
Abstract
Capture of acid gases holds crucial importance for addressing air pollution and climate change, where achieving a molar ratio for adsorption and separation of acid gases on an active site higher than 1.0 remains challenging. Herein, we demonstrate that three nitrogen-bonded one Zn sites within a single-crystalline-like porous carbon (Zn-N3@SC-PC) derived from controlled carbonization of ZIF-8-C ≡ N with KCl, exhibit supra-multi-molar adsorption for CO2, COS, and H2S, even to 1:6 ratio for SO2 on the Zn-N3. This exceptional performance is attributed to the protruded structure in the Zn-N3@SC-PC for more coordination between Zn vacant orbital and acid gases evidenced by DFT calculation and in situ EXAFS. The high capacity for capturing acid gases on this adsorbent is crucial for future in carbon neutrality and environment protection.
Similar content being viewed by others
Introduction
Industrial processes like natural gas exploitation, gasoline desulfurization, coal utilization, iron smelting, and flue gas discharging1,2 release huge amounts of acid gases, including SO2, CO2, COS, and H2S. These emissions contribute to serious environmental and health issues such as climate change, air pollution, catalyst poisoning, equipment corrosion, and health hazards3,4. Traditionally, capturing acid gases has relied on amine scrubbing, which suffer from low efficiency, corrosion, and complex regeneration processes5. Although newer liquid scrubbing agents (such as ionic liquids and polyethyleneimine) have shown promise, they often struggle with thermal stability and volatilization losses, limiting their widespread use6,7.
Enter the realm of porous adsorbents, which offer a promising solution by reducing corrosion, enhancing stability, and allowing for customizable frameworks and adsorption sites. The capture of acid gases over porous adsorbents shows low energy consumption and facile operating procedures, these materials maintain their performance through multiple adsorption-desorption cycles8,9,10. Identifying and optimizing adsorption sites of porous adsorbents like carbons, POPs, COFs, zeolites, and MOFs11,12,13 play critical role for their performance for acid gases capture. Unfortunately, the reported adsorption sites (e.g., base groups) often fall short due to their adherence to a 1:1 stoichiometric interaction rule with acid gases14,15, limiting their adsorption capacity, selectivity and recognition accuracy.
Inspired by the multi-molar coordination principle between acid-base groups and transition metal16, which presents a special advantage as they can break this stoichiometric limitation. Transition metal sites usually possess abundant coordinative vacant orbitals with adjustable coordination numbers (C.N.), which allow them to capture multiple acid gases, especially gaseous sulfides, with enhanced coordination ability. However, challenges remain only atomic dispersing and exposing transition metal sites into stable porous materials, rational regulating their bonding structure and coordinative environment, high-performed adsorption active sites higher than 1:1 stoichiometric interaction with acid gases can be created.
Our research addresses these challenges by designing three nitrogen-bonded one zinc (Zn-N3) on a single-crystalline-like porous carbon framework. This material can be rationally designed from controllable carbonization of ZIF-8-C ≡ N, with KCl included to enhance porosity and adjust the C.N. of Zn (Fig. 1a). The resulting Zn-N3@SC-PC boasts large BET surface area, abundant nanoporosity, and high density of Zn-N3 (7.3 wt%) with excellent accessibility. Remarkably, the Zn-N3 exhibits intensified interaction with acid gases, far exceeding the 1:1 stoichiometry derived from its protruded structure for vacant orbital deformation. This allows Zn-N3 to sequentially capture more than six SO2 molecules, displaying typical supra-multi-molar adsorption functionality. This breakthrough imparts the Zn-N3@SC-PC with superior properties for SO2 capture, including high capacity (21.2 mmol/g, 25 °C, 1.0 bar), improved Ideal Adsorption Solution Theory (IAST) selectivity, and approximate 100% Zn-N3 efficiency. Under flue gas conditions, the Zn-N3@SC-PC achieves excellent performance for SO2 high-precise separation.

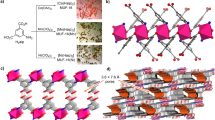
a Synthetic scheme of Zn-Nx@SC-PCs. b XANES spectra and (c) FT-EXAFS spectra of Zn-Nx@SC-PCs, ZnPc (zinc phthalocyanine), Zn foil and ZnO. WT-EXAFS of (d) Zn-N3@SC-PC and (e) Zn-N4@SC-PC. EXAFS R space fitted curve of (f) Zn-N3@SC-PC and (h) Zn-N4@SC-PC (data-red and fit-black). Schematic model of (g) Zn-N3@SC-PC and (i) Zn-N4@SC-PC. Zn (purple), N (blue) and C (gray). All the FT-EXAFS spectra are plotted without phase correction.
