
Abstract
Sulfadoxine-pyrimethamine (SP) is recommended for perennial malaria chemoprevention in young children in high burden areas across Africa. Mutations in the dihydropteroate synthase (dhps) gene (437 G/540E/581 G) associated with sulfadoxine resistance vary regionally, but their effect on SP protective efficacy is unclear. We retrospectively analyse time to microscopy and PCR-confirmed re-infection in seven efficacy trials including 1639 participants in 12 sites across Africa. We estimate the duration of SP protection against parasites with different genotypes using a Bayesian mathematical model that accounts for variation in transmission intensity and genotype frequencies. The longest duration of SP protection is >42 days against dhps sulfadoxine-susceptible parasites and 30.3 days (95%Credible Interval (CrI):17.1-45.1) against the West-African genotype dhps GKA (437G-K540-A581). A shorter duration of protection is estimated against parasites with additional mutations in the dhps gene, with 16.5 days (95%CrI:11.2-37.4) protection against parasites with the east-African genotype dhps GEA (437G-540E-A581) and 11.7 days (95%CrI:8.0-21.9) against highly resistant parasites carrying the dhps GEG (437G-540E−581G) genotype. Using these estimates and modelled genotype frequencies we map SP protection across Africa. This approach and our estimated parameters can be directly applied to any setting using local genomic surveillance data to inform decision-making on where to scale-up SP-based chemoprevention or consider alternatives.
Similar content being viewed by others

Introduction
Infants and young children are at highest risk of severe malaria, with 76% of all malaria deaths occurring in children under five1. Sulfadoxine-pyrimethamine (SP) is recommended by the World Health Organisation (WHO) in perennial malaria chemoprevention (PMC), aiming to reduce malaria cases and deaths in young children2. PMC, which now includes the formerly known intermittent preventative treatment in infants (IPTi), involves administering a single SP dose to children without malaria symptoms at predefined intervals, in areas of moderate-to-high perennial transmission2. The latest WHO guidelines encourage tailored PMC implementation, allowing countries flexibility to expand target age groups, adapt delivery platforms, drugs, and dosing schedules to site-specific resistance profiles, malaria endemicity, and seasonality2. A recent meta-analysis estimated a pooled efficacy of IPTi-SP across nine trials to be 22% against clinical malaria, 18% against anaemia and 15% against hospital admission3. However, only a few countries have adopted IPTi-SP, in part due to concerns about dosage and administration to infants, and perceived lack of protective efficacy.
An important determinant of antimalarial therapeutic and protective efficacy is the prevalence of mutations associated with drug resistance4,5,6,7. SP resistance is conferred by mutations in the dihydropteroate synthase (Pfdhps) and dihydrofolate reductase (Pfdhfr) genes and these are associated with clinical and parasitological drug failure8. The dhfr genotype IRN (51I-59R-108N), which is associated with partially reduced efficacy of pyrimethamine, spread quickly following the change of first-line treatment to SP and prevalence of these mutations remain high across sub-Saharan Africa9,10. In contrast, the prevalence of mutations in the dhps gene, which affect the sulfadoxine component of SP, varies by country and region9. In West and Central Africa, the dhps GKA (437G-K540-A581) in combination with dhfr IRN genotype confers partial resistance to SP10. In East Africa, the combination of dhfr IRN mutant with the dhps genotype GEA (437G-540E-A581), has been associated with SP treatment failure8. The further addition of dhps A581G generates the genotype GEG (437G-540E-581G), present in limited geographical foci in East and Southern Africa11, and thought to confer even higher SP resistance12,13.
Trials of IPTi and IPT in pregnant women (IPTp) suggest reduced effectiveness in areas with high prevalence of SP resistance markers, though data on resistance markers typically come from separate studies. In Korogwe, Tanzania (2004-2008), there was no significant protective efficacy conferred by IPTi-SP after 21 days12,14,15. Evidence from a trial done in the same area and period as the IPTi trial, showed that the GEG genotype was present in approximately half of the samples collected on the day of enrolment13. Similarly, IPTp-SP studies reported an association between the dhps GEG genotype and loss of protection from infection16,17, and low birthweight18. An IPTi trial conducted in Uganda, reported an SP efficacy of just 7% against clinical malaria compared to the control arm. In the same district, a trial found almost all episodes of malaria on the day of recruitment were with parasites carrying the GEA genotype with no 581G19. However, IPTi-SP efficacy was sustained in a trial conducted in Maputo, Mozambique20, where approximately half of the parasites harboured the dhps GEA genotype and half were dhps AKA (sulfadoxine-susceptible)21. In the absence of the 540E and 581G mutations, SP protective efficacy appears higher. In an area of low SP resistance in Navrongo, Ghana (2000–2002), a significant protective efficacy of IPTi with SP was estimated for the first 42 days following the last IPTi-SP dose14,22,23. The protective efficacy of SP is affected by the frequency of mutation-carrying parasites, but the exact impact of different dhps mutant combinations has never been systematically characterised.
Understanding the effect of different combinations of dhps mutations on the length of protection is important to inform chemoprevention strategies with SP, and SP-containing antimalarials, such as SP with amodiaquine (SPAQ)2. With renewed interest in PMC-SP adoption, national malaria programmes require evidence of protective efficacy in the presence of different resistance profiles to inform decisions on where PMC-SP can be implemented. Historical therapeutic efficacy studies (TES) of SP (or SP with artesunate, SPAS) with dhps and reinfection genotyping can be used to provide insights into the length of protection offered by SP against each genotype. Individually, these TES are not powered to assess protective efficacy and genotype effects. Here, we pool individual-level data from seven therapeutic efficacy studies in 1639 patients with a Plasmodium falciparum infection, collected from Malawi, Tanzania, Benin, Mozambique and South Africa, where new infections and detailed genotype data were reported13,24,25,26,27,28,29,30. We quantify SP protective efficacy and mean duration of protection against new infections with each of the main dhps genotypes that are common in Africa: dhps AKA(dhps sulfadoxine-susceptible), GKA, GEA, and GEG, by fitting to trial data using an existing modelling framework that accounts for the underlying risk of infection and underlying genotype frequencies31. Further, we validate these findings using data from IPTi-SP studies.
Results
Study characteristics
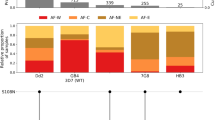
We systematically searched the Worldwide Antimalarial Resistance Network (WWARN) Clinical Trials Publication Library to identify trials of SP or SPAS efficacy using PCR methods to distinguish new infections from recrudescent infections. A total of seven eligible studies were identified across 12 sites, in Malawi, Northern Tanzania, Benin, Mozambique and South Africa13,24,25,26,27,28,30 between 2000 and 2006 (Table 1). The 1639 participants included in these studies were symptomatic malaria patients, consisting of children under five years for the studies conducted in Malawi, Northern Tanzania and Benin, or any ages >1 or >2 years for the studies in Mozambique and South Africa, respectively. Treatment failures were identified via positive blood smears at follow-up visits and classified as recrudescence or reinfection using PCR (Supplementary Table 1). Individual-participant data on time to new infection with each genotype since the time of drug administration were either obtained directly from the research groups (six studies) or extracted from publications where unavailable (one study25). Data from a total of 21 trial arms were available, including SP, SPAS, SPAQ, chloroquine (CQ) and SPCQ. Across sites, all dhps parasite genotypes (AKA, GKA, GEA, GEG) were found (Fig. 1 and Table 1). Genotyping information on day 0 before SP treatment and on the day of failure was extracted where available. However, many samples contained missing information on the full dhps genotype (Supplementary Table 2), but these were still included in the analysis.

