
Abstract
Chronic pain and opioid overdose deaths highlight the need for non-addictive analgesics with novel mechanisms. The δ opioid receptor (δOR) is a promising target, as it lacks the respiratory depression associated with µ opioid receptor (µOR) agonists. However, early δOR full agonists caused seizures, limiting their clinical use. Partial δOR agonists may offer more controlled receptor activation than full agonists, but their development has been hindered by uncertainty regarding the molecular mechanism of partial agonism. Here we show that C6-Quino, a bitopic ligand developed through structure-based design, acts as a selective δOR partial agonist. Functional studies reveal that C6-Quino shows differential activity at G-protein and arrestin pathways and interacts with the sodium binding pocket, confirmed through cryo-EM analysis. C6-Quino demonstrates oral activity, analgesic activity in chronic pain models without causing δOR-related seizures and µOR-related adverse effects which have limited opioid usage in recent times. This discovery outlines a new strategy for developing δOR-targeted analgesics and provides a framework for optimizing signaling profiles of other Class A GPCRs.
Similar content being viewed by others

Introduction
Opioid-based pain management has targeted µ opioid receptor (µOR) agonism, a highly effective approach in the treatment of acute pain, but the overuse of µOR agonists in both clinical and illicit use has led to a major public health crisis due to their severely aversive side effects, including impaired GI transit, antinociceptive tolerance, potential for abuse and addiction, and a potentially lethal respiratory depression1. Alternative strategies employed to safely harness the potential of diverse human opioid systems (κ opioid receptor (κOR), δ opioid receptor (δOR), and µOR) remain elusive, despite their great appeal. Studies of the δ-opioid receptor (δOR) identify three unique features recommending it as an ideal target for pain management: (1) The expression level of δORs is upregulated in chronic pain states2; (2) δOR agonists are devoid of most of the negative side effects associated with µOR agonists3; and (3) δOR agonists are effective against headache disorders and migraine4. Unfortunately, first generation δOR agonists like BW373U86, SNC80, and SNC162 exhibited anti-hyperalgesic properties and convulsions at higher doses5. Later generation molecules, including ARM390, and ADL5859, showed no seizure-inducing effects6 but present other limitations. Like other δOR agonists, ARM390 developed analgesic tolerance following chronic administration although it displayed low internalization capability at δOR7. ADL5859 did not meet primary endpoints in phase 2 clinical trials targeting osteoarthritis in the knee. (https://classic.clinicaltrials.gov/ct2/show/NCT00979953) Another promising candidate, (±)-TAN67, proved inconsistent due to polypharmacology at other targets (e.g., MRGPRX2)8 and unexpected off-target effects9,10,11. Two recently developed G protein-biased δOR agonists, TRV250 and PN6047, have both completed a phase I clinical trial for neuropathic pain12,13. The correlations between G-protein and arrestin signaling with in vivo pharmacology at δOR have not been fully elucidated. It appears that adverse effects are multifactorial, possibly caused by the activation of specific G protein subtypes14,15 in addition to activation of GRK subtypes and distinct Gβγ subunits16.
Emerging evidence suggests that reduced intrinsic efficacy for G protein activation could lead to improved side effect profiles for opioids (as demonstrated by μOR-focused studies)17,18,19,20,21, antipsychotics22,23 and non-hallucinogenic psychedelic analogs24. While it remains unclear whether partial agonism at δORs confers comparable benefits across different pain modalities, these findings highlight the potential of δOR partial agonists as safer therapeutic agents. Partial agonists produce a submaximal response compared to full agonists and have been associated with fewer side effects including opioid physical dependence. Unfortunately, the molecular mechanism mediating partial agonism and its structural basis remain unclear, severely hindering the rational design of such ligands.
Recently, structural advances focusing on the conserved sodium site in a highly diverse array of class A GPCRs have proposed this site as an “efficacy-switch” controlling ligand efficacy25,26,27,28,29. Sodium acts as a negative allosteric modulator30 and is critical for the control of signaling in a number of GPCRs at physiological concentrations. Specifically, the sodium pocket is shown to undergo dramatic conformational changes upon receptor activation, with recent studies proving residues in the sodium pocket control the basal activity of the receptor and differentially modulate GPCR activation towards signaling at either G-protein or β-arrestin pathways31,32,33,34,35. Consistent with these findings, the high-resolution inactive state structure of δOR revealed a sodium ion in a pocket at the bottom of the orthosteric site36. Several mutations in the δOR sodium pocket converted the action of the δOR antagonist, naltrindole (NTI), into partial or full agonism in the β-arrestin2 pathway, further validating the sodium site as an attractive target to control ligand efficacy and modulate signaling activity through the δOR. Furthermore, recent cryo-EM studies of δOR agonist complexes have provided some structural insights into ligand-induced receptor activation, suggesting a possible role of sodium pocket interactions in modulating signaling bias37.
In the present study, we used a structure-based approach and rationally designed a highly selective δOR partial agonist (C6-Quino) adapted from NTI, intended to function as a bitopic ligand by targeting both the orthosteric site and, with a polar head group, the sodium site (Fig. 1A). A transition from partial to full agonism was observed with a shorter length of the carbon-chain linker between the groups targeting the orthosteric core and the polar head group (C5-Quino). In both cell lines transfected with human δOR and whole-cell electrophysiological recordings from rat neurons in ventral tegmental area (VTA), C6-Quino displayed partial agonist activity compared to the full agonist C5-Quino and other typical δOR agonists. We then obtained single particle cryo-EM structures of C5-Quino (2.6 Å) and C6-Quino (2.8 Å) bound to δOR, confirming their interaction with the sodium site. The cryo-EM structures coupled with molecular dynamics simulations revealed water-mediated interactions between the ligand functional groups and key residues in the sodium site, which control efficacy at both G-protein and β-arrestin signaling pathways. In mice, C6-Quino exhibited antinociceptive activity in chronic pain models of neuropathic pain, inflammatory pain, and migraine. Unlike many existing δOR agonists, C6-Quino does not cause convulsions. C6-Quino also shows reduced hyperlocomotor activity and reduced respiratory depression compared to morphine. Together, these properties suggest the utility of developing pharmacological bitopic entities for the treatment of chronic pain with limited undesired adverse effects.

A Ligand efficacy can be modulated through the allosteric site, while the orthosteric site controls potency and efficacy. Created in BioRender. CHE, T. (2025) https://BioRender.com/u29m598B Design of δOR partial agonist C6-Quino. C Binding and D functional selectivity of C6-Quino at μOR, κOR, δOR and E Gi1 and β-arrestin 2 signaling of C6-Quino at δOR, referenced to DPDPE. All assays were tested with three independent biological replicates and are represented as mean \(\pm \) SEM. Affinity, potency and efficacy values for (C, D and E) are shown in Supplementary Tables 1, 2 and 3.
Results
Developing selective δOR bitopic ligands by targeting the orthosteric site
To achieve selective activation of the human delta opioid receptor (δOR), we explored the design of agonists starting with the indole structure adapted from NTI, a known selective δOR antagonist. The morphinan skeleton is known to yield both δOR agonists and antagonists, with N-substituents switching the efficacy profile. For example, NTI (with N-methyl-cyclopropane) is an antagonist, while oxymorphindole (with N-methyl) is an agonist. NTI was chosen as our starting point because it was resolved in a high-resolution δOR X-ray crystal structure (PDB: 4N6H) with the sodium ion in a highly conserved and functionally critical sub-pocket36. This structure shows the distance between the basic amine of NTI and the carboxy group of D952.50 (residues numbered according to Ballesteros-Weinstein numbering) residue of the allosteric sodium-binding site to be 11.2 Å. To engage the allosteric sodium binding site, we swapped the cyclopropylmethyl group of the NTI core (Fig. 1B) starting from the basic nitrogen using an aliphatic chain linker (Cn where n = 3, 5, 6, and 7) connected to a positively charged guanidine group (C3- to C7-guano) as a functional “warhead”. The guanidine group was chosen as the polar head group based on its demonstrated utility in creating bitopic ligands that can engage both the orthosteric binding site and the sodium-binding pocket in opioid receptors, as evidenced by recent studies14,37.
We first identified two indole derivatives, C5-Indole and C6-Indole, as possessing the optimal linker lengths (5-carbon and 6-carbon, respectively, see Supplementary Fig. 1 through 6 for structures and synthesis of all analogs, and Supplementary Fig. 4 in particular for indole core structures) to engage the sodium binding pocket. We examined Gi1 signaling for these derivatives at κOR and μOR and found that C5-Indole showed approximately 50-fold selectivity for δOR over κOR while C6-Indole showed about 90-fold selectivity (Supplementary Fig. 7A and 7B). Neither of the two compounds showed agonist activity at μOR (Supplementary Table 1). However, since C6-Indole maintained 35% partial efficacy at κOR (Supplementary Fig. 7B), we decided to modify the chemical structure from indole to quinoline (see Supplementary Fig. 5 for structures and synthesis), aiming to increase selectivity for δOR over κOR. In our δOR computational model, the hydrophobic indole moiety is nestled within a highly hydrophobic pocket lined by V6.55, W6.58, and a flexible L7.35 residue (Supplementary Fig. 7C), which can accommodate the increased ligand size upon substitution of indole with quinoline. In contrast, the corresponding pockets in μOR and κOR exhibit polar characteristics due to non-conserved basic (K6.58) and acidic (E6.58) residues, respectively, as well as less flexible and bulkier polar side chains at position 7.35 (W7.35 in μOR and Y7.35 in κOR, also see Supplementary Fig. 838 for sequence alignment). These polar residues increase the sub-pocket’s polarity and reduce its volume, leading to unfavorable interactions and steric clashes with the larger quinoline ring. Confirming this prediction, C6-Quino maintained high potency for δOR in signaling assays (Supplementary Fig. 7D) but did not show measurable signaling at κOR (Supplementary Fig. 7E) or μOR (Supplementary Table 1). δOR subtype selectivity was further confirmed using binding affinity assays (Fig. 1C, Supplementary Fig. 9 and Supplementary Table 2). We also confirmed selectivity over NOP, at which C6-Quino did not show binding and neither of the C5/C6 derivatives showed agonist or antagonist activity (Supplementary Fig. 10). In summary, improved subtype selectivity in both functional and binding affinity assays (Fig. 1C–E, Supplementary Fig. 9 and Supplementary Table 2) was attained through the indole-quinoline modifications interacting with the orthosteric binding site.
C6-Quino was screened across a ~ 317 target panel in the PRESTO TANGO assays using β-arrestin2 as the read out through the Psychoactive Drug Screening Program at the National Institute of Mental Health39. In this platform, the signal increased >threefold above basal levels only at cholinergic receptor muscarinic 5 (CHRM5) and approached this level for HC2A, GRPG, GPR35 and GPR65. However, when a dose response analysis was carried out at these targets, C6-Quino displayed no agonist activity, strongly suggesting this result to be a false positive (Supplementary Fig. 11).