Moreover, the Zn-N3 breaks the 1:1 stoichiometric rule with other acid gases (CO2, H2S, COS) as well, enabling direct air capture of CO2 with approximate 100% separation precision. Its overall performance for acid gas capture and sieving surpasses any other reported porous adsorbents, which have been strictly limited by the 1:1 stoichiometric rule. To our knowledge, the Zn-N3 represents the first adsorption site that prominently breaks this rule, offering an opportunity for designing highly efficient adsorbents for selective capture and sieving of acid gases.
Results
Structure and coordination of Zn-Nx
The successful preparation of the ZIF-8-C ≡ N precursor can be confirmed by X-ray diffraction (XRD) and Fourier transform infrared spectroscopy (FT-IR, Supplementary Fig. 1). XRD and Raman spectroscopy were used to study framework crystallinity and defects in the derived Zn-Nx@SC-PCs. The XRD results revealed two broad peaks at around 26.1° and 44.3°, corresponding to (002) and (101) planes, indicating the amorphous frameworks of the Zn-Nx@SC-PCs (Supplementary Fig. 2a). Raman spectra showed peaks at around 1348 and 1589 cm−¹, corresponding to the D and G bands, which are associated with structural defects and graphitic layers, respectively (Supplementary Fig. 2b). The ID/IG values, ranging from 1.00 to 1.05, further confirmed the amorphous nature and the presence of abundant defects.
The electronic state and coordination structures of Zn-Nx were studied by X-ray absorption near-edge structure spectroscopy (XANES) and extended X-ray absorption fine structure (EXAFS) spectroscopy. The Zn-Nx@SC-PCs showed a unique XANES spectra compared with references (Fig. 1b). The intensive edge height indicates the valence state of Zn in Zn-Nx@SC-PCs is cationic state17. The symmetric edge shape suggests the coordination geometry is tetrahedral. The Fourier transform-EXAFS (FT-EXAFS) spectra showed a strong peak at 1.44 Å, similar to zinc phthalocyanine (ZnPc), which is assigned to the Zn-N bond (Fig. 1c). Meanwhile, the absence of Zn-Zn peaks at 2.30 Å (corresponding to Zn foil) and 2.87 Å (corresponding to ZnO) on the Zn-Nx@SC-PCs excludes the formation of Zn or ZnO in the samples18.
Compared with Zn-N4@SC-PC, the Zn-N bond intensity in Zn-N3@SC-PC decreased, with a noticeable left shift in the R-space. This shift likely results from a reduced number of nitrogen atoms coordinated with Zn. Additionally, wavelet transforms of Zn K-edge EXAFS (WT-EXAFS) oscillations revealed differences: ZnO reference showed two intensity maxima at 4.5 Å−1 and 6.0 Å−1 for Zn-O and Zn-(O)-Zn coordination path, respectively, whereas the Zn-Nx@SC-PCs and ZnPc showed only one at 3.9 Å−1 for Zn-N path, differing from the 6.0 Å−1 peak of Zn foil for metallic Zn-Zn path(Fig. 1d, e and Supplementary Fig. 3). This confirmed the existence of Zn-N coordination bonds in Zn-Nx@SC-PCs19. The EXAFS fitting results revealed that Zn-N3 is the dominant coordination structure in Zn-Nx@SC-PCs carbonized at 900 °C, while Zn-N4 predominates in those carbonized at 800 °C (Fig. 1f ~ 1i, Supplementary Table 1). This suggests that the Zn-Nx structures can be easily tuned by adjusting the carbonization temperature.
The X-ray photoelectron spectroscopy (XPS) of Zn-Nx@SC-PCs revealed the presence of C, N, Zn, and O, with the ratios of N and Zn decreasing as the carbonization temperature increased (Supplementary Fig. 4). The N 1 s spectra were divided into five peaks at approximately 398.4, 399.0, 400.3, 401.7, and 404.2 eV, corresponding to pyridinic nitrogen (N-6), nitrogen bonded with Zn (Zn-N), pyrrolic nitrogen (N-5), graphitic nitrogen (N-G), and oxidized nitrogen (N-O), respectively (Fig. 2a)20. The content of these nitrogen species, calculated from the XPS spectra, matched the elemental analysis results (Supplementary Table 2, 3). The Zn 2p signals displayed two peaks at 1021.4 and 1045.0 eV, assigned to the 2p3/2 and 2p1/2 doublet of Zn2+ (Supplementary Fig. 4). The binding energy of Zn 2p3/2 (1021.4 eV) was lower than that of ZnO (1022.0 eV), indicating that Zn species were primarily bonded with nitrogen in Zn-Nx@SC-PCs, leading to the formation of Zn-Nx21,22. Furthermore, the intensity of EPR signal of the Zn-N3@SC-PC is observed to be much stronger than that of the Zn-N4@SC-PC (Fig. 2b), indicating a higher number of unpaired electrons in the Zn-N3@SC-PC, which results in the formation of increased amount of coordination vacant orbitals in the sample.

