
Abstract
Cells utilize protein disaggregases to avoid abnormal protein aggregation that causes many diseases. Among these, caseinolytic peptidase B protein homolog (CLPB) is localized in the mitochondrial intermembrane space and linked to human disease. Upon CLPB loss, MICU1 and MICU2, regulators of the mitochondrial calcium uniporter complex (mtCU), and OPA1, a main mediator of mitochondrial fusion, become insoluble but the functional outcome remains unclear. In this work we demonstrate that CLPB is required to maintain mitochondrial calcium signalling and fusion dynamics. CLPB loss results in altered mtCU composition, interfering with mitochondrial calcium uptake independently of cytosolic calcium and mitochondrial membrane potential. Additionally, OPA1 decreases, and aggregation occurs, accompanied by mitochondrial fragmentation. Disease-associated mutations in the CLPB gene present in skin fibroblasts from patients also display mitochondrial calcium and structural changes. Thus, mtCU and fusion activity are dependent on CLPB, and their impairments might contribute to the disease caused by CLPB variants.
Similar content being viewed by others
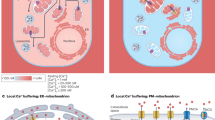
Introduction
Abnormal protein aggregates are causative of a toxic cell environment, underlying several pathological conditions1,2. To reverse protein aggregation, cells express specialized protein disaggregases to solubilize proteins restoring their native structure and function3,4. Recently, the Caseinolytic peptidase B protein homolog (human CLPB, also known as SKD3), located in the mitochondrial intermembrane space (IMS), was identified as the first mitochondrial disaggregase3. CLPB acts as a chaperone that couples ATP hydrolysis to protein disaggregation and reactivation3. Notably, pathogenic variants in the CLPB gene (NM_001258392.3) are associated with human disease. Biallelic variants and specific monoallelic variants have been associated with a neurodevelopmental disorder characterized by progressive brain atrophy, intellectual disability/developmental delay, movement disorder, neutropenia and 3-methylglutaconic aciduria (MIM#616271, 619813,5,6,7,8,9). Additionally, cataract is observed only in biallelic cases. Also, patients with isolated neutropenia (MIM#619813) have been reported10. Furthermore, CLPB upregulation is associated with anti-cancer drug resistance in acute myeloid leukemia (AML)11.
Unexpectedly, upon CLPB knockout, MICU1 and MICU2 were among the top hits with increased insolubility3. In addition, unbiased identification of the mtCU interactome found that EMRE interacts with CLPB12. MICU1 and MICU2 localize in the IMS and interact with MCU and EMRE, which form and stabilize, respectively, the pore of the mitochondrial calcium uniporter complex (mtCU). MICUs control the channel opening by sensing increases in cytosolic Ca2+ concentrations ([Ca2+]c) through EF-hand Ca2+-binding domains13,14. MICU1-MICU2 heterodimers cooperatively translate increases in [Ca2+]c into a sigmoidal response of mitochondrial Ca2+ uptake13,14,15. The turnover of the mtCU components should be finely regulated to avoid mitochondrial Ca2+ dysregulation and mitochondrial functional impairment. Aberrant NADH oxidation and ROS production were reported to lead to slower MCU turnover16. Furthermore, excessive levels of EMRE are targeted for degradation by m-AAA-proteases such as AFG3L2 and SPG7. This prevents the over-incorporation of EMRE into the mtCU, which could lead to the formation of uncontrolled MCU channels17,18,19. Evidence of proteolytic control of the MICU gatekeepers has also been emerging. Degradation of MICU1 through the Ubiquitin Proteosome System has been reported, whereas MICU2 was not modulated by this mechanism20. Additionally, the i-AAA protease YME1L has been shown to regulate MICU1 by degrading MICU1 monomers21,22. Thus, the dimerization of MICU1 with MICU2 is needed not only for gatekeeping of the mtCU but also to protect the MICUs from degradation22,23. Overall, these regulatory mechanisms ensure precise control over the levels of the mtCU, contributing to the fine-tuned regulation of mitochondrial Ca2+ uptake17,18,19.
CLPB directly interacts with OPA1, an inner mitochondrial membrane (IMM) protein, and CLPB loss increases OPA1 insolubility3,11,24. OPA1 is a dynamin-like guanosine triphosphatase, which is critical for mitochondrial fusion dynamics and cristae structure. Loss of OPA1 causes mitochondrial fusion loss, fragmentation, cristae remodeling, Ca2+ uptake impairment, and cell death susceptibility25,26,27,28,29. OPA1 activity is controlled by proteolytic cleavage mediated by the mitochondrial metallopeptidase OMA1 and YME1L26,30,31,32,33. Dysfunction of mitochondria triggers OPA1 processing, resulting in mitochondrial fragmentation, which facilitates the targeted removal of damaged mitochondria.
Recently, CLPB was reported to alter the functionality of the mitochondrial protease complex SPY (STOML2, PARL, YME1L)34. Because YME1L was shown to be central to the proteolytic control of MICU1/MICU2 and OPA121,22, we hypothesized that CLPB could affect the mtCU and OPA1 both directly via chaperone/disaggregase activity and indirectly by altering its regulatory proteases. We also reasoned that the effect of CLPB on the mtCU complex and OPA1 might have functional relevance in mitochondrial Ca2+ signaling and fusion dynamics. Because both the components of mtCU and OPA1 are linked to human pathogenesis35,36, their alterations might be relevant for the disease caused by CLPB variants.
In this work, we have combined chronic and acute loss models for CLPB together with cells harboring disease-associated CLPB mutations. We have found that upon CLPB loss, the mtCU-forming proteins were altered, showing downregulation and aggregation of MICU1, MICU2, and the precursor form of EMRE. Loss of CLPB also resulted in decreased mitochondrial Ca2+ uptake, without altering cytosolic Ca2+ transients. Furthermore, we documented aggregation, reduced abundance, and impaired processing of OPA1, leading to mitochondrial fusion inhibition and fragmentation. We therefore conclude that CLPB is a critical regulator of the mtCU complex abundance and activity, mitochondrial Ca2+ signaling, and fusion dynamics.
Results
CLPB deletion leads to changes in the abundance of the mitochondrial Ca2+ uniporter components
Removal of CLPB has been reported to cause heightened insolubility of the regulatory components of the mtCU, MICU1 and MICU23,24. However, the effect of the lack of CLPB on mitochondrial Ca2+ signaling has not yet been evaluated. Therefore, we set out to study the mtCU composition and mitochondrial Ca2+ handling in wild type (WT) and CLPB knockout (KO) HAP1 cells.
Initially, we assessed the impact of CLPB loss on the abundance of each component of the mtCU by immunoblotting whole cell lysates of WT and KO HAP1 cells (Fig. 1a). Whole cell lysates include all proteins solubilized by RIPA and likely exclude the insoluble protein aggregates. Thus, whole cell lysates might show decreased level for the aggregated proteins. As expected, CLPB was present in the WT cells and undetectable in the CLPB KO cells (Fig. 1b). Immunoblots revealed that the levels of the mtCU components, MICU1, MICU2, and the mature form of EMRE (m-EMRE) were decreased in CLPB KO cells as compared with WT cells (Fig. 1b, c). Conversely, the levels of the pore-forming subunit, MCU were higher in CLPB KO cells than in WT cells (Fig. 1b, c). Given that the mtCU gatekeeping is mediated by dimeric MICUs, we also evaluated the abundance of the MICU1-MICU2 dimers13,14. Consistent with the findings for MICU1 and MICU2 monomers, MICU1-MICU2 dimers were decreased in the absence of CLPB (Fig. 1d, e). These changes in the abundance of the mtCU components raise the possibility that their stability might be influenced by the loss of CLPB.

a Scheme depicting the assembly of the mtCU (MCU, EMRE, MICU1, MICU2). Graphics Created in BioRender. Cartes Saavedra, B. (2025) https://BioRender.com/v91z573. b, c Representative Western blot and quantification of the mtCU components and CLPB obtained from whole cell lysates of wild type (WT) and CLPB knockout (KO) HAP1 cells. GRP75 was used as a loading control. d, e Representative Western blot and quantification of MICU1 and MICU2 dimers from WT and CLPB KO HAP1 cell lysates. TOM20 was used as a loading control. In c and e data represent the mean ± SEM. n = 5 and 4 for WT and CLPB KO, respectively. For analysis, an unpaired Student T-test was used. *p ≤ 0.05 and **p ≤ 0.01. HEK MICU1 KO cells in panels b and d were used as a control for MICU1 loss. f, g Representative Western blot and quantification of the mtCU components and CLPB in CLPB-silenced HeLa cells for 96 h. GRP75 was used as a loading control. Data are presented as mean ± SEM. n = 3 replicates for each condition. For analysis, an unpaired Student T-test was used. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. h, i Representative Western blot and quantification of the mtCU components and CLPB from cell lysates of CLPB-overexpressed HeLa cells for 24 or 48 h. GRP75 was used as a loading control. Data are presented as mean ± SEM. n = 3 replicates for each condition. For analysis, one-way ANOVA was used with post hoc Bonferroni tests for each sample. *p ≤ 0.05 and **p ≤ 0.01. j Aggregation assay from isolated mitochondria of CLPB-silenced HeLa cells for 96 h. Mitochondrial isolation was performed and the total proteins (T), along with the soluble (S) and insoluble fractions (P), were purified and analyzed via Western blotting. PDH served as a negative control. n = 2. k Representative Western blot of the mtCU components, CLPB and the AAA+ proteases YME1L and AFG3L2 subjected to CLPB, YME1L or AFG3L2 silencing in HeLa cells after 96 h. GRP75 was used as a loading control. n = 3. Source data are provided as a Source Data file.
To address potential compensatory mechanisms resulting from the stable deletion of CLPB, we evaluated the impact of its acute silencing and overexpression on the levels of the mtCU components in whole cell lysates of HeLa cells. First, we investigated whether the acute silencing of CLPB could also influence the protein expression of the mtCU components. Consistent with the findings from the stable CLPB deletion in HAP1 cells, its acute silencing resulted in reduced protein levels of MICU2 (Fig. 1f, g). There was no discernible effect of CLPB silencing on MICU1, and the impact on MCU was mild (Fig. 1f, g). On the other hand, the precursor form of EMRE (p-EMRE) was reduced by CLPB silencing (Fig. 1f, g). However, the effect on the m-EMRE was mild (Fig. 1f, g), as observed in CLPB KO HAP1 cells.
To address how increased levels of CLPB influence the mtCU components, we overexpressed CLPB in HeLa cells for either 24 or 48 h, and we performed immunoblotting analysis of the mtCU components. As expected, CLPB protein levels exhibited a progressive increase at both 24 and 48 h, accompanied by a mild elevation in MICU1 only at the later time point and an increase in MICU2 (Fig. 1h, i). The expression of MCU remained unaltered and the levels of p-EMRE were increased, while m-EMRE remained unchanged (Fig. 1h, i).
We also explored whether CLPB silencing affected other IMM ion channels. Specifically, we tested the ATP-sensitive K+ channel, MITOK37, and LETM1, an EF-hand-containing protein, whose function is still debated38,39. Consistent with a specific role in the regulation of mitochondrial Ca2+ homeostasis, the abundance of these two IMM proteins was unaffected by the silencing of CLPB (Supplementary Fig. 1).