Efficacy modulation of δOR ligands by targeting the allosteric sodium site
With the engagement of the allosteric sodium site of δOR, we were aiming for potent partial agonist activity in the Gi1 protein signaling pathway and low efficacy in arrestin pathways. We found that, by varying the linker length, the potency and efficacy of bitopic ligands could be significantly changed. For example, C3-, C5-, and C6-Quino displayed high potency, while C7-Quino showed diminished potency for Gi1 at human δOR (EC50 = 1.2 nM, 2.4 nM, 9.9 nM, and 28 nM, respectively). While C3-Quino efficacy was close to that of a full agonist (Emax = 90 ± 3%), C5-, C6-, and C7-Quino were partial agonists with reduced efficacies for Gi1 (78 ± 2,% 69 ± 2% and 78 ± 2%, respectively) (Fig. 2A, B and Supplementary Table 3). For β-arrestin1 recruitment, the potency of C5-, C6-, and C7-Quino was gradually reduced with increased linker length (EC50 = 28 nM, 190 nM, 600 nM; Emax = 65 ± 2, 31 ± 2, 45 ± 3%, respectively). For β-arrestin2 recruitment, the potency and efficacy of C3-, C5, C6-, and C7-Quino were EC50 = 3.1 nM, 20 nM, 81 nM, 500 nM; Emax = 91 ± 8, 82 ± 2, 43 ± 2, 72 ± 3%, respectively) (Supplementary Fig. 12 and Supplementary Table 3). The increase in efficacy of C7-Quino compared to C6-Quino is unclear, which again emphasizes the interactions with the sodium site could achieve efficacy modulation. While signaling profiles differed, the C5–C7 derivatives all maintained similar binding affinity for the δOR (Fig. 2C and Supplementary Table 4).

A General structures of Quino and Indole scaffolds. B Gαi-1 signaling of C3, C5, C6, and C7 quino analogs at δOR using TRUPATH BRET assays. Ligand efficacy can be modulated through the allosteric site and is dependent on linker. Potency and efficacy values are shown in Supplementary Table 3. C Radioligand binding of Quino compounds with different linker lengths. Figures contain mean \(\pm \) SEM grouped from three independent biological replicates. Quantification of data can be found in Supplementary Table 4. D Gαi-1 signaling of C3, C4, C5, C6, C7 indole analogs, and NTI at δOR using TRUPATH BRET assays. Potency and efficacy values are shown in Supplementary Table 12. Figures contain mean \(\pm \) SEM grouped from three independent biological replicates. E Summary of whole cell electrophysiological recordings from neurons in the ventral tegmental area (VTA) of acute rat brain slices, showing partial agonism of C6-Quino.
Our assays also revealed that the potency and efficacy trends among the C5, C6, and C7 derivatives were consistent across the indole core (Fig. 2A and D, Supplementary Table 3), indicating that these properties are influenced by factors beyond the orthosteric site, such as the linker length. Namely, bitopics with C3 (EC50 = 1.8 nM), C4 (EC50 = 3.5 nM), C5 (2.8 nM) and C6 (5.9 nM) linkers were potent agonists preferentially recruiting Gi1, while ligands with the C7 linker showed tenfold reduced G-protein potency (EC50 = 53 nM). Similarly, we saw a loss of efficacy across the series (84 ± 2%, 80 ± 5%, 76 ± 3%, 52 ± 2% and 40 ± 2%, respectively) when compared to the reference DPDPE. Collectively, the potency and efficacy of guanidine derivatives showed a diminishing trend with the aliphatic linker chain length from C3, C4, C5, C6 to C7 while maintaining similar binding affinity (Fig. 2A and C, Supplementary Tables 3 and 4).
Based on these data, our preferred lead partial agonist was C6-Quino, which has a higher δOR selectivity, reasonable potency, and lower intrinsic efficacy at both arrestin subtypes. To further evaluate C6-Quino, we first confirmed its partial agonism and comparable potency across species by demonstrating similar efficacy at both human and murine δORs, with Emax values of 68% and 70%, respectively, while retaining nanomolar potency (Supplementary Fig. 12) in accordance with high sequence similarity (Supplementary Fig. 1338) before progressing to physiological studies in native neuronal systems. To assess partial agonism within a physiologically native and endogenous system, we conducted whole-cell electrophysiological recordings from neurons in the ventral tegmental area (VTA) in acute rat brain slices. Full δOR agonists like DPDPE and deltorphin have robust somatodendritic effects on VTA neurons40. We used voltage clamp experiments to measure changes in the holding current (Iholding) induced by bath application of 10 μM C6-Quino. To establish a proper control, we performed similar experiments in separate brain slices from the same rats, measuring responses to 10 μM DPDPE. The distribution of responses to C6-Quino varied from responses to DPDPE, with the mean change in Iholding being close to 0 pA, suggesting that the partial agonism of C6-Quino maintains in vivo (Fig. 2E).
To better understand the signaling profile of our newly identified partial agonist, C6-Quino was next compared against other known δOR ligands across various chemical classes using the TRUPATH-based G protein activation and arrestin recruitment assays41. Structurally and pharmacologically distinct ligands include peptides DPDPE, deltorphin II and Leu-Enkephalin (Leu-Enk); the diarylmethylpiperidines SNC80, SNC162, ARM390 and closely related ADL5859, and morphinan (±)-TAN67 (SB205607), in addition to C5-Quino. At Gi1, C6-Quino showed the lowest efficacy among all ligands profiled. A similar pattern was seen at both arrestin subtypes as well, with the exception of (±)-TAN67 (Supplementary Fig. 14). While efficacies of (±)-TAN67 are comparable with C6-Quino, C6-Quino displays a much lower potency (EC50 = 31 nM, Emax = 45 ± 3%) in the β-arrestin2 pathway compared to (±)-TAN67 (EC50 = 1.1 nM, Emax = 26 ± 3%) (Supplementary Table 5). A similar tendency towards decreased intrinsic efficacy was seen at other Gα-subtypes for C6-Quino compared to other known δOR ligands though in this case both C5-Quino as well as C6-Quino efficacies were similar (Supplementary Fig. 14 and Supplementary Table 5). Overall, we conclude that bitopic engagement with the sodium site leads to reduced intrinsic efficacy at G-protein and even more at arrestin signaling pathways.
CryoEM structures of δOR bound to bitopic ligands
To further confirm the interaction of the bitopic ligands with the δOR sodium site, we solved cryo-EM structures of C5- and C6-Quino (Fig. 3A) bound to δOR at a global 2.62 Å and 2.80 Å resolution, respectively (Supplementary Fig. 15 and Supplementary Table 6). The complex consists of δOR, C5- or C6-Quino, and Gαi1, Gβ1, and Gγ2 heterotrimers stabilized by scFv16. It is worth pointing out that our active δOR-G protein structures do not include any thermostabilized mutations of the receptor as presented in previous active-like δOR structures with mutations in key motifs42,43, enabling more reliable analysis of the conformational changes responsible for receptor activation.

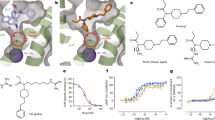
A Overall architecture of δOR-C5-Quino Gαi1Gβ1Gγ2 complex and δOR-C6-Quino Gαi1Gβ1Gγ2 complex assembly. Density maps of the ligands are zoomed in. B Comparison of C6-Quino bound δOR with previous inactive- and active-state δOR structure. δOR-naltrindole (PDB ID 4N6H), δOR-deltorphin (PDB ID 8F7S) C Comparison of ligand binding pose between C6-Quino, naltrindole, and deltorphin. D δOR-C6-Quino interactions in the orthosteric binding pocket. E Residues with distinct effects on C6-Quino and DPDPE were characterized via mutation in BRET-G protein activation or arrestin recruitment assays. Potency and efficacy values are shown in Supplementary Table 9. Figures contain data as mean \(\pm \) SEM grouped from three independent biological replicates. Statistical significance analyses of potency changes between groups are compared using one-way ANOVA with Dunnett’s multiple-comparison test. Compared to WT, Gi1 Q105A: p = 0.003, K214A: p = 0.002; β-arrestin 2 Q105A: p = 0.002, K214A: p = 0.02.
Both the δOR-C5-Quino and δOR-C6-Quino complex structures display a fully active-state and similar conformation in all the protein subunits with a root mean square deviation (r.m.s.d of 0.6 Å), despite C6-Quino being a partial agonist (Fig. 3B). This is largely attributed to the binding of intracellular G protein heterotrimer that stabilizes the receptor conformation in this specific state. Both receptor conformations show a typical outward movement of the intracellular region of transmembrane helices VI (TM6) by 12 Å compared to the NTI bound δOR structure (Cα distance of S2696.23 compared to NTI-bound inactive state, PDB: 4N6H) (Fig. 3B). This outward TM6 movement is a prominent feature of active state GPCR structures opening the intracellular site for G-protein binding. TM6 in C6-Quino structure also had additional outward movement compared to the 10 Å in the KGCHM07 agonist-bound δOR structure (PDB: 6PT3) (Fig. 3B). The additional outward TM6 movement is likely a result of G-protein binding in δOR-C6-Quino. Interestingly, the active-state conformation of the receptor pocket, lacking Na+, may have D2.50 protonated, as suggested by pKa calculations and the facilitation of Na+ egress44 by protonated D2.50. However, the presence of the cationic guanidinium group of the bitopic ligands interacting with D2.50 is likely to preclude its protonation. This guanidinium group also impacts the conformation of the pocket, preventing its full collapse. Nevertheless, this conformation differs from the inactive Na+- bound form, as it allows - or even facilitates - the activation-related outward movement of TM6.
Compared with the full agonist δOR-deltorphin-Gi1 protein structure, both TM5 and TM6 are in a similar position, although ICL3 appears to adopt a different conformation (Fig. 3B). The density map is at high resolution, providing an unambiguous placement of the ICL3 in our structure. The more ordered ICL3 in the C6-Quino compared to deltorphin-bound δOR may contribute to the stability of the ligand-specific ternary complex, because ICL3’s dynamic conformational equilibrium acts as an autoregulatory mechanism that impacts G-protein coupling to the receptor45.
As we hypothesized, the structures clearly show that both bitopic ligands occupy two pockets in δOR: the orthosteric ligand pocket and the allosteric sodium pocket (Fig. 3C). Both C5-Quino and C6-Quino bind similarly as NTI in the orthosteric site (Fig. 3C), which is expected since they were designed based on the NTI scaffold. Interestingly, C6-Quino and deltorphin barely share the binding site, with the exception of the phenol group present in the first tyrosine of deltorphin and the morphinan portion of C6-Quino, each pointing toward TM5 (Fig. 3C). This difference in binding pose is notable because both C6-Quino and deltorphin are highly selective for δOR over other opioid receptor subtypes. One observation is that the quinoline ‘address group’ of C6-Quino forms strong hydrophobic interactions with ECL3, particularly π–π interactions with W2846.58 (Fig. 3D), whereas deltorphin forms extensive interactions with ECL242. Both are consistent with findings from structures of all four opioid receptors bound to their endogenous peptides, showing that the extracellular loops of opioid receptors act as filters for selectivity43.