a N 1 s XPS spectra, (b) EPR spectra, (c) N2 adsorption-desorption isotherms of Zn-Nx@SC-PCs. d SEM, (e, f) TEM, (g) HAADF-STEM, (h) HAADF images, and (i–l) elemental maps of Zn-N3@SC-PC.
The porosity of Zn-Nx@SC-PCs was analyzed using N2 adsorption-desorption isotherms at −196 °C (Fig. 2c and Supplementary Fig. 5). The textural parameters are listed in Supplementary Table 4. Zn-Nx@SC-PCs exhibit high volume adsorption ( ~ 600 cm3/g) at low relative pressure (p/p0 < 0.2), with an adsorption-desorption platform observed at p/p0 ≈ 0.3, indicating the formation of abundant micro-supermicroporosity23. These materials showcased large BET surface areas (1398–1847 m2/g) and pore volumes (0.35–0.64 cm3/g). The pore size distribution, estimated from the adsorption branches, showed dual peaks centered at 0.5–1.0 nm and 1.5–3.0 nm, confirming the micro-supermicroporous structures. During carbonization, the intrinsic microporosity of ZIF-8-C ≡ N was preserved, while the flowing of KCl, evaporation of Zn, and releasing of gas templates (e.g., CO2, NH3) created additional micro-supermicroporosity and structure defects. This process promoted the recombination of Zn to form abundant and highly accessible Zn-Nx.
Scanning electron microscopy (SEM), transmission electron microscopy (TEM), high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM), and elemental mapping were used to study the crystallinity, inner porosity, and dispersion of Zn-N3 in Zn-N3@SC-PC. ZIF-8-C ≡ N exhibits single crystallinity with smooth surface and uniform sizes at around 5 μm (Supplementary Fig. 6). The derived Zn-N3@SC-PC maintained this single crystallinity, albeit with size reduction and surface roughening (Fig. 2d and Supplementary Fig. 6). TEM images confirmed the single crystallinity and revealed abundant nanoporosity and structural defects (Fig. 2e and f). HAADF-STEM images of Zn-N3@SC-PC indicated a high density of atomic Zn dispersed throughout entire sample (Fig. 2g and Supplementary Fig. 7), with an inductively coupled plasma optical emission spectroscopy (ICP-OES) measured Zn content of 7.3 wt%. The corresponding elemental maps showed the presence of carbon, nitrogen, and zinc with a homogeneous distribution (Fig. 2h–l). These results confirmed the successful creation of Zn-N3 in Zn-N3@SC-PC.
Acid gases capture
Given the high density of Zn-Nx with tunable coordination structures and strong interaction with acid waste gases, the performance of Zn-Nx@SC-PCs for adsorbing and sieving acid gases (SO2, CO2, H2S and COS) was investigated. Supplementary Fig. 8 shows the SO2 isotherms of Zn-Nx@SC-PCs, ZnO/AC, N-PC-900 and Zn/N-PC-900 at 25 °C, 1.0 bar. The SO2 capacities of Zn-Nx@SC-PCs rise sharply at low relative pressures ( ≤ 20 kPa) and slow down at higher pressures ( ≥20 kPa), indicating a strong interaction between SO2 and Zn-Nx@SC-PCs. The SO2 capacity of Zn-N3@SC-PC (21.2 mmol/g) surpasses that of Zn-N4@SC-PC (17.2 mmol/g). Despite similar BET surface areas but higher content of Zn-N4, the higher activity of Zn-N3 over Zn-N4 for SO2 adsorption is evident.
To highlight the importance of Zn-N3 for SO2 capture, we removed Zn-N3 from Zn-N3@SC-PC to create N-PC-900, which was then impregnated with an equal mass of Zn2+ to form Zn/N-PC-900. Similarly, activated carbon was impregnated with the same amount of Zn2+ to produce Zn/AC. The SO2 capacities of N-PC-900, Zn/N-PC-900 and Zn/AC were 14.5, 15.2 and 9.4 mmol/g, respectively, all much lower than Zn-N3@SC-PC under identical conditions, confirming the critical role of Zn-N3 in SO2 capture. At 80 °C, Zn-N3@SC-PC still demonstrated high SO2 capacity (8.8 mmol/g), outperforming N-PC and Zn-N4@SC-PC (Supplementary Fig. 8). The Zn-N3@SC-PC gave high low-pressure SO2 capacities to date (Fig. 3a, Supplementary Table 5), which was more important for real-world applications. Supplementary Fig. 9 showed the adsorption kinetic experiments and corresponding fitted adsorption kinetic model of Zn-Nx@SC-PCs for SO2, which exhibit typical pseudo-second order kinetic model with the fitting degree (R2) higher than 0.999 (Supplementary Table 6), suggesting the chemical interaction dominated the SO2 adsorption on the Zn-Nx@SC-PCs.