As mentioned above, the regulatory components of the mtCU, MICU1 and MICU2, were identified among the mitochondrial proteins with reduced solubility in CLPB KO HAP1 cells3. This prompted us to investigate whether the reduction in the levels of the mtCU components upon CLPB silencing was due to an increase in their insolubility. To address this, we performed an aggregation experiment with the total protein of isolated mitochondria (fraction T) and extracted with 1% Triton both soluble (fraction S) and insoluble proteins in the pellet (fraction P). The experiment revealed that the main components of the mtCU complex (MCU, EMRE, MICU1, MICU2) were present in the insoluble fraction of CLPB-silenced cells, further highlighting the critical role of CLPB in preserving their solubility (Fig. 1j). Furthermore, OPA1 was also detected in the insoluble fraction of CLPB-silenced cells (Fig. 1j), consistent with previous reports3, while the matrix protein PDH was not influenced by CLPB loss (Fig. 1j). We then investigated whether the alterations in the abundance of the mtCU components upon CLPB perturbations could be attributed solely to changes in their solubility or also to variations in their proteolysis. Given that CLPB does not exhibit any proteolytic activity, we focused our attention on the mitochondrial AAA proteases known to degrade IMS and IMM proteins, the i-AAA protease YME1L and the m-AAA protease AFG3L240. In neurons, EMRE has been confirmed as a substrate of these proteases18 and recently, an interplay between CLPB and the IMM quality control proteins such as PARL and YME1L has been shown34. We performed concomitant silencing of either the i-AAA protease YME1L or the m-AAA protease AFG3L2 together with CLPB (Fig. 1k, Supplementary Fig. 2). This experiment shows that while CLPB silencing alone reduces the protein content of MICU2 (Fig. 1f, k), the concomitant silencing of either YME1L or AFG3L2 restores MICU2, whereas m-EMRE expression levels were only restored upon concomitant AFG3L2 silencing (Fig. 1k, Supplementary Fig. 2). Furthermore, upon YME1L silencing, MICU1 protein levels increase consistently with previous reports21 (Fig. 1k and Supplementary Fig. 2). Curiously, knockdown of AFG3L2 increases CLPB abundance, and this might be explained by a complex mechanism, as AFG3L2 and CLPB are located in spatially distinct mitochondrial compartments. Overall, these data suggest that CLPB exerts its activity on the mtCU by a dual mechanism, by preventing both aggregation and proteolytic degradation.
Given the role of CLPB in maintaining the solubility of IMS proteins, we asked whether silencing CLPB could impact the integrated stress response (ISR) pathway. This pathway is an essential cellular signaling network that helps organisms to adapt to various stressors, supporting homeostasis and survival41. This evolutionarily conserved mechanism activates a series of responses to specific stress signals, primarily through the phosphorylation of eukaryotic translation initiation factor 2 alpha (eIF2α)41. This phosphorylation selectively enhances the translation of particular mRNAs, especially those encoding stress-responsive transcription factors such as ATF4 and CHOP42. First, we examined the phosphorylation levels of eIF2α but observed no difference in CLPB-silenced cells compared to controls (Supplementary Fig. 3a). We then assessed whether the levels of ATF4, CHOP, and other eIF2α-induced genes were upregulated in the absence of CLPB. In the same direction, no differences were observed (Supplementary Fig. 3b). Furthermore, we also checked the activation of the mitochondrial unfolded protein response (UPRmt)43. This stress response is triggered by the accumulation of unfolded or misfolded proteins within the mitochondrial matrix, leading to a retrograde signal sent to the nucleus. This signal increases the expression of proteases and chaperones to manage the abnormal protein load in the mitochondria44. We assessed the activation of the UPRmt pathway in CLPB-deficient cells by performing qPCR on CLPB-silenced HeLa cells, as previously described45. We observed a small increase in the expression of the two mitochondrial chaperones HSPA9 and HSPD1, but not in that of the transcription factor ATG5 and of the mitochondrial peptidase Lonp1 (Supplementary Fig. 3c), suggesting that the acute downregulation of CLPB may mildly induce the UPRmt response.
Mitochondrial Ca2+ signaling is suppressed upon the loss of CLPB
Having observed that CLPB alters the protein content of the mtCU subunits, we investigated whether the cytoplasmic and mitochondrial matrix Ca2+ signals in intact cells were altered in CLPB KO HAP1 cells. In intact WT, CLPB KO and CLPB rescued cells, we simultaneously imaged both cytoplasmic and mitochondrial matrix [Ca2+] ([Ca2+]c and [Ca2+]m, respectively) (Fig. 2a, b). The cells were transfected for 48 h with the Ca2+ sensor mtGCaMP6f to follow [Ca2+]m, and with either an empty vector (WT and CLPB KO cells) or with the WT CLPB protein to rescue its expression (Supplementary Fig. 4a). To monitor the [Ca2+]c, cells were loaded with 2 μM of Fura-2/AM. First, we tested the effect of the endoplasmic reticulum (ER) store depletion by adding 2 µM of thapsigargin (Tg) in Ca2+-free extracellular buffer, and then added back 1 mM CaCl2 to stimulate Store Operated Ca2+ Entry (SOCE). In WT cells, Tg induced a transient [Ca2+]c increase, which was coupled with a slow and small [Ca2+]m rise, whereas SOCE caused a larger [Ca2+]c and [Ca2+]m (Fig. 2a, b, black traces). The lack of CLPB did not alter the [Ca2+]c signals induced either by the depletion of the ER Ca2+ store or SOCE (Fig. 2a, red trace and left panels in Fig. 2c, d); however, the [Ca2+]m rise was suppressed in both conditions (Fig. 2b, red trace and right panels in c and d) and, it was re-established by acute CLPB rescue (Fig. 2b, blue trace and right panel in c). Based on the [Ca2+]m decay, the mitochondrial Ca2+ extrusion was not affected by the absence of CLPB (Supplementary Fig. 5a). Since [Ca2+]m was monitored by a non-ratiometric reporter protein, the resting [Ca2+]m could not be reliably assessed. To address this issue, we took advantage of the fluorescence lifetime imaging microscopy (FLIM) which eliminates intensity-related artifacts and measures an absolute parameter of the probe allowing straightforward quantification. We used the mitochondrial matrix Ca2+ sensor, mtRCaMP that shows [Ca2+]-dependent lifetime changes46. CLPB KO cells showed a significantly shorter lifetime compared with WT HAP1 cells indicating that the absence of CLPB leads to reduced resting [Ca2+]m (Supplementary Fig. 6a). The difference was also maintained upon Tg stimulation (Supplementary Fig. 6a). Overall, these findings show the dependence of mitochondrial Ca2+ homeostasis on CLPB.

a, b Average traces of cytosolic Ca2+ concentration ([Ca2+]c) and mitochondrial matrix Ca2+ concentration ([Ca2+]m) during a Store Operated Ca2+ Entry (SOCE) protocol in WT), CLPB knockout and rescued cells. All the cells co-expressed the mitochondrial Ca2+ sensor mtGCaMP6f together with either the empty vector (WT and CLPB KO cells) or CLPB WT expressing vector (rescued cells) for 48 h. Cells were loaded with 2 μM Fura-2/AM and were stimulated with 2 μM Tg to deplete the ER Ca2+ store in a Ca2+-free medium, followed by the readdition of 1 mM of CaCl2 to induce SOCE. The inset in b shows the amplification of the [Ca2+]m during Ca2+ depletion of the ER. c Quantification of [Ca2+]c and [Ca2+]m rise in response to the ER Ca2+ store depletion. d Quantification of cytoplasmic and mitochondrial Ca2+ peak in response to Ca2+ readdition. The values in c and d represent the Area under the Curve (AUC) calculated from 30 to 150 s for c and from 370 to 570 s for d. Data are presented as mean ± SEM. WT cells, n = 127 cells; CLPB KO cells, n = 78 cells; CLPB rescued cells, n = 68 cells, from 12 independent experiments. For analysis, one-way ANOVA was used with post hoc Bonferroni tests. *p ≤ 0.05; ***p ≤ 0.001. Source data are provided as a Source Data file.
Since we observed differences in the levels of mtCU components between stable CLPB deletion (Fig. 1b, c) and acute CLPB silencing (Fig. 1f, g), we decided to analyze [Ca2+]c and [Ca2+]m also in the latter model. As observed in CLPB KO HAP1 cells, [Ca2+]c was not affected by acute CLPB silencing in HeLa cells (Fig. 3a). As for [Ca2+]m, aequorin measurements in HeLa cells showed a marked reduction of the transient rise evoked by stimulation with the inositol 1,4,5-triphosphate (IP3)-generating agonist histamine upon acutely silencing CLPB (Fig. 3b), while the rate of Ca2+ extrusion was unaffected (Supplementary Fig. 5b). The concomitant overexpression of CLPB with its silencing rescued the wild type HeLa [Ca2+]m phenotype (Fig. 3b, Supplementary Fig. 4b), confirming the dependence of mitochondrial Ca2+ dynamics on CLPB. The resting [Ca2+]m, measured by the GFP-based fluorescent Ca2+ indicator (4mtGCaMP6f), was increased by CLPB silencing in HeLa cells (Supplementary Fig. 6b).

a, b Cytosolic Ca2+ concentration ([Ca2+]c) and mitochondrial matrix Ca2+ concentration ([Ca2+]m) measurements, respectively, in intact HeLa cells transfected with the indicated siRNAs and constructs for 96 h together with cytosolic and mitochondrial aequorin transfected 48 h before Ca2+ measurements and challenged with maximal histamine stimulation. Left panel: representative traces; right panel: bar diagram representing the mean [Ca2+] peak upon stimulation. n = 3 for each condition for [Ca2+]c measurements. siCTR, n = 3; siCLPB, n = 3; siCLPB+CLPB n = 4 for [Ca2+]m measurements. c [Ca2+]m measurements in permeabilized HeLa cells transfected with the indicated siRNAs and constructs for 96 h together with mitochondrial targeted aequorin transfected 48 h before Ca2+ measurements and perfused with 1 μM Ca2+. Left panel: representative traces; middle panel: bar diagram representing the mean [Ca2+]m peak upon stimulation; right panel: bar diagram representing the mean [Ca2+]m speed. siCTR, n = 4; siCLPB, n = 5; siCLPB+CLPB, n = 4. In all panels, data are presented as mean ± SEM. For data analysis, one-way ANOVA was used with post hoc Bonferroni tests for each sample. *p ≤ 0.05. Source data are provided as a Source Data file.
Effect of CLPB targeting on mitochondrial Ca2+ uptake and mitochondrial membrane potential (ΔΨm)
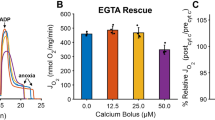
The mitochondrial Ca2+ uptake activity of CLPB-silenced HeLa cells was tested in permeabilized cells. After permeabilization in EGTA-containing Ca2+-free intracellular buffer (IB/EGTA), Ca2+ accumulation was initiated by switching the perfusion buffer to IB containing 1 μM [Ca2+] buffered by EGTA. CLPB-silenced cells showed a lower rate and speed of Ca2+ accumulation than the WT (Fig. 3c). Thus, suppression of Ca2+ uptake occurred independently of [Ca2+]c transients and the high [Ca2+] microdomains between ER and mitochondria. Importantly, as observed in intact cells, the overexpression of CLPB in the silenced cells can rescue the Ca2+ phenotype (Fig. 3c, Supplementary Fig. 4b).
To specifically investigate Ca2+ uptake by mitochondria, we also carried out fluorometric assays. WT and CLPB KO cell suspensions were permeabilized in an intracellular-like buffer also containing Tg to inhibit ER Ca2+ uptake and succinate to energize mitochondria. Upon the addition of Ca2+ boluses (3 μM and 20 μM CaCl2), we evaluated Ca2+ clearance (Fig. 4a). This reflected mtCU-mediated mitochondrial Ca2+ uptake as it was entirely inhibited by Ru26547 (Supplementary Fig. 7a). Upon the submicromolar [Ca2+] elevation evoked by 3 μM CaCl2, mitochondrial Ca2+ uptake was observed in neither WT nor CLPB KO cells (Fig. 4a). However, upon raising [Ca2+] to several micromolar, mitochondrial Ca2+ uptake was apparent in both WT and CLPB KO, albeit less in the CLPB KO (Fig. 4a). These findings confirm that the absence of CLPB impairs mitochondrial Ca2+ uptake.