C5-Quino and C6-Quino form conserved interactions with orthosteric pocket residues but display unique functional activity. The basic tertiary amine of C6-Quino interacts directly with the acidic residue D1283.32 through a salt bridge at 2.9 Å, a conserved interaction observed in other ligands bound to δOR (Fig. 3D).
To obtain insights into dynamics, eight independent molecular dynamics (MD) simulations of 1000 ns each were performed for each complex. While both ligands remained bound in the pocket for all trajectories (Supplementary Fig. 17A–D and Supplementary Table 8), we observed different ratios of direct versus water-mediated-only interactions. (See Materials and Methods for additional details). C5-Quino maintained slightly closer contact to D1283.32 (C5-Quino 3.3 Å versus C6-Quino 3.5 Å on average) in the MD simulations; in both cases, direct interactions with D1283.32 were due to the protonated tertiary amine of the ligands, which can be accompanied by water-mediated interaction with D1283.32 through the tertiary hydroxyl group near the protonated amine of the ligands. (Supplementary Fig. 14A and Supplementary Figs. 18A and D). By measuring autocorrelation C(t) of each water-mediated interaction, we also showed that water-mediated interaction at Y1293.33 was significantly longer-lived for C5-Quino (939 ± 52 ns) than C6-Quino (646 ± 34 ns). (Supplementary Figs. 18B–F). Otherwise, both C5- and C6-Quino showed substantial hydrophobic interactions at M1323.36, V2175.42, I2776.51, and W2846.58 (Supplementary Fig. 19).
We performed mutagenesis screening on residues that potentially interact with C6-Quino (Fig. 3E, Supplementary Fig. 20 and Supplementary Fig. 21). Whereas several mutations reduced the agonist activity of C6-Quino in G protein activation or arrestin recruitment, the mutations Q1052.60A and K2145.39A significantly increased the potency for Gi1 activation and β-arrestin2 recruitment (Fig. 3E, Supplementary Fig. 20 and Supplementary Table 9). This effect appears to be specific for C6-Quino as the two mutations almost abolished the activity and binding affinity of reference DPDPE (Fig. 3E, Supplementary Fig. 20, Supplementary Tables 9 and 10). The binding affinity of C6-Quino increases with a Q1052.60A mutation while it remains unchanged with the K2145.39A mutation (Supplementary Fig. 20 and Supplementary Table 10). The mechanism by which Q1052.60A and K2145.39A are increasing signaling potency is difficult to determine but may involve the removal of steric clash from the side chains after mutation, although molecular dynamics simulations found no direct contact at Q1052.60 with C6-Quino (Supplementary Fig. 18). In contrast, the mutation V2816.55A shows the opposite effect to Q1052.60A or K2145.39A, i.e., it leads to a loss (~11-fold) of activity for C6-Quino, but a 3-fold increase of activity of DPDPE. The binding affinity of DPDPE is increased in the V2816.55A mutation, suggesting the increase of activity could be attributed to better binding, potentially due to less steric clash (Supplementary Fig. 20, Supplementary Table 10). The binding affinity of C6-Quino remains the same with the V2816.55A mutation, indicating that C6-Quino and DPDPE interact with this residue differently.
Direct interactions with sodium site confer unique active-state like conformational changes
The extension of the guanidine group to the sodium site leads to a re-arrangement of the sodium-binding pocket residues (Fig. 4A), yet to different extents for C5 and C6 because of the linker length. The sodium site of δOR consists of several acidic and polar residues, D952.50, N1313.35, S1353.39, N3107.45, and S3117.46, that coordinate the positively charged sodium ion in the inactive state δOR. Upon activation, these pocket residues undergo re-arrangements, leading to the disruption of the sodium-interacting networks and expulsion of the sodium ion. Specifically, C6-Quino forms a hydrogen bond with D952.50 at 2.9 Å, and with S1353.39 at 3.9 Å, while C5-Quino that has a shorter linker forms a weak interaction with the D952.50 (4.3 Å) and no direct interactions with other sodium-site residues.

A δOR-C6-Quino interactions in the sodium binding site. B Statistics on Polar Contact between D95 and Ligands in MD Trajectories. Three coarse categories of polar contacts are presented in (up) for C5 and in (bottom) for C6; they are “No Polar Contact” (i.e., D95 does not participate in direct nor water-mediated interaction with the ligand), “WaterMediatedOnly” (i.e., there are water bridge(s) formed between ligand and residues in the sodium pocket without direct interaction with D95), and “Direct Interactions” (i.e., D95 is directly involved in the polar contact, which may or may not be supplemented by additional water bridge(s)). Data are represented as mean. Error bars in SEM (n = 8 trajectories) are shown. C Conformational changes of sodium pockets residues between C6-Quino, naltrindole- and deltorphin-bound δOR. D The NPxxY motif, located at the bottom of the sodium site, undergoes unique conformational changes upon C6-Quino binding. E Structures of quino derivatives with modified “warheads” F Signaling of derivatives with different warheads demonstrates that polar interactions between ligand and allosteric site residues are crucial for ligand activity. Figures contain data as mean \(\pm \) SEM grouped from three independent biological replicates.
In MD simulations the guanidine group was predicted to almost always interact with D952.50, with “direct contact” dominating over “water-mediated-only” interactions in both C5-Quino (70% versus 27%) and C6-Quino (84% versus 14%) (Fig. 4B). The “direct contact” in C6-Quino is slightly (not statistically significantly) more frequent than that in C5-Quino, though stronger direct contact is also supported by the slightly shorter average salt bridge distance to D952.50 in C6-Quino (3.0 Å) than in C5-Quino (3.2 Å) (Supplementary Fig. 17). Interestingly, in C5-Quino, the direct salt bridge formed between the guanidine group of C5-Quino and D952.50 was often supported by an additional water bridge with D952.50 itself as well as water bridges with N3107.45 or S1353.39 (Fig. 4B, Supplementary Fig. 22). We also showed that while rapid exchange with the bulk solvent is common for those bridging waters in both ligands, C5-Quino had somewhat longer-lived water-mediated D952.50 interaction than C6-Quino (correlation time 800 ± 71 ns versus 579 ± 79 ns). (Supplementary Fig. 22B, E).
Mutation of sodium site residues D952.50, N1313.35, S1353.39, and S3117.46 to alanine lead to non-functional receptors, making it difficult to study the effects of these residues on C6-Quino function (Supplementary Fig. 21 and Supplementary Table 11). The structural comparison between C6-Quino bound and NTI-bound δOR shows several significant conformational displacements of residues, including N1313.35, S1353.39, N3107.45, and S3117.46 which display 2.8 Å side-chain movement from inactive to active states (Fig. 4C). Interestingly, when compared with the deltorphin-bound active-state δOR structure, N1313.35, N3107.45, and S3117.46, display further displacement in the C6-bound δOR structure. This is likely due to the disruption of charged interactions by the guanidine head of C6-Quino. As a direct effect of the altered arrangement in the sodium site, the NPxxY motif, adjacent to the sodium site, also displays unique conformations between C6-Quino and NTI or deltorphin bound δOR (Fig. 4D). However, this large displacement is not observed in another highly conserved DRY motif located in the intracellular end of TM3 that has been implicated in mediating receptor activation and interactions with G proteins (Supplementary Fig. 23). These conserved sites have been implicated as important regulators in transducing the signal from the extracellular pocket to intracellular G protein coupling.
The idea that interactions with residues in the sodium-binding pocket play roles in conformational dynamics and subsequent signaling and transducer coupling was also corroborated by the differences caused solely by the length of the linker for guanidine compounds we presented earlier. To confirm this, we synthesized compounds with neutral warheads like urea, a polar warhead such as amino, and with no warhead at all (Fig. 4E and Supplementary Fig. 6) to change the interaction patterns in the sodium site. As expected, we observed a drastic loss of potency and efficacy for C6-urea (EC50 = 1.4 μM and Emax = 53 ± 3% at Gi1, arrestin not detectable), C6-quino-CH2CH2NH2 (EC50 = 25 nM and Emax = 60 ± 2% at Gi1, EC50 = 180 nM and 170 nM with an Emax of 27 ± 2% and 46 ± 2% at β-arrestin1 and β-arrestin2), and all-carbon C6-quino-CH2CH3 (EC50 = 390 nM and Emax = 55 ± 2% at Gi1, EC50 = 2.1 and 1.0 μM with an Emax of 23% and 64 ± 2% at β-arrestin1 and β-arrestin2) compounds (Fig. 4F, see Supplementary Fig. 24 and Supplementary Table 12 for arrestin data). However, the C6-urea also had a reduction in binding affinity, making it difficult to determine if reduced activity is due to sodium pocket interactions or binding affinity (Supplementary Fig. 25 and Supplementary Table 13).
The binding of these bitopic analogs was examined by molecular docking. Altering the guanidine moiety to carbamide in the context of the C6-quinoline-modified NTI scaffold yields a noticeable reduction in its interaction with the allosteric pocket in δOR. In the case of the guanidine structure, the positively charged guanidine effectively establishes a strong, end-on salt bridge interaction with the negatively charged D952.50 residue46. Moreover, a robust hydrogen bond network is formed through guanidine, involving the basic carbonyl oxygen with S1353.39 and the amide hydrogen with S3117.46. In contrast, the neutral carbamide functionality only exhibits hydrogen bonding with D952.50, alongside a hydrogen bond network similar to that formed by guanidine (Supplementary Fig. 26A and C).
For the C8-quinoline derivative featuring primary amine terminal group (Supplementary Fig. 26B), while a salt bridge interaction is present between the positively charged amine and D952.50, this interaction is weaker than that of the guanidine warhead (Supplementary Fig. 26C, fewer ion-dipole interactions and less hydrogen bonding in the case of the amine). The C8-amino-based scaffold is further stabilized by two hydrogen bonds, one with S1353.39 and another with N3107.45, contributing to its enhanced potency. Although it outperforms the C6-urea variant, it still falls short when compared to C6-guanidine quinoline-modified NTI-based scaffolds in terms of functional potency.