a SO2 capacities of Zn-N3@SC-PC and literature-reported high-performed adsorbents at 25 °C, 0.1 and 1.0 bar. b Cycling of Zn-N3@SC-PC for SO2 adsorption at 25 °C and 1.0 bar. c In situ DRIFTS spectra of SO2 adsorption on Zn-N3@SC-PC at 25 °C for 60 min and SO2 desorption at 40 ~ 150 °C. d SO2 adsorption curves of various samples for different times at 25 °C and 1.0 bar. e SO2:Zn-N3 stoichiometry at different temperatures and pressure.
The cycling stability of Zn-N3@SC-PC was also examined, maintaining its SO2 capacity after 50 cycles (Fig. 3b), indicating superior stability and good reversible interaction with SO2. The adsorbed SO2 on the Zn-N3@SC-PC could be completely removed to achieve its regeneration at the temperature higher than 110 °C, as verified by in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS, Fig. 3c). Additionally, Zn-N3@SC-PC exhibited a much faster SO2 adsorption rate than N-PC-900 and PC-900 under simulated flue gas conditions (Supplementary Fig. 10), due to enhanced interaction between Zn-N3 and SO2, making it highly suitable for flue gas desulfurization.
SO2 selectivity is a crucial parameter for evaluating the performance of the adsorbents24,25,26. The Ideal Adsorption Solution Theory (IAST) selectivities of Zn-Nx@SC-PCs for SO2/CO2/N2 were studied, considering the widespread presence of CO2 and N2 in flue gas. CO2 and N2 isotherms of Zn-Nx@SC-PCs were also provided (Supplementary Figs. 11 and 12). The Zn-Nx@SC-PCs showed lower CO2 and N2 capacities (3.1–3.5 and 0.20–0.23 mmol/g) compared to SO2. The corresponding SO2/CO2, SO2/N2 and CO2/N2 IAST (0.1/0.9) selectivities ranged from 22.4 to 18.2, 347 to 287 and 31.5 to 29.8 at 25 °C (Supplementary Fig. 13–15), which remained high (141–207, 15.9–19.5 and 10.9–13.9) at 80 °C, outperforming many reported adsorbents (Supplementary Table 7).
Combining high SO2 capacities and excellent SO2/CO2/N2 IAST selectivities, breakthrough tests of Zn-N3@SC-PC for simulated flue gas (0.85 v%SO2/15 v%CO2/84.15 v%N2) with the flow rate of 10 mL/min were conducted. N2 broke through instantly upon contact with Zn-N3@SC-PC, while CO2 and SO2 had breakthrough times of 290.5–352.2 min/g and 2065.2–4271.9 min/g, respectively (Supplementary Fig. 16). The corresponding SO2 saturated breakthrough capacity reached 11.4 mmol/g, aligning with the SO2 isotherms results. During the dormant stage of Zn-N3@SC-PC, SO2 was undetectable by the sensor, indicating precise separation of SO2 without any escaping, surpassing many high-performance porous materials in the field (Supplementary Figs. 17 and 18).
To understand the pivotal role of Zn-N3 in SO2 capture, we investigate the dependence of SO2 capacity of Zn-N3@SC-PC on adsorption time at 25 °C and 1.0 bar (Fig. 3d). The comparison with N-PC-900 revealed that the capacity difference, attributed to Zn-N3, was 6.7 mmol/g, with a Zn-N3 content of 1.1 mmol/g, far exceeding the 1:1 stoichiometric rule. Nearly 6.1 mol of SO2 can be adsorbed per mol of Zn-N3, demonstrating a supra-multi-molar adsorption effect. In contrast, the molar ratio of SO2 to Zn-N4 and nitrogen sites were 0.9 and 0.8, respectively (Supplementary Fig. 19). The SO2 supra-multi-molar adsorption on Zn-N3 was also observed at different pressures and temperatures, with SO2 stoichiometry ranging from 4.0 to 6.1 (Fig. 3e and Supplementary Figs. 20–22).
Besides SO2, Zn-N3 also achieved supra-multi-molar adsorption for CO2, COS, and H2S, with about 2.1 mol CO2, 4.9 mol COS, and 3.7 mol H2S adsorbed per mol Zn-N3 at 25 °C, 1.0 bar (Supplementary Fig. 23). These results were very different from Zn-N4 (1.2 mol COS, 0.4 mol CO2 and 1.0 mol H2S/mol Zn-N4, Supplementary Fig. 24) and N-PC-900 (0.2 mol COS, 0.1 mol CO2 and 0.3 mol H2S/mol N, Supplementary Fig. 25). This indicates the excellent universality of Zn-N3 for supra-multi-molar acid gases capture, thus endowing high CO2, COS and H2S capacities (3.5 mmol/g, 9.9 mmol/g and 10.5 mmol/g, Supplementary Fig. 26). Interestingly, CO2 in the air can be efficiently and precisely separated over Zn-N3@SC-PC, showing great significance for controlling climate change, a feat not achieved by other high-performance porous materials with different adsorption sites (Supplementary Fig. 27).