a, b Simultaneous measurements of Ca2+ clearance and mitochondrial membrane potential (ΔΨm) in WT and CLPB KO HAP1 permeabilized cells following consecutive additions of 3 μM and 20 μM CaCl2. Two μM FCCP + 5 μM Oligomycin was added to induce complete mitochondrial membrane depolarization. n = 15 and 14 technical replicates for WT and CLPB KO, respectively. c ΔΨm was tested in intact WT and CLPB KO cells by measuring tetramethyl rhodamine methyl ester (TMRM) uptake (25 nM). Five μM of Oligomycin was used to induce maximal TMRM uptake, while 5 μM FCCP to fully discharge it. For quantification TMRM fluorescence was obtained during the 30-s period prior to the addition of oligomycin. Data are presented as the mean ± SEM from 9 experiments for each condition. For analysis, an unpaired Student T-test was used. d HeLa cells silenced for 96 h for CLPB were loaded with TMRM and TMRM fluorescence measurements were performed. Where indicated 10 μM carbonyl cyanide 3-chlorophenylhydrazone (CCCP) was added. a.u., arbitrary units. n = 52 and n = 49 cells for siCTR and siCLPB respectively. Data are presented as mean ± SEM. e Representative Western blot of the (oxidative phosphorylation) OXPHOS complexes in control and CLPB-silenced HeLa cells for 96 h. Whole cell lysates were subjected to Western blotting analysis with a cocktail of primary antibodies for the specific components of the OXPHOS complexes. Left panel: low exposure image. Right panel: high exposure image. n = 3. f Oxygen consumption rate (OCR) in HeLa cells transfected with the indicated siRNAs and constructs for 96 h (left panel). Components of OCR accounting for basal, ATP-linked, and maximal respiration, spare capacity, non-mitochondrial OCR and proton leak are quantified in the right panel. Data are normalized on mean Calcein fluorescence. Data are presented as mean ± SEM. n = 15 replicates for each condition. For analysis, one-way ANOVA was used with post hoc Bonferroni tests for each sample. *p ≤ 0.05; **p ≤ 0.01. Source data are provided as a Source Data file.
Because Cupo et al. showed that proteins involved in mitochondrial electron transport and mitochondrial respiratory chain complex assembly are also more insoluble in cells lacking CLPB3, we assessed whether the Ca2+ phenotypes might reflect a change in the driving force of mitochondrial Ca2+ accumulation. We measured the ΔΨm using a potentiometric fluorescent dye, tetramethyl rhodamine (TMRM) simultaneously with [Ca2+] (Fig. 4b). Smaller succinate-induced TMRM uptake was observed in CLPB KO cells compared to WT, suggesting lesser ΔΨm when CLPB expression is ablated (Fig. 4b). However, by checking individual runs, we observed that the mitochondrial Ca2+ clearance in CLPB KO cells was still suppressed even when the corresponding WT showed comparable ΔΨm (Supplementary Fig. 7b). Furthermore, since Ca2+ imaging was performed in intact adherent cells, we decided to evaluate the ΔΨm by testing the TMRM uptake in intact cells and we did not find differences in the ΔΨm comparing intact WT and CLPB KO cells (Fig. 4c). Similar results were obtained by acutely knocking down CLPB in HeLa cells (Fig. 4d). Overall, these findings suggest that the ΔΨm is unlikely to be the main cause of both the mitochondrial Ca2+ clearance suppression and the reduction of the mitochondrial Ca2+ signaling, indicating that impairment of the mtCU Ca2+ channel activity must be considered in CLPB KO-deficient cells.
As mitochondrial Ca2+ plays a well-established role in regulating oxidative metabolism48 and since CLPB is responsible for the solubility of components of the respiratory chain complexes3, we explored the potential effect of CLPB silencing on oxidative phosphorylation (OXPHOS). Whole cell lysates from CLPB-silenced HeLa cells were subjected to Western blotting analysis with a Total OXPHOS Antibody Cocktail kit that includes antibodies against specific subunits of the five complexes of the OXPHOS (NDUFB8, SDHB, UQCRC2, MTCO1, and ATP5a). Our results indicate that CLPB silencing does not affect the protein content of the analyzed OXPHOS subunits (Fig. 4e). To further test whether oxidative metabolism was affected by CLPB silencing, we measured OCR in the same conditions. Cells were treated consecutively with oligomycin, to measure ATP-linked respiration and proton leak; FCCP, to measure maximal oxygen consumption and the spare capacity; and finally with rotenone and antimycin, to verify the extent of non-mitochondrial respiration. No changes were detected in CLPB silencing cells compared to control and to silenced cells concomitantly overexpressing CLPB, except in non-mitochondrial oxygen consumption rate (OCR), which suggests increased cellular oxidative stress (Fig. 4f).
Loss of CLPB causes mitochondrial fragmentation and mitochondrial fusion instability
CLPB has been demonstrated to play a critical role in the solubility of OPA1, the main player in IMM fusion3,24. Consistently, we observed reduced levels of OPA1 and by confocal imaging, mitochondrial fragmentation in CLPB KO as compared to WT HAP1 cells (Fig. 5a, b). OPA1 has different isoform variants which are proteolytically cleaved by OMA1 and YME1L to produce short forms of the protein49. The balance between short and long forms has been extensively reported to be relevant in the mitochondrial fusion/fission balance27. To evaluate whether the mitochondrial fragmentation observed in CLPB KO cells might involve changes in the OPA1 proteolytic process besides the reduced abundance of OPA1, we tested the abundance of the different forms of OPA1, classified as a and b for the long forms and, c, d, and e for the short forms. As a reference, we silenced YME1L or OMA1 in HAP1 WT cells, and compared the OPA1 forms with those present in CLPB KO cells (Supplementary Fig. 8). The Western blot of the CLPB KO HAP1 cells revealed the lack of the short form d of OPA1 that was also decreased by YME1L silencing in WT cells (Supplementary Fig. 8), demonstrating that OPA1 processing is altered by the absence of CLPB likely by a defect on YME1L. These findings are consistent with observations in acute myeloid leukemia (AML) cells lacking CLPB11.

a Representative Western blot and quantification of OPA1 under reducing conditions from WT and CLPB KO HAP1 cells. The relative changes in the OPA1 abundance were normalized by TOM20 levels. Data are presented as mean ± SEM. n = 7 and 6 for WT and CLPB KO cells, respectively. For data analysis, an unpaired Student T-test was used. ***p ≤ 0.001. b Representative confocal images of mitochondrial morphology in WT and CLPB KO HAP1 cells stained with 20 nM tetramethyl rhodamine methyl ester (TMRM). The percentage of elongated, intermediate and fragmented mitochondria was quantified and plotted for each condition. n = 20 cells. c Representative confocal images of mitochondrial network continuity and mtDendra2 spreading before photoactivation (PA), 50 s and 450 s after PA. d Mitochondrial fusion events frequency, left upper panel. Mitochondrial fusion classification scheme, right upper panel. Complete mitochondrial fusion events, left bottom panel. Kiss-and-run fusion events, right bottom panel. Data are presented as mean ± SEM. n = 20 cells for WT and CLPB KO. For data analysis, unpaired T-test was used for each pair of data. *p ≤ 0.05; ***p ≤ 0.001. Graphics created in BioRender. Sanchez Vazquez, V. (2025) https://BioRender.com/y87b112. e Representative images of mitochondrial morphology in HeLa cells transfected for 96 h with the indicated siRNAs and constructs together with the mitochondrially targeted fluorescent protein 4mtEMERALD. Cells were fixed and mitochondrial morphology was analyzed. Scale bar = 10 μm. f Quantification of the percentage of elongated, intermediate, and fragmented mitochondria of the experiment as in e. Data are presented as mean ± SEM. n = 30 cells for siCTR, siCLPB and siCLPB+CLPB. For data analysis, one-way ANOVA was used with post hoc Bonferroni tests for each sample. *p ≤ 0.05; ns: non-significant. Source data are provided as a Source Data file.
Next, we assessed mitochondrial network continuity and fusion activity using a mitochondrial matrix-targeted photoconvertible fluorescent protein, mtDendra2. A 3×3 µm2 ROI area was selected and illuminated with a 405 nm laser (photoconversion, PC) to switch the excitation/emission of mtDendra2. Compared with WT cells, CLPB KO cells displayed a more restricted spreading of the photoconverted mtDendra2 50 s and 450 s after PC, confirming reduced mitochondrial matrix continuity and mitochondrial dynamics (Fig. 5c). The mtDendra2 spreading kinetic can be affected by mitochondrial network continuity, fusion and mitochondrial motility. To selectively evaluate mitochondrial fusion, we measured the exchange of the mtDendra2 between photoconverted and non-photoconverted mitochondria occurring within 7 min after mtDendra2 photoconversion. The total mitochondrial fusion activity was decreased by almost 50% in the CLPB KO compared to WT (Fig. 5d, left upper panel). Given that OPA1 abundance affects mitochondrial fusion stability50, we classified the mitochondrial fusion events as complete fusion and kiss-and-run events, where complete fusion occurs when the content is completely mixed with no separation of the two organelles, and kiss-and-run events occur when a mitochondrial fusion event is followed by a fission event (Fig. 5d, right upper panel). Our findings showed a decrease in complete mitochondrial fusion events and an increase in kiss-and-run fusion events (Fig. 5d, lower panels), which are less sensitive to partial decreases in OPA150. Overall, the decrease in mitochondrial fusion is consistent with decreased functional OPA1 and the increase in kiss-and-run fusion events is consistent with decreased YME1L-mediated cleavage of OPA1.
Collectively, these results indicate that loss of CLPB results in reduced levels of the total OPA1 combined with altered OPA1 processing, which correlates with impaired mitochondrial fusion dynamics. Similar to the stable KO of CLPB in HAP1 cells, acute silencing of CLPB in HeLa cells caused mitochondrial fragmentation, which was rescued by exogenous CLPB expression, extending our observations on mitochondrial morphology to both acute and prolonged CLPB loss (Fig. 5e, f). These results highlight the relevance of CLPB in mitochondrial morphology and fusion dynamics, likely through the regulation of OPA1 levels, solubility and processing.
Skin fibroblasts from patients with mutations in the CLPB gene show alterations in mitochondrial Ca2+ homeostasis and cristae morphology
To determine whether the alterations in mitochondrial Ca2+ homeostasis in HAP1 and HeLa cells are also present in cells from individuals carrying pathogenic variants in the CLPB gene, we analyzed three distinct patient-derived skin fibroblast cell lines.
Fibroblasts from Ala591Val, Pro427Leu and Arg561Trp mutated patients were described in8,9. Briefly, the Ala591Val patient presents a homozygous autosomal recessive mutation of the C-terminal D2 ___domain which is conserved among AAA+ proteins involved51. This patient is characterized by chronic severe neutropenia and 3-MGA-uria. Patients Pro427Leu and Arg561Trp are characterized by de novo heterozygous autosomal dominant mutations and present moderate/severe neutropenia and mild/moderate 3-MGA-uria, respectively. Each of the mutations lie in the AAA+ ___domain52. Ala591Val abolishes both ATPase and disaggregase activity3. The Pro427Leu mutation mildly decreases ATPase activity while more appreciably decreasing disaggregase activity9. Arg561Trp has not been directly tested in vitro; however, other mutations at the Arg561 position abolish both ATPase and disaggregase activity10,53.
A reduction in the protein content of MICU2 is observed in fibroblasts harboring the Ala591Val and Pro427Leu mutations, whereas CLPB itself, MCU, MICU1, MICU2 and EMRE increase is associated with Arg561Trp (Fig. 6a, Supplementary Fig. 9a). Similar to the approach used with CLPB-silenced HeLa cells (Fig. 1j), we conducted an aggregation experiment on patient fibroblasts. In fibroblasts harboring the Ala591Val and Pro427Leu mutations, we observed aggregation of OPA1 and MICU2 (Fig. 6b), which aligns with the reduced MICU2 levels found in whole cell lysates from these cells (Fig. 6a). Unexpectedly, fibroblasts with the Arg561Trp mutation did not show aggregation of OPA1 or mtCU components (Fig. 6b). Considering the changes in the expression of the mtCU components (Fig. 6a, b, Supplementary Fig. 9a) and the alterations in the mitochondrial Ca2+ signaling observed in CLPB-silenced HeLa cells and CLPB KO HAP1 cells (Figs. 2, 3), we proceeded to a similar analysis in patient fibroblasts. Differently from what was observed in HeLa cells silencing CLPB, we did not observe any difference in mitochondrial Ca2+ uptake upon maximal histamine stimulation (Fig. 6c). To elucidate the apparent discrepancy between the alteration in the expression of mtCU components (Fig. 6a, Supplementary Fig. 9a), and the unchanged mitochondrial Ca2+ uptake upon stimulation in the fibroblasts from the patients (Fig. 6c), we analyzed basal [Ca2+]m by measuring Ca2+ loading of non-stimulated cells expressing 4mtGCaMP6f. We observed an increase in [Ca2+]m levels under resting conditions in the Ala591Val and Pro427Leu lines of fibroblasts (Fig. 6d), where MICU2 protein content is strongly decreased (Fig. 6a, Supplementary Fig. 9a), suggesting that these cells are characterized by alterations in the gating properties of the channel under basal conditions. No change in basal [Ca2+]m levels is associated with Arg561Trp (Fig. 6d), which also increases the mtCU components abundance (Fig. 6a, Supplementary Fig. 9a). This result might be explained by increased mitochondrial Ca2+ clearance in Arg561Trp (Supplementary Fig. 5c), because a significant increase in the abundance of TMEM65, the newly identified component of the mitochondrial efflux pathway54,55, was identified in the Arg561Trp cells (Supplementary Fig. 9b, c).

