract
Scavenger receptor class A member 3 (SR-A3) is implicated in metabolic diseases; however, the relationship between SR-A3 and metabolic dysfunction-associated fatty liver disease (MAFLD) has not been documented. Here, we show that hepatic SR-A3 expression is significantly reduced in human and animal models in the context of MAFLD. Genetic inhibition of SR-A3 in hamsters elicits hyperlipidemia, hyperglycemia, insulin resistance, and hepatic steatosis under chow-diet condition, yet escalates in diet-induced MAFLD. Mechanistically, SR-A3 ablation enhances E3 ligase XIAP-mediated proteasomal ubiquitination of PTEN, leading to AKT hyperactivation. By contrast, hepatic overexpression of human SR-A3 is sufficient to attenuate metabolic disorders in WT hamsters fed a high-fat-high-cholesterol diet and ob/ob mice via suppressing the XIAP/PTEN/AKT axis. In parallel, pharmacological intervention by PTEN agonist oroxin B or lipid lowering agent ezetimibe differentially corrects MAFLD in hamsters.
Similar content being viewed by others


Introduction
Metabolic dysfunction-associated fatty liver disease (MAFLD)1, which is characterized by excess lipid accumulation in the liver, is the most common chronic liver disease, with an increasing prevalence accounting for nearly 30% of the general population2. MAFLD is a hepatic manifestation of metabolic syndrome, including elevated plasma triglycerides, low high density lipoprotein cholesterol (HDL-C), hyperglycemia or high blood pressure. It encompasses a wide spectrum of diseases, ranging from simple non-alcoholic fatty liver (MAFL) to metabolic dysfunction-associated steatohepatitis (MASH), then progressing to worsening phenotypes of fibrosis, cirrhosis, and eventually hepatocellular carcinoma (HCC). Although a thyroid hormone receptor agonist has recently been approved by FDA for the treatment of MASH, developing new drugs has become an urgent task to solve the MAFLD issue, afflicting a large population over the world due to the heterogeneity and complexity of MAFLD-related etiologies and pathologies.
Scavenger receptor class A (SR-A) consists of 5 members with ubiquitous expression, which is originally discovered for their capacity of binding with modified low density lipoprotein (LDL) at the cell membrane to regulate lipid and tissue homeostasis3. Unlike other SR-A family members, SR-A member 3 (SR-A3) lacking the globular binding ___domain of scavenger receptor cysteine-rich (SRCR) mainly localizes at the endoplasmic reticulum (ER) and Golgi apparatus, which is responsible for scavenging excess reactive oxygen species (ROS)4,5. SR-A3 has been implicated in multiple biological activities, including oxidative stress6, human malignancies7,8,9,10,11, viral infections12,13, and Alzheimer’s disease14. Recently, emerging evidence demonstrates that SR-A3 may play a crucial role in metabolic disorders, such as type 2 diabetes (T2D) and obesity because both an increase in DNA methylation of SR-A3 in patients with T2D and a reduction of SR-A3 protein levels in the white adipose tissues of genetically obese and diabetic mice have been observed15. Given that the ER and Golgi apparatus are two key organelles regulating cellular lipid metabolism16,17 and ROS generation, and that T2D and obesity are important risk factors of MAFLD, we thus proposed that SR-A3 might also play a role in MAFLD development. However, the relationship between SR-A3 and MAFLD has not been well established and how SR-A3 governs MAFLD progression remains elusive. Since Syrian golden hamsters and humans share the analogous metabolic traits18, we sought to investigate the influence of SR-A3 on MAFLD using hamsters.
Here, we use SR-A3-deficient (SR-A3−/−) hamsters constructed by CRISPR/Cas9 gene editing, hepatic overexpression of human SR-A3 (hSR-A3) in hamsters and ob/ob mice to further explore the molecular mechanism by which SR-A3 controls MAFLD development. We identify decreased hepatic SR-A3 expression in both MAFLD patients and various animal models of MAFLD. SR-A3 deletion enhances serine/threonine kinase AKT activation to induce hepatic steatosis in male hamsters under the chow diet (CD) condition and further exacerbates diet-induced MAFLD by promoting E3 ligase X-linked inhibitor of apoptosis (XIAP)-mediated phosphatase and tensin homolog (PTEN) ubiquitination and degradation. Conversely, hepatic overexpression of hSR-A3 by adeno-associated virus 8 (AAV8) is sufficient to abrogate fatty liver in high-fat-high-cholesterol diet (HFHCD)-fed wild type (WT) hamsters and CD-fed ob/ob mice. Furthermore, the pharmacological intervention of PTEN agonist oroxin B (OB) and lipid lowering agent ezetimibe possessing AKT inhibition property, both alleviate diet-induced MAFLD phenotypes in hamsters through the XIAP/PTEN/AKT axis. Thus, our results unravel SR-A3 as an important modulator of hepatic energy metabolism and confer a potential therapeutic target against MAFLD.
Results
Hepatic SR-A3 expression level is decreased in patients and rodent animals with MAFLD
To evaluate the expression of hepatic SR-A3 in MAFLD patients, we analyzed a publicly available dataset (GSE126848) comprising 12 normal controls (NC), 15 MAFL patients, and 16 MASH patients from the gene expression omnibus (GEO) database (Fig. 1a). Compared with NC, hepatic Sr-a3 mRNA levels were significantly lower in patients with MAFL and MASH (Fig. 1b, c), while no significant difference was observed between patients with MAFL and MASH (Fig. 1d). The following correlation analysis also suggested that SR-A3 was closely involved in glucose metabolism, lipid metabolism, inflammation, and fibrosis (Fig. 1e). Likewise, decreased mRNA and protein levels of SR-A3 was detected in the livers of MAFLD patients verified by liver biopsy results relative to normal individuals (Fig. 1f–h), which may be attributable to the fact that SR-A3 DNA was hypermethylated in MAFLD patients compared with normal healthy controls (Fig. 1i). Furthermore, hepatic Sr-a3 mRNA expression was consistently reduced in high-fat-diet (HFD)-fed mice and ob/ob mice (Fig. 1j). Similar results were also found in hamsters challenged by a HFHCD or a methionine-choline-deficient diet (MCDD) for 8 weeks, respectively (Fig. 1j). Moreover, the mRNA level of Sr-a3 was reduced in HepG2 cells treated with oleic acid (OA) and palmitic acid (PA) (Fig. 1k). Collectively, these results imply the inverse correlation between hepatic SR-A3 expression level and MAFLD development, suggesting the potential protective effect of hepatic SR-A3 against MAFLD.

a Principal component analysis (PCA) plot of liver samples of normal controls (NC, n = 12) and patients with metabolic dysfunction-associated fatty liver (MAFL, n = 15) and metabolic dysfunction-associated steatohepatitis (MASH, n = 16) from a GEO dataset (GSE126848). b–d Volcano plots of hepatic gene expressions in MAFL vs NC (b), NASH, MASH vs NC (c), and MASH vs MAFL from the same dataset in (a). e Correlation analysis of Sr-a3 expression related to glucose metabolism, lipid metabolism, inflammation, and fibrosis from the same dataset in (a). f Representative images of HE, oil red O and immunohistochemistry (IHC) staining of human SR-A3 (hSR-A3) in liver tissues from normal individuals as NC and metabolic dysfunction-associated fatty liver disease (MAFLD) patients (HE: NC, n = 3; MAFLD, n = 7; oil red O: NC, n = 3; MAFLD, n = 7; IHC: NC, n = 5; MAFLD, n = 19). Scale bars, 60 μm for HE and IHC, 100 μm for oil red O. g IHC staining quantification (average optical density, AOD) from (f). h The relative mRNA expression levels of Sr-a3 in liver tissues from the same individuals in (f). i Methylation levels was detected in the gene body (cg00839333) and promoter (cg11046502) sites of SR-A3 DNA in normal healthy controls (NC, n = 106) and MAFLD patients (n = 87) based on six independent GEO datasets (GSE48325, GSE49542, GSE61258, GSE61278, GSE65057, and GSE69852). j Relative mRNA expression levels of Sr-a3 in the livers of WT and ob/ob mice with indicated diets for 8 weeks (WT/CD: n = 3; WT/HFD: n = 6; ob/ob: n = 5); relative mRNA expression levels in the livers of WT hamsters fed with a chow diet (CD), a high-fat-high-cholesterol diet (HFHCD), and a methionine-choline-deficient diet (MCDD) for 8 weeks, respectively (WT/CD: n = 7; WT/HFHCD: n = 7; ob/ob: n = 8). k Relative mRNA expression of Sr-a3 in HepG2 cells treated with 1% bovine serum albumin (BSA) and 1 mM oleic acid (OA) for 24 h (left) and 0.2% bovine serum albumin (BSA) and 200 μM palmitic acid (PA) for 24 h (right) from 6 independent experiments. Data were presented as mean ± SEM. Statistical significance was determined by likelihood ratio test (b–d), unpaired two-sided Student’s t-test (g, k) or Mann Whiney test (h, i) or one-way ANOVA (j) with Bonferroni multiple-comparison analysis. Source data are provided as a Source Data file.
Genetic deletion of SR-A3 induces hypertriglyceridemia, hyperglycemia, and hepatic steatosis in hamsters on a chow diet
To further investigate how SR-A3 participates in MAFLD, we utilized CRISPR/Cas9 gene editing to target the exon 2 of hamster Sr-a3 with a deletion of 26 base pairs (bps), leading to a reduction in Sr-a3 mRNA expression by 97% and undetectable protein level (Fig. S1a–d). Under chow diet (CD) condition, 20 week-old male SR-A3-deficient (SR-A3−/−) hamsters exhibited a notable increase in plasma triglyceride (TG), total cholesterol (TC), and non-esterified fatty acid (NEFA) without affecting body weight (Fig. 2a–c). However, female SR-A3−/− hamsters only displayed higher plasma TG (Fig. S2a–c). Furthermore, to clarify the lipoprotein profile of male hamsters, we performed the fast protein liquid chromatography (FPLC) and found a marked increase in the contents of TG carried on very low-density lipoprotein (VLDL) or chylomicron (CM) particles accompanied by elevated low density lipoprotein cholesterol (LDL-C) in male SR-A3−/− hamsters (Fig. 2d). Meanwhile, we found no significant changes in lipoprotein lipase (LPL) activity and hepatic VLDL secretion between the two genotypes (Fig. 2e, f), while plasma TG was steadily elevated and remained higher in the SR-A3−/− group than their WT counterparts throughout the 8-h experimental course in the oral fat load test (Fig. 2g, Fig. S2d). Additionally, more neutral lipids were also observed in the jejunum of the male SR-A3−/− hamsters (Fig. 2h), indicating a cooccurrence of increased intestinal lipid absorption and retention that synergistically contribute to higher plasma TG levels in the SR-A3−/− hamsters. Next, we examined SR-A3’s role in glucose metabolism and found that male SR-A3−/− hamsters showed higher fasting glycemia compared to the WT group under CD, whereas no significant changes were detected in female hamsters (Fig. 2i, Fig. S2e). In alignment, plasma insulin level was higher in the male SR-A3−/− hamsters 2 h after the initial glucose challenge (Fig. 2j). We also observed impaired glucose tolerance and insulin resistance (IR) in both male and female SR-A3−/− hamsters relative to their respective WT controls (Fig. 2k, l, Fig. S2f, g). Together, these results indicate that SR-A3 deficiency impairs glucose tolerance and insulin sensitivity in a sex-independent manner.