As far as we know, no previous studies have achieved supra-multi-molar adsorption of versatile acid gases using one single site. Although Zn-N3@SC-PC shows slightly lower SO2 capacities compared to a few high-performance MOFs under ideal conditions, Zn-N3 demonstrates much enhanced interactions with acid gases. This results in improved adsorption efficiency and recognizability. Consequently, Zn-N3@SC-PC exhibits high flue gas SO2 capacities and approximate 100% precision for direct CO2 capture from air (Supplementary Fig. 28). These characteristics are especially valuable for applications in air pollution control and carbon neutralization27,28.
Understanding superior performance of Zn-N3@SC-PC
To unravel the superior performance of Zn-N3@SC-PC, the interaction between SO2 and Zn-Nx@SC-PCs was explored by using theoretical calculation. Two structural models were constructed to represent the Zn-N3 and Zn-N4 (Supplementary Fig. 29), which align with XAFS findings. An enormous induced deformation of Zn-N3 substrate occurred during its adsorption of guest molecules (e.g., SO2, CO2, COS, H2S, H2O, and O2), and the planar configuration transit into a non-planar one (Supplementary Fig. 30). The deformation energy of −184.6 kJ/mol indicates the thermodynamically favorable transition and the stability of Zn-N3-protruded configuration.
Consequently, adsorption energies of six SO2 molecules on the protruded structure were calculated (Fig. 4a and Supplementary Fig. 31). The first three SO2 molecules were adsorbed by strong chemisorption interactions with adsorption energies of −127.5, −109.3, and −71.7 kJ/mol via Zn-O coordination bonds of 2.12 ~ 2.16 Å. However, this saturated coordination Zn in the protruded structure cannot directly allow the chemisorption with the subsequent three SO2 molecules. Instead, the adsorption is dominated by electrostatic interactions with the pre-adsorbed three SO2 molecules of adsorption energies of −56.1, −51.7, and −49.6 kJ/mol, respectively. Interestingly, a substantial adsorption energy of −49.6 kJ/mol can still be achieved even with the sixth SO2 adsorbed on Zn-N3, indicating a potential supra-multi-molar adsorption configuration between SO2 and Zn-N3. In contrast, the adsorption of the first SO2 molecule on Zn-N4 was attributed a weak adsorption with the adsorption energy of −61.4 kJ/mol and Zn…O distance of 2.29 Å, and the energies for subsequent SO2 molecules were also lower than those for Zn-N3 ( < − 50.0 kJ/mol, Supplementary Fig. 32). Additionally, SO2 adsorption energies on naked nitrogen sites were much lower than on Zn-Nx (Supplementary Figs. 33 and 34), highlighting the crucial role of the structure of Zn in SO2 supra-multi-molar adsorption. Notably, the Zn-N3 shows the strongest adsorption energies for SO2 among the reported adsorbents16,17,18.

a Energy diagram depicting adsorption process of SO2 on Zn-N3, ΔE (kJ/mol) represents the adsorption energy of each SO2. b Analysis of the localized orbitals of Zn-N3-Protruded and Zn-N3-Plane. NBO analysis of (c) ZnN3-Protruded and (d) ZnN3-Protruded-3SO2. e IGMH analysis of 6SO2 adsorbed on ZnN3-Protruded. (f) XANES spectra of fresh, used and regenerated Zn-N3@SC-PC. Corresponding EXAFS R space fitted curve of (g) fresh, (h) used and (i) regenerated Zn-N3@SC-PC.
To understand this supra-multi-molar adsorption of Zn-N3 for SO2, analysis of the localized molecular orbitals (LMOs) energy was employed to explore the electronic nature of this adsorption (Fig. 4b, Supplementary Fig. 35 and Supplementary Table 8). Zn 3 d orbitals are fully occupied, i.e., [Ar]3d10, but the orbitals of two different Zn-N3 configurations demonstrate the different coordination environments of Zn in the Zn-N3-protruded and Zn-N3-plane, the Zn-N3-protruded can provides more contact area to bind three SO2 molecules via coordinated bonds, leading the hexa-coordinated Zn. Moreover, the Zn-N3-plane structure exhibits a greater distortion in the substrate’s geometric arrangement compared to Zn-N3-protruded, as evidenced by the C-N-C bond angles (Supplementary Fig. 36). This is likely one of the inherent causes of deformation. In summary, the influence of the guest molecules, the distortion of the substrate, and the reduction in orbital energy drive the alterations in the local environment of Zn atom. This deformed Zn site plays a pivotal role for SO2 supra-multi-molar adsorption.