a Whole cell lysates extracted from skin fibroblasts from patients carrying mutations in the CLPB gene were subjected to Western blotting analysis for the different mtCU complex components. GRP75 was used as a loading control. n = 3. b Aggregation assay from isolated mitochondria from skin fibroblasts from patients carrying mutations in the CLPB gene. After mitochondrial isolation, the total proteins (T), along with the soluble (S) and insoluble fractions (P), were purified and analyzed via Western blotting with the indicated antibodies. PDH and TOM20 served as negative controls. n = 2. c [Ca2+]m measurements in intact skin fibroblasts of patients carrying the indicated mutations transfected with mitochondrial targeted aequorin for 48 h and challenged with maximal histamine stimulation. Left panel: representative traces; right panel: bar diagram representing the mean [Ca2+]m peak upon stimulation ± SEM. n = 6 replicates for each condition. d Resting [Ca2+]m in skin fibroblasts from patients carrying mutations in the CLPB gene, evaluated through ratiometric imaging (ex490/410) of the mitochondrial-targeted GCaMP6f transfected 48 h before the measurements. Data are presented as mean ± SEM. CTR, n = 16; CLPB A[591]V, n = 17; CLPB P[427]L, n = 16; CLPB R[561]W, n = 12. For data analysis, one-way ANOVA was used with post hoc Bonferroni tests for each sample. *p ≤ 0.05; **p ≤ 0.01. Source data are provided as a Source Data file.
Given the aggregation of OPA1 in fibroblasts carrying the CLPB mutations Ala591Val and Pro427Leu, we investigated potential alterations in mitochondrial structure by performing electron microscopy–based morphometric analysis on patient fibroblasts. Mitochondria from all three patient lines exhibited abnormal ultrastructure, with preserved mitochondrial area but increased circularity, indicative of mitochondrial fragmentation (Fig. 7a, b, f). Additionally, cristae abnormalities were observed, including increased number, decreased length, and increased width (Fig. 7c–f). Since normal mitochondrial structure is essential for maintaining electron transport chain function and ATP production, we measured OCR in the patient cells, similar to the analysis performed in CLPB-silenced HeLa cells (Fig. 4f). This analysis revealed impaired basal respiration in fibroblasts harboring the Ala591Val and Pro427Leu mutations, as well as reduced maximal mitochondrial respiration and spare capacity in all three mutations (Fig. 7g), suggesting that CLPB mutations impact both mitochondrial structure and function.

a–e Quantification of mitochondrial area a, circularity b, cristae number c, length d, and width e. Data are presented as mean ± SEM. n = 30 cells analyzed per condition. For data analysis, one-way ANOVA was used with post hoc Bonferroni tests for each sample. *p ≤ 0.05; ***p ≤ 0.001. f Representative electron microscopy images of mitochondria from control and skin fibroblasts from patients carrying mutations in the CLPB gene. g Oxygen consumption rate (OCR) in skin fibroblasts from patients with mutations in the CLPB gene (left panel). Components of OCR accounting for basal, ATP-linked, and maximal respiration, spare capacity, non-mitochondrial OCR and proton leak are quantified in panel in the right panel. Data are normalized on mean Calcein fluorescence. Data are presented as mean ± SEM. n = 10 replicates for each condition. For data analysis, one-way ANOVA was used with post hoc Bonferroni tests for each sample. *p ≤ 0.05; **p ≤ 0.01. Source data are provided as a Source Data file.
CLPB disaggregase activity is critical for mtCU and OPA1 homeostasis
To test the outcome of the different patient mutations in the same cellular background, we generated plasmids encoding the specific CLPB mutations identified in the patients studied. These plasmids, containing the Ala591Val, Pro427Leu, and Arg561Trp mutations, were overexpressed in HeLa cells for 48 h. Following this, we examined the expression levels of mtCU components. Our analysis showed that the Ala591Val and Pro427Leu mutations lead to a significant reduction in the regulatory channel isoforms MICU1 and MICU2 (Fig. 8a, Supplementary Fig. 9d). In contrast, the Arg561Trp mutation does not alter complex composition (Fig. 8a, Supplementary Fig. 9d), possibly due to the transient nature of the experiment. Furthermore, overexpression of all the mutated forms of CLPB induced aggregation of OPA1, accompanied by mild aggregation of MICU2 and m-EMRE only in Ala591Val and Pro427Leu overexpressing cells (Fig. 8b). Consistent with findings in fibroblasts from patients with CLPB mutations, these alterations in mtCU levels and stability were associated with elevated basal [Ca2+]m in Ala591Val and Pro427Leu, and no change of [Ca2+]m in Arg561Trp mutant overexpressing HeLa cells (Fig. 8c, d). Furthermore, in all three mutants, no changes in mitochondrial efflux rates were observed (Supplementary Fig. 5d).

a Representative Western blots of the mtCU components from whole cell lysates of HeLa cells transfected for 48 h with the indicated constructs. GRP75 was used as a loading control. n = 3. b Aggregation assay from isolated mitochondria from HeLa cells transfected for 48 h with the indicated constructs. PDH and TOM20 served as negative controls. n = 2. c, d Mitochondrial matrix [Ca2+] ([Ca2+]m) peak and resting [Ca2+]m, respectively in HeLa cells expressing the indicated constructs for 48 h. The ratiometric imaging (ex490/410) was performed using the mitochondrial-targeted GCaMP6f. Data are presented as mean ± SEM. n = 10 cells for all the conditions. For analysis, one-way ANOVA was used with post hoc Bonferroni tests. **p ≤ 0.01; ***p ≤ 0.001. e Resting [Ca2+]m in HAP1 CLPB KO cells and CLPB KO cells expressing the CLPB WT or the CLPB mutants for 48 h. The resting [Ca2+]m was obtained by Fluorescence Lifetime imaging using the mitochondrial-targeted sensor mtRCaMP. Data are presented as mean ± SEM. CLPB KO, n = 56 cells; CLPB WT, n = 75 cells; CLPB A[591]V, n = 27 cells; CLPB P[427]L, n = 74 cells; CLPB R[561]W, n = 80 cells, from 3 experiments. For analysis, one-way ANOVA was used with post hoc Bonferroni tests for each sample. *p ≤ 0.05; **p ≤ 0.01. f, g Average traces for cytosolic Ca2+ concentration ([Ca2+]c) and [Ca2+]m signaling in CLPB KO cells and CLPB KO cells expressing the indicated constructs for 48 h. The cytoplasmic and mitochondrial Ca2+ increases were induced by adding 1 mM CaCl2 and previously stimulated with 2 μM Tg. h, i Quantification of the [Ca2+]c and [Ca2+]m responses from f and g, respectively. Data are presented as mean ± SEM of the AUC from 370 to 570 s. CLPB KO, n = 27 cells; CLPB WT, n = 16 cells; CLPB A[591]V, n = 11 cells; CLPB P[427]L, n = 30 cells; CLPB R[561]W, n = 24 cells, from at least 5 experiments. For analysis, one-way ANOVA was used with post hoc Bonferroni tests. *p ≤ 0.05; **p ≤ 0.01. Source data are provided as a Source Data file.
Finally, we compared side-by-side WT CLPB and the patients’ mutants in the same null background by overexpressing the plasmids in HAP1 CLPB KO cells (Supplementary Fig. 10a). In line with the previous finding seen in fibroblast and HeLa cells, HAP1 CLPB KO cells expressing the mutants Ala591V and Pro427Leu showed elevated basal [Ca2+]m levels without affecting the SOCE-induced [Ca2+]c peak, [Ca2+]m rise or decay (Fig. 8e–i, Supplementary Fig. 5e, Supplementary Fig. 10b–g). No significant change in [Ca2+] was found in Arg561Trp mutant overexpressing CLPB KO cells (Fig. 8e–i, Supplementary Fig. 5e, Supplementary Fig. 10b–g). Overall, the mtCU component changes seem to correlate with the changes in basal [Ca2+]m for the various mutants in the original fibroblast background as well as in HeLa overexpression and HAP1 rescue models.
Discussion
CLPB, the first described IMS protein disaggregase in mammals, has emerged as a potential regulator of mitochondrial function. Here, we show that mitochondrial Ca2+ handling and mitochondrial fusion dynamics are impaired in different models with chronic or acute loss of CLPB and patients’ harboring mutations in the CLPB gene. These observations extend the reports that CLPB loss caused reduced solubility of the mtCU regulatory components, MICU1 and MICU2, and the mitochondrial dynamics protein OPA13 and provide the first direct evidence for the dependence of mitochondrial Ca2+ signaling and dynamics on CLPB. Our results also point to a potential cellular pathomechanism in patients harboring mutations in the CLPB gene.
Our data extend recent evidence3,18,56 that CLPB is a major regulator of the mtCU complex stability in 3 different cell types. Indeed, both chronic and acute loss of CLPB results in reduced levels of the MICU regulatory subunits and of both p-EMRE and m-EMRE. The reduction in MICU protein content aligns with the localization of CLPB in the IMS and the short half-life of MICUs proteins20. Importantly, we show that this effect is specific to the mtCU since CLPB does not interfere with the stability of MITOK and LETM1, though CLPB has been shown to solubilize several members of the SLC25 family, including the Ca2+-binding SLC25A25 and SLC25A133. Moreover, our data extends the support for the role of CLPB in preserving the solubility of components of the mtCU. In addition to the MICUs, EMRE and MCU were also detected in the insoluble mitochondrial fraction of CLPB-silenced cells, indicating a decrease in the availability of the functional mtCU channels. These changes can come together to account for the decrease in mitochondrial Ca2+ uptake capacity revealed by our study.
Another possible contributor to the reduced mitochondrial Ca2+ uptake is an alteration in the proteolysis of the mtCU components. In this regard, EMRE has been confirmed as a substrate of YME1L and AFG3L217,18,19, two AAA proteases that degrade IMS and IMM proteins40. Furthermore, recent data demonstrated that dysfunction in the disaggregase activity of CLPB impairs the integrity and function of the proteolytic SPY (STOML2, PARL, YME1L) complex34, suggesting that the control of the mtCU components could be regulated directly by CLPB or by function impairment of the proteolytic control. Therefore, we focused on investigating whether alterations in the protein content of the mtCU components are solely due to changes in their solubility and not to alterations in their proteolysis. The concomitant silencing of either YME1L or AFG3L2 together with CLPB showed restored protein levels of MICU2 and EMRE, indicating that CLPB exerts its activity on the mtCU through a dual mechanism: (1) the loss of the disaggregase activity of CLPB leads to the insolubility of the mtCU components; (2) the absence of CLPB enhances the activity of the mitochondrial AAA proteases, thus promoting the degradation of specific mtCU components. It is possible that the absence CLPB enhances the activity of the mitochondria AAA proteases through some direct mechanism or that the loss of CLPB destabilizes soluble forms of certain IMS proteins that then become more susceptible substrates for proteolysis.