a Schematic representation of 12 week-old male WT and SR-A3−/− hamsters fed a CD for 8 weeks and then fasted for 16 h before sacrifice. b, c Body weight (b), plasma triglyceride (TG), total cholesterol (TC), and non-esterified fatty acid (NEFA) (c) (n = 7/group). d Plasma lipoprotein profile analysis by fast protein liquid chromatography (FPLC) (n = 7/group). e Determination of plasma lipoprotein lipase (LPL) activity (n = 7/group). f Analysis of very-low-density lipoprotein (VLDL) secretion rates (n = 7/group). g Determination of plasma TG levels after olive oil gavage (n = 6/group). h Oil red O staining of jejunal epithelium after olive oil gavage. (WT: n = 5; SR-A3−/−: n = 7). Scale bars, 200 μm. i Plasma glucose level at sacrifice (n = 7/group). j Plasma insulin level was measured 2 h after glucose i.p. injection (2 g/kg) (n = 5/group). k Glucose tolerance test (GTT) and area under curve (AUC) quantification (n = 6/group). l Insulin tolerance test (ITT) and AUC analysis (n = 6/group). m Representative images of gross morphology, Hematoxylin and Eosin (HE), oil red O, and sirius red staining of the livers at sacrifice (n = 7 /group). Scale bars, 100 μm for HE and oil red O, 200 μm for sirius red. n The ratio of liver to body weight (BW), and quantifications of oil red O and sirius red staining (n = 7/group). o Plasma ALT and AST levels (n = 7/group). p Liver TG and TC content (n = 7/group). q PCA analysis of liver lipidome from (a) (n = 3/group). r Three significantly increased lipid species in the livers analyzed by targeted lipidome (WT: n = 4; SR-A3−/−: n = 3). s, t Top 10 differentially changed species in liver triglyceride (TAG), diglyceride (DAG), free fatty acid (FFA) (n = 3/group). u Relative expression levels of genes in livers from (a) (WT: n = 7; SR-A3−/−: n = 8). Data were presented as the mean ± SEM. Statistical significance was determined by unpaired two-sided Student’s t-test (b, c-middle/right, i–k-right, n-middle, o-right, p-right, u), or two-sided Mann–Whitney test (c-left, e, l-right, n-left/right, o-left, p-left) or two-way ANOVA (f–k-left, l-left) with Bonferroni multiple-comparison analysis. Source data are provided as a Source Data file.
In agreement with the findings described above, male SR-A3−/− hamsters exhibited increased the ratio of liver to body weight, lipid accumulation, and hepatic TG content (Fig. 2m–p), but no obvious lipid accumulation and fibrosis in livers were observed in female groups (Fig. S2h, i). To further determine the changes in lipid species associated with these metabolic phenotypes caused by SR-A3 deletion, we then performed a targeted lipidome from liver tissues of CD-fed male WT and SR-A3−/− hamsters. The principal component analysis (PCA) showed a distinct separation of these two groups (Fig. 2o). Triglyceride (TAG), diglyceride (DAG), and free fatty acid (FFA) were the significantly increased lipid species in the livers of male SR-A3−/− hamsters compared to WT controls (Fig. 2r–t). Consistently, targeting SR-A3 in male hamsters promoted hepatic de novo lipogenesis (DNL) and dampened fatty acid oxidation (FAO), but did not affect fatty acid uptake, and VLDL secretion (Fig. 2u). Thus, these data demonstrate that genetic deletion of SR-A3 in male hamsters results in hepatic steatosis by the combined action of increased DNL and decreased FAO.
Inactivation of SR-A3 aggravates HFHCD-induced IR, obesity and MAFLD
To explore the influence of SR-A3 on diet-induced MAFLD, we then subjected 12 week-old WT and SR-A3−/− hamsters of both sexes on HFHCD for 8 weeks (Fig. 3a and Fig. S3a). Surprisingly, only male SR-A3−/− hamsters had a significant increase in body weight, whole-body fat mass, and the ratio of epididymal white adipose tissue (eWAT) and inguinal white adipose tissue (iWAT) mass to body weight (Figs. 3b–e, S3b, 4a, 4f). As expected, the Hematoxylin and Eosin (HE) staining of eWAT and iWAT revealed adipocyte hypertrophy, characterized by an elevation in adipocyte size and a remarkable shift of adipocyte area distribution to larger sizes (Figs. S4b, c, S4g, h), with more CD68 signals and lower Ucp1 mRNA expression (Fig. S4d, e, S4i), suggesting increased macrophage infiltration and reduced browning in eWAT in male SR-A3−/− hamsters.

a Experimental design. Male WT and SR-A3−/− hamsters at the age of 12 weeks were fed an HFHCD for 8 weeks (n = 7/group) and then fasted for 16 h before sacrifice. b, c Gross view (b) and body weight (c) of hamsters at sacrifice (n = 7/group). d, e The whole-body fat mass measured by magnetic resonance imaging (MRI, d) and corresponding quantification (e) (n = 5/group). f, g Representative images of plasma samples (f), plasma TG and TC (g) from indicated hamsters at sacrifice (n = 7/group). h Plasma lipoprotein profiles analyzed by FPLC (n = 7/group). i–k Plasma NEFA (i), glucose (j), and insulin (k) levels from indicated hamsters at sacrifice (n = 7/group). l GTT and AUC quantification was determined (WT: n = 7; SR-A3−/−: n = 8). m ITT and AUC analysis (WT: n = 8; SR-A3−/−: n = 9). n Representative images of gross morphology, HE, oil red O, BODIPY staining, and electron microscopy (EM) of liver sections from indicated hamsters in (a) (n = 7/group). LD in EM indicates lipid droplet; scale bars, 100 μm for HE, oil red O and BODIPY staining, and 2 μm for EM. o The ratio of liver/BW (n = 7/group) and the quantification of oil red O staining (n = 7/group) and BODIPY staining (WT: n = 6; SR-A3−/−: n = 7). BW indicates body weight. p Liver TG and TC content of indicated hamsters at sacrifice (n = 7/group). q Plasma ALT (n = 7/group) and AST levels (WT: n = 8; SR-A3−/−: n = 7). r The hepatic expression levels of genes in livers from (a) (n = 7/group). s, t Representative images and quantifications of sirius red (s) (n = 7/group) and immunofluorescent staining of CD68 (red) (t) in livers (WT: n = 7; SR-A3−/−: n = 6). Scale bars, 100 μm. White arrows indicate CD68 positive area. u The expression levels of genes in livers at sacrifice (n = 7/group). Data were presented as the mean ± SEM. Statistical significance was determined by unpaired two-sided Student’s t-test (c–l-right, (o–u), or two-sided Mann–Whitney test (k, m-right) or two-way ANOVA (l-left, m-left) with Bonferroni multiple-comparison analysis. Source data are provided as a Source Data file.
As shown in Fig. 3f, we observed milky plasma in male SR-A3−/− hamsters, corresponding to a significant increase in plasma TG, TC, and NEFA levels compared to their WT controls (Fig. 3g, i). Consistent with the findings shown in CD-fed animals, the contents of both TG and TC distributed in VLDL and CM particles were profoundly increased, accompanied by a modest elevation of lipids in LDL fractions (Fig. 3h). However, the HFHCD challenge did not affect plasma TG, TC, and NEFA levels in female hamsters between the two genotypes (Fig. S3c). Furthermore, male SR-A3−/− hamsters fasted for 16 h showed significant hyperglycemia and hyperinsulinemia as well as exacerbated glucose intolerance and IR relative to controls (Fig. 3j–m). To explore the underlying mechanism by which SR-A3 regulates glucose metabolism, we analyzed gene expression profiling using RNA-sequencing in the livers of male WT and SR-A3−/− hamsters HFHCD-fed for 8 weeks. A total of 219 differential expression genes (DEGs) were measured (Fig. S5a). We found that upregulated DEGs were predominantly clustered on carbohydrate metabolism in the livers of SR-A3−/− by Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis, among which glycolysis and gluconeogenesis were ranked as the top strikingly changed pathways (Figs. S5b, 5c). These results were further validated by qPCR, showing that the expression levels of genes regulating gluconeogenesis and glycolysis were significantly increased in the livers of male SR-A3−/− hamsters relative to WT animals (Fig. S5d). Although female SR-A3−/− hamsters had hyperglycemia, glucose tolerance was not greatly altered, but insulin sensitivity was still impaired to some extent (Fig. S3d–f).
When fed an HFHCD, male SR-A3−/− hamsters had higher ratio of liver to body weight, elevated liver TG and TC content, aggravated hepatic steatosis as well as larger lipid droplets compared to WT hamsters, as shown in Fig. 3n–p. In agreement, male SR-A3−/− hamsters showed significantly increased Srebp1c-mediated DNL and VLDL secretion in the liver without affecting fatty acid uptake and FAO (Fig. 3r). Furthermore, plasma levels of ALT and AST, which are indicative of liver injury, were elevated in these hamsters in comparison to WT controls (Fig. 3q). Additionally, the livers of male SR-A3−/− hamsters exhibited enhanced inflammatory and fibrotic responses, as evidenced by increased clustering of the inflammatory marker CD68/F4/80 (pan-macrophage), no significant difference in CD206 (M2 marker), and significantly elevated expression of genes associated with inflammation and fibrosis (Fig. 3s–u, Fig. S6a, b). In contrast, female hamsters did not display overt changes in hepatic lipid accumulation and fibrosis (Fig. S3g, h). Additionally, a notable elevation in TUNEL-positive cells was discerned in the livers of SR-A3 male hamsters relative to WT controls, indicating that SR-A3 deficiency may contribute to an augmented incidence of apoptotic cells in the liver (Fig. S6c). Taken together, these findings demonstrate that genetic inactivation of SR-A3 promotes metabolic syndrome, including obesity, combined hyperlipidemia, worsening glucose intolerance and IR, which, in turn, contributes to MAFLD only in male hamsters under HFHCD condition.
SR-A3 ablation exacerbates diet-induced MASH
To assess whether SR-A3 ablation could exacerbate the progression of MASH and fibrosis, hamsters were given a prolonged HFHCD or a MCD challenge. Initially, we fed 12 week-old male WT and SR-A3−/− hamsters with HFHCD for 16 weeks (Fig. S7a). SR-A3−/− hamsters exhibited elevated body weight, the ratio of eWAT weight to body weight, and the ratio of liver weight to body weight (Fig. S7b). Despite no changes in plasma TG and TC, fasting glucose level and plasma ALT and AST contents were significantly increased in SR-A3−/− hamsters (Fig. S7c–f), accompanied by more liver TG accumulation, severe hepatic steatosis, inflammation, and fibrosis when compared to the control group (Fig. S7g–m). We then challenged 12 week-old male WT and SR-A3−/− hamsters with MCDD for 8 weeks (Fig. S8a). In agreement with the observations in mice19,20, MCDD also caused significant body weight loss in two groups to the same extent (Fig. S8b). Additionally, plasma TG and TC levels were identical between the two genotypes, while SR-A3−/− hamsters had higher plasma NEFA concentration than WT hamsters (Fig. S8c). Furthermore, no changes in fasting glucose level and glucose intolerance were found, but insulin sensitivity was impaired in SR-A3−/− hamsters (Fig. S8d–f). Despite no changes in the ratio of liver to body weight, SR-A3−/− hamsters exhibited more severe hepatic steatosis and fibrosis accompanied by elevated plasma AST, increased hepatic TAG, DAG, FFA and TC (Fig. S8g–n), corresponding to elevated Srebp1c-mediated DNL, unaltered fatty acid uptake, FAO and VLDL secretion (Fig. S8r). Immunofluorescence staining demonstrated a notable increase in pan-macrophage infiltration and apoptotic cell numbers within the livers of SR-A3−/− hamsters, accompanied by a significant reduction in anti-inflammatory M2 macrophages (Fig. S8o–q). Altogether, these data imply that SR-A3 deletion escalates lipid accumulation and MASH upon various diet intervention.