To elucidate the electronic nature of these strong adsorption interactions of Zn-N3-protruded to the first three SO2 molecules, we conducted a comprehensive investigation using natural bond orbital (NBO) analysis (Fig. 4c, d and Supplementary Figs. 37–40 and Supplementary Table 9). The study indicates that the outer layer of Zn atom possesses four lone pair antibonding orbitals (LP*) derived from the hybridization of Zn 4 s and 4p atomic orbitals, which can accommodate the oxygen lone pairs (LP) of SO2. These unoccupied orbitals of Zn atom in Zn-N3-protruded with larger spatial accessibility facilitates the coordination with SO2. The natural bond orbitals of each component before and after SO2 adsorption reveal that the unoccupied orbitals of the Zn atom in the protruded structure undergo significant deformation, indicating its strong binding with the lone pairs of SO2 (Supplementary Table 10). Furthermore, these strong chemical interactions also greatly alter the polarity of the adsorbed SO2, providing a stronger non-covalent interaction to more SO2 molecules as indicated by the independent gradient model based on the Hirshfeld partition (IGMH) analysis (Fig. 4e and Supplementary Fig. 41). These findings suggest that SO2 molecules are more orderly arranged and tightly bound, forming an impressive ribbon structure. This strong interaction is also evident through NBO analysis, demonstrating the interaction between the lone pair (LP) of SO2 and the LP* orbitals (Supplementary Fig. 42). This supra-multi-molar interaction with COS, H2S, and CO2 can also be realized on the Zn-N3 due to their strong adsorption energies, as verified by DFT optimized structures (Supplementary Figs. 43–46), suggesting its good universality for acid gases capture.
The proposed structure of the adsorption sites could be validated by the XANES and EXAFS spectra of Zn-N3@SC-PC under various conditions. As shown in the XANES spectra of the Zn-N3@SC-PC (Fig. 4f), the adsorption edge is higher and more symmetric after SO2 molecules were adsorbed, suggesting the coordination geometry of Zn changes from protruded trigonal in the fresh sample to the octahedral in the used sample. The FT-EXAFS spectra of Zn K edge are fitted (Fig. 4g–i, Supplementary Table 11) to confirm the coordination environment of the Zn-N3 under different conditions. The C.N. of Zn-N is 3 in the fresh sample, and the coordination environment around Zn atoms contains Zn-N and Zn-O paths after adsorption of SO2. The Zn atoms coordinated with the three O atoms in multiple SO2 to form multi-molar adsorption. After regeneration, the Zn atoms can be restored to the Zn-N path again. The spectroscopic results match well with the theoretical models. In situ XPS spectra confirmed the formation of [Zn-N3]-xSO2 as SO2 adsorbed on Zn-N3@SC-PC (Supplementary Fig. 47), resulting in a more positive charge state of the Zn atom29. The decrease in intensity and blue shift of the Zn-N signal suggested a reduction in electron donor capacity, altering the intrinsic vacant orbital of Zn atom. Overall, the C.N. analysis of Zn sites in fresh, adsorbed, and regenerated states matches well with DFT results, highlighting the enhanced but reversible interaction between Zn-N3 and SO2.
Discussion
We developed a protruded Zn-N3-anchored single-crystalline-like porous carbon from KCl-assisted carbonization of ZIF-8-C ≡ N. This material, Zn-N3@SC-PC, possesses large BET surface areas, abundant nanoporosity, high density of Zn-N3 with tunable coordination environment, and good accessibility. The Zn-N3 exhibits unique deformed vacant orbital, resulting in enhanced coordination interactions with acid gases like SO2, CO2, H2S, and COS, which far surpass the classical 1:1 stoichiometric rule. Approximately 6.1 mol of SO2, 1.9 mol of CO2, 4.5 mol of COS, or 3.4 mol of H2S can be captured by the Zn-N3, demonstrating supra-multi-molar adsorption. Particularly, the Zn-N3@SC-PC showcases excellent breakthrough performance and approximate 100% separation precision for both SO2 capture from flue gas and direct air capture of CO2, a feat unachievable on any other porous adsorbents. This work presents a rational methodology for designing porous adsorbents with well-defined adsorption sites and improved functionalities, highlighting their potential for the acid gases precisely capture and sieving from various industrial processes.
Methods
Samples preparation
Synthesis of ZIF-8-C ≡ N and Zn-Nx@SC-PCs
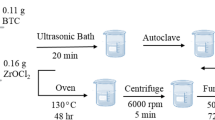
Zinc nitrate heptahydrate (1.3 g) was dissolved into 50 mL of methanol, denoted as solution A. 2-methylimidazole (2.76 g) and 4,5-dicyanoimidazole (0.15 g) were dissolved in another 50 mL of methanol, denoted as solution B. Then, the solution A was added rapidly into solution B. The resultant mixture was vigorously stirred at room temperature for 2 h and further aged for 24 h; the white precipitate was formed, which was centrifugated and washed with methanol for three times and dried at 60 °C overnight, giving ZIF-8-C ≡ N precursor.