We reasoned that the regulation of the stability/solubility of the mtCU components leads to alterations in the Ca2+ homeostasis. Indeed, we found that every CLPB perturbation that affected the abundance or aggregation of an mtCU component also resulted in a mitochondrial Ca2+ phenotype, a change in the resting or stimulation associated [Ca2+]m and in the underlying Ca2+ uptake. Both acute and chronic CLPB loss resulted in decreased mitochondrial Ca2+ uptake and attenuation of the IP3-, ER Ca2+ depletion and SOCE-induced [Ca2+]m rise. Stable CLPB KO also caused lower resting [Ca2+]m. However, neither [Ca2+]c nor ΔΨm was affected by CLPB deletion. Unaltered ΔΨm has also been observed in CLPB KO cells from acute myeloid leukemia (AML)11. Mitochondrial Ca2+ efflux could also contribute to the observed Ca2+ phenotypes but it is unlikely because of [Ca2+]m decay kinetic was unaltered. These findings converge on the mtCU as the source of the CLPB loss-induced Ca2+ phenotype. Notably, selective loss of MICU1/2 would be predicted to enhance mitochondrial Ca2+ uptake at submicromolar [Ca2+]c increases, which we did not find. Thus, we propose that the MICU1/2 decrease/aggregation come together with the changes in the other mtCU components e.g. EMRE to lessen mitochondrial Ca2+ uptake.
Our study revealed that CLPB not only affects mitochondrial Ca2+ homeostasis but also mitochondrial morphology and fusion activity. Mechanistically, our experiments highlighted that CLPB loss led to OPA1 downregulation. OPA1 is a key player in mitochondrial fusion dynamics and cristae junction stabilization26,50. Previous studies demonstrated decreased solubility of OPA1 in CLPB KO HAP1 cells3, and a direct interaction between CLPB and OPA1 in AML cells11. We showed that proteolytic cleavage of the long forms of OPA1 is altered since a short OPA1 form, which is processed by the protease activity of YME1L, is highly reduced in CLPB KO HAP1 cells. Similar OPA1 phenotype has been observed, and reproduced here, in cells lacking YME1L that also display fragmented mitochondria57. However, it is also possible that the decrease in the short form of OPA1 could be mediated by an increased instability of the short form and subsequent proteolytic degradation. In addition, CLPB has emerged as an important regulator of the proteolytic SPY complex, which includes YME1L34. Indeed, CLPB KO cells displayed a reduction of the short form d, whereas forms c and e were highly accumulated34. Together, our data indicate that CLPB is central to OPA1 processing, likely by modifying the activity of YME1L but also by preventing OPA1 aggregation. Furthermore, reduction of the OMA1 protein levels was found in CLPB KO cells34, indicating that CLPB might also affect the activity of other proteases. In terms of mitochondrial fusion mechanics, upon CLPB loss, the overall fusion activity decreased, and the remaining fusion events are often unstable, kiss-and-run fusion events. This aligns with our previous findings that if OPA1 decreases, first, the stable fusion events go down, whereas the kiss-and-run fusion events are spared50. Notably, OPA1 abundance has also been linked to mitochondrial Ca2+ uptake but OPA1 decrease was found to enhance the IP3-mediated [Ca2+]m rise25,58 which is the opposite of what we showed for the CLPB loss.
CLPB is associated with human disease3,59. To explore whether mitochondrial Ca2+ handling is part of the cellular pathogenesis, we studied patient-derived fibroblasts from individuals with monoallelic (Pro427Leu and Arg561Trp) or biallelic (Ala591Val) variants in the ATPase ___domain of the CLPB gene8,9. To minimize the impact of the diverse fibroblast background, we also introduced the CLPB mutations into WT HeLa cells and CLPB KO HAP1 cells. For Pro427Leu and Ala591Val, our study revealed reduced mtCU component protein levels and aggregation of the same proteins, similar to the observations made in CLPB KO HAP1 and HeLa cells but the basal [Ca2+]m was elevated and the histamine-evoked [Ca2+]m was unchanged. Unexpectedly, the Arg561Trp mutant did not affect the mtCU proteins and the [Ca2+]m phenotype. The explanation for the different [Ca2+]m alterations for CLPB KO and for Pro427Leu and Ala591Val, and for the lack of mtCU phenotype for Arg561Trp remains elusive. However, in harmony with the mtCU differences with the different mutations, the clinical course of the Pro427Leu and Ala591Val patients is more severe than that of the Arg561Trp patient. Determining the exact cause of the different mitochondrial biochemistry and function outcomes of the different patient mutations will require future studies.
In summary, by combining different cellular models and measurements, we have characterized here the functional impact of CLPB in mitochondrial Ca2+ signaling and fusion dynamics by maintaining the stability of the mtCU and OPA1 and the AAA proteases that process MICUs, OPA1 and other IMS proteins. Of note, the absence of CLPB results in a pathological phenotype sharing common features with patients carrying mutations in mtCU and OPA1 (e.g., neurological impairment and cataracts)35,36, albeit with distinctive characteristics consistent with its specific role as a disaggregase for numerous IMS proteins. Overall, these findings reveal a new potential mechanism of CLPB pathology that acts in the quality control of key IMS proteins and potentially regulates mitochondrial signaling and dynamics. Therapeutic targeting of this mechanism might be beneficial in diseases displaying impaired handling of several IMS proteins, and not limited to MICUs or OPA1 proteins.
Methods
Ethics declaration
The study was performed in compliance with the Declaration of Helsinki and conforming to the laws and regulations of the respective countries and institutes. All individuals gave written informed consent for genetic investigations and functional investigations in fibroblasts and for publication of data; this part of the study was approved by the local ethics committees, either Technical University Munich, Germany (5360/12 S) or Land Salzburg, Austria (415-E/2552/10-2019) as appropriate. Patients gave additional written consent for publication of their photos.
Patients skin fibroblasts
Skin fibroblasts were kindly provided by Dr. Saskia Wortmann (University Children’s Hospital, PMU Salzburg, Austria) and Prof. Ron Wevers (Radboud University, Nijmegen, The Netherlands).
Individual 1 (P#98, NM_001258392.3(CLPB):c.[1682 C > T]; c.[1682 C > T], p.[Ala591Val]; p.[Ala591Val]): This patient showed a phenotype at the severe end of the spectrum with severe developmental delay (unable to roll over or sit, nonverbal), truncal hypotonia, severe tetraspasticity and dystonia, severe neutropenia and recurrent infections, bilateral cataracts and death by the age of 3 years. The variant is located in a highly conserved helix in the small subdomain of the nucleotide binding ___domain (NBD) and abolishes the disaggregase activity of Skd3 in vitro3,52,53.
Individual 2 (P#2 in9, NM_030813.4 c.[1280 C > T]; [=], p.[Pro427Leu]; [=]. This patient showed a phenotype at the severe end of the spectrum with severe developmental delay (unable to roll over, nonverbal), truncal hypotonia, severe tetraspasticity, severe neonatal onset epilepsy, neutropenia during recurrent infections, and death by the age of 19 months. This variant is located adjacent to the pore-loop motif of Skd3 which contracts substrate to enable disaggregase activity9,52,53.
Individual 3 (P#5 in9, NM_030813.4 c.[1681 C > T];[=], p[Arg561Trp]. This patient (12.4 years at last follow-up) showed a phenotype of moderate severity with developmental delay (able to walk assisted with walking frame but lost this at age 9.5 years, able to express 10–20 words), severe neutropenia with recurrent infections, epilepsy. This variant mutates the key arginine residue in the R-finger motif, which helps form the ATP binding pocket between two adjacent AAA+ motifs within the hexamer. Mutations at the R561 position most potently inhibit Skd3 disaggregase activity and are strongly dominant negative9,10,52,53.
Of note, all 3 individuals had 3-methylglutaconic aciduria.
Cell culture and transfection
HeLa cells and skin fibroblasts from patients with mutations in the CLPB gene were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Thermo Fisher Scientific), supplemented with 10% FBS (Fetal Bovine Serum, Thermo Fisher Scientific), 1% penicillin (100 U/ml), and streptomycin (100 μg/ml) (Euroclone) at 37 °C under 5% CO2. HAP1 WT and HAP1 CLPB KO cells were were grown in Iscove’s Modified Dulbecco’s Medium (IMDM) with L-Glutamine and 25 mM HEPES (Gibco, #12440053) supplemented with 10% FBS (Gibco, #16000044), MEM Non-Essential Amino acids 1X (Gibco, #11140050), and 100 U/mL Penicillin/Streptomycin (Gibco, #15149121) at 37 °C under 5% CO2. HeLa cells and skin fibroblasts were transfected with a standard Ca2+-phosphate method, as already performed60. In all overexpressing experiments, an empty vector (pcDNA3.1) served as a control, while a non-targeting siRNA was used as a control in all silencing experiments. For HAP1 cells, transient transfection was done using the standard Lipofectamine 3000 reagent protocol (Invitrogen, #L3000001) with 1–2 μg DNA. For mitochondrial Ca2+ measurements, HAP1 WT and HAP1 CLPB KO cells were transfected with the mitochondrial matrix-targeted Ca2+ probes, mtGCaMP6f or mtRCaMP, and with the empty vector or with mammalian CLPB WT or mutants (A591V, P427L or R561W) for acute rescue for 48 h. In the case of confocal microscopy experiments, WT and CLPB KO cells were transfected with 1 μg of mtDendra2 for tracking the mitochondrial fusion events.
siRNA and constructs
The siRNA for CLPB was purchased from Sigma-Aldrich (cat. n° SASI_Hs01_00166516); while the siRNA for AFG3L2, YME1L and OMA1 were purchased from Invitrogen (cat. n° HSS116885; HSS116546; HSS174400, respectively). The non-targeting siRNA (MISSION siRNA Universal Negative Control #2, cat n° SIC002) was purchased from Sigma-Aldrich. pcDNA3.1-4mtGCaMP6f was previously described14,61. The CLPB (myc-Flag-tagged) construct (NM_009191) was purchased from Origene (CAT#: MR209968L3V).
Western blotting and antibodies
HeLa cells and skin fibroblasts were lysed in RIPA buffer (150 mM NaCl, 50 mM Tris-HCl pH=8, 1 mM EGTA, 1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% SDS), supplemented with Complete EDTA-free protease inhibitor cocktail (Roche Applied Science), and Phosphatase inhibitor cocktail (Roche Applied Science). Following incubation on ice for 30 min, crude extracts were centrifuged at 15000 × g for 10 min at 4 °C to remove debris, aggregates and insoluble proteins. The resulting supernatants, containing the extracted proteins were quantified using the BCA Protein Assay Kit (Thermo Fisher Scientific), and are referred as whole cell lysates. For western blotting, 20 μg of whole cell protein extracts were dissolved in LDS sample buffer (Life Technologies) supplemented with 100 mM dithiothreitol (DTT), and heated for 5 min at 95 °C. Proteins were then separated via SDS-page electrophoresis on 4–12% Bis-Tris NuPage gels (Thermo Fisher Scientific) and transferred to a nitrocellulose membrane (Thermo Fisher Scientific) using semi-dry electrophoretic transfer (Bio-Rad). Membranes were subsequently blocked for 1 h at RT with either 5% non-fat dry milk in TBS-tween (50 mM Tris-HCl, 150 mM NaCl, 0.1% Tween) or 5% Bovine Serum Albumin (BSA) (Sigma-Aldrich) in TBS-tween, based on the primary antibody used. The membranes were then incubated with the primary antibody overnight at 4 °C. Secondary HRP-conjugated antibodies, obtained from Bio-Rad, were incubated for 1 h at RT followed by detection by chemiluminescence (SuperSignal Pico, Pierce). The following primary antibodies were used: anti-MCU (1:1000, Sigma-Aldrich HPA016480), anti-MICU1 (1:1000, Sigma-Aldrich HPA037480), anti-MICU2 (1:1000, Sigma-Aldrich HPA045511), anti-EMRE (1:1000, Santa Cruz sc-86337), anti-CLPB (1:1000, ProteinTech 15743-1-AP), anti-GRP75 (1:1000, Santa Cruz sc-133137), anti-LETM1 (1:1000, Abnova M03), anti-MITOK (1:10000, Sigma-Aldrich Cat #HPA010980), anti-OPA1 (1:1000, BD biosciences Cat #612606), anti-OXPHOS (1:1000, Abcam Cat #ab110413), anti-GAPDH (1:1000, Santa Cruz, Cat #sc-25778), anti-PDH (1:1000, Cell Signaling Technology Cat #2784), anti-AFG3L2 (1:1000, Thermo Fisher Scientific Cat #PA5-95347), anti-YME1L (1:1000, Sigma-Aldrich Cat #SAB1301252), anti-eIF2-α (1:1000, Cell Signaling Technology, Cat #5324), anti-phospho-eIF2-α (Ser51) (1:1000, Cell Signaling Technology, Cat #3597), anti-TMEM65 (1:1000, Thermo Fisher Scientific, Cat #PA5-112762, anti-Tom20 (1:1000, Cell Signaling Technology, Cat #42406).