SR-A3 maintains lipid homeostasis by suppressing XIAP-mediated PTEN ubiquitination and AKT phosphorylation
To explore how SR-A3 regulates glucose and lipid metabolism, we performed RNA-sequencing from the livers of male WT and SR-A3−/− hamsters on CD or HFHCD for 8 weeks. A total of 1169 and 2570 DEGs were measured, of which 329 overlapped DEGs (Fig. 4a) were used as input for further KEGG analysis. Among the top 15 pathways in RNA-sequencing, PI3K-AKT signaling pathway was significantly enriched that is closely involved in both glucose and lipid metabolism (Fig. 4b). Gene Set Enrichment Analysis (GSEA) from the same input of CD-fed hamsters as shown in Fig. 4a further confirmed that PI3K-AKT-MTOR pathway was critically upregulated in the context of SR-A3 deficiency (Fig. 4c). Next, to further demonstrate the relationship between SR-A3 and AKT signaling, we conducted a protein-protein-interaction (PPI) network analysis based on STRING and found potential intermediate linkers of XIAP/PTEN between SR-A3 and AKT (Fig. 4d). In our in vivo models, SR-A3−/− hamsters exhibited elevated levels of XIAP, reduced PTEN, and heightened phosphorylation of AKT (p-AKT) (Fig. 4e, f). This was accompanied by increased expression of AKT-mediated downstream lipogenic proteins such as mTOR and its phosphorylated form (p-mTOR), as well as the precursor and the mature nuclear form of SREBP-1c (p/n-SREBP-1c) (Fig. 4e, f).

a Venn diagram of hepatic differentially expressed genes (DEGs) from RNA sequencing in 12 week-old male WT and SR-A3−/− hamsters fed a CD (n = 3/group) or HFHCD for 8 weeks (n = 5/group). b Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis based on the overlapped DEGs in (a). c Gene Set Enrichment Analysis (GSEA) analysis from the same CD-fed hamsters in (a). d Protein-protein interaction (PPI) network analysis based on STRING and genes involved in indicated pathways. e, f Immunoblot (IB) analysis of indicated proteins in livers from 20 week-old CD- (e) or HFHCD-fed (f) male WT and SR-A3−/− hamsters (n = 4/group). Relative normalized protein ratios (SR-A3−/−/WT) as fold change and p-values are shown on the right. g–j Oil red O staining (g, i) and IB analysis of indicated proteins (h, j) in HepG2 cells transfected with indicated vectors for 24 h followed by 0.2% BSA or 200 μM PA incubation for another 24 h in three independent experiments. Scale bars, 200 μm. k-l siSR-A3- (k) or hSR-A3 -transfected (l) HepG2 cells were treated with or without MG132 (10 μM) and cycloheximide (CHX, 50 μg/ml), and IB analysis of proteins at indicated timepoints in three independent experiments. m, n IB analysis of input and immunoprecipitation (IP) analysis using anti-PTEN antibody in HepG2 cells. o-p IB analysis of input and anti-FLAG (o) and anti-PTEN (p) immunoprecipitates from HepG2 cells. q, r Oil red O staining (q) and IB analysis of indicated proteins (r) in HepG2 cells transfected with indicated vectors for 24 h, followed by incubation with 200 μM PA for 24 h in three independent experiments. Scale bars, 200 μm. s, t Oil red O staining (s) and IB analysis of indicated proteins (t) in HepG2 cells transfected siSR-A3 or NC siRNA for 6 h, followed by treatment with oroxin B (OB, 200 mg/ml) for 12 h and incubation with 200 μM PA for another 24 h in three independent experiments. Scale bars, 200 μm. Data were expressed as the mean ± SEM. Significance was determined by unpaired two-sided Student’s t-test (e, f). Source data are provided as a Source Data file.
Next, to further clarify the molecular mechanism by which SR-A3 alleviated lipogenesis, HepG2 cells were transfected with vectors expressing small interfering SR-A3 (siSR-A3) or human SR-A3 (hSR-A3) to knock down or overexpress SR-A3, respectively (Fig. S9a, b). SR-A3 silencing significantly increased lipid accumulation (Fig. 4g), while hSR-A3 overexpression decreased lipid content in HepG2 cells treated with PA (Fig. 4i). Despite no overt differences in mTOR and p-SREBP-1c, similar changes of increased XIAP, decreased PTEN, enhanced p-AKT and p-mTOR, and n-SREBP-1c as reported in vivo models were also observed in siSR-A3 transfected HepG2 cells in the absence or presence of PA versus control groups (Fig. 4h). Conversely, hSR-A3 overexpression reversed the above changes in protein levels caused by SR-A3 silencing (Fig. 4j). To further explore the mechanism by which SR-A3 modulates the regulation of XIAP, we have conducted in vitro experiments by performing cycloheximide (CHX) assay and observed that silencing SR-A3 led to a longer half-life of XIAP (Fig. S10a), whereas overexpression of SR-A3 significantly accelerated the degradation of XIAP (Fig. S10b). Our results indicated that SR-A3 mediated the proteasomal degradation of XIAP protein.
Of note, the mRNA level of Pten was unaffected by SR-A3 ablation in vivo (Fig. S11a, b), while PTEN protein degradation promoted by SR-A3 deficiency was validated both in vivo and in vitro, and vice versa, indicating a post-transcriptional regulation of PTEN by SR-A3 (Fig. 4e, f, h, and j). To further test this possibility, we then silenced or overexpressed SR-A3 in HepG2 cells with the treatment of proteasome inhibitor MG132 in the presence of protein synthesis inhibitor cycloheximide (CHX) in a time course, and these results suggested that SR-A3 could stabilize PTEN via blocking proteasome-mediated PTEN degradation (Fig. 4k, l). Next, we explored how SR-A3 orchestrated the process of PTEN ubiquitination. As shown in Fig. 4m and n, PTEN ubiquitination was significantly strengthened by SR-A3 silencing but weakened by SR-A3 overexpression in HepG2 cells. Given SR-A3’s role as a scavenging receptor without ubiquitinating capacity and based on the prediction by PPI, XIAP, an E3 ligase, might be the potential molecule regulating PTEN ubiquitination in the setting of SR-A3 deficiency. To corroborate the interrelations among SR-A3, XIAP, and PTEN, we conducted Co-immunoprecipitation (Co-IP) assays and found that SR-A3, XIAP, and PTEN exhibited a capacity to directly engage with one another (Fig. 4o, p). After silencing XIAP, HepG2 cells incubated with PA showed less lipid deposition together with the blockade of XIAP-mediated downstream signaling events (PTEN/p-AKT/p-mTOR/n-SREBP-1c) caused by SR-A3 deletion, supporting XIAP as the downstream effector of SR-A3 (Fig. 4q, r). Likewise, PTEN agonist OB could also reduce lipid content in PA-treated HepG2 cells caused by SR-A3 loss, and abrogate PTEN loss-induced AKT hyperactivation, along with decreased AKT phosphorylation cascade response without changing total XIAP/mTOR/AKT/p-SREBP-1c levels (Fig. 4s, t). Hence, our data indicate that SR-A3 protects against hepatic steatosis by inhibiting XIAP-mediated PTEN ubiquitination to block AKT-mediated hepatic lipogenesis.
Hepatic SR-A3 deficiency aggravates hepatic steatosis in HFD-fed mice
In order to ascertain whether the implication of SR-A3 alation in MAFLD development is liver-specific, we attempted to utilize AAV-mediated shRNA to delete Sr-a3 gene from the liver of hamster. However, due to the technical issue, the knockdown efficacy of our AAV-shRNA system was not ideal in hamsters. As a result, we proceeded to construct liver-specific SR-A3 knockout (SR-A3△hep) mice through the Cre-Loxp system. Aged matched male SR-A3F/F and SR-A3△hep mice were fed a high-fat-diet (HFD) for 12 weeks to induce hepatic steatosis (Fig. S12a). It is noteworthy that the absence of hepatic SR-A3 did not result in any discernible impact on body weight, plasma TG levels, or NEFA levels. However, there was a notable elevation in circulating TC levels (Fig. S12b, c). Despite no significant change in fasting glucose levels, SR-A3△hep mice exhibited impaired glucose tolerance and worsened insulin sensitivity (Fig. S12d–f). Moreover, higher ratios of liver to body weight, elevated liver TG content and more pronounced hepatic steatosis was observed in SR-A3△hep mice (Fig. S12g–i). Consistently, hepatic SR-A3 ablation promoted hepatic DNL and fatty acid uptake, but did not influence FAO or VLDL secretion (Fig. S12j). The histological analysis and qPCR results indicated that hepatic SR-A3 deficiency did not influence the severity of inflammation and fibrosis (Fig. S12g, k). In conclusion, these findings suggest that liver-specific deletion of SR-A3 is sufficient to cause lipid metabolism disturbances without signs of exacerbating inflammation or fibrosis in HFD-fed mice. Mechanically, similar to our discoveries in hamsters, the accumulated hepatic lipids in SR-A3△hep mice was probably a consequence of AKT hyperactivation by impairing XIAP/PTEN axis, fueling Srebp1c-induced lipid synthesis (Fig. S12l). Therefore, our data suggests that SR-A3 deletion in hepatocyte pomotes hepatic steatosis in male mice.
AAV8-mediated hepatic overexpression of hSR-A3 mitigates hepatic steatosis in HFHCD-fed hamsters and ob/ob mice
To address the therapeutic potential of hepatic SR-A3, liver-specific overexpression of hSR-A3 by adeno-associated virus 8 (AAV8) was conducted in male WT hamsters, while control hamsters were administered with a null vector (Null AAV8) (Fig. 5a). Two weeks after the virus injection, CD-fed animals were switched to an HFHCD for another 4 weeks (Fig. 5a). To confirm the expression efficiency, we measured the mRNA level of hepatic hSr-a3 and found that hSr-a3 was exponentially expressed in the hSR-A3 AAV8-injected hamsters compared to the Null AAV8-injected hamsters (Fig. 5b). Surprisingly, body weight, the ratio of liver to body weight ratio, the ratio of eWAT to body weight, and adipocyte area of eWAT were not altered in both groups (Fig. S13a–e). Interestingly, overexpression of hSR-A3 led to a marked decrease in plasma TG and NEFA levels, but not plasma TC levels in hamsters (Fig. 5c), which were further confirmed by the FPLC analysis, showing a significant decrease in the contents of TG and TC distributed in VLDL/CM particles, and a modest increase in HDL-C (Fig. 5d). Meanwhile, we found that hSR-A3 AAV8-injected hamsters had lower plasma glucose levels with improved glucose tolerance and IR relative to their controls (Fig. 5e–g). Further, hepatic steatosis but not fibrosis was significantly attenuated in the livers of hSR-A3 AAV8-treated hamsters (Fig. 5h, i, Fig. S14a, b). Consistently, hSR-A3 overexpression indeed blunted Srebp1c-mediated DNL by suppressing XIAP activity to promote PTEN stability, thereby decreasing downstream effectors of p-AKT, p-mTOR, mTOR and p/n-SREBP-1c protein levels, without affecting fatty acid uptake, FAO, and VLDL secretion at the transcriptional level (Fig. 5j, k).