Zn-Nx@SC-PCs were obtained by vacuum thermal pyrolysis of ZIF-8-C ≡ N in the presence of KCl additive at different temperatures (T = 800 °C and 900 °C). Firstly, ZIF-8-C ≡ N and KCl with a mass ratio of 1:2 was ground well at room temperature, the resulted solid mixture was then transferred into the tube furnace and carbonized at the target temperature for 3 h with the heating rate of 2 °C/min. Afterwards, the natural cooling was performed until to room temperature, the as-obtained product was collected from the furnace, washed with abundant water and ethanol to remove any impurities. Finally, the Zn-Nx@SC-PCs was dried under vacuum at 60 °C in an oven overnight. The structures of Zn-Nx can be controlled by adjusting the final carbonization temperature. It is noteworthy that Zn-N3 is the dominant coordination structure in the Zn-Nx@SC-PCs carbonized at 900 °C, and Zn-N4 predominates in the Zn-Nx@SC-PCs carbonized at 800 °C, which can be confirmed by the EXAFS fitting results. The Zn content of Zn-N4@SC-PC and Zn-N3@SC-PC were measured to be up to 15.1 wt% and 7.3 wt% based on inductively coupled plasma (ICP) analysis.
Gas adsorption and separation
SO2 adsorption isotherm tests of various samples were performed on a corrosion-resistant apparatus, which includes a storage chamber and an adsorption chamber. The temperature of the apparatus was regulated by a thermostatic water bath. Before tests, the samples were degassed under vacuum at 150 °C for 12 h to remove any volatile impurities. The separation properties of Zn-Nx@SC-PCs for mixed gas were carried out on the Micromeritics Autochem 2920 system connected with a Hiden HPR-20 mass spectrometer. The adsorbents were loaded in a U-type quartz tube (6 mm × 190 mm) and installed to the experimental system. Typically, the adsorbent was firstly purged with He flow (30 mL/min) and treated at 200 °C for 3 h, then the sample was cooled down to the experimental temperature. After that, the airflow of the system was changed into the gas mixture containing SO2/CO2/N2 (0.85 v%/15 v%/84.15 v%) with flowing rate of 10 mL/min. The composition of the outlet gas was monitored online by using the mass spectrometer. The detailed instruments testing principles were illustrated in the Supplementary Information.
Theoretical calculations
Theoretical calculations were performed to understand the supra-multi-molar adsorption mechanism of Zn-Nx@SC-PCs for acid gases capture. Density functional theory (DFT) calculations, including geometry optimizations and frequency calculations, were performed using the ωB97XD hybrid exchange-correlation functional in conjunction with a def2-SVPP basis set. Natural Bond Orbital (NBO) analysis was carried out at the ωB97XD/def2-TZVP level of theory. Localized Molecular Orbital (LMO) analysis and the independent gradient model based on Hirshfeld partition (IGMH) were conducted using the Multiwfn and VMD software package. All theoretical calculations were carried out using the Gaussian 16 software package. The detailed computational procedures were summarized in the Supplementary Information.
Data availability
All data supporting the findings of this study are available from the corresponding authors upon request. Source data are provided with this paper.
References
Lin, J.-B. et al. A scalable metal-organic framework as a durable physisorbent for carbon dioxide capture. Science 374, 1464–1469 (2021).
Han, X., Yang, S. & Schröder, M. Porous metal-organic frameworks as emerging sorbents for clean air. Nat. Rev. Chem. 3, 108–118 (2019).
Cui, X. et al. Ultrahigh and selective SO2 uptake in inorganic anion‐pillared hybrid porous materials. Adv. Mater. 29, 1606929 (2017).
Morris, R. E., Čejka, J. & Čejka Exploiting chemically selective weakness in solids as a route to new porous materials. Nat. Chem. 7, 381–388 (2015).
Li, W. et al. Adsorption of sulfur dioxide in Cu(II)-carboxylate framework materials: the role of ligand functionalization and open metal sites. J. Am. Chem. Soc. 144, 13196–13204 (2022).
Li, X., Chen, K., Guo, R. & Wei, Z. Ionic liquids functionalized MOFs for adsorption. Chem. Rev. 123, 10432–10467 (2023).
Kan, X. et al. Multiscale co‐assembly to meso‐macroporous foamed single‐crystal metal-organic frameworks for the supported capture of sulfur dioxide. Adv. Funct. Mater. 34, 2312044 (2024).
Snyder, B. E. et al. A ligand insertion mechanism for cooperative NH3 capture in metal-organic frameworks. Nature 613, 287–291 (2023).
Chen, F. et al. Deep desulfurization with record SO2 adsorption on the metal-organic frameworks. J. Am. Chem. Soc. 143, 9040–9047 (2021).