C6-Quino displays δOR dependent anti-allodynia without adverse effects
To probe the possible utility of the partial agonist C6-Quino in vivo, we examined its effects in mice. First, we tested the anti-allodynic effects of C6-Quino in the chronic constriction injury (CCI) model of neuropathic pain. C6-Quino displayed dose-dependent inhibition of CCI-induced mechanical allodynia and a long-lasting anti-allodynic effect when administered subcutaneously (sc.) at 30 mg/kg (Fig. 5A). C6-Quino was also effective at alleviating Complete Freund’s Adjuvant (CFA)-induced peripheral hyperalgesia and nitroglycerin (NTG)-induced cephalic allodynia at the same subcutaneous dose (Fig. 5B, C). The efficacy was comparable to the prototypic δOR agonist SNC80 administered at 10 mg/kg, sc. C6-Quino was also orally active in the CCI assay after administration of a 30 mg/kg, p.o. dose, showing no significant difference in efficacy compared to sc. administration of the same dose (Fig. 5D). To assess the contribution of δOR to C6-Quino mediated anti-allodynia, mice were pretreated for 10 min with the δOR selective antagonist naltriben (NTB; 3.2 mg/kg sc.) prior to testing with C6-Quino (30 mg/kg sc.) in the CCI assay. Consistent with δOR being the major target of C6-Quino, NTB significantly antagonized C6-Quino mediated anti-allodynia (Fig. 5D).

A Mice were subjected to chronic constriction injury (CCI), a model of neuropathic pain. They demonstrate reductions in paw withdrawal 7 days after surgery (second leftmost points). This effect was not reversed by the subcutaneous administration of the vehicle (5% DMSO/95% saline, n = 14 mice) and C6-Quino at 5 mg/kg dose (n = 8 mice). However, C6-Quino displays significant anti-allodynic effects at either 10 mg/kg (n = 9 mice) or 30 mg/kg (n = 10 mice, with 7 mice at 100 and 140 min, and 3 mice at 180 min) doses similar to a one-hour pretreatment with gabapentin administered at a 50 mg/kg dose IP (n = 11 mice). B C6-Quino (30 mg/kg, IP) shows comparable anti-hyperalgesic effects to SNC80 (10 mg/kg, IP) in the Complete Freund’s Adjuvant (CFA) model of inflammatory pain. C In the nitroglycerin-induced chronic migraine model, C6-Quino (30 mg/kg, IP) completely reversed cephalic allodynia. D C6-Quino was effective after both oral and subcutaneous administration at 30 mg/kg, with its effects blocked by the δ-opioid receptor antagonist NTB in the CCI model. E Pharmacokinetic analysis confirmed reasonable brain and plasma exposure of C6-Quino following an 80-min pretreatment. F Unlike the full δ-opioid agonist SNC80, C6-Quino (30 mg/kg, sc) did not induce convulsions. G C6-Quino, unlike morphine, did not cause hyperlocomotion. H Mice were administered either saline (n = 20), vehicle (n = 12), morphine (30 mg/kg, sc; n = 12), or C6 Quino (30 mg/kg, sc; n = 12), and the breath rate was measured every 20 min for 120 min. Morphine significantly reduced the breath rate with respect to saline at 20 min, 40 min, 60 min (****p < 0.0001) and 80 min (*p = 0.0225) post-drug administration. C6-Quino was not significantly different from vehicle control as determined by 2-way ANOVA followed by Dunnett’s multiple-comparison test. All data are represented as mean ± SEM. For detailed statistical analyses of (A–D and F, G), see Supplementary Tables 15–21.
These results prompted us to evaluate the pharmacokinetics of C6-Quino after subcutaneous administration of a 30 mg/kg dose. We measured concentrations of the intact drug in brain and plasma samples at peak effect (80 min), revealing plasma concentrations of ~1200 ng/ml and a brain-plasma ratio of 1:6. At the 80 min time point, >100-fold higher drug concentrations were obtained in the brain compared to the δOR Gi1 EC50 concentration (Fig. 5E). In human liver microsomes, C6-Quino had a half-life >2 h, while sunitinib, a kinase inhibitor used as a reference, had a half-life of 27 min. C6-Quino also had a half-life >8 h in human plasma (Supplementary Table 14). C6-Quino, carbamazepine and ritonavir had 88%, 65%, and 99.2% protein binding, respectively, in initial studies in human plasma, suggesting our current lead bitopic ligand has >5% free drug available for binding its target (Supplementary Table 14).
Since δOR agonists are known to produce seizures47, we tested for the induction of pro-convulsant effects by C6-Quino at our highest analgesic dose (30 mg/kg, sc.), comparing it to the full δOR agonist SNC80 (10 mg/kg, sc.). While SNC80 produced robust seizures lasting 20 min, C6-Quino showed no signs of seizures in mice (Fig. 5F). Moreover, as seizures are attributed to a central δOR activity, we also administered C6-Quino intracerebroventricularly at a 100 nmol dose, with C6-Quino again showing no pro-convulsant activity. In contrast, intracerebroventricular administration of SNC80 and (±)-TAN67 at the equivalent dose caused robust seizures (Supplementary Fig. 27).
Pretreatment with C6-Quino (30 mg/kg, s.c.) produced neither hyper- nor hypolocomotion, while morphine at equianalgesic doses (10 or 30 mg/kg, s.c.) showed a significant increase in locomotion above baseline (Fig. 5G). In the continuous laboratory animal monitoring systems (CLAMS) assay as described before20, morphine showed respiratory depression while C6-Quino showed no decrease in breath rate after administration of a 30 mg/kg, sc. dose, although a non-significant decrease in breath rates was observed shortly after administration (Fig. 5H).
In summary, our approach to target an allosteric sodium site with a charged polar head group in bitopic ligands yielded in vivo analgesic efficacy without typically observed opioid adverse effects. Most importantly, for a ligand showing δOR agonism, C6-Quino did not cause seizures when administered either peripherally or centrally.
Discussion
In this study focusing on δOR, we employed a structure-based approach to design partial agonists by simultaneously targeting the orthosteric site and the sodium-binding pocket. This bitopic approach allows us to modulate signaling efficacy at G-proteins as well as β-arrestins to develop safer pharmacological options for the treatment of chronic pain.
Chronic pain poses a significant and complex health challenge, necessitating the development of safe and effective analgesics that avoid the adverse effects and addictive potential of conventional opioids. While µOR agonists have demonstrated effectiveness for acute pain, they are accompanied by severe side effects and abuse liabilities. In contrast, δOR agonists have shown promise in rodent models of chronic pain and generally lack the undesired effects of µOR agonists3, including respiratory depression, addiction or constipation. Therefore, they hold significant promise as targets for pain-relieving therapies in the treatment of neuropathic and inflammatory pain, conditions that currently lack effective treatment options48. However, the major limitation to δOR agonist development in the past has been that selective ligands such as SNC80 and SNC162 have produced convulsions, rendering them unsuitable for clinical use3.
Recently, two competing theories have been proposed to explain why certain drugs have beneficial pharmacological profiles: biased signaling and low efficacy signaling49,50,51,52. Biased signaling, where a ligand can activate specific signaling pathways or cellular responses through a receptor while not fully activating all possible pathways, has been proposed as a method to achieve safe therapeutics by fine-tuning the pharmacological effects. However, achieving biased signaling poses a challenge as it requires a deep understanding of the receptor’s molecular mechanisms and the ligand’s interaction with it. Low-efficacy signaling, on the other hand, refers to the partial agonist’s reduced ability to activate a receptor compared to a full agonist. Developing partial agonists with the right balance of efficacy necessitates finding a pharmacological “sweet spot”, where the ligand provides therapeutic benefits while avoiding overstimulation or inadequate activation of the receptor.
In line with this emerging need, our investigation focused on the transition from full to partial agonism. Intriguingly, as we engaged the sodium binding site, we observed such a transition between C5-Quino and C6-Quino. This difference in efficacy was even more pronounced in arrestin activity than in G-protein signaling. To better understand this difference in efficacy, we combined structural and pharmacological approaches to confirm that interactions with the sodium site play important roles in ligand efficacy modulation.
How receptor efficacy is modulated has been previously unclear. A recent study uncovered the mechanism responsible for xanomeline’s efficacy-driven selectivity among closely related muscarinic acetylcholine receptors (mAChRs)53. Xanomeline binds similarly to the inactive states of all five mAChR subtypes but differently to their active states, due to steric clashes of its tail with the second extracellular loop. Changes in efficacy are driven by the contraction of the binding pockets upon activation, a mechanism that could apply to other G-protein-coupled receptors (GPCRs) where binding pockets change size during activation. These findings indicate that small structural changes, such as addition of a few atoms to a drug molecule, can alter efficacy. These changes can be explained through classical thermodynamic principles of binding at the orthosteric site, without considering factors like binding kinetics, receptor internalization, or receptor oligomerization.
In summary, C6-Quino displayed potent analgesic effects without typical δOR adverse effects. Although our data strongly suggest that the distinct pharmacological profiles of SNC80 and C6-Quino arise primarily from their differing efficacies, we cannot exclude the possibility that structural differences between these two ligands also contribute to the observed in vivo effects. C6-Quino’s unique properties, including the absence of seizures, locomotor impairment, and reduced respiratory depression, highlight its potential as a candidate for chronic pain treatment. These findings also suggest that targeting the sodium site offers a new approach towards modulating signaling activity, thereby opening up new opportunities for the development of analgesics with enhanced safety profiles and efficacy in treating chronic pain, particularly in neuropathic and inflammatory conditions where current treatment options are limited. Further research and clinical trials will help validate the potential of δOR partial agonists as safer therapeutic agents for chronic pain. Moreover, the approach presented here serves as a versatile strategy for enhancing the signaling characteristics of other Class A GPCRs, making it a valuable tool for optimizing their performance.
Methods
Chemistry
Reagents purchased from Alfa Aesar, Fisher Scientific, and Sigma-Aldrich Chemicals were used without further purification. Reaction mixtures were purified by silica gel flash chromatography on E. Merck 230–400 mesh silica gel 60 using a Teledyne ISCO CombiFlash Rf instrument with UV detection at 280 and 254 nm. RediSep Rf silica gel normal phase columns were used with MeOH in DCM or EtOAc in Hexane solvent systems with gradients as indicated. Reversed-phase RediSep columns (C18, 100 Å, 5 micron) were used with H2O and MeCN containing 0.05% TFA. Reported yields are isolated yields upon purification of each intermediate. Final purified (purity ≥95%, LC-MS Agilent 1100 Series LC/MSD) compounds were used for the study. NMR spectra were collected using Varian 400 MHz NMR instrument at the NMR facility of Washington University School of Medicine in St. Louis. Chemical shifts are reported in parts per million (ppm) relative to residual solvent peaks at the nearest 0.01 for proton and 0.1 for carbon: CDCl3 1H: 7.26, 13C: 77.1; and CD3OD 1H: 3.31, 13C: 49.0). Peak multiplicity in NMR spectra are apparent peaks as reported by MestreNova software, namely s – singlet, d – doublet, t – triplet, q – quartet, m – multiplet for example. Coupling constant (J) values are expressed in Hz. Mass spectra were obtained at the St. Louis College of Pharmacy using the Agilent 1100 Series LC/MSD by electrospray (ESI) ionization with a gradient elution program (Ascentis Express Peptide C18 column, acetonitrile/water 5/95/95/5, 5 min, 0.05% formic acid) and UV detection (214 nm/254 nm). High-resolution mass spectra were obtained using a Bruker 10T APEX -Qe FTICR-MS and the accurate masses are reported for the molecular ion [M + H]+. Detailed experimental descriptions and characterization of the new compounds are included in the synthesis section.