a Scheme of the experiment. 8 week-old WT hamsters were administered with Null adeno-associated virus 8 (AAV8) or hSR-A3 AAV8 via jugular vein and then fed a CD for 2 weeks before switching to an HFHCD for 4 weeks at sacrifice (n = 7/group). b Determination of human Sr-a3 (hSr-a3) mRNA expression in the livers at sacrifice (n = 7/group). c–e Plasma TG, TC, NEFA levels (c), lipoprotein profiles analyzed by FPLC (d) and plasma glucose levels (e) at sacrifice (n = 7/group). f GTT and AUC measurement (n = 5/group). g ITT and AUC analysis (n = 5/group). h Representative images and quantitative analysis of HE and oil red O staining in livers from indicated hamsters in (a) (n = 7/group). Scale bars,100 μm. i Liver TG and TC contents (n = 6/group). j IB analysis of proteins in livers from Null AAV8 and hSR-A3 AAV8 injected hamsters. Densitometric quantification of relative normalized protein ratios were measured (hSR-A3/Null, n = 3/group), and p-values were shown on the right. k The expression levels of genes in livers from the above hamsters in (a) (n = 7/group). l Scheme showing the procedure of CD-fed ob/ob mice administered with hSR-A3 AAV8 or Null AAV8 (n = 5/group). m Validation of hSr-a3 expression in livers from indicated mice in (l). n–p Determination of plasma TG, TC (n), glucose (o), and insulin (p) at sacrifice (n = 5/group). q GTT and AUC analysis (n = 4/group). r ITT and AUC quantification (n = 4/group). s Representative images and quantification of HE and oil red O staining of the livers from the indicated mice in (l) (n = 5/group). Scale bars, 100 μm. t Liver TG and TC levels of livers from ob/ob mice in (l) (n = 5/group). u IB of indicated proteins in livers of ob/ob mice (n = 3/group). Analysis of the relative protein level of hSR-A3/Null ratio and p-values were shown on the right. v The expression levels of genes in livers from the above mice (n = 5/group). Data were presented as the mean ± SEM. Significance was determined by unpaired two-sided Student’s t-test (c, e, f-right, g-right, (h–p) q-right, r-right, (s–v), or two-sided Mann–Whitney test (b, i) or two-way ANOVA (f-left, g-left, q-left, r-left) with Bonferroni multiple-comparison analysis. Source data are provided as a Source Data file.
Given that hepatic SR-A3 levels were significantly reduced in ob/ob mice compared to the lean WT controls (Fig. 1j), we extended our evaluation of the beneficial effect of hSR-A3 overexpression to this genetically obese model with MAFLD. As shown in Fig. 5l, 9 week-old male ob/ob mice were administered hSR-A3 or Null AAV8 and then were maintained on CD for 8 weeks. The hepatic hSr-a3 overexpression efficiency was validated (Fig. 5m). In line with the data obtained from HFHCD-fed WT hamsters, hSR-A3 overexpression did not change body weight, liver/body weight ratio, and eWAT/body weight (Fig. S13f–h). The levels of plasma TG and TC were unchanged between the two groups, which is inconsistent with our findings in hamsters (Fig. 5c, n). In addition, hSR-A3 AAV8-injected ob/ob mice had a significant reduction in plasma glucose levels and a reduced trend in plasma insulin levels versus the Null AAV8-injected controls (Fig. 5o, p). Correspondingly, glucose intolerance and IR were alleviated (Fig. 5q, r), further favoring the key role of hSR-A3 in maintaining glucose homeostasis. Moreover, hSR-A3 overexpression attenuated hepatic lipid accumulation but not fibrosis in ob/ob mice (Fig. 5s, t, Fig. S14c, d). Also, its downstream XIAP/PTEN/AKT signaling axis was changed accordingly similar to Fig. 5j except for the mTOR protein (Fig. 5u). However, hSR-A3 overexpression in ob/ob mice only upregulated the mRNA expression of genes involved in FAO, but not in DNL, fatty acid uptake and VLDL secretion (Fig. 5v).
Overall, these findings indicate that liver-specific hSR-A3 overexpression, whether in HFHCD-induced WT hamsters or genetically obese ob/ob mice, exerts a protective effect on maintaining glucose and lipid homeostasis, blunting hepatic steatosis without affecting body weight.
AKT inhibition ameliorates dyslipidemia, hyperglycemia, and hepatic steatosis in HFHCD-fed hamsters
To determine whether increased PTEN or decreased AKT activity improved the metabolic abnormalities in SR-A3−/− hamsters, we then treated the HFHCD-fed hamsters with PTEN agonist Oroxin B (OB) or lipid-lowering agent ezetimibe that also inhibits AKT activity via daily gavage for 4 weeks (Fig. 6a). Both agents significantly reversed the phenotypes of increased body weight, the ratio of liver to body weight, plasma TG, TC, and glucose levels caused by SR-A3 deficiency, but only ezetimibe reduced plasma NEFA levels (Fig. 6b–f) and showed more potent effects on restoring lipid and glucose homeostasis. In addition, both OB and ezetimibe improved liver dysfunction in the context of SR-A3 ablation, while ezetimibe exhibited more potent effects against liver impairment (Fig. 6g). Consistently, OB only partially abrogated hepatic steatosis, while ezetimibe intervention completely corrected the severe hepatic lipid accumulation in SR-A3−/− hamsters (Fig. 6h–j). As expected, PTEN activity was increased by OB; however, its downstream effectors of p-AKT, p-mTOR, and p/n-srebp1c were blocked by both drug agents (Fig. 6k, l). In summary, these results show the metabolic abnormalities induced by an HFHCD could be reversed by OB and ezetimibe, to some extent, through blocking AKT hyperactivation caused by SR-A3 deficiency.

a Schematic illustration of 12 week-old male WT and SR-A3−/− hamsters fed an HFHCD for 4 weeks along with daily oral gavage of saline (WT: n = 5; SR-A3−/−: n = 5) or oroxin B (OB, 30 mg/kg) (WT: n = 5; SR-A3−/−: n = 5)or ezetimibe (Eze, 2 mg/kg) (WT: n = 6; SR-A3−/−: n = 6). b–f Body weight (b), liver/BW (c), plasma TG, TC (d), NEFA (e), and glucose (f) levels were measured at the endpoint of the experiment in (a). BW indicates body weight. g Plasma ALT and AST levels at the endpoint of the experiment (n = 5/group). h Liver TG and TC levels from hamsters at sacrifice (n = 5/group). i Representative images of HE and oil red O staining of liver sections (n = 5/group). Scale bars, 100 μm. j Quantification of oil red O staining (n = 5/group). k, l Representative Western blotting of indicated proteins in livers from the indicated groups in (a). The relative normalized protein ratios were calculated and p-values were shown on the right (n = 4/group). Data were presented as the mean ± SEM. Significance between groups was determined by one-way ANOVA test followed by Tukey’s multiple comparison (b–h, j–l). Source data are provided as a Source Data file.
Discussion
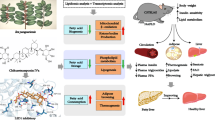
The molecular mechanisms underlying MAFLD are complex and the pathophysiological basis still largely remains elusive so far. In the present study, we identified that the SR-A3 expression was negatively associated with human MAFLD in line with our observations in in vivo and in vitro models. Genetic deletion of SR-A3 elicited hyperlipidemia, hyperglycemia, glucose intolerance, IR, and hepatic steatosis in CD-fed male hamsters, which were further aggravated upon HFHCD feeding through enhancing Srebp1c-mediated DNL and impairing FAO. In addition, SR-A3 deficiency also caused hepatic steatosis and IR when hamsters were fed an MCDD. Mechanistic study revealed that SR-A3 suppressed hepatic AKT activation by inhibiting XIAP-mediated PTEN ubiquitination and degradation to prevent hepatic lipid accumulation. Hepatic overexpression of hSR-A3 by AAV8 or PTEN agonist OB partially alleviated fatty liver in hamsters on HFHCD; however, ezetimibe with the dual effects of lipid-lowering and AKT inhibition fully restored the lipid and glucose homeostasis in HFHCD-fed both WT and SR-A3−/− hamsters and then completely corrected the phenotypes of MAFLD, suggesting that the XIAP/PTEN/AKT axis plays an essential role in the MAFLD progression caused by SR-A3 deficiency (Fig. 7). Our novel findings uncovered that SR-A3 is an important molecule maintaining metabolic homeostasis in the liver.

SR-A3 effectively blocks XIAP-mediated ubiquitination and degradation of PTEN by promoting protein degradation of the E3 ubiquitin ligase XIAP. Mechanistically, PTEN can significantly improve metabolic disorders such as insulin resistance, impaired glucose tolerance, dyslipidemia and MAFLD by inhibiting the overactivation of AKT and lipid synthesis pathway regulated by its downstream transcription factor SREBP1c. In contrast, in the SR-A3-deficient state, XIAP protein stability is significantly enhanced and its E3 ubiquitin ligase activity can promote the ubiquitination and degradation of PTEN, which in turn leads to aberrant activation of the AKT pathway and increased SREBP1c-mediated de novo lipid synthesis. This cascade ultimately exacerbates pathological processes such as insulin resistance, abnormal glucose metabolism, dyslipidemia and MAFLD.
AKT is a key master regulator of glucose and lipid in the liver. Under the physiological condition, activation of AKT suppresses hepatic glucose production by inhibiting FOXO-1 phosphorylation, meanwhile AKT activation promotes Srebp1c-mediated DNL21,22. Recently, however, independent studies reported that hyperactivation of liver AKT was one of the key contributors to MAFLD. Mini P. Sajan et al. found that ob/ob mice characterized by hyperinsulinemia, IR, and obesity, have higher AKT activity at the basal level compared with their lean WT controls23, whereas suppression of hepatic AKT signaling could effectively attenuate MAFLD in mice24,25, suggesting that in the context of metabolic diseases, AKT hyperactivation, instead of suppressing hepatic glucose production, but rather stimulates Srebp1c-mediated hepatic DNL, ultimately resulting in hyperlipidemia and hepatic steatosis26. In agreement with previous findings, our RNA-sequencing analysis revealed that the PI3K-AKT signaling pathway was significantly activated in both CD and HFHCD-fed SR-A3−/− hamsters, suggesting that loss of SR-A3 probably contributes to hepatic steatosis due to AKT hyperactivation. Moreover, CD or HFHCD-fed hamsters with SR-A3 deficiency consistently showed an increase in the phosphorylated AKT levels in the livers, thus leading to hyperinsulinemia, hyperglycemia, and hypertriglyceridemia. It was noted that, although the detailed evidence to demonstrate why the depletion of SR-A3 from female hamsters had no effect on MAFLD could not be provided in the present study, it was found that the expression level of hepatic Sr-a3 mRNA in female hamsters was markedly decreased when compared to male hamsters (Fig. S15a). We postulated that the knockout of the Sr-a3 gene at such a low expression level in the presence of estrogen would not elicit a MAFLD phenotype, given that estrogen has already been demonstrated to exert an anti-MAFLD effect27,28. Future study is needed to explore the hormone action on the regulation of SR-A3 function.