Li, J. et al. Structural and dynamic analysis of sulphur dioxide adsorption in a series of zirconium‐based metal-organic frameworks. Angew. Chem. Int. Ed. 61, e202207259 (2022).
Guo, D. et al. Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts. Science 351, 361–365 (2016).
Suo, X. et al. Tailoring the pore size and chemistry of ionic ultramicroporous polymers for trace sulfur dioxide capture with high capacity and selectivity. Angew. Chem. Int. Ed. 133, 7062–7067 (2021).
Smith, G. L. et al. Reversible coordinative binding and separation of sulfur dioxide in a robust metal-organic framework with open copper sites. Nat. Mater. 18, 1358–1365 (2019).
Wang, C. et al. Highly efficient and reversible SO2 capture by tunable azole-based ionic liquids through multiple-site chemical absorption. J. Am. Chem. Soc. 133, 11916–11919 (2011).
Chen, F. F. et al. Multi‐molar absorption of CO2 by the activation of carboxylate groups in amino acid ionic liquids. Angew. Chem. Int. Ed. 128, 7282–7286 (2016).
Li, J. et al. Guest-controlled incommensurate modulation in a meta-rigid metal-organic framework material. J. Am. Chem. Soc. 142, 19189–19197 (2020).
Li, S. et al. Low-valence Znδ+(0<δ<2) single-atom material as highly efficient electrocatalyst for CO2 reduction. Angew. Chem. Int. Ed. 60, 22826–22832 (2021).
Lu, Z. et al. Zinc single-atom-regulated hard carbons for high-rate and low-temperature sodium-ion batteries. Adv. Mater. 35, 2211461 (2023).
Han, L. et al. Stable and efficient single-atom Zn catalyst for CO2 reduction to CH4. J. Am. Chem. Soc. 142, 12563–12567 (2020).
Chen, J. et al. Promoting CO2 electroreduction kinetics on atomically dispersed monovalent ZnI sites by rationally engineering proton-feeding centers. Angew. Chem. Int. Ed. 61, e202111683 (2022).
Masuda, R. et al. Heterogeneous single-atom zinc on nitrogen-doped carbon catalyzed electrochemical allylation of imines. J. Am. Chem. Soc. 145, 11939–11944 (2023).
Yang, Q. et al. Single carbon vacancy traps atomic platinum for hydrogen evolution catalysis. J. Am. Chem. Soc. 144, 2171–2178 (2022).
Gellrich, C. et al. A precursor‐derived ultramicroporous carbon for printing iontronic logic gates and super‐varactors. Adv. Mater. 36, 2401336 (2024).
Yang, S. et al. Irreversible network transformation in a dynamic porous host catalyzed by sulfur dioxide. J. Am. Chem. Soc. 135, 4954–4957 (2013).
Han, Z. et al. A {Ni12}-wheel-based metal-organic framework for coordinative binding of sulphur dioxide and nitrogen dioxide. Angew. Chem. Int. Ed. 61, e202115585 (2022).
Yang, S. et al. Selectivity and direct visualization of carbon dioxide and sulfur dioxide in a decorated porous host. Nat. Chem. 4, 887–894 (2012).
Carter, J. H. et al. Exceptional adsorption and binding of sulfur dioxide in a robust zirconium-based metal-organic framework. J. Am. Chem. Soc. 140, 15564–15567 (2018).
Nugent, P. et al. Porous materials with optimal adsorption thermodynamics and kinetics for CO2 separation. Nature 495, 80–84 (2013).
Wei, G. et al. Single-atom zinc sites with synergetic multiple coordination shells for electrochemical H2O2 production. Angew. Chem. Int. Ed. 62, e202313914 (2023).
Acknowledgements
This work was supported by the National Science Foundation of China (22221005, U24A20528, 22378064, 22022804, 21978052), the Natural Science Foundation of Fujian Province (2024J011012, 2024J08035), the National Key R&D Program of China (2022YFB4002402), Major Program of Qingyuan Innovation Laboratory (00121003). We thank the staff at the BL14W1 beamline of the Shanghai Synchrotron Radiation Facility for the measurements of EXAFS and XANES.
Author information
Authors and Affiliations
Contributions
Conceptualization: Fujian Liu, Lilong Jiang. Methodology: Guanqing Zhang, Fengqing Liu, Qiliang Zhu, Jingyi Tan, Shouchao Zhong. Funding acquisition: Fujian Liu, Lilong Jiang. Writing-original draft: Guanqing Zhang, Shouchao Zhong, Fujian Liu, Yu Tang. Writing-review & editing: Anmin Zheng, Lilong Jiang, Feng-Shou Xiao
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yuanbin Zhang and the other anonymous reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, G., Liu, F., Zhong, S. et al. Surpassing stoichiometric limitation for supra-multi-molar adsorption and separation of acid gases. Nat Commun 16, 2861 (2025). https://doi.org/10.1038/s41467-025-58148-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58148-z