Model building and refinement
Ligand models and restraints were generated by eLBOW in Phenix54. Models were first docked into the cryo-EM map in Chimera55, followed by iterative manual adjustment in COOT56, and real space refinement in Phenix. Ligand coordination was also optimized by GemSpot57. The refinement statistics were provided in Supplementary Table 6.
Molecular docking
The receptor protein δOR was extracted from the RCSB server for human (PDBID: 4N6H) representing antagonist-bound (NTI) inactive state of the receptor. The stabilizing single-chain variable fragments (triethylene glycol, L(+)-tartaric acid, oleic acids) objects, as well as sodium ions and water molecules were removed from the δOR structure leaving the receptor protein subunit and the crystallized NTI. The asparagine N131 was mutated to glycine G131 and the protein was processed via the addition and optimization of hydrogens and optimization of the side chain residues. In the final refinement of the δOR-C6-Guano complexes the N131 side chain was restored.
For comparative modeling, the mouse µOR was extracted from the RCSB server (PDBID: 5C1M) representing the agonist-bound (morphinan agonist BU72) active state of the receptor. The G protein mimetic camelid antibody object was deleted, leaving the receptor protein subunit and the crystallized BU72, and the protein was prepared by addition and optimization of hydrogens and optimization of the side chain residues. The human κOR was extracted from the RCSB server (PDBID: 6B73) representing the agonist-bound (epoxymorphinan opioid agonist MP1104) active state of the receptor. The active-state-stabilizing nanobody was deleted, leaving the receptor protein subunit, and the crystallized MP1104, and the protein was prepared by addition and optimization of hydrogens and optimization of the side chain residues.
Before performing molecular docking, ligands were conceptualized with chiral definitions, given formal charges, and their energies were optimized. All-atom docking was carried out using the energy-minimized structures for all ligands with an effort value of 10, and the ligand docking box for possible grid docking was selected to encompass the extracellular half of the protein.
A variety of rigid-protein docking combinations were used while docking into the active-state δOR. The top-scoring docking solutions from these docking experiments were further improved using successive rounds of minimization and Monte Carlo sampling of the ligand conformation, which included side-chain residues close to the ligand (within 5 Å) in the receptor orthosteric sites.
These docking studies were also performed employing C5-Quino and C6-Quino δOR bound resolved cryoEM structures, particularly with C5-Quino to be used as an initial model for docking C5-Indole bitopic ligand.
All the above-mentioned molecular modeling operations were performed in the ICM-Pro v3.9-2b molecular modeling and drug discovery suite (Molsoft LLC).
Expression and purification of scFv16 (CryoEM)
The Bac-to-Bac Baculovirus Expression System (Invitrogen) was used in Spodoptera frugiperda Sf9 insect cells to express scFv16 with a His tag attached to the C-terminus. NiCl2 (final concentration 1 mM) and CaCl2 (final concentration 5 mM) were added to cells and incubated for 6 h at 4 °C. The cells were pelleted at 4000 rpm for 30 min and the supernatant was filtered through a 0.2 μm pore size. The supernatant was then incubated with Ni-NTA resin at 4 °C overnight. The beads were washed 15–20 times the bead volume with washing buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 0.00075% MNG/CHS (10:1), 0.00025% GDN, 30 mM imidazole). Proteins were eluted with elution buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 0.00075% MNG/CHS (10:1), 0.00025% GDN, 300 mM imidazole). The proteins were then concentrated using a 10-kDa molecular weight cutoff Vivaspin concentrator to 500 μL. The sample was purified using a Superdex 200 Increase 10/300 gel filtration column in 20 mM HEPES pH 7.5, 100 mM NaCl, 0.00075% MNG/CHS (10:1), 0.00025% GDN. Fractions were combined, concentrated, and frozen.
Expression and purification of δOR with heterotrimeric Gi1 and scFv16 (CryoEM)
Expression and purification of scFv16 are described above. δOR and heterotrimeric Gi1 were expressed in Spodoptera frugiperda Sf9 insect cells using the Bac-to-Bac Baculovirus Expression System (Invitrogen). The cells were centrifuged at 4000 rpm for 15 min at 4 °C. The cells were resuspended in resuspension buffer (20 mM HEPES pH 7.5, 50 mM NaCl, 1 mM MgCl2, 2.5 units Apyrase, 1× protease inhibitor, 10 μM agonist (C5-Quino or C6-Quino)) and rotated at room temperature for 1.5 h. Following centrifugation at 30,000 rpm for 30 min at 4 °C, the cells were resuspended in solubilization buffer (40 mM HEPES pH 7.5, 100 mM NaCl, 1 mM MgCl2, 5% glycerol, 0.6% MNG/CHS (10:1), 1× protease inhibitor, scFv16 (0.2 mg), 10 μM agonist (C5-Quino or C6-Quino). Cells were incubated with solubilization buffer for 5 h at 4 °C and then centrifuged at 30,000 rpm for 30 min at 4 °C. TALON Superflow Metal Affinity Resin and imidazole (20 mM final concentration) were added to the supernatant and incubated overnight at 4 °C. The resin was washed 15–20 cv with wash buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 1 mM MgCl2, 5% glycerol, 30 mM imidazole, 0.01% MNG/CHS (10:1), 10 μM agonist (C5-Quino or C6-Quino)). Protein was eluted with elution buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 1 mM MgCl2, 5% glycerol, 250 mM imidazole, 0.01% MNG/CHS (10:1), 10 μM agonist (C5-Quino or C6-Quino)) and concentrated using a 100 kDa molecular weight cut-off Vivaspin concentrator to 500 μL. The sample was purified using a Superdex 200 Increase 10/300 gel filtration column in running buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 0.00075% MNG/CHS (10:1), 0.00025% GDN, 100 μM TCEP, 2.5 μM agonist). Fractions were combined and concentrated. His-tagged TEV protease, PNGase, Precision Protease, 0.06 mg scFv16, agonist (final concentration 5 μM), 0.005% MNG/CHS (10:1) were added and incubated overnight at 4 °C. The sample was purified again using a Superdex 200 Increase 10/300 gel filtration column in running buffer. The sample was concentrated and sent for cryo-EM analysis.
Cryo-EM sample preparation and image acquisition
Images were collected at 45,000 × g magnification resulting in a 0.88 Å pixel size. A total of 2526 movies were collected for C5-Quino (0.4–2.4 μm defocus range) and 3090 movies for C6-Quino (0.1–2.5 μm defocus range). The total dose was 54.2 e−/Å2.
Cryo-EM data collection and 3D reconstruction
The purified samples (3–4 μL) were applied to glow-discharged 300-mesh Au grids (Quantifoil R1.2/1.3) individually and vitrified using a Vitrobot mark IV (ThermoFisher). Cryo-EM imaging was performed on a Talos Artica operated at 200 kV at a nominal magnification of 45,000-times using a Gatan K3 direct electron detector at a physical pixel size of 0.88 Å. Each movie stack was recorded for 2 to 2.7 s in 60 frames at a dose rate of ~15 e−/pixel/s. Movies were collected automatically with SerialEM (version 63) using an optimized multishot array procedure58. Dose-fractionated image stacks were subjected to the beam-induced motion correction followed by contrast transfer function estimation. Particles were selected by the Blob Picker, extracted from micrographs and then used for 2D classification, 3D classification followed by non-uniform refinement. All these steps were performed in cryoSPARC59,60.
Model building and refinement
Maps from cryoSPARC were used for map building, refinement and subsequent structural interpretation. The dominant-negative Gαi1 trimer model and scFv16 model were adapted from the cryo-EM structure of the KOR–Gαi1 complex (PDB: 8DZP)15.The receptor DOR model was taken from the inactive-state DOR-Naltrindole structure (PDB: 4N6H)36. The receptor, Gαi1 protein, and scFv16 were docked into the cryo-EM map using Chimera55. The complex model (DOR-Gαi1 protein-scFv16) were manually built in Coot56, followed by several rounds of real-space refinement using Phenix54. The model statistics was validated using Molprobity61. Structural figures were prepared using Chimera or Pymol (https://pymol.org/2/).
Molecular dynamics simulations
All Molecular Dynamics (MD) simulations were conducted using the Gromacs (v.2020.3) simulation engine62 under CHARMM36 force field parameters and topologies63. Initial coordinates of the δOR-C5-Gi complex and the δOR-C6-Gi complex were derived from the corresponding cryo-EM structures wherever available; missing side chains, minor loops, and the helical lobe in Gi1 were grafted and optimized from a Gi subunit alpha-1 structure (PDBID: 5KDO) using the ICM-Pro v3.9-2d followed by structure regularization in the same software64. The full atomic systems for MD simulations were prepared on the CHARMM-GUI Interface (Version 3.8)65. The molecular content of the system is listed in Supplementary Table 9. Both complexes were embedded in a lipid bilayer made of 1,2-Dipalmitoylphosphatidylcholine (DPPC), Dioleoylphosphatidylcholine (DOPC) and Cholesterol (CHL1) with ratio DPPC:DOPC:CHOL 0.55:0.15:0.30 referencing simulations done for studies of activations in G-protein-coupled receptors66, where the ratio is within the liquid-liquid coexistence region of the phase diagram of the ternary mixture67. Initial membrane coordinates were assigned by the PPM server (Version 2.0)68 via the CHARMM-GUI Membrane Builder69. The system was solvated with TIP3P water resulting in 293,559 atoms and 286,470 atoms in total for δOR-C5-Gi complex and δOR-C6-Gi complex respectively. Both C5 and C6 ligands have a net charge of +2 as they were both protonated at the tertiary nitrogen of NTI and the guanidine group. All ligands were parametrized accordingly using the Ligand Reader & Modeller tool in CHARMM-GUI Interface (Version 3.8)65, which uses CGenFF70 as a backend to generate the necessary CHARMM general force-field parameters. For each complex, eight independent trajectories were simulated starting from the assembled system. After initial energy minimizations not exceeding 50,000 steps, six equilibration stages were performed for all systems with gradually decreased restraint force constants to various components as provided by the CHARMM-GUI membrane builder69,70. In the last two equilibration stages totaling 60 ns, temperature and pressure were controlled with the Berendsen Thermostat71 at 303.15 K and the Berendsen Barostat72 at 1 Bar respectively, where, in the first 20 ns, dihedral restraints on the protein and position restraints on the heavy atoms of protein and lipids were applied; in the last 40 ns, only position restraints on protein backbone were applied. After equilibration, production runs of up to 1000 ns under NVT condition were then conducted with a V-rescale thermostat73 at 303.15 K. The LINCS algorithm was applied to constrain bond lengths in all simulations74. All MD simulations were performed with the GPU cluster at the Center for Advanced Research Computing of the University of Southern California. MD trajectories were analyzed using the MDTraj Python package75.