To date, AKT activation has been reported to be regulated by many factors. Two independent studies pointed out that SR-A3 could regulate the proliferation and migration of different cancer cells by inhibiting AKT activity10,11. However, neither studies mentioned the mechanism by which SR-A3 regulates the phosphorylation of AKT, nor the impact of SR-A3-mediated AKT inhibition on cellular glucose and lipid metabolism in liver cells. To investigate the precise molecular mechanism underlying the pathogenesis of MAFLD caused by SR-A3 deficiency, we applied PPI network for deep prediction and found that SR-A3 might regulate the AKT signaling through PTEN and XIAP. PTEN is a negative regulator of AKT, triggering AKT dephosphorylation29, while XIAP, an E3 ubiquitin ligase, can directly ubiquitinate PTEN to regulate PTEN content and compartmentalization in primary mouse embryonic fibroblasts30. In the present study, we found that SR-A3 deficiency caused PTEN loss at the protein level but not mRNA, suggesting SR-A3 modulates PTEN degradation by post-transcriptional modification. Previously, Zheng et al. demonstrated that the C-terminus of SR-A3 could bind to XIAP with high affinity in RWPE1 cells, an immortalized prostate epithelial cell line, and SR-A3 inhibited the function of XIAP to induce the apoptosis of PC3 cells, a prostate cancer cell line31. Thus, we proposed that SR-A3 might maintain PTEN stability by suppressing XIAP activity. As expected, we discovered increased XIAP protein levels and decreased PTEN protein levels in SR-A3−/− hamsters. Meanwhile, Co-IP results further confirmed the interactions of SR-A3, XIAP, and PTEN. RNA interference experiments further showed that SR-A3 silencing-induced ubiquitination of PTEN is XIAP-dependent. Of note, depleting PTEN from mouse liver caused fatty liver with improved glucose tolerance because AKT activation can inhibit FOXO1-mediated gluconeogenesis32, but unlike mice, hamsters with reduced hepatic PTEN level differentially exhibited an upregulation of genes involved in gluconeogenesis. This discrepancy may be explained by the animal species and the difference should be further validated by a head-to-head analysis.
Importantly, SR-A3 deficiency aggravated HFHCD-induced obesity and adipocyte hypertrophy, accompanied by elevated peripheral NEFA content, increased hepatic VLDL secretion. This is consistent with the observations reported in MAFLD patients with obesity, hyperinsulinemia and hypertriglyceridemia. In these individuals, elevated peripheral NEFA by 59% and DNL by 26% are held accountable for the hepatic lipid accumulation and VLDL secretion33. PTEN has been reported to modulate adipose homeostasis. Adipose-specific deletion of PTEN from mice significantly reduced UCP-1 levels in the corresponding adipose tissues, including white and brown adipose tissues, to impair energy expenditure34. Of note, SR-A3 is also highly expressed in the adipose tissue, where it mainly controls adipogenic differentiation35. Consistently, although SR-A3 deficiency had no obvious impact on obesity in CD-fed hamsters, HFHCD-aggravated adipocyte hypertrophy as well as more severe inflammation and reduced UCP-1 expression in adipocytes were observed in knockout animals. We postulated that loss of SR-A3 also disrupted PTEN activity in adipose tissue to cause this metabolic phenotype. In addition, our data from liver mRNA-qPCR analysis demonstrated that excess lipid export from the liver through the VLDL secretory pathway might lead to ectopic lipid deposition in adipose tissue, which then also triggered local inflammation and impaired thermogenesis, eventually causing increased hypertrophy of eWAT in HFHCD-fed SR-A3−/− hamsters. However, future investigations should be warranted to obtain direct evidence demonstrating the possible mechanism by which SR-A3 influences adipose function in an organ cross-talk dependent manner.
As SR-A3 is highly conserved among humans, hamsters, and mice, to further validate the beneficial effects of SR-A3 in MAFLD on multi-species and advance SR-A3 into the clinic to treat MAFLD patients, we then overexpressed human SR-A3 (hSR-A3) via AAV8 in the livers of HFHCD-fed WT hamsters and ob/ob mice. We found that hepatic hSR-A3 overexpression was sufficient to put a brake on MAFLD progression, with improved metabolic parameters such as lower levels of plasma lipids and glucose, blunted glucose intolerance and IR, and dampened hepatic steatosis, suggesting that hepatic SR-A3 is an essential molecule regulating metabolic homeostasis in the liver. Notably, increasing lines of evidence show that AAV-based gene therapy as a once-and-for-all treatment only requires a single injection and provides more desirable patient compliance. There are concerns, however, that AAV injection induces carcinoma in mice with obesity and diabetes36, which in turn limits its application in the MAFLD population. Thus, we sought for pharmacological agents to treat MAFLD. Since we had already verified that SR-A3 deficiency could excessively activate AKT by enhancing PTEN degradation, we activated PTEN in SR-A3−/− hamsters with OB. Unexpectedly, OB could only partially reverse the MAFLD phenotype induced by SR-A3 deficiency. Considering that hamsters lacking SR-A3 showed fatty liver with severe dyslipidemia, hence, targeting AKT together with lipid-lowering against MAFLD might be a better choice.
Furthermore, it is important to pay attention that complete suppression of AKT has been reported to paradoxically trigger HCC and other glucose/insulin-related adverse effects, rather than slow disease progression37. Indeed, MK2206, a pan-AKT inhibitor, is commonly used in cancer therapy and has proven anti-MAFLD potency in animal models38,39; however, the concomitant induction of hyperglycemia and other toxic effects associated with MK2206 in cancer clinical trials40,41, prompting us to rule out its application in our animal models. Thus, we searched for pharmacological agents with partial AKT-inhibition effect and found ezetimibe is a favorable surrogate for pan-AKT inhibitors42,43. Other than its known capacity to lower lipids, ezetimibe also has pleiotropic properties, such as anti-inflammation, DNL inhibition, and IR alleviation44,45, which might be more effective against MAFLD. As expected, periodic oral gavage of ezetimibe indeed decreased hepatic AKT phosphorylation, which in turn improved hepatomegaly, and markedly normalized lipid and glucose metabolism disturbances, ultimately reversing the liver pathology of HFHCD-fed SR-A3−/− hamsters. Strikingly, in contrast to results in LDLR−/− hamsters, where ezetimibe could merely lower the lipid levels of HFHCD-fed LDLR−/− hamsters back to their normal ranges46, which were still higher than WT hamsters, we found a complete remission of plasma lipid levels in HFHCD-fed SR-A3−/− hamsters close to that of CD-fed WT hamsters. Overall, our data suggests that ezetimibe possessing lipid-lowering and AKT inhibition is a more effective therapeutic agent than OB for MAFLD patients with SR-A3 deficiency.
Collectively, in the present study, our new findings for the first time demonstrate that SR-A3 plays an essential role in orchestrating hepatic lipid and glucose metabolism to protect against MAFLD, and provides new insights into the therapeutic approach to treat MAFLD in the setting of SR-A3 deficiency in future clinical trials.
Methods
Human sample collection
All the biospecimens of nonalcoholic fatty liver disease (NAFLD) patients used in the present study were collected from the Department of Infectious Diseases at Shengjing Hospital affiliated to China Medical University. Patients who met the diagnostic criteria of the updated NAFLD guidelines were recruited based on liver biopsy results47,48. Briefly, individuals with liver histology (steatosis in >5% of hepatocytes) and alcohol consumption <20 g per day for women and 30 g per day for men are defined as NAFLD patients. Meanwhile, secondary causes that could result in fatty liver were excluded from this study, including alcoholic hepatitis, genotype 3 hepatitis C virus, drug-induced hepatitis, autoimmune hepatitis, Wilson’s disease, etc. A total of 24 patients were separated into two groups: Normal controls (NC, n = 5), and NAFLD (n = 19) based on Steatosis Activity Score (SAF). Written informed consent was obtained from all patients prior to the investigation. Liver samples were collected in accordance with our approved protocol from Medical Ethics Committee of Shengjing Hospital Affiliated to China Medical University (2023PS913K). All procedures were implemented abiding by the tenets of the Helsinki Declaration of 1975.
Animal models
Generation of SR-A3 deficient hamsters
The sgRNA was designed by targeting the exon 2 of Golden Syrian hamster SR-A3 via Optimized CRISPR Design (http://crispr.mit.edu/) and the sequences were as follows, GAGATCAGCTGGCGGTGACAGGG. After amplification and transcription of sgRNA in vitro using Megascript T7 Kit (Ambion), phenol/chloroform extraction was applied for the purification of SR-A3 sgRNA. Then sgRNA was diluted in RNase-free water with a measured concentration close to 200 ng/ul and stored at -80 °C for further use. Then, Xbal digestion was performed to linearize the humanized cas9 cDNA in the plasmid. The mMESSAGE mMACHINE T7 kit (Ambion) was used for Cas9 mRNA transcription. Next, the end product was diluted with RNase-free water at a concentration of 500 ng/ul and quickly removed into -80 °C for storage. All the following procedures were performed under a microscope. Firstly, prior to microinjections, mineral oil was applied in M2 medium (Sigma-Aldrich) as injection medium, sgRNA (20 ng/ul) and Cas9 mRNA (50 ng/ul) were simultaneously injected into the cytoplasm of fertilized eggs, which displayed a clear shape of pronuclei. When the injected zygotes were incubated in HECM-10 medium for 30 min at 37.5 °C, we then injected embryos with normal morphology into bilateral oviducts, about 15 embryos/ oviduct.
WT hamsters
Wild type (WT) Syrian golden hamsters were purchased from Vital River Laboratory (Beijing, China).
Mice
Adult male leptin-deficient, ob/ob mice (8 week-old) were purchased from Beijing Huafukang Biotechnology (Beijing, China). C57BL/6 mice (WT mice) aged 8 weeks were purchased from Vital River Laboratories (Beijing, China). After acclimation for 1 week, WT mice were fed with CD (4% fat, Beijing Keao–Xieli, 1016706714625204224) and HFD (60% fat, Research Diet, D12492) for 8 weeks. After euthanasia, livers from WT mice were collected for further analysis, as indicated in Fig. 1.
All animals were housed in a specific pathogen-free (SPF) room with constant 20–26 °C temperature and 40%–70% humidity, in a 12 h light/12 h dark cycle and had ad libitum access to water and CD unless stated otherwise. Euthanasia was conducted via carbon dioxide asphyxiation prior to cervical dislocation in accordance with American Veterinary Medical Association (AVMA) guidelines. The care and use of mice and hamsters (e.g. tissue collections of liver, adipose tissue and intestine) were reviewed and approved by the Institutional Animal Care and Use Committee of Peking University and were in accordance with protocol (LA2021334).
AAV8-mediated overexpression of hSR-A3
WT hamsters and ob/ob mice were injected with AAV8 vectors (hSR-A3/Null) at a dose of 1*10^13vg/kg via the jugular vein, or the tail vein, respectively. The total injection volume was 500 ul, as the AAV8 stock solution was further diluted by saline. After feeding with CD for 2 weeks, hamsters were switched to HFHCD for another 4 weeks, and ob/ob mice were kept on CD for another 6 weeks.