HAP1 WT and HAP1 CLPB KO cells were washed two times with PBS and then harvested with 0.05% trypsin-EDTA (Gibco, #25300054). Immediately, the cells were lysed with RIPA buffer supplemented with protease inhibitors (leupeptin, antipain, pepstatin, 1 μg/ml of each) and PMSF 1 mM. Lysates were centrifuged at 12,000 rpm for 10 min at 4 °C to remove debris, aggregates, and insoluble proteins. The resulting supernatants, containing the extracted proteins and indicated as whole cell lysates were quantified using the DC protein assay kit (Bio-Rad, #5000113), and 25 μg of protein was loaded in a NuPAGE 10% and 4–12% Bis-Tris gels (Invitrogen, #NP0322BOX) and then separated electrophoretically in reducing and non-reducing conditions. After electrophoretic separation, proteins were transferred onto LF PVDF membranes (Bio-Rad, #1704275) and those were blocked for 1 h at room temperature, following the incubation overnight at 4 °C with the primary antibodies. The following antibodies were used: anti-CLPB (Proteintech, #15743-1-AP, 1:1000), anti-MCU (Sigma, #HPA016480, 1:500), anti-MICU1 (Sigma, #HPA037480, 1:500), anti-MICU2 (Abcam, #ab101465, 1:1000), anti-EMRE (Bethyl, #A300-BL19208, 1:1000), anti-TOM20 (Proteintech, #667771, 1: 1000), anti-HSP70 (Thermo Scientific, #MA3-028, 1:1000), and anti-OPA1 (BD Lab, #612606, 1:1000). After antibodies incubation, membranes were washed three times with TBS-Tween for 10 min and then incubated with fluorescence secondary antibodies (Licor, 1:5000) for 1 h in shaking at room temperature in the dark. Proteins were detected with chemiluminescence using the Azura Biosystems software. Proteins of interest as well as loading control bands were quantified by densitometry using ImageJ software (NIH, Bethesda, USA). Western blots shown throughout the paper are representative of at least three independent experiments.
Aggregation assay
Isolation of mitochondria from HeLa cells was performed through differential centrifugation, as previously described62. To process the soluble and insoluble mitochondrial fractions, 100 μg of crude mitochondrial extracts from each sample were centrifuged for 5 min at 20,000 × g at 4 °C, in two separate tubes. In one tube the pellet was resuspended in 30 μl of LDS sample buffer (Life Technologies) and 5 μl of 50% glycerol, representing the total protein fraction (marked as T). The pellet in the other tube was resuspended in 25 μl of fractionation buffer (10 mM Tris-HCl pH=8, 1 mM EDTA, 1% Triton X-100), supplemented with Complete EDTA-free protease inhibitor cocktail (Roche Applied Science). After 10 min of incubation on ice, the sample was centrifuged for 10 min at 20,000 × g, at 4 °C. Subsequently, 25 μl of supernatant were transferred to a new tube and LDS sample buffer, and 5 μl of 50% glycerol was added. This sample represents the soluble fraction (S). The remaining pellet was resuspended in LDS sample buffer diluted in 8 M urea and supplemented with 5 μl of 50% glycerol. The latter represents the insoluble fraction (P). Samples were shaken at 40 °C for 10 min and then subjected to Western blotting analysis.
RNA extraction, reverse transcription, and qPCR
Total RNA was extracted from HeLa cells using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. RNA concentration was quantified using a NanoDrop spectrophotometer (Thermo Fisher Scientific). For cDNA synthesis, 1 μg of RNA from each sample was reverse transcribed using the SuperScript II cDNA synthesis kit (Thermo Fisher Scientific), following the manufacturer’s protocol. Oligo(dT)12–18 primer (Thermo Fisher Scientific) was used for first-strand cDNA synthesis, along with dNTPs (deoxynucleotide triphosphates, Thermo Fisher Scientific) and reverse transcriptase.
The resulting cDNA was analyzed by quantitative PCR (qPCR) using the QuantStudio 5 thermocycler (Thermo Fisher Scientific) and SYBR Green chemistry (Bio-Rad). β-Actin was used as a housekeeping gene for normalization. Gene expression levels were quantified using the 2 − ΔΔCT method63.
qPCR primer sequences were as follows:
hsATF4:
Fw 5ʹ-GAGGTGGCCAAGCACTTCAA-3ʹ
Rv 5ʹ-CAATCTGTCCCGGAGAAGGC-3ʹ
hsCHOP:
Fw 5ʹ-TTCACCACTCTTGACCCTGC-3ʹ
Rv 5ʹ-GGAATCTGGAGAGTGAGGGC-3ʹ
hsASNS:
Fw 5ʹ-GCGACCAAAAGAAGCCTTCA-3ʹ
Rv 5ʹ-AGGGAAATTTCTGGGCTGCA-3ʹ
hsChaC1:
Fw 5ʹ-CTGACCAACCACTGAAGGCA-3ʹ
Rv 5ʹ-ATTCAAGGTTGTGGCCGGAG-3ʹ
hsPCK2
Fw 5ʹ-TGCAGCAGAACACAAAGGGA-3ʹ
Rv 5ʹ-TTCCATGCTCAGCCAGTGTT-3ʹ
hsATF5:
Fw 5ʹ-GGAGCTTGGAAACCTGGTCT-3ʹ
Rv 5ʹ-TCCCTTAGCGTAGTGTCCCA-3ʹ
hsHspd1:
Fw 5ʹ-GACCAAAGACGATGCCATGC-3ʹ
Rv 5ʹ-GCACAGCCACTCCATCTGAA-3ʹ
hsHspa9:
Fw 5ʹ-CCACTGCCTCTACTGTCTCC-3ʹ
Rv 5ʹ-GGCTCCTCTCTCTGATCCCA-3ʹ
hsLonP1:
Fw 5ʹ-CCTCACACATCCACCTGCAT-3ʹ
Rv 5ʹ-AGTCATGGCCAGATTCTGCC-3ʹ
hsCLPB:
Fw 5ʹ-CCATCCAGATGTGCTCACCA-3ʹ
Rv 5ʹ-CTGGCCACATGGGAGGTCAT-3ʹ
hsACTIN:
Fw 5ʹ-CCTTTTATGGCTCGAGCGGC-3ʹ
Rv 5ʹ-CATCCATGGTGAGCTGGC-3ʹ
Measurements of mitochondrial membrane potential
HeLa cells were loaded with 20 nM tetramethyl rhodamine methyl ester (TMRM, Thermo Scientific) for 30 min at 37 °C and then transferred to the imaging system. The probe was excited at 560 nm and emission light was recorded in the 590–650 nm range. Image acquisition occurred every 1 s with a fixed 100 msec exposure time. Carbonyl cyanide 3-chlorophenylhydrazone (CCCP, 10 μM), an uncoupler of oxidative phosphorylation, was added after 20 acquisitions to completely collapse the ΔΨ. TMRM imaging was performed on an inverted microscope (Zeiss Axiovert 200) equipped with a 40×/1.3 N.A. PlanFluor objective. Excitation was performed with a Deltaram V high-speed monochromator (Photon Technology International) paired with a 75 W Xenon Arc lamp. Images were captured using an Evolve 512 Delta EMCCD (Photometrics), and the system was controlled by MetaMorph 7.5 (Molecular Devices), assembled by Crisel Instruments. Data are presented as the average background-corrected fluorescence under resting conditions and analyzed with the Fiji distribution of ImageJ.
HAP1 WT and HAP1 CLPB KO cells plated on Poly-D-Lysine coated round (25 mm) coverslips were transferred to the thermostated stage at 37 °C in presence of extracellular medium (ECM, 120 mM NaCl, 5 mM NaHCO3, 10 mM Na-HEPES, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2 mM CaCl2, 10 mM glucose and 10 mM Na-HEPES, pH 7.4) containing 0.25 % BSA. Immediately, fluorescence signal was recorded at 555/15 nm in a Leica DMI 6000B epifluorescence microscope equipped with a EM-CCD camera (Princeton Instruments) and a 40×/1.35 N.A. oil immersion objective. The images were acquired every 3 s with an exposure time of 10 msec. After 1 min of basal recording, 25 nM TMRM was added to the cells and the TMRM uptake was monitored for 25 min. Oligomycin 5 μM was added to induce the maximal TMRM uptake and 5 μM Trifluoromethoxy carbonylcyanide phenylhydrazone (FCCP) was applied at the end of the experiment to produce complete mitochondrial depolarization. Images were analyzed using Spectralyzer Software. For quantification, we measured the average TMRM fluorescence during the 30-s period prior to the addition of oligomycin.
Aequorin [Ca2+] measurements
HeLa cells were seeded on 13 mm glass coverslips at 60% confluence in a 24-well plate. Cells were then transfected with the cytosolic (cytAEQ) or the low-affinity mitochondrial (mtAEQmut) aequorin probes together with the indicated siRNA or plasmid with a standard Ca2+-phosphate procedure, as previously described60. Ca2+ measurements were performed as previously described14. Alternatively, [Ca2+] measurements were performed using a PerkinElmer EnVision plate reader with a two-injector unit. Cells were transfected with a standard Ca2+ phosphate procedure60 in 24-well plates and then transferred to a 96-well plate (1:5 dilution) the day before the experiment. One hour before the experiment, the apo-aequorin was reconstituted with 5 μM coelenterazine in 70 μL of modified KRB buffer, and luminescence for each well was measured for 1 min. The experiment was performed by first injecting 100 μM histamine to activate Ca2+ transients and then 100 μM digitonin in a hypotonic Ca2+-rich medium to discharge the remaining aequorin pool. The data were then analyzed and calibrated with a custom-made macro-enabled Excel workbook. The mitochondrial Ca2+ efflux was calculated as maximal mitochondrial Ca2+ efflux speed divided by the peak [Ca2+] after histamine stimulation.
Mitochondrial [Ca2+] measurements in permeabilized cells
HeLa cells were grown on 13 mm glass coverslips at 60% confluence and co-transfected with mtAEQmut together with the indicated constructs. One hour before the experiment, the aequorin reconstitution was performed by incubating the cells with 5 μM coelenterazine at 37 °C in a modified KRB buffer and then transferred to the perfusion chamber of a custom-built luminometer. The experiments were performed in an intracellular-like buffer (IB: 130 mM KCl, 10 mM NaCl, 2 mM K2HPO4, 5 mM succinic acid, 5 mM malic acid, 1 mM MgCl2, 20 mM HEPES pH=7.0, 1 mM pyruvate) supplemented with 100 μM EGTA (IB/EGTA). Cells were perfused for 1 min with 100 μM digitonin in IB/EGTA to permeabilize the plasma membrane. Mitochondrial Ca2+ uptake in permeabilized cells was measured by perfusing IB containing 1 μM free-[Ca2+]. Calculated free-[Ca2+] was predicted with CHELATOR software, and confirmed fluorometrically with the Fura2 free acid form64. The experiment was terminated by lysing the cells with 100 μM digitonin added to IB/EGTA, discharging the unbound aequorin pool. Mitochondrial Ca2+ uptake speed was calculated as the first derivative by using the SLOPE Excel function and smoothed for three time points. The maximal Ca2+ uptake speed is represented by the higher value reached during Ca2+ addition. All the materials used were from Sigma-Aldrich unless otherwise specified.