Radioligand binding assay
Membranes used for the radioligand binding assay were prepared from HEK293T cells from ATCC which transiently expressed human wildtype δOR, μOR, κOR, or δOR mutants. Standard binding buffer (50 mM Tris, 0.1 mM EDTA, 10 mM MgCl2, 0.3% BSA, pH 7.4) was used to perform the binding assay in 96-well plates. For competition binding, 50 μl of 3× drug was added to 50 μl HEK239T membrane and radioligand ([3H]-DPDPE, [3H]-DAMGO or [3H]-U69,593, 1 nM final concentration). Binding incubation was at room temperature for 2 hours in the dark. After incubation, rapid vacuum filtration was applied to the reaction onto GF/A filtermat which presoaked with 0.3% polyethylenimine at 4 °C using a cell harvester. The filtermat was washed three times with 4 °C washing buffer (50 mM Tris HCl, pH 7.4) to terminate binding reaction. A scintillation sheet was melted onto the filtermat and radioligand activity was quantified using a MicroBeta counter. GraphPad Prism 10.1.1 software was used to analyze the binding results that aligned to one-site models.
Receptor cell surface expression
Expression of human wild-type δOR and mutants was determined using an enzyme-linked immunosorbent assay (ELISA). To measure expression, HEK293T cells from ATCC were transiently transfected with equal amounts of human wild-type δOR or mutant δOR DNA. After 24 h, cells were plated in a poly-L-lysine-coated 96-well white plate. Cells were plated in DMEM with 1% dialyzed FBS at a density of 60,000 cells per well and incubated overnight. The following day, plates were decanted, washed with 1\(\times\) PBS (pH 7.4), and cells were fixed with 4% (w/v) paraformaldehyde for 20 min at room temperature. Cells were washed two times with 1\(\times\) PBS and incubated with 3% BSA for one hour at room temperature. Following 3% BSA treatment, cells were incubated with anti-FLAG (M2)-horseradish peroxidase-conjugated antibody diluted (1:20,000) in 3% BSA for one hour at room temperature. Cells were then washed three times with 1\(\times\) PBS and 1-Step Ultra TMB ELISA substrate was added and incubated for 15–30 min at 37 °C. The reaction was stopped by adding 2 M sulfuric acid (H2SO4). Plates were read at 450 nm wavelength using the PHERAstar FSX plate reader. Data were analyzed using GraphPad Prism (version 10.1.1).
Bioluminescence resonance energy transfer (BRET) assay
HEK293T cells obtained from ATCC were transfected with a mixture containing the same ratio of individual human or murine opioid receptors (κOR, δOR, μOR, NOP or mδOR), Gα-RLuc8, Gβ, and Gγ-GFP2 DNA, all diluted in Opti-MEM (Gibco-ThermoFisher) to assess the G protein activation upon ligand binding. To measure ligand-induced recruitment of arrestin, HEK293T cells were subjected to transfection using a 1:5 DNA ratio of individual human opioid receptors (κOR, δOR, or μOR) fused to BRET donor Renilla luciferase (RLuc8) at the cytoplasmic C-terminus of receptors and β-arrestin2 fused to acceptor (mVenus) at the N-terminus. A ratio of 2 µl transfection reagent (Transit 2020, Mirus Biosciences):1 µg DNA was combined and incubated for 40 min for transient transfection. DMEM containing 1% dialyzed FBS was used to seed the 18–24 h-transfected cells at a density of 40,000–50,000 cells per 200 μl per well in 96-well white, clear bottom cell culture plates previously coated with poly-L-lysine. The following day, after media aspiration and cell washing with 60 µL per well of a drug buffer (1× HBSS and 20 mM HEPES, pH 7.4), 60 µL of the RLuc substrate was added per well and incubated in the dark at room temperature for 5 min. Coelenterazine 400a (5 µM) and coelenterazine h (5 µM) were used for Gα protein activation and arrestin recruitment, respectively. Subsequently, cells were treated with 30 µL of (3×) of multiple drug concentrations using a drug buffer (1× HBSS and 20 mM HEPES, pH 7.4) containing 0.3% BSA and incubated for 5 min in the dark at room temperature. Using the Mithras LB940 multimode microplate reader, BRET ratios represented by the detected GFP2 emission at 510 nm to Rluc emission at 395 nm and the detected mVenus emission at 485 nm to Rluc emission at 530 nm were used to assess the Gα protein activation and β-arrestin2 recruitment, respectively. The BRET ratios were normalized to the maximal response of the reference compound. The dose-response curve was generated by plotting the normalized data against different drug concentrations using GraphPad Prism (version 10.4.0) software to measure the ligand potency (EC50) and efficacy (Emax).
Mice
C57BL/6J mice (24–38 g) were purchased from Jackson Laboratories (Bar Harbor, ME) or CD1 mice (26–40 g) were purchased from Charles River Laboratories (Wilmington, MA). All mice were used at 8–12 weeks of age throughout the manuscript and were opioid naïve. All mice were maintained on a 12 h light/dark cycle with Purina rodent chow and water available ad libitum and housed in groups of five until testing. All animal studies reported adhere to the ARRIVE guidelines76. All procedures were preapproved by the Institutional Animal Care and Use Committee (University of Florida Gainsville and Washington University in Saint Louis) and conducted according to the 2011 NIH Guide for the Care and Use of Laboratory Animals. C57BL/6J mice were used in assays of locomotor and respiration14. CD-1 mice were used to examine anti-allodynic effects in the chronic constriction nerve injury assay of neuropathic pain.
Pharmacokinetic Study
C6-Quino was administered to male CD-1 mice through the subcutaneous route at a 30 mg/kg dose. At 80 min post administration, mice were anesthetized under isoflurane and sacrificed for tissue harvest. Blood was removed by intracardial puncture and collected in K2EDTA tubes. The tubes were centrifuged, and the supernatant (plasma) was recovered and stored at −80 °C until processing and analysis. Brains were quickly rinsed off with PBS and blot-dried, then snap frozen in liquid nitrogen and stored at −80 °C until processing and analysis.
Analytical Sample Preparations: Tissue samples were then weighed and placed into Navy bead lysis kit tubes. Naïve tissue was used to prepare Standard, Quality control (QC) and Blank samples in tissue matrix. To each sample tube was added the appropriate volume of cold acetonitrile:water (3:1) to achieve a tissue concentration of 200 mg/mL. Tubes were placed in a bead beater for 3 min, then centrifuged at 3200 rpm for 5 min at 4 °C. The supernatants were transferred in Eppendorf tubes and stored at −80 °C until the day of analysis. The day of the analysis, the samples were thawed on ice, mixed vigorously then centrifuged at 3200 rpm for 5 minutes at 4 °C. Standards and QCs were freshly prepared by spiking different concentration in naïve matrix (plasma or brain). Supernatants (30 ml) were collected and transferred into a 96-well plate. Then cold acetonitrile (150 μl) spiked with internal standard (IS) was added to blanks, standards, QCs and unknown samples. Only cold acetonitrile (150 μl) was added to the double blanks (DB). Samples were mixed vigorously for 10 min then centrifuged at 3200 rpm for 10 minutes at 4 °C. Supernatants were transferred into a 96-well plate, evaporated to dryness under nitrogen at RT. Samples were reconstituted in 100 µL of 0.1% v/v formic acid in water:acetonitrile (90:10). Plate was sealed, vortexed during 5 min, briefly centrifuged, then submitted for LC/MS analysis.
LC-MS/MS analysis of samples were completed using a mass spectrometer (SCIEX Triple Quad 5500+ system—QTRAP Ready) link to a LC system (ExionLC AD—HPLC system). Data analyses were performed using SCIEX OS software. HPLC and MS/MS parameters are provided in the accompanying tables.
Mechanical sensitivity assessment
Mechanical sensitivity was determined using manual von Frey hair stimulation of the hindpaw28,31. For all behavioral experiments, mice were randomly assigned to groups following the first basal test for mechanical sensitivity. The experimenter was blinded to the drug or condition being tested. No adverse effects were observed in any of the experiments. All mice were tested in a separate behavioral testing room with low-light (~ 35–50 lux) and low-noise conditions, between 09:00 and 16:00. For all behavioral tests, mice were habituated to the testing rack for 2 days prior to the first test day, and on each test day for 60 min prior to the first measurement. During the habituation days, a medium-force von Frey filament was used to accustom the mice to the filaments. To assess mechanical sensitivity, the threshold for responses to punctate mechanical stimuli (mechanical allodynia) was tested according to the up-and-down method.
Chronic constriction injury (CCI)
Male CD-1 mice anesthetized with isoflurane were subjected to CCI, as described previously77,78 to induce mechanical allodynia and hyperalgesia. Briefly, after anesthetization, mice were subjected to surgery in which an incision was made along the surface of the biceps femoris of the right hind paw77. Blunt forceps were used to split the muscle and expose the right sciatic nerve. The tips of the two 0.1–10 µL pipette tips facing opposite directions were passed under the sciatic nerve to allow for the easy passage of two sutures under the nerve, 1 mm apart. The sutures were tied loosely around the nerve and knotted twice, and the skin was closed with two 9 mm skin staples. The mice were allowed to recover for 7 days prior to baseline von Frey testing, as described above, to confirm the induction of mechanical allodynia in each mouse. A response to von Frey fibers of lower force, otherwise not observed in naïve mice, was an indication of mechanical allodynia, consistent with the demonstration of neuropathic pain. The mice confirmed as allodynic were then administered either the control vehicle (5:95 DMSO:saline), or Gabapentin (50 mg/kg, IP), or the test compound C6-Quino (5, 10, mg/kg, sc or 30 mg/kg sc/po). To evaluate the potential antagonistic effects of naltriben (NTB), a selective δ-opioid receptor antagonist, mice were pretreated with NTB (3.2 mg/kg, sc) or vehicle (5:95% DMSO:saline) 10 min prior to the administration of C6-Quino (30 mg/kg, sc). Each mouse was then tested for the threshold for mechanical allodynia every 20 min up to 180 min post-treatment with the use of calibrated von Frey filaments as described above, until the threshold that induced paw withdrawal was determined as a measure of nocifensive behavior77.