Ezetimibe and oroxin B administration
WT and SR-A3−/− male hamsters (8 week-old) were randomly assigned to saline or ezetimibe or orxin B groups while challenged with HFHCD for 4 weeks before euthanasia. Ezetimibe, 10 mg/tablet (MSD Pharma) was diluted in 50 ml saline (20% ezetimibe) and then orally administered in hamsters at a dose of 2 mg/kg body weight. Oroxin B was added in DMSO and kept in -80°C as stocking solution (150 mg/ml), which was further diluted by saline into working solution (3 mg/ml) on a daily basis. Then, hamsters were treated with daily gavage of oroxin B at a dose of 30 mg/kg body weight.
HepG2 cell line
The HepG2 cells used in this study were generously provided by Professor Jichun Yang, who had originally obtained these cells from the American Type Culture Collection (ATCC; Catalog No. HB-8065). Detailed information on the cell line is accessed through the link https://www.atcc.org/products/hb-8065. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, 11965092) containing 10% FBS (TransGen, FS401) and 1% penicillin-streptomycin (Gibco, 15140122) at 37 °C with 5% CO2 in a humidified chamber. Cells were routinely checked and mycoplasma-free. Cells were seeded in 6-well and reached 60%–70% confluence when the medium was replaced with serum-free DMEM medium and treated with indicated fatty acids or BSA. Cells were incubated for 24 h at 37 °C incubator before they were harvested for qPCR analysis.
Genotyping
DNA was extracted from ears of WT, and SR-A3−/− hamsters by proteinase K (MERCK, 1245680100) with tissue lysis buffer. Genotyping was implemented by PCR; WT hamsters resulted in a 288-base pair (bp) product, and a 262-bp product for SR-A3−/− hamsters.
Plasma biochemical characteristics
Plasma samples were collected from the retro-orbital plexus of hamsters or mice after overnight fasting. The levels of plasma TG, TC, glucose, and NEFA were measured using the following kits: Triglyceride Assay Kit (BioSino, 100000220), Cholesterol Assay Kit (BioSino, 100000180), Glucose Assay Kit (BioSino, 100000240), and LabAssay™ NEFA Kit (FUJIFILM Wako, 633-52001). All measurements were performed in strict accordance with the manufacturers’ protocols. Additionally, the degree of liver injury was assessed based on the plasma levels of alanine aminotransferase (ALT) and aspartate transaminase (AST), using reagents from the Nanjing Jiancheng Bioengineering Institute (Nanjing, China), following the manufacturers’ instructions.
FPLC
After overnight fasting, blood samples were collected from indicated hamsters and kept on ice before they were centrifuged at 4000 rpm, 4 °C for 10 min. Plasma from each experimental group was pooled together (at least 100ul) and loaded in the fast-protein-liquid-chromatography (GE Healthcare, AKTA) for lipoprotein analysis. Plasma was fractionated into 40 fractions with elution buffer (0.15 M NaCl, 1 mM EDTA, 0.01 M Na2HPO4, pH = 7.5) at a flow rate of 0.5 ml/min. TG and TC content of each eluting fraction were determined as indicated above.
Protein extraction and western blotting
Tissues or cells were harvested and lysed in RIPA buffer (APPLYGEN, C1053-500) supplemented with protease inhibitor (Roche, 04693132001) and phosphatase inhibitor (Roche, 4906845001). Lysates were placed on ice and vortexed every 10 min for half an hour before centrifugation for 30 min at 12,000 g, 4 °C. The supernatants were collected and protein concentration was measured by Pierce BCA Protein Assay (Thermo Fisher, 23225). Protein lysates were heated in 1X loading buffer at 37 °C for 1 h. 20–30 ug proteins were loaded in SDS-PAGE, transferred to nitrocellulose (NC) membrane, and blocked in skim milk for 1 h before incubating with indicated primary antibodies overnight. After washing with 1X TBST for three times (10 min each time), the second antibodies were added for incubation for another 1 h. Finally, after another three washes with 1X TBST, the protein bands were detected with ECL Kit (Solarbio, PE0010) by iBright 1500 (Invitrogen).
Plasma LPL activity
Sixteen week-old WT and SR-A3−/− hamsters fasting overnright were intraperitoneally injected with 2 U/g body weight heparin, and blood samples were collected 30 min after injection and instantly centrifuged at 4000 rpm for 10 min to produce plasma. Plasma LPL activity was measured by the LPL Activity Kit (Abcam, ab204721) following the manufacturer’s procedures with minor modifications.
VLDL secretion
Hamsters aged 20 week were fasted for 12 h, and injected with Poloxomer P407 (i.p. injection, 1500 mg/kg). Blood was taken at 0 min, 30 min, 1 h, 2 h, 3 h, 4 h post-injection. Plasma TG was measured as indicated.
Oral fat load and intestine tissue section
CD-fed hamsters were starved for 12 h and weighed before fasting blood was taken. Then these hamsters were gavaged with olive oil (10ul/g, La Sicilia, 8011845004871), and blood was collected from intra-orbital vein accordingly, at 30 min, 1 h, 2 h, 4 h, and 8 h after the olive oil challenge. Moreover, jejunums from both CD-fed WT and SR-A3−/− hamsters were dissected 8 h after oil gavage and quickly removed into 4% paraformaldehyde (PFA) for fixation.
GTT and ITT
GTT was conducted in hamsters or ob/ob mice after an overnight fast (21:00-9:00), following an intraperitoneal (i.p.) injection of 2 g/kg glucose (20% glucose, D-(+)-Glucose, Sigma-Aldrich, G8270) for hamsters or 1 g/kg glucose (10% glucose) for ob/ob mice. ITT was performed in 4–6 h fasted hamsters or ob/ob mice by injecting either 0.75 U/kg boy weight (for hamsters) or 1 U/kg (for ob/ob mice) insulin (Humulin R, Lilly France, HI0219). Plasma glucose levels were determined at 0, 15, 30, 60, and 120 min by Glucose Assay Kit in hamsters or by glucometer (Roche) in mice. Blood samples were taken from hamsters via the intro-orbital vein or from mice via the tail vein at indicated time points. Plasma insulin was determined by Ultrasensitive Rat Insulin Elisa Kit (Mercodia, 10-1251-01) in hamsters or by LBIS Mouse Insulin Elisa Kit (FUJIFILM Wako, 638-01489) in mice following manufacturers’ instructions.
Histological analysis of liver and eWAT
Livers and eWAT from indicated hamsters/mice were dissected, fixed in 4% PFA overnight, and dehydrated in 20% sucrose for another 12–24 h prior to being embedded in paraffin. 5 um thick tissue sections were stained with hematoxylin and eosin (HE, Sigma, HHS128-4L, HT110232-1L) or sirus red (Saint-Bio, R21890) in accordance with the manufacturers’ procedure.
Immunohistochemistry (IHC) staining
Paraffin-embedded liver tissue sections from human NAFLD patients underwent dewaxing, rehydration, antigen unmasking, and endogenous peroxidase blockade. Then goat serum (ZSGBBIO, ZLI-9022) was added for incubation for 1 h. Removed blocking solution and added anti-SR-A3 (1:200, Novus, NBP2-13286) for overnight incubation. As subsequent incubation with secondary antibody (ZSGB-BIO, ZB2305) for 1 h at 37 °C, then 1X diaminobenzidine (DAB, ZSGB-BIO, ZLI-9018) for 90–120 s, sections were finally counterstained with hematoxylin, dehydrated, mounted with coverslips. ImageScope was used for image capture. Average optical density (AOD) was then calculated by image J.
Immunofluorescence (IF) staining
Paraffin-embedded liver/eWAT tissue sections from hamsters/mice were dewaxed, rehydrated, antigen-unmasked and blocked with 5% BSA diluted in TBST for 30 min before incubating with rabbit anti-CD68 or anti-F4/80 or CD206 diluted in 5% BSA (1:200, Boster, BA3638) in 4 °C lightproof container. After rewarming at room temperature for 1 h, sections were washed with PBS for three times and a secondary antibody Goat anti-rabbit 555 (abcam, ab150062) diluted in 5% BSA was added. After several washes, sections were mounted with Mounting Medium with DAPI (ZSBIO, ALI-9557) and were visualized by All-in-One Fluorescence Microscope BZ-X800 (Keyence, Japan).
Oil red O staining
Hamsters were anesthetized by 3% pentobarbital sodium followed by 4% PFA. Livers/jejunums were harvested, post-fixed for 12–24 h, embedded with OCT (Sakura, 4583), cryosectioned into 7 um sections and stored at -20°C. Tissue sections were rewarmed at room temperature and washed by distilled water three times. Slides were soaked in 60% isopropanol for 10 s and then stained with 0.3% freshly prepared oil red O solution (Sigma-Aldrich, O0625) for 30 min. 60% isopropanol was used for washing away floating color and cell nuclei were counterstained with hematoxylin. Next, 90% glycerin was mounted in the center of tissue sections for slide sealing. For cells, HepG2 cells were fixed in 4% PFA for 10 min prior to being stained with 60% oil red O for 30 min.
TUNEL staining
The frozen liver sections were fixed in 4% paraformaldehyde for 30 min. Subsequently, an in-situ cell death detection kit (Roche, 11684817910) was employed for the immunofluorescence-based detection and quantification of apoptosis (programmed cell death) at the single-cell level. This method relies on the labeling of DNA strand breaks, and the procedure was strictly conducted in accordance with the manufacturer’s protocols.
Transmission Electron Microscope (TEM)
Hamsters were anesthetized with 3% pentobarbital and euthanized. Livers were perfused with normal saline before being fixed with 1% glutaraldehyde (G5882, Sigma-Aldrich) with 2% paraformaldehyde (P6148, Sigma-Aldrich), all of which were diluted in 0.1 M phosphate buffer (PB) (PH7.4). Fixed livers were then sectioned into small blocks (~1 mm3) and quickly removed into the fixative (2.5% glutaraldehyde + 2% paraformaldehyde) in 0.1 M PB (pH 7.4) for another 1 h at room temperature before transferring to 4 °C container for storage. Next, these samples were washed 5 times (15 min per time) in 0.1 M PB (pH 7.4) and incubated with 0.1 M imidazole (diluted by 0.1 M PB with a pH of 7.4) for 1 h at room temperature in a lightproof space. Followed by 5 washes in distilled water (8 min/wash), these samples were stained with 2% aqueous uranyl acetate at 4 °C for >12 h. After several washes with distilled water, liver samples underwent multistep gradient dehydration of ethanol (30%, 50%, 70%, 85%, 95%, 100%) and exchanges with pure acetone two times, respectively, each step lasting for 8 min. Finally, specimens were prepared by a gradual permeation and embedding with epoxy resin EMbed 812 (14120, Electron Microscopy Sciences). Diamond knives (Diatome, Switzerland) were used to trim and section tissue blocks under a Leica Microsystem UC7 ultramicrotome. Ultra-sections with a thickness of 75 nm were counterstained with uranyl acetate and lead citrate and were viewed by Tecnai G2 Spirit BioTWIN transmission electron microscope (FEI) with a CCD camera (Orius 832, Gatan) at 120 kV.