Basal [Ca2+]m measurements
HeLa cells and patient fibroblasts were plated on 24 mm glass coverslips and transfected with 4mtGCaMP6f. 48 h later, coverslips were transferred to an imaging chamber and incubated in 1 ml of modified KRB buffer (5.5 M glucose, 1 mM CaCl2). Imaging was performed on an inverted microscope (Zeiss Axiovert 200), equipped with a 40x /1.3 N.A. PlanFluor objective. Excitation was performed with a Deltaram V high-speed monochromator (Photon Technology International) equipped with a 75 W Xenon Arc lamp. Images were captured using an Evolve 512 Delta EMCCD (Photometrics), and the system was controlled by MetaMorph 7.5 (Molecular Devices), assembled by Crisel Instruments. To measure resting [Ca2+]m a GCaMP probe targeted to the mitochondrial matrix was generated (4mtGCaMP6f)65. This probe is ratiometric and cells were alternatively illuminated every second at 490 and 410 nm, and fluorescence was collected through a 525/50 filter (Chroma) mounted on an OptoSpin25 wheel (Cairn research). The exposure time was 100 mseconds. 100 μM Histamine was added to stimulate maximal mitochondrial Ca2+ uptake. Analysis was performed with the Fiji distribution of ImageJ. Data are presented as fluorescent ratio (490/410 nm) after frame-by-frame background correction. The mitochondrial Ca2+ efflux was calculated expressed as residual mitochondrial Ca2+ after 90 s divided by the peak [Ca2+] after histamine stimulation.
Epifluorescence Ca2+ imaging
Transfected HAP1 WT and HAP1 CLPB KO cells were plated on Poly-D-Lysine coated round (25 mm) coverslips the day before the experiment and loaded with 2 μM Fura-2/AM in extracellular medium (ECM, 120 mM NaCl, 5 mM NaHCO3, 10 mM Na-HEPES, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2 mM CaCl2, 10 mM glucose and 10 mM Na-HEPES, pH 7.4) containing 2% BSA in the presence of 0.003% Pluronic F-127 and 100 μM sulfinpyrazone for 20 min at 37 °C in the dark. After that, cells were washed three times with a Ca2+-free medium containing 0.25 % BSA. For simultaneous Ca2+ measurements (cytoplasm and mitochondria), Fura-2 was recorded at 340 and 380 nm (high and low Ca2+, respectively), while mtGCaMP6f was excited at 485/15 nm, using dual-band dichroic and emission filters, and the images were acquired every 0.5 s. For the Store Operated Ca2+ Entry (SOCE) protocol, thapsigargin was applied to block SERCA pump, discharging the ER Ca2+ store, while Ca2+ was added to the medium in order to trigger Ca2+ entry in the cytoplasm. Fura-2 fluorescence values were transformed in Ca2+ concentration (μM) after calibration in vitro by adding 1 mM CaCl2 (maximal fluorescence), followed by 10 mM EGTA/Tris, pH 8.5 to reach the minimum fluorescence. Then, using the following formula, we calculated the [Ca2+]:
Were Kd is the Ca2+ dissociation constant (0.224 μM for Fura-2), R is the ratio of the fluorescence intensities (340/380), Rmin is the fluorescence ratio in Ca2+-free conditions (EGTA), Rmax is the fluorescence ratio in Ca2+ saturated conditions (1 mM Ca2+), Sb2 is the fluorescence intensity at 380 nm in Ca2+ saturated conditions, and Sf2 is the fluorescence intensity at 380 nm in Ca2+-free conditions.
Fluorometric assay
Cell suspensions (2.4 mg total protein) of HAP1 WT and HAP1 CLPB KO cells were permeabilized with saponin (40 μg/mL) in intracellular medium (ICM, 120 mM KCl, 10 mM NaCl, 1 mM KH2PO4, 20 mM Tris-HEPES at pH 7.2) at 37 °C for 5 min with stirring in the presence of proteases inhibitors (leupeptin, antipain, pepstatin, 1 μg/ml of each), 2 mM MgATP and 2 μM thapsigargin (Enzo Life Sciences, BML-PE180-0005). After the permeabilization, 1 μM Fura-2-FF (TefLabs) and 1.5 μM TMRM were added to the cells to simultaneously measure mitochondrial Ca2+ uptake and ΔΨm, respectively, and the experiments were performed in presence of 2 mM MgATP, 20 μM CGP-37157 (Enzo Life Sciences, BML-CM119-0005) and 2 mM succinate, using a multiwavelength excitation dual-wavelength emission fluorimeter (DeltaRAM, PTI), using 340 to 380 nm excitation and 500 nm emission for Fura-FF, and 545 nm excitation and 580 nm emission for TMRM. After the addition of 20 μM Ca2+ which was used for measuring the mitochondrial Ca2+ clearance, the combination of 2 μM of the protonophore FCCP and 5 μM Oligomycin was used to produce the complete depolarization (TMRM de-quench). To inhibit mitochondrial Ca2+ uptake, we used 3 µM Ru265 (gift from Justin J. Wilson). In the case of the ΔΨm traces, these were normalized to the TMRM fluorescence after the addition of FCCP multiplied by 1.16 (TMRMFCCPx1.16). For Ca2+ measurements, calibration of Fura-2-FF was done at the end of each experiment, adding 1 mM CaCl2, followed by 10 mM EGTA/Tris, pH 8.5.
Fluorescence lifetime imaging
Fluorescence lifetime was recorded in HAP1 WT and HAP1 CLPB KO expressing the mitochondrial-targeted Ca2+ probe mtRCaMP. For those experiments in which CLPB KO cells were rescued, cells expressed the WT or mutants CLPB (A[591]V, P[427]L and R[561]W) besides mtRCaMP. Imaging acquisition was performed on an Olympus IX73 inverted microscope equipped with a 40×/1.35 N.A. UApo/340 oil objective and a monochrome camera to capture fluorescent images. During the imaging acquisition, cells were kept in 0.25% BSA ECM containing 120 mM NaCl, 5 mM NaHCO3, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2 mM CaCl2, 10 mM glucose, and 10 mM Na-HEPES, pH 7.4, at 37 °C. Lifetime measurements were done by using a Lambert Instruments LIFA system with a Lambert Instruments Multi-LED, a multicolor laser sources (L6Cc, Oxxius), and a LIFA signal generator controlled by LIFA software (version1.3.0). For excitation of the mtRCaMP, 561 nm DPSS laser was used with a 50 to 80% power and modulated at 40 MHz with an exposure time of 500 milliseconds. Rhodamine was used as a reference for calibration of the equipment with a reference lifetime of 2.7 ns.
Lifetime data were obtained using the LIFA software (version 1.3.0) and the background fluorescence signal was avoid by drawing a mask in a region of the field in absence of cell. Only cells with lifetime values above the background were used for calculating the average lifetime.
Mitochondrial morphology analysis
HeLa cells were plated on 24 mm coverslips until 50% confluence and transfected with a plasmid encoding 4mtEMERALD66, a green fluorescent protein targeted to mitochondria, together with the indicated constructs, using a standard Ca2+-phosphate method, as previously described60. 24 h after transfection, cells were washed 3 times with PBS, fixed in 4% formaldehyde solution for 10 min, and coverslips were mounted with ProLong Gold Antifade reagent (Thermo Fisher Scientific). Images were acquired on a Leica TCS-SP5-II-RS-WLL equipped with a 100×/1.4 N.A. The pinhole was set to 1 airy unit, pixel size was about 100 nm. Plan-apochromat objective. 4mtEmerald was excited by the 488 nm laser line and images were collected in the 495–535 nm range. The acquired images were then analyzed using a custom-made ImageJ script. Briefly, images were background-corrected and mitochondria were counted using the “Analyze particles” function, using 0.2 μm2 as the lower cutoff. Mitochondria were then classified according to their size as fragmented (circularity > 0.8 or length <3 μm), elongated (circularity <0.2 or length > 6 μm), and intermediate (other cases). For each cell analyzed the area occupied by elongated, intermediate, and fragmented mitochondria was normalized on the mitochondrial area, and expressed as a percentage.
Mitochondrial fusion analysis
Mitochondrial fusion on HAP1 cells was studied using a Carl Zeiss confocal Laser Scanning Microscope (LSM) 880 with AiryScan detectors at the MitoCare Center at Thomas Jefferson University. A 3×3 um ROI was used for the studies. The acquisition involved a 405 nm laser to photoactivate the mtDendra2, followed by the excitation with a 488 nm and 561 nm line laser to follow the two emissions of mtDendra2. Images were acquired with a frequency of 1 s for 450 s. During the imaging acquisition, cells were kept in 0.25% BSA ECM containing 120 mM NaCl, 5 mM NaHCO3, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2 mM CaCl2, 10 mM glucose, and 10 mM Na-HEPES, pH 7.4, at 37 °C. Individual mitochondrial fusion events were quantified and classified as previously described50. Briefly, mitochondrial fusion events were quantified by the exchange of photoactivated mtDendra2 between a donor mitochondrion (photoconverted) and an acceptor mitochondrial (non-photactivated). Complete mitochondrial fusion events were considered when, after a fusion event, there was no separation of the two organelles, while a kiss-and-run fusion event was considered when an evident separation of the two organelles was observed after a fusion event. The frequency of mitochondrial fusion events was analyzed as previously described50.
Oxygen consumption rate (OCR) measurements
To measure the oxygen consumption rate in CLPB-silenced Hela cells and patient fibroblasts, cells were cultured overnight on an XF24 microplate (Agilent, #100777-004). On the day of the experiment, cells were incubated for 2 h at 37 °C in a non-CO2 incubator in DMEM, supplemented with 1 mM sodium pyruvate, 5 mM glucose, 33 mM NaCl, 15 mg phenol red, 25 mM HEPES, and 1 mM of L-glutamine. Oxygen consumption was measured in real-time using the XF24 Extracellular Flux Analyzer (Agilent). For the measurement protocol, basal respiration was first recorded, followed by sequential injection of 1 μM oligomycin (Sigma Aldrich, #75351), 1 μM FCCP, and 1 μM rotenone (Sigma Aldrich, #45656) plus 1 μM antimycin A (Sigma Aldrich, #A8674). The results were normalized to the fluorescence of Calcein (Sigma-Aldrich, #80011-3). To determine fluorescence, cells were incubated with 2 μM Calcein for 30 min at 37 °C before measuring using a Perkin Elmer EnVision plate reader in well-scan mode, with excitation at 480/20 nm and emission at 535/20 nm.
Transmission electron microscopy
Patient fibroblasts were cultured in 24-well plates, fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) for 1 h at 4 °C, and then post-fixed with a solution containing 1% osmium tetroxide and 1% potassium ferrocyanide in 0.1 M sodium cacodylate buffer for 1 h at 4 °C. The samples were subsequently incubated overnight with 0.25% uranyl acetate at 4 °C. After three rinses in water, the cells were dehydrated through a graded ethanol series and embedded in epoxy resin (Sigma). Ultrathin sections (60–70 nm) were cut using an Ultratom V (LKB) ultramicrotome, counterstained with uranyl acetate and lead citrate, and examined with a Tecnai G2 (FEI) transmission electron microscope at 100 kV. Images were captured using a Veleta (Olympus Soft Imaging System) digital camera. For structural quantification, measurements were performed on 30 cells per condition.
Statistical analysis of data
All data were analyzed for statistical significance using GraphPad Prism software. Statistical data are presented as mean ± SEM unless otherwise specified. Depending on the experiments, a student’s t-test, Mann-Whitney t-test, and one or two-way ANOVA with Bonferroni or Dunnett’s post hoc test were applied. p ≤ 0.05 was considered significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided with this paper.
References
Chuang, E., Hori, A. M., Hesketh, C. D. & Shorter, J. Amyloid assembly and disassembly. J Cell Sci 13, jcs189928 (2018).
Eisele, Y. S. et al. Targeting protein aggregation for the treatment of degenerative diseases. Nat. Rev. Drug Discov. 14, 759–780 (2015).
Cupo, R. R. & Shorter, J. Skd3 (human ClpB) is a potent mitochondrial protein disaggregase that is inactivated by 3-methylglutaconic aciduria-linked mutations. Elife 9, e55279 (2020).
Louros, N., Schymkowitz, J. & Rousseau, F. Mechanisms and pathology of protein misfolding and aggregation. Nat. Rev. Mol. Cell Biol. 24, 912–933 (2023).
Capo-Chichi, J. M. et al. Disruption of CLPB is associated with congenital microcephaly, severe encephalopathy and 3-methylglutaconic aciduria. J. Med Genet 52, 303–311 (2015).