Complete Freund’s Adjuvant (CFA) allodynia
Complete Freund Adjuvant is an established model of inflammatory pain that induces mechanical sensitization2. C57BL/6J male and female mice had pre-CFA mechanical threshold baseline recorded prior to being injected with a 15-µL of CFA cell suspension (Sigma-Aldrich, St. Louis, MO) into the left hind paw using a Hamilton syringe. Seventy-two hours after Complete Freund’s Adjuvant (CFA) injection to the left hind-paw, peripheral von Frey mechanical thresholds were taken and the mice were separated into four groups: mice that were given vehicle (1:1:8 DMSO/cremophor/saline; IP), C6-Quino (30 mg/kg; IP, dissolved in 1:1:8 DMSO/cremophor/saline), gabapentin (50 mg/kg, IP dissolved in saline), and SNC80 (10 mg/kg; IP dissolved in saline). Post-drug von-Frey measurements were taken 1 h post administration.
Cephalic mechanical thresholds
C57BL/6J mice were treated with vehicle (saline(SAL)) or nitroglycerine (NTG) every other day for 9 days in the NTG model of chronic migraine. Cephalic mechanical thresholds were assessed on days 1, 5, and 9. On each test day, basal thresholds were first measured before the mice received the NTG or vehicle injection. After the basal measurement, NTG was administered, and cephalic mechanical thresholds were retested 2 h later to evaluate post-treatment effects. On day 10, baseline measurements were again taken, and NTG-treated mice continued to show significant cephalic allodynia. Mice were then administered either vehicle, SNC80 (10 mg/kg, IP) or C6-Quino (30 mg/kg, IP) and tested 80 min later.
Respiratory and locomotor effects
Respiration rates and spontaneous ambulation rates were monitored using the automated, computer-controlled Comprehensive Lab Animal Monitoring System (CLAMS, Columbus Instruments, Columbus, OH) as described previously28,31. Awake, freely moving adult male C57BL6/J mice were habituated in closed, sealed individual apparatus cages (23.5 cm × 11.5 cm × 13 cm) for 60 min before testing. A baseline for each animal was obtained over the 60-min period before drug injection, and testing began immediately post-injection. Saline, Vehicle (5:95 DMSO: saline), morphine (30 mg/kg, sc), or C6-Quino (30 mg/kg, sc) were administered, and five minutes later, mice were placed in the CLAMS testing cages for 200 min. Respiration rate (breaths/min) of each mouse was measured using a pressure transducer built into the sealed CLAMS cage. Simultaneously, locomotion was assessed using infrared beams located in the floor of the cage. Ambulation was determined by counting the number of sequential breaks of adjacent beams. Data are expressed as a percentage of the vehicle control response for respiratory depression and as average locomotion (XAMB)—defined as the total number of beam breaks during each 5-min time period.
Seizure assay
To assess drug-induced seizurogenic activity79, C57BL/6J male mice were placed in a clear plastic cylinder (25 cm diameter, 35 cm height) immediately following drug injection and their activity was recorded in a well-lit, quiet room. A recording time of 90 min was chosen for the tested compounds based on previous observations of seizures time lengths in experiments with 30 mg/kg, sc C6-Quino and SNC80 at 10 mg/kg, sc along with vehicle. If animals were not presenting seizure activity after 30 min, the recording time was shortened accordingly. Seizure severity was scored based on the modified Racine scale (half-scores allowed) in bins of 3–5 min. Onset to first seizure symptom, onset to highest Racine score, and highest Racine score were also assessed. In additional testing, SNC80, (±)-TAN67 and C6-Quino were each examined for seizure activity after i.c.v. administration of a 100 nmol dose. Additional control mice received only i.c.v. vehicle (50% DMSO:saline).
Plasma binding
Plasma protein binding was determined using equilibrium dialysis in the Thermo Scientific™ RED Device. The plate was incubated with shaking at 37 °C for 6 h. The concentration of the drug in the plasma vs buffer compartments was determined by LC-MS/MS. The fraction bound in plasma was calculated as ([plasma] – [buffer])/[plasma]).
Plasma stability
Plasma stability was evaluated by incubating 10 µM test compound with undiluted plasma at 37 °C with aliquots removed at multiple time points out to four hours. Aliquots were added to acetonitrile (5X, v: v) to stop any enzymatic activity and held on ice. Samples were centrifuged through a Millipore Multiscreen Solvinter 0.45-micron low binding PTFE hydrophilic filter plate and analyzed by LC-MS/MS. Data were log transformed and represented as half-life.
Liver microsomal assays
Liver microsomal assays were performed following the previously reported protocols80. Briefly, microsome stability was evaluated by incubating C6-Quino (1 μM) with 1 mg/mL hepatic microsomes (human, rat, or mouse) in a 100 mM potassium phosphate buffer, pH 7.4. The reactions were held at 37 °C with continuous shaking. The reaction was initiated by adding NADPH to a final concentration of 1 mM. The final incubation volume was 300 μL, and 40 μL aliquots were removed at 0, 5, 10, 20, 40, and 60 min. Each removed aliquot was added to 160 μL acetonitrile to stop the reaction and precipitate the protein. NADPH dependence of the reaction was evaluated in parallel incubations without NADPH. C6-Quino metabolism by CYP3A4 was verified using a validated 3A4 inhibitor assay, in which 1 μM C6-Quino in the presence of 1 μM ketoconazole for selective 3A4 blockade. At the end of the assay, the samples were centrifuged through a 0.45-μm filter plate (Millipore Solventer low binding hydrophilic plates, cat# MSRLN0450) and analyzed by LC–MS/MS. The data were log-transformed, and the results were reported as half-life. Note: Protein concentration for both liver microsomes as well as S9 was 1 mg/mL. For S9 fraction experiments, we simply swapped microsomes with S9. S9 was bought from XenoTech. Verapamil was used as control for S9 experiments. The half-life for verapamil was 24.14 min (data not shown).
Assessment of off–target activity using PRESTO-Tango GPCR-ome
To identify potential off-target activity of C6-Quino, we used the National Institutes of Mental Health Psychoactive Drug Screen Program. C6-Quino was first tested for activity against 330 non-olfactory GPCRs using the PRESTO-Tango GPCRome screening β-arrestin2 recruitment assay39 at 10 μM. The activity at each receptor was measured in quadruplicate. Screening of compounds was accomplished using previously described methods with several modifications (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4424118/)53. First, HTLA cells were plated in DMEM with 2% dialyzed FBS and 10 U/mL penicillin–streptomycin. Next, the cells were transfected using an in-plate PEI method (https://www.ncbi.nlm.nih.gov/pmc/articlesPMC4012321/). PRESTO-Tango receptor DNA constructs were resuspended in OptiMEM and hybridized with PEI prior to dilution and distribution into 384-well plates and subsequent addition to cells. After overnight incubation, diluted drugs in DMEM with 1% dialyzed FBS were added to cells without replacement of the medium. The remaining steps of the PRESTO-Tango protocol were followed as previously described39.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Characterization data of all compounds is available in the Supporting Information and the biological data are available in the Supporting information and Source Data file provided with this paper. The coordinate and cryo-EM map of DOR-Gi1-C5-Quino and DOR-Gi1-C6-Quino have been deposited to Protein Data Bank (PDB) and Electron Microscopy Data Bank (EMDB) with accession code 9CGK (EMD-45582 [https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-45582]) and 9CGJ (EMD-45581 [https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-45581]), respectively (https://www.rcsb.org/). The authors declare that all the data supporting the findings of this study are available within the article, extended data and supplementary information files. The trajectories for the Molecular Dynamics simulations have been deposited in Zenodo. https://doi.org/10.5281/zenodo.13840672 for simulations with C5-Quino and in Zenodo. https://doi.org/10.5281/zenodo.13840648 for simulations with C6-Quino. Source data are provided with this paper.
References
Blanco, C. & Volkow, N. D. Management of opioid use disorder in the USA: present status and future directions. Lancet 393, 1760–1772 (2019).
Cahill, C. M. et al. Delta opioid receptor activation modulates affective pain and modality‐specific pain hypersensitivity associated with chronic neuropathic pain. J. Neurosci. Res. 100, 129–148 (2022).
Quirion, B., Bergeron, F., Blais, V. & Gendron, L. The Delta-Opioid receptor; a target for the treatment of pain. Front. Mol. Neurosci. 13, 52 (2020).
Pradhan, A. A., Smith, M. L., Zyuzin, J. & Charles, A. Opioid receptor agonists inhibit migraine‐related hyperalgesia, aversive state and cortical spreading depression in mice. Br. J. Pharmacol. 171, 2375–2384 (2014).
Jutkiewicz, E. M. et al. δ-Opioid agonists: differential efficacy and potency of SNC80, Its 3-OH (SNC86) and 3-Desoxy (SNC162) derivatives in sprague-dawley rats. J. Pharm. Exp. Ther. 309, 173–181 (2004).
Chu Sin Chung, P. et al. Delta opioid receptors expressed in forebrain GABAergic neurons are responsible for SNC80-induced seizures. Behav. Brain Res. 278, 429–434 (2015).
Pradhan, A. A. et al. Ligand-directed trafficking of the δ-opioid receptor in vivo: two paths toward analgesic tolerance. J. Neurosci. 30, 16459–16468 (2010).
Lansu, K. et al. In silico design of novel probes for the atypical opioid receptor MRGPRX2. Nat. Chem. Biol. 13, 529–536 (2017).
Yajima, Y. et al. Modulation of NMDA- and (+)TAN-67-induced nociception by GABA(B) receptors in the mouse spinal cord. Life Sci. 68, 719–25 (2000).
Nielsen, C. K. et al. δ-Opioid Receptor Function in the Dorsal Striatum Plays a Role in High Levels of Ethanol Consumption in Rats. J. Neurosci. 32, 4540–4552 (2012).
Yajima, Y. et al. Effects of differential modulation of μ-, δ- and κ-opioid systems on bicuculline-induced convulsions in the mouse. Brain Res. 862, 120–6 (2000).
Conibear, A. E. et al. A novel G protein–biased agonist at the δ opioid receptor with analgesic efficacy in models of chronic pain. J. Pharm. Exp. Ther. 372, 224–236 (2020).
Fossler, M. J. et al. A phase I, randomized, single‑blind, placebo‑controlled, single ascending dose study of the safety, tolerability, and pharmacokinetics of subcutaneous and oral TRV250, a G protein-selective delta receptor agonist, in healthy subjects. CNS Drugs 34, 853–865 (2020).
Faouzi, A. et al. Structure-based design of bitopic ligands for the µ-opioid receptor. Nature 613, 767–774 (2023).
Han, J. et al. Ligand and G-protein selectivity in the κ-opioid receptor. Nature 617, 417–425 (2023).
Che, T. & Roth, B. L. Molecular basis of opioid receptor signaling. Cell 186, 5203–5219 (2023).
Gillis, A. et al. Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci. Signal. 13, eaaz3140 (2020).