Magnetic resonance imaging (MRI) and analysis of fat volume
The fat volume was measured in hamsters fed with HFHCD for 8 weeks by magnetic resonance imaging (MRI). MRI was performed using a 3.0 T MAGNETOM Trio (Siemens, Germany), with a rat body coil (CG, China). Hamsters were anesthetized with 1.5% isoflurane (R510-22, RWD, China) in 1 L/min of compressed air. The MRI protocol contains three sequences. Firstly, a T2-weighted rapid acquisition relaxation enhanced sequence was acquired (TR = 4190 ms, TE = 78 ms, NEX = 1, slice thickness = 2 mm, slices = 22, FOV = 13 × 13 cm2, matrix = 384 × 384, acquisition time = 3:30 min). Secondly, a t2-weighted rapid acquisition relaxation enhanced sequence with water saturation (TR = 4190 ms, TE = 78 ms, NEX = 1, slice thickness = 2 mm, slices = 22, FOV = 13 × 13 cm2, matrix = 384 × 384, acquisition time = 3:30 min) was acquired to evaluate the fat distribution. Lastly, a t1 vibe 3D sequence with Dixon water and fat separation (TR = 6.7 ms, TE = 3.675 ms, NEX = 12, slice thickness = 2 mm, slices = 22, FOV = 13×13 cm2, matrix = 256 × 256, acquisition time = 3:48 min) was acquired to evaluate the proton density fat fraction (PDFF). Whole body fat volumes of hamsters were analyzed by threshold segmentation of the fat distribution images, as calculated by multiplying the number of voxels above the threshold and the volume of each voxel.
Lipid extraction
Lipids were extracted from ~30 mg of frozen tissues using a modified version of the Bligh and Dyer’s method as described previously49. Briefly, tissues were homogenized in 900 µL of chloroform: methanol: MilliQ H2O (3:6:1) (v/v/v). The homogenate was then incubated at 1500 rpm for 30 min at 4 °C. At the end of the incubation, 350 µL of deionized water and 300 µL of chloroform were added to induce phase separation. The samples were then centrifuged and the lower organic phase containing lipids was extracted into a clean tube. Lipid extraction was repeated once by adding 500 µL of chloroform to the remaining aqueous phase, and the lipid extracts were pooled into a single tube and dried in the SpeedVac under OH mode. Samples were stored at -80 °C until further analysis.
Lipidomics analyses
All lipidomic analyses were conducted at LipidALL Technologies using a Shimadzu Nexera 20AD-HPLC coupled with Sciex QTRAP 6500 PLUS as reported previously50. Separation of individual lipid classes of polar lipids by normal phase (NP)-HPLC was carried out using a TUP-HB silica column (i.d. 150×2.1 mm, 3 µm) with the following conditions: mobile phase A (chloroform: methanol:ammonium hydroxide, 89.5:10:0.5) and mobile phase B (chloroform:methanol:ammonium hydroxide:water, 55:39:0.5:5.5). MRM transitions were set up for comparative analysis of various polar lipids. Individual lipid species were quantified by referencing to spiked internal standards. d9-PC32:0(16:0/16:0), d9-PC36:1p(18:0p/18:1), d7-PE33:1(15:0/18:1), d9-PE36:1p(18:0p/18:1), d31-PS(d31-16:0/18:1), d7-PA33:1(15:0/18:1), d7-PG33:1(15:0/18:1), d7-PI33:1(15:0/18:1), C17-SL, d5-CL72:8(18:2)4, Cer d18:1/15:0-d7, d9-SM d18:1/18:1, C8-GluCer, C8-GalCer, d3-LacCer d18:1/16:0, Gb3 d18:1/17:0, d7-LPC18:1, d7-LPE18:1, C17-LPI, C17-LPA, C17-LPS, C17-LPG, d17:1 Sph, d17:1 S1P, C14-BMP, d3-16:0-carnitine were obtained from Avanti Polar Lipids. GM3-d18:1/18:0-d3 was purchased from Matreya LLC. Free fatty acids were quantitated using d31-16:0 (Sigma-Aldrich) and d8-20:4 (Cayman Chemicals).
Glycerol lipids including diacylglycerols (DAG) and triacylglycerols (TAG) were quantified using a modified version of reverse phase HPLC/MRM51. Separation of neutral lipids was achieved on a Phenomenex Kinetex-C18 column (i.d. 4.6 × 100 mm, 2.6 µm) using an isocratic mobile phase containing chloroform:methanol:0.1 M ammonium acetate 100:100:4 (v/v/v) at a flow rate of 300 µL for 10 min. Levels of short-, medium-, and long-chain TAGs were calculated by referencing to spiked internal standards of TAG(14:0)3-d5,TAG(16:0)3-d5 and TAG(18:0)3-d5 obtained from CDN isotopes, respectively. DAGs were quantified using d5-DAG17:0/17:0 and d5-DAG18:1/18:1 as internal standards (Avanti Polar Lipids)52.
RNA isolation and mRNA analysis
Total RNA was extracted from tissues (liver/eWAT) or cells (HepG2 cells) with TransZol UP (TransGen, ET111-01-V2). Reverse transcription PCR was performed using a First-Strand cDNA Synthesis Kit (TransGen, AT301-03), and qPCR was carried out using a SYBR PCR kit (TransGen, AQ132-24) with QuantStudio 3 (Thermo Fisher) in accordance with the manufacturers’ instructions. Gapdh or β-Actin was used as internal controls for the qPCR.
Transcriptome analysis
RNA was extracted from liver tissues of WT and SR-A3−/− hamsters CD-fed (n = 3/group) or HFHCD-fed for 8 weeks (n = 5/group). After assessment of RNA integrity using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, USA), the cDNA library was prepared following the Illumina protocol. The library preparations were sequenced on an Illumina Novaseq platform and 150 bp paired-end reads were generated before clustering of the index-coded samples via cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumia). Raw data (raw reads) of fastq format were firstly processed through fastp software and clean data with high quality were obtained. Next, the clean reads were mapped with Mesocricetus auratus genome using Hisat2 v2.0.5. FPKM (Fragments Per Kilobase of transcript sequence per Millions base pairs) was used to estimate gene expression levels. Differential expression genes (DEGs) analysis was performed using DESeq2 R package (1.20.0). Genes with an adjusted p-value < 0.05 and |log2 (fold change) | > 0.264 found by DESeq2 were considered significance. KEGG enrichment analysis of DEGs was implemented in accordance with (http://www.genome.jp/kegg/) and clusterProfiler R package.
AAV8 construct
AAV vectors were generated by the triple-plasmid system with HEK193 cells. Briefly, one plasmid contains the gene of interest (human SR-A3, hSR-A3), another plasmid encodes repcap, and the third helper plasmid (pHelper) in combination transfected HEK293 cells with PEI, thus hSR-A3 was cloned in AAV vectors. 48–72 h post-transduction, highly purified AAV8 carrying hSR-A3 was obtained from supernatants after several steps of digesting, filtration, affinity chromatography, anion exchange chromatography, ultrafiltration, and condensation.
Transfection of plasmids
Plasmids carrying hSR-A3 or null vectors were purchased from lncbio and were transduced into HepG2 cells using Liposomal Transfection Reagent (YEASEN, 40802ES03) following the manufacturer’s protocol. Cells were seeded in 6-well and reached 90–95% confluence at transfection. 6 h after transfection, the Opti-MEM medium (Gibco, 31985070) was changed, and fresh serum-free DMEM-high glucose plus 1% penicillin/streptomycin was added. After fasting for 18 h, cells were incubated with serum-free DMEM-high glucose supplemented with 500uM PA (Sigma, P0500) or 0.5% BSA (Sigma-Aldrich, 9048468) for 24 h. After several gentle washes with PBS, cells were incubated with serum-free DMEM plus 100 nM/L insulin (Humulin R, Lilly France, HI0219) at 37 °C for 15 min. Finally, cells were put on ice, washed with ice-cold PBS, and collected for qPCR and Western Blotting.
RNA interference
HepG2 cells were transfected with siSR-A3 or siRNA control by Lipofectamine RNAiMAX (Invitrogen, 13778150) as described by the manufacturer. Cells were seeded in 6-well and reached 60-70% confluence at transfection. 24 h post-transfection, the Opti-MEM medium was replaced with serum free DMEM-high glucose supplemented with 200 uM PA or 0.2% BSA and cells were incubated for another 24 h. Cells were then treated with insulin as indicated above.
Coimmunoprecipitation (Co-IP) assay
For coimmunoprecipitation (Co-IP) assays, HepG2 cells were transfected with null vectors, hSR-A3 plasmid, or siSR-A3 for 48 h. Then, HepG2 cells were lysed with RIPA lysis buffer containing a protease inhibitor. After centrifugation, the supernatant containing proteins was subjected to IP with indicated antibodies and then protein A/G agarose beads overnight at 4 °C. The beads were washed with NaCl buffer and boiled with 2X SDS loading buffer prior to analysis by Western blots.
Hepatic cholesterol/triglyceride contents
30-60 mg of liver tissue was homogenized in 1 mL cold phosphate buffer solution (PBS), and then 4 mL of chloroform/methanol (v:v = 2:1) was added. The mixture was vortexed for 2 min, and then kept on ice for 20 min. After a centrifugation at 3000 rpm for 30 min, the chloroform layer was transferred to a new glass tube with a glass syringe (labeled as Tube 1). Another 3 mL of chloroform/methanol was added into the remaining mixture tube, vortexed, centrifuged as the 1st round procedure. The chloroform layer was added into the Tube 1 and dried under nitrogen. The contents of cholesterol and triglyceride were measured by dissolving lipids with 500 μL 3% triton X-100.
Quantification and statistical analysis
Mice were age- and body-weight-matched before randomly assigned to groups. Data were presented as mean ± SEM. Shapiro-Wilk test was used for data normalization analysis, if data conforms to normal distribution, unpaired two-sided Student’s t-test was used for comparisons between the two groups, otherwise Mann–Whitney test was performed. Multi-group comparisons were determined by one-way ANOVA. Groups that are mixed by time factors were calculated by two-way ANOVA. Data were analyzed by GraphPad Prism 9.0. Significance was demonstrated as follows, *p < 0.05, **p < 0.01, ***p < 0.001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided with this paper. Public RNA-seq data are available in the GSE database under the accession number GSE289495 and the lipidomics data are deposited in the database of China National Center for Bioinformation (OMIX009203), which are publicly available via the link https://ngdc.cncb.ac.cn/omix/preview/Mq4Mblfg. All data supporting the findings described in this manuscript are available in the article and in the Supplementary Information and from the corresponding author upon request. Source data are provided with this paper.
References
Eslam, M., Sanyal, A. J., George, J. & International Consensus P. MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 158, 1999–2014 e1991 (2020).
Younossi, Z. M. et al. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): a systematic review. Hepatology 77, 1335–1347 (2023).
Goldstein, J. L., Ho, Y. K., Basu, S. K. & Brown, M. S. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc. Natl Acad. Sci. USA 76, 333–337 (1979).
Cheng, C. et al. Recognition of lipoproteins by scavenger receptor class a members. J. Biol. Chem. 297, 100948 (2021).
Zani, I. A. et al. Scavenger receptor structure and function in health and disease. Cells 4, 178–201 (2015).
Han, H. J., Tokino, T. & Nakamura, Y. CSR, a scavenger receptor-like protein with a protective role against cellular damage causedby UV irradiation and oxidative stress. Hum. Mol. Genet 7, 1039–1046 (1998).
Yu, G. et al. CSR1 suppresses tumor growth and metastasis of prostate cancer. Am. J. Pathol. 168, 597–607 (2006).
Bock, A. J., Nymoen, D. A., Brenne, K., Kaern, J. & Davidson, B. SCARA3 mRNA is overexpressed in ovarian carcinoma compared with breast carcinoma effusions. Hum. Pathol. 43, 669–674 (2012).