Saunders, C. et al. CLPB variants associated with autosomal-recessive mitochondrial disorder with cataract, neutropenia, epilepsy, and methylglutaconic aciduria. Am. J. Hum. Genet 96, 258–265 (2015).
Tucker, E. J. et al. Premature ovarian insufficiency in CLPB deficiency: transcriptomic, proteomic and phenotypic insights. J. Clin. Endocrinol. Metab. 107, 3328–3340 (2022).
Wortmann, S. B. et al. CLPB mutations cause 3-methylglutaconic aciduria, progressive brain atrophy, intellectual disability, congenital neutropenia, cataracts, movement disorder. Am. J. Hum. Genet 96, 245–257 (2015).
Wortmann, S. B. et al. Neutropenia and intellectual disability are hallmarks of biallelic and de novo CLPB deficiency. Genet Med 23, 1705–1714 (2021).
Warren, J. T. et al. Heterozygous variants of CLPB are a cause of severe congenital neutropenia. Blood 139, 779–791 (2022).
Chen, X. et al. Targeting mitochondrial structure sensitizes acute myeloid leukemia to venetoclax treatment. Cancer Discov. 9, 890–909 (2019).
Delgado de la Herran, H. et al. Systematic mapping of mitochondrial calcium uniporter channel (MCUC)-mediated calcium signaling networks. EMBO J 43, 5288-5326 (2024).
Csordas, G. et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell Metab. 17, 976–987 (2013).
Patron, M. et al. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 53, 726–737 (2014).
Kamer, K. J., Grabarek, Z. & Mootha, V. K. High-affinity cooperative Ca(2+) binding by MICU1-MICU2 serves as an on-off switch for the uniporter. EMBO Rep. 18, 1397–1411 (2017).
Balderas, E. et al. Mitochondrial calcium uniporter stabilization preserves energetic homeostasis during Complex I impairment. Nature Communications 13, 2769 (2022).
Hurst, S., Baggett, A., Csordas, G. & Sheu, S.-S. SPG7 targets the m-AAA protease complex to process MCU for uniporter assembly, Ca2+ influx, and regulation of mitochondrial permeability transition pore opening. J. Biol. Chem. 294, 10807–10818 (2019).
Konig, T. et al. The m-AAA protease associated with neurodegeneration limits MCU activity in mitochondria. Mol. Cell 64, 148–162 (2016).
Tsai, C.-W. et al. Proteolytic control of the mitochondrial calcium uniporter complex. Proc. Natl Acad. Sci. 114, 4388–4393 (2017).
Matteucci, A. et al. Parkin-dependent regulation of the MCU complex component MICU1. Sci. Rep.-Uk 8, 14199 (2018).
MacVicar, T. et al. Lipid signalling drives proteolytic rewiring of mitochondria by YME1L. Nature 575, 361–365 (2019).
Tsai, C.-W. et al. Mechanisms and significance of tissue-specific MICU regulation of the mitochondrial calcium uniporter complex. Mol. Cell 82, 3661–3676.e3668 (2022).
Petrungaro, C. et al. The Ca2+-Dependent Release of the Mia40-Induced MICU1-MICU2 Dimer from MCU Regulates Mitochondrial Ca2+ Uptake. Cell Metab. 22, 721–733 (2015).
Wu, D. et al. Comprehensive structural characterization of the human AAA+ disaggregase CLPB in the apo- and substrate-bound states reveals a unique mode of action driven by oligomerization. PLoS Biol. 21, e3001987 (2023).
Cartes-Saavedra, B. et al. OPA1 Modulates Mitochondrial Ca2+ Uptake Through ER-Mitochondria Coupling. Frontiers in Cell and Developmental Biology 9, 774108 (2022).
Frezza, C. et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126, 177–189 (2006).
Fry, M. Y. et al. In situ architecture of Opa1-dependent mitochondrial cristae remodeling. EMBO J. 43, 391–413 (2024).
Zhivotovsky, B. et al. The Effect of OPA1 on Mitochondrial Ca2+ Signaling. PLoS ONE 6, e25199 (2011).
Song, Z., Ghochani, M., McCaffery, J. M., Frey, T. G. & Chan, D. C. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 20, 3525–3532 (2009).
Ehses, S. et al. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 187, 1023–1036 (2009).
Griparic, L., van der Wel, N. N., Orozco, I. J., Peters, P. J. & van der Bliek, A. M. Loss of the intermembrane space protein mgm1/opa1 induces swelling and localized constrictions along the lengths of mitochondria. J. Biol. Chem. 279, 18792–18798 (2004).
Meeusen, S. et al. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase MGM1. Cell 127, 383–395 (2006).
Olichon, A. et al. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 278, 7743–7746 (2003).
Baker, M. J. et al. CLPB disaggregase dysfunction impacts the functional integrity of the proteolytic SPY complex. J Cell Biol 223, e202305087 (2024).
Amati-Bonneau, P. et al. OPA1-associated disorders: phenotypes and pathophysiology. Int J. Biochem Cell Biol. 41, 1855–1865 (2009).
Vecellio Reane, D., Serna, J. D. C. & Raffaello, A. Unravelling the complexity of the mitochondrial Ca2+ uniporter: regulation, tissue specificity, and physiological implications. Cell Calcium 121, 102907 (2024).
Paggio, A. et al. Identification of an ATP-sensitive potassium channel in mitochondria. Nature 572, 609–613 (2019).
Froschauer, E., Nowikovsky, K. & Schweyen, R. J. Electroneutral K+/H+ exchange in mitochondrial membrane vesicles involves Yol027/Letm1 proteins. Biochim Biophys. Acta 1711, 41–48 (2005).
Jiang, D., Zhao, L., Clish, C. B. & Clapham, D. E. Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf-Hirschhorn syndrome. Proc. Natl Acad. Sci. USA 110, E2249–E2254 (2013).
Opalinska, M. & Janska, H. AAA Proteases: Guardians of Mitochondrial Function and Homeostasis. Cells 7, 163 (2018).
Costa-Mattioli, M. & Walter, P. The integrated stress response: From mechanism to disease. Science 368, eaat5314 (2020).
Pakos-Zebrucka, K. et al. The integrated stress response. EMBO Rep. 17, 1374–1395 (2016).
Zhao, Q. et al. A mitochondrial specific stress response in mammalian cells. EMBO J. 21, 4411–4419 (2002).
Lu, H. et al. Mitochondrial Unfolded Protein Response and Integrated Stress Response as Promising Therapeutic Targets for Mitochondrial Diseases. Cells 12, 20 (2022).
Fiorese, C. J. et al. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr. Biol. 26, 2037–2043 (2016).
van der Linden, F. H. et al. A turquoise fluorescence lifetime-based biosensor for quantitative imaging of intracellular calcium. Nat. Commun. 12, 7159 (2021).
Marta, K., Hasan, P., Rodriguez-Prados, M., Paillard, M. & Hajnoczky, G. Pharmacological inhibition of the mitochondrial Ca(2+) uniporter: Relevance for pathophysiology and human therapy. J. Mol. Cell Cardiol. 151, 135–144 (2021).
D’Angelo, D., Vecellio Reane, D. & Raffaello, A. Neither too much nor too little: mitochondrial calcium concentration as a balance between physiological and pathological conditions. Front Mol. Biosci. 10, 1336416 (2023).
Song, Z., Chen, H., Fiket, M., Alexander, C. & Chan, D. C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 178, 749–755 (2007).
Liu, X., Weaver, D., Shirihai, O. & Hajnoczky, G. Mitochondrial ‘kiss-and-run’: interplay between mitochondrial motility and fusion-fission dynamics. EMBO J. 28, 3074–3089 (2009).
Zolkiewski, M. A camel passes through the eye of a needle: protein unfolding activity of Clp ATPases. Mol. Microbiol. 61, 1094–1100 (2006).
Cupo, R. R. & Shorter, J. Decoding Skd3 (Human CLPB): a Mitochondrial Protein Disaggregase Critical for Human Health. Isr. J. Chem. 64, https://doi.org/10.1002/ijch.202300153 (2024).
Cupo, R. R. et al. Unique structural features govern the activity of a human mitochondrial AAA+ disaggregase, Skd3. Cell Rep 40, 111408 (2022).
Garbincius, J. F. S. et al. TMEM65 regulates NCLX-dependent mitochondrial calcium efflux. bioRxiv https://doi.org/10.1101/2023.10.06.561062 (2023).
Vetralla, M. W., et al. TMEM65-dependent Ca2+ extrusion safeguards mitochondrial homeostasis. bioRxiv https://doi.org/10.1101/2023.10.10.561661 (2023).
Plovanich, M. et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One 8, e55785 (2013).
Wang, R. et al. Identification of new OPA1 cleavage site reveals that short isoforms regulate mitochondrial fusion. Mol. Biol. Cell 32, 157–168 (2021).
Fulop, L., Szanda, G., Enyedi, B., Varnai, P. & Spat, A. The effect of OPA1 on mitochondrial Ca(2)(+) signaling. PLoS One 6, e25199 (2011).
Mroz, D. et al. CLPB (caseinolytic peptidase B homolog), the first mitochondrial protein refoldase associated with human disease. Biochim Biophys. Acta Gen. Subj. 1864, 129512 (2020).
Tosatto, A., Rizzuto, R. & Mammucari, C. Ca(2+) Measurements in Mammalian Cells with Aequorin-based Probes. Bio Protoc 7, e2155 (2017).
Vecellio Reane, D. et al. A MICU1 splice variant confers high sensitivity to the mitochondrial Ca2+ uptake machinery of skeletal muscle. Mol. Cell 64, 760–773 (2016).
Wieckowski, M. R., Giorgi, C., Lebiedzinska, M., Duszynski, J. & Pinton, P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 4, 1582–1590 (2009).
Rao, X., Huang, X., Zhou, Z. & Lin, X. An improvement of the 2^(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinforma. Biomath. 3, 71–85 (2013).
Schoenmakers, T. J., Visser, G. J., Flik, G. & Theuvenet, A. P. CHELATOR: an improved method for computing metal ion concentrations in physiological solutions. Biotechniques 12, 870–874 (1992). 876-879.
Chen, T. W. et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 (2013).
Scarpelli, P. H. et al. Melatonin activates FIS1, DYN1, and DYN2 related-genes for mitochondria fission: Mitoemerald-GFP as a tool to visualize mitochondria structure. J. Pineal Res. 66, e12484 (2019).
Acknowledgements
The authors are grateful to Dr. Federico Caicci and Dr. Francesco Boldrin for electron microscopy. The schemes in Figs. 1a and 5d were created with BioRender, which we acknowledge. This research was supported with funding from the Italian Ministry of Health (RF-2016-02363566 to R.R.), the Italian Ministry of University and Research (PRIN 2020R2BP2E_002 to R.R., PRIN 20207P85MH to A.R.), the CARIPARO foundation (to R.R.), National Center for Gene Therapy and Drugs Based on RNA Technology, funded in the framework of the National Recovery and Resilience Plan (NRRP), Mission 4, Component 2, Investment 1.4, funded by the European Union - Next Generation EU, Project CN00000041, CUP C93C22002780006, Spoke n. 4 “Metabolic and Cardiovascular diseases” to R.R., and by NIH grants (RO1-NS132056, RO1-GM151536 to G.H.). D.V.R. was supported by European Union HORIZON-MSCA-2021-PF 101065790.
Author information
Authors and Affiliations
Contributions
D.D.A., V.H.S.V., B.C.S. Investigation, Visualization and Writing – Original Draft; D.V.R. and G.G. Investigation, R.R.C. Resources, Writing – Review & Editing and Conceptualization; D.V.R. Investigation; J.S., R.W. and S.W. Resources; F.P., H.D.H. Resources and Review & Editing; G.H., A.R. and R.R Conceptualization, Writing – Original Draft, Writing – Review & Editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Dipayan Chaudhuri and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
D’Angelo, D., Sánchez-Vázquez, V.H., Cartes-Saavedra, B. et al. Dependence of mitochondrial calcium signalling and dynamics on the disaggregase, CLPB. Nat Commun 16, 2810 (2025). https://doi.org/10.1038/s41467-025-57641-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57641-9