Chakraborty, S. et al. A novel mitragynine analog with low-efficacy mu opioid receptor agonism displays antinociception with attenuated adverse effects. J. Med. Chem. 64, 13873–13892 (2021).
Bhowmik, S. et al. Site selective C–H functionalization of Mitragyna alkaloids reveals a molecular switch for tuning opioid receptor signaling efficacy. Nat. Commun. 12, 3858 (2021).
Uprety, R. et al. Controlling opioid receptor functional selectivity by targeting distinct subpockets of the orthosteric site. eLife 10, e56519 (2021).
Ople, R. S. et al. Signaling modulation mediated by ligand water interactions with the sodium site at μOR. ACS Cent. Sci. https://doi.org/10.1021/acscentsci.4c00525 (2024).
Peris, L. & Szerman, N. Partial agonists and dual disorders: focus on dual schizophrenia. Front. Psychiatry 12, 769623 (2021).
Lieberman, J. A. Dopamine partial agonists: a new class of antipsychotic. CNS Drugs 18, 251–267 (2004).
Cunningham, M. J. et al. Pharmacological mechanism of the non-hallucinogenic 5-HT 2A agonist ariadne and analogs. ACS Chem. Neurosci. 14, 119–135 (2023).
Gutiérrez-de-Terán, H. et al. The role of a sodium ion binding site in the allosteric modulation of the A2A adenosine G protein-coupled receptor. Structure 21, 2175–2185 (2013).
Miller-Gallacher, J. L. et al. The 2.1 Å resolution structure of cyanopindolol-bound β1-Adrenoceptor identifies an intramembrane Na+ Ion that stabilises the ligand-free receptor. PLoS ONE 9, e92727 (2014).
Luginina, A. et al. Structure-based mechanism of cysteinyl leukotriene receptor inhibition by antiasthmatic drugs. Sci. Adv. 5, eaax2518 (2019).
Wang, S. et al. D 4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science 358, 381–386 (2017).
Zarzycka, B., Zaidi, S. A., Roth, B. L. & Katritch, V. Harnessing ion-binding sites for GPCR pharmacology. Pharmacol. Rev. 71, 571–595 (2019).
Pert, C. B., Pasternak, G. & Snyder, S. H. Opiate agonists and antagonists discriminated by receptor binding in brain. Science 182, 1359–1361 (1973).
Katritch, V. et al. Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci. 39, 233–244 (2014).
Liu, W. et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337, 232–236 (2012).
Schöppe, J. et al. Crystal structures of the human neurokinin 1 receptor in complex with clinically used antagonists. Nat. Commun. 10, 17 (2019).
Massink, A. et al. Sodium ion binding pocket mutations and adenosine A 2A receptor function. Mol. Pharm. 87, 305–313 (2015).
Capaldi, S. et al. Allosteric sodium binding cavity in GPR3: a novel player in modulation of Aβ production. Sci. Rep. 8, 11102 (2018).
Fenalti, G. et al. Molecular control of δ-opioid receptor signalling. Nature 506, 191–196 (2014).
Cheng, L. et al. Cryo-EM structure of small-molecule agonist bound delta opioid receptor-Gi complex enables discovery of biased compound. Nat. Commun. 15, 8284 (2024).
Robert, X. & Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014).
Kroeze, W. K. et al. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 22, 362–369 (2015).
Margolis, E. B., Fujita, W., Devi, L. A. & Fields, H. L. Two delta opioid receptor subtypes are functional in single ventral tegmental area neurons, and can interact with the mu opioid receptor. Neuropharmacology 123, 420–432 (2017).
Olsen, R. H. J. et al. TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nat. Chem. Biol. 16, 841–849 (2020).
Claff, T. et al. Elucidating the active δ-opioid receptor crystal structure with peptide and small-molecule agonists. Sci. Adv. 5, eaax9115 (2019).
Wang, Y. et al. Structures of the entire human opioid receptor family. Cell 186, 413–427.e17 (2023).
Vickery, O. N. et al. Intracellular transfer of Na+ in an active-state G-protein-coupled receptor. Structure 26, 171–180.e2 (2018).
Sadler, F. et al. Autoregulation of GPCR signalling through the third intracellular loop. Nature 615, 734–741 (2023).
Donald, J. E., Kulp, D. W. & DeGrado, W. F. Salt bridges: geometrically specific, designable interactions. Proteins 79, 898–915 (2011).
Broom, D. et al. Convulsant activity of a non-peptidic δ-opioid receptor agonist is not required for its antidepressant-like effects in Sprague-Dawley rats. Psychopharmacology 164, 42–48 (2002).
Gavériaux-Ruff, C., Karchewski, L. A., Hever, X., Matifas, A. & Kieffer, B. L. Inflammatory pain is enhanced in delta opioid receptor-knockout mice. Eur. J. Neurosci. 27, 2558–2567 (2008).
Faouzi, A., Varga, B. R. & Majumdar, S. Biased opioid ligands. Molecules 25, 4257 (2020).
Ramos-Gonzalez, N., Paul, B. & Majumdar, S. IUPHAR themed review: Opioid efficacy, bias, and selectivity. Pharmacol. Res. 197, 106961 (2023).
Qu, Q. et al. Insights into distinct signaling profiles of the µOR activated by diverse agonists. Nat. Chem. Biol. 19, 423–430 (2023).
Varga, B. R., Streicher, J. M. & Majumdar, S. Strategies towards safer opioid analgesics—A review of old and upcoming targets. Br. J. Pharmacol. https://doi.org/10.1111/bph.15760 (2022).
Powers, A. S. et al. Structural basis of efficacy-driven ligand selectivity at GPCRs. Nat. Chem. Biol. 19, 805–814 (2023).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D: Biol. Crystallogr. 66, 213 (2010).
Pettersen, E. F. et al. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D: Biol. Crystallogr. 66, 486 (2010).
Robertson, M. J., van Zundert, G. C. P., Borrelli, K. & Skiniotis, G. GemSpot: a pipeline for robust modeling of ligands into Cryo-EM maps. Structure 28, 707–716.e3 (2020).
Peck, J. V., Fay, J. F. & Strauss, J. D. High-speed high-resolution data collection on a 200 keV cryo-TEM. IUCrJ 9, 243–252 (2022).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Chen, V. B. et al. ıt MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D. 66, 12–21 (2010).
Abraham, M. J. et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017).
Abagyan, R., Totrov, M. & Kuznetsov, D. ICM—A new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J. Comput Chem. 15, 488–506 (1994).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM‐GUI: a web‐based graphical user interface for CHARMM. J. Comput Chem. 29, 1859–1865 (2008).
Leonard, A. N. & Lyman, E. Activation of G-protein-coupled receptors is thermodynamically linked to lipid solvation. Biophys. J. 120, 1777–1787 (2021).
Davis, J. H., Clair, J. J. & Juhasz, J. Phase equilibria in DOPC/DPPC-d62/cholesterol mixtures. Biophys. J. 96, 521–539 (2009).
Lomize, M. A., Pogozheva, I. D., Joo, H., Mosberg, H. I. & Lomize, A. L. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 40, D370–D376 (2012).
Wu, E. L. et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem. 35, 1997–2004 (2014).
Vanommeslaeghe, K. et al. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 31, 671–690 (2010).
Berendsen, H. J. C. Transport properties computed by linear response through weak coupling to a bath. In Computer Simulation in Materials Science: Interatomic Potentials, Simulation Techniques and Applications (eds. Meyer, M. & Pontikis, V.) 139–155 https://doi.org/10.1007/978-94-011-3546-7_7 (Springer Netherlands, Dordrecht, 1991).
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A. & Haak, J. R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. G. E. M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997).
McGibbon, R. T. et al. MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys. J. 109, 1528–1532 (2015).
Percie Du Sert, N. et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. PLoS Biol. 18, e3000410 (2020).
Wilson, L. L. et al. Characterization of CM-398, a novel selective sigma-2 receptor ligand, as a potential therapeutic for neuropathic pain. Molecules 27, 3617 (2022).
Cirino, T. J. et al. Characterization of sigma 1 receptor antagonist CM-304 and its analog, AZ-66: novel therapeutics against allodynia and induced pain. Front. Pharmacol. 10, 678 (2019).
Gutridge, A. M. et al. Evaluation of kratom opioid derivatives as potential treatment option for alcohol use disorder. Front. Pharmacol. 12, 764885 (2021).
Kennedy, N. M. et al. Optimization of a series of Mu opioid receptor (MOR) agonists with high G protein signaling bias. J. Med. Chem. 61, 8895–8907 (2018).
Acknowledgements
This work was supported by NIH grant RO1DA057790 to S.M., T.C., V.K., J.P.M., UG3DA053094 and R01NS130882 to A.P., R01NS134981 to E.B.M. and R35GM153437 grant to V.K., S.M.B. thanks the PhRMA Foundation Predoctoral Fellowship in Drug Discovery (ISNI ID 0000 0000 9959 8153, Crossref Funder ID 100001797). We thank the staff at the Washington University Center for Cellular Imaging for sample screening; the staff of the NIDA Drug Supply Program for providing the 3H-Naltrindole; Huang, XP and the members of the Psychoactive Drug Screening Program (PDSP) at UNC-Chapel Hill for the GPCRome screening analysis. MD simulations were performed with the GPU cluster at the Center for Advanced Research Computing of the University of Southern California.
Author information
Authors and Affiliations
Contributions
S.M. and V.K. conceived the hypothesis. B.R.V., S.M.B., J.H.L., V.K., J.P.M., S.M., and T.C. wrote and edited the article with contributions from all co-authors. B.R.V. designed and synthesized the compounds under S.M.’s supervision. S.M.B. obtained cryo-EM structures and conducted mutational and binding studies under T.C.’s supervision. A.E.D., K.A., and A.K. performed BRET assays under T.C. and S.M.’s supervision. V.W. performed off-target screening under X.-P.H.’s supervision. S.A.Z. and A.L.N. provided molecular docking, and J.H.L. contributed MD simulation studies under V.K.’s supervision. J.A. conducted CFA studies under A.P.’s supervision, S.O.E., R.S. and H.R.H. performed anti-allodynia and other opioid adverse studies under JPM’s supervision. E.B.M. carried out electrophysiology studies, J.F.F. aided in structural characterization of bitopics.
Corresponding authors
Ethics declarations
Competing interests
S.M. is a cofounder of Sparian biosciences. S.M., V.K., S.A.Z., T.C., J.P.M., and B.R.V. have filed patents on delta opioid bitopics developed in this manuscript. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Marc Landry and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Varga, B.R., Bernhard, S.M., El Daibani, A. et al. Structure-guided design of partial agonists at an opioid receptor. Nat Commun 16, 2518 (2025). https://doi.org/10.1038/s41467-025-57734-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57734-5