Brown, C. O. et al. Scavenger receptor class A member 3 (SCARA3) in disease progression and therapy resistance in multiple myeloma. Leuk. Res. 37, 963–969 (2013).
Jiang, L. et al. CSR1 suppresses tumor growth and metastasis of human hepatocellular carcinoma via inhibition of HPIP. Eur. Rev. Med. Pharm. Sci. 21, 3813–3820 (2017).
Kim, J., You, H. J. & Youn, C. SCARA3 inhibits cell proliferation and EMT through AKT signaling pathway in lung cancer. BMC Cancer 22, 552 (2022).
Xu, K. et al. Transcriptome analysis of HepG2 cells expressing ORF3 from swine hepatitis E virus to determine the effects of ORF3 on host cells. Biomed. Res. Int. 2016, 1648030 (2016).
Tian, Y. et al. Scavenger receptor class a, member 3 is associated with severity of hand, foot, and mouth disease in a case-control study. Medicine 98, e17471 (2019).
Adewuyi, E. O., O’Brien, E. K., Nyholt, D. R., Porter, T. & Laws, S. M. A large-scale genome-wide cross-trait analysis reveals shared genetic architecture between Alzheimer’s disease and gastrointestinal tract disorders. Commun. Biol. 5, 691 (2022).
Karachanak-Yankova, S. et al. Epigenetic alterations in patients with type 2 diabetes mellitus. Balk. J. Med. Genet. 18, 15–24 (2015).
Gao, R. et al. Comprehensive analysis of endoplasmic reticulum-related and secretome gene expression profiles in the progression of non-alcoholic fatty liver disease. Front Endocrinol. (Lausanne) 13, 967016 (2022).
Cakir, I. & Nillni, E. A. Endoplasmic reticulum stress, the hypothalamus, and energy balance. Trends Endocrinol. Metab. 30, 163–176 (2019).
Liu, G., Lai, P., Guo, J., Wang, Y. & Xian, X. Genetically-engineered hamster models: applications and perspective in dyslipidemia and atherosclerosis-related cardiovascular disease. Med. Rev. 1, 92–110 (2021).
Haczeyni, F. et al. Mouse models of non-alcoholic steatohepatitis: a reflection on recent literature. J. Gastroenterol. Hepatol. 33, 1312–1320 (2018).
Pan, J. et al. Insulin-like growth factor binding protein 1 ameliorates lipid accumulation and inflammation in nonalcoholic fatty liver disease. J. Gastroenterol. Hepatol. 36, 3438–3447 (2021).
Li, S., Brown, M. S. & Goldstein, J. L. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc. Natl Acad. Sci. USA 107, 3441–3446 (2010).
Onyango A. N. Excessive gluconeogenesis causes the hepatic insulin resistance paradox and its sequelae. Heliyon 8, e12294 (2022).
Sajan, M. P., Ivey, R. A., Lee, M. C. & Farese, R. V. Hepatic insulin resistance in ob/ob mice involves increases in ceramide, aPKC activity, and selective impairment of Akt-dependent FoxO1 phosphorylation. J. Lipid Res. 56, 70–80 (2015).
Tan, X. et al. Caffeine ameliorates AKT-driven nonalcoholic steatohepatitis by suppressing de novo lipogenesis and MyD88 palmitoylation. J. Agric. Food Chem. 70, 6108–6122 (2022).
Jeong, S. H. et al. Hippo-mediated suppression of IRS2/AKT signaling prevents hepatic steatosis and liver cancer. J. Clin. Invest. 128, 1010–1025 (2018).
Hagiwara, A. et al. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab. 15, 725–738 (2012).
Della Torre, S. Non-alcoholic fatty liver disease as a canonical example of metabolic inflammatory-based liver disease showing a sex-specific prevalence: relevance of estrogen signaling. Front Endocrinol. (Lausanne) 11, 572490 (2020).
Lonardo, A. et al. Sex differences in nonalcoholic fatty liver disease: state of the art and identification of research gaps. Hepatology 70, 1457–1469 (2019).
Osborne T. F.& Espenshade P. J. Lipid balance must be just right to prevent development of severe liver damage. J. Clin. Invest. 132, e160326 (2022).
Van Themsche, C., Leblanc, V., Parent, S. & Asselin, E. X-linked inhibitor of apoptosis protein (XIAP) regulates PTEN ubiquitination, content, and compartmentalization. J. Biol. Chem. 284, 20462–20466 (2009).
Zheng, Z. L., Tan, L. Z., Yu, Y. P., Michalopoulos, G. & Luo, J. H. Interaction of CSR1 with XIAP reverses inhibition of caspases and accelerates cell death. Am. J. Pathol. 181, 463–471 (2012).
Horie, Y. et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J. Clin. Investig. 113, 1774–1783 (2004).
Donnelly, K. L. et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 115, 1343–1351 (2005).
Huang, W., Queen, N. J., McMurphy, T. B., Ali, S. & Cao, L. Adipose PTEN regulates adult adipose tissue homeostasis and redistribution via a PTEN-leptin-sympathetic loop. Mol. Metab. 30, 48–60 (2019).
Chen, P. et al. Scara3 regulates bone marrow mesenchymal stem cell fate switch between osteoblasts and adipocytes by promoting Foxo1. Cell Prolif. 54, e13095 (2021).
Cheng, Y. et al. AAV induces hepatic necroptosis and carcinoma in diabetic and obese mice dependent on Pebp1 pathway. EMBO Mol. Med. 15, e17230 (2023).
Wang, Q. et al. Spontaneous hepatocellular carcinoma after the combined deletion of Akt isoforms. Cancer Cell 29, 523–535 (2016).
Ye, Q. et al. Deficiency of gluconeogenic enzyme PCK1 promotes metabolic-associated fatty liver disease through PI3K/AKT/PDGF axis activation in male mice. Nat. Commun. 14, 1402 (2023).
Gu, L. et al. Fructose-1,6-bisphosphatase is a nonenzymatic safety valve that curtails AKT activation to prevent insulin hyperresponsiveness. Cell Metab. 35, 1009–1021 e1009 (2023).
Xing, Y. et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 21, 78 (2019).
Yap, T. A. et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J. Clin. Oncol. 29, 4688–4695 (2011).
Mostafa, R. E. & Abdel-Rahman, R. F. Ezetimibe alleviates acetic acid-induced ulcerative colitis in rats: targeting the Akt/NF-κB/STAT3/CXCL10 signaling axis. J. Pharm. Pharmacol. 75, 533–543 (2023).
Wang, Y. et al. Cholesterol-lowering intervention decreases mTOR complex 2 signaling and enhances antitumor immunity. Clin. Cancer Res. 28, 414–424 (2022).
Kalogirou, M., Tsimihodimos, V. & Elisaf, M. Pleiotropic effects of ezetimibe: do they really exist? Eur. J. Pharm. 633, 62–70 (2010).
Muraoka, T. et al. Ezetimibe decreases SREBP-1c expression in liver and reverses hepatic insulin resistance in mice fed a high-fat diet. Metabolism 60, 617–628 (2011).
Lin, X. et al. Dietary-induced elevations of triglyceride-rich lipoproteins promote atherosclerosis in the low-density lipoprotein receptor knockout Syrian golden hamster. Front Cardiovasc. Med. 8, 738060 (2021).
[Guidelines of prevention and treatment for nonalcoholic fatty liver disease: a 2018 update]. Zhonghua. Gan. Zang. Bing. Za. Zhi. 26, 195–203 (2018).
Cotter, T. G. & Rinella, M. Nonalcoholic fatty liver disease 2020: the state of the disease. Gastroenterology 158, 1851–1864 (2020).
Lam S. M. et al. Quantitative lipidomics and spatial MS-imaging uncovered neurological and systemic lipid metabolic pathways underlying troglomorphic adaptations in cave-dwelling fish. Mol. Biol. Evol. 39, msac050 (2022).
Lam, S. M. et al. A multi-omics investigation of the composition and function of extracellular vesicles along the temporal trajectory of COVID-19. Nat. Metab. 3, 909–922 (2021).
Shui, G. et al. Toward one step analysis of cellular lipidomes using liquid chromatography coupled with mass spectrometry: application to Saccharomyces cerevisiae and Schizosaccharomyces pombe lipidomics. Mol. Biosyst. 6, 1008–1017 (2010).
Shui, G. et al. Derivatization-independent cholesterol analysis in crude lipid extracts by liquid chromatography/mass spectrometry: applications to a rabbit model for atherosclerosis. J. Chromatogr. A 1218, 4357–4365 (2011).
Acknowledgements
We are indebted to Dr. Ying-Chun Hu, Zhen-Yang Kong and Hong-Zhang Zhou for their professional technical assistance in EM sample preparation and image analysis at the Core Facilities of School of Life Sciences, Peking University, and Qiang Shen and Ling Zhang at the Institute of Cardiovascular Sciences, State Key Laboratory of Vascular Homeostasis and Remodeling, Peking University, for her technical support in HE and sirius red stainings. We thank Professor Jichun Yang at the Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Peking University, for providing HepG2 cells; Aiden Xian from Shepton High School for discussing and proofing the manuscript. Icons of hamsters, mice, and diets in experimental illustrations were by Figdraw. This work was supported by the National Natural Science Foundation of China (NSFC) 82270479, 82070460 and HY2021-1 to X.X.; the Beijing Natural Science Foundation 7242084 to X.X.; the Fundamental Research Funds for the Central Universities to X.X.; Peking University Medicine plus X Pilot Program-Platform Construction Project 2024YXXLHPT010 to X.X.; the National Key Research and Development Program of China from the Ministry of Science and Technology 2021YFF0702802 to Y.W.; the NSFC 8240033347 to G.M.
Author information
Authors and Affiliations
Contributions
X.X. conceived the study, designed experiments, secured funding and critically revised the paper. P.L., and Y.H. performed in vivo experiments, G.M. and Y.Z. conducted in vitro experiments. P.L., G.M, Y. Z. and Y. H. analyzed the results. Y.L.(Yanwei Li), C.Z., Y.D. and X.D. were responsible for human sample collection and explanation. Y.L.(Yiran Liu) and D.Z. provided data analysis and visualization support in RNA-sequencing. J.G., W.Z., Y.Z., Y.X., L.Z., G.C., Z.Z., Y.H., S.M., J.C.(Jingxuan Chen), and J.C.(Jinxuan Chen) helped with tissue collection and provided support in animal experiments. X.G. constructed the SR-A3−/− hamster. G.C. helped with AAV8 tail vein injection. L.X. assisted in the MRI experiment and the ensuing data analysis. S.W. designed, constructed, and packaged the AAV8. S.L. and G.S. offered data analysis and technical support in targeted liver lipidome. P.L., G.M., J.G., W.H., Y.W., and X.X. discussed and interpreted the results from the study. Z.Z., X.X., and P.L. designed the graphical abstract. P. L. and G.M. drafted the manuscript and all the Figures. All authors agreed with the final submitted manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declared no competing interests.
Peer review
Peer review information
Nature Communications thanks Liang Guo and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lai, P., Miao, G., Zhao, Y. et al. SR-A3 suppresses AKT activation to protect against MAFLD by inhibiting XIAP-mediated PTEN degradation. Nat Commun 16, 2430 (2025). https://doi.org/10.1038/s41467-025-57585-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57585-0