Purple stars indicate the included sites. Annotations and the table denote the main dihydropteroate synthase (dhps) genotype profiles in those studies (with circles showing the presence of a mutation). The triple, quadruple, quintuple, and sextuple definitions indicated with the asterisk assume that the triple mutation in the dihydrofolate reductase (dhfr) gene (52I/ 59R/108N) is ubiquitous. The observed dhps GKA (437G-K540-A581) genotype may be considered as a proxy for the quadruple mutant, and the dhps GEA (437G-540E-A581) and GEG (437G-540E-581G) genotypes as proxies for the quintuple and sextuple mutants, respectively. ZAF=South Africa, MOZ= Mozambique, SWZ= Eswatini. Genotypes: dhps AKA (A437-K540-A581), dhps GKA (437G-K540-A581), dhps GEA (437G-540E-A581), dhps GEG (437G-540E-581G).
Duration of protection against different genotypes
Data from the identified efficacy trials of SP and SPAS were used to estimate the protection against the sulfadoxine-susceptible dhps AKA or mutant genotypes (GKA, GEA, GEG). We use a deterministic multi-strain model31 to model the probability of new infection with each genotype by fitting Weibull survival curves to the reinfection data using Hamiltonian Monte Carlo (HMC) methods (Supplementary Note 1). We fit the model to all data across 12 trial sites simultaneously accounting for the site-specific underlying malaria incidence and genotype frequency in the parasite population. The mean duration of protection against new infection was calculated using the estimated shape and scale parameters of the Weibull survival curve (Supplementary Note 1). In our modelling approach we model partial protection, rather than using a step function where someone is either protected or not on a particular day. Partial protection is expressed as a probability of protection over time since dose and this curve is constant across settings, irrespective of transmission.
Analysing all trial sites and drug arms together (Fig. 2), our model was able to fit the data well, with model-predicted values all within the 95% confidence intervals of the data with one exception of the SP arm in Malawi. Additionally, the model-predicted values for the genotype frequencies in each site closely matched the frequencies observed on day 0 (Supplementary Fig 1). Protection against sulfadoxine-susceptible parasites (dhps AKA) was significantly longer (55.7 days, 95% Credible Interval (CrI): 46.9–71.6) compared to most dhps mutant genotypes (Table 2, Supplementary Fig. 2 and Supplementary Table 3). The dhps GKA mutant reduced the duration of protection to 33.9 days (95% CrI: 16.8–56.8, p = 0.143), and the GEA mutant reduced protection further to 10.7 days (95% CrI: 8.9–21.9, p < 0.001). SP protection against the highly resistant dhps GEG genotype) was estimated to be similar to the GEA mutant, at 11.7 days (95% CrI: 8.0–21.9, p = 0.003), but was based on only one study with shorter duration of follow-up.

Markers denote the observed proportion infected across trial follow-up among those at risk of new infection, along with error bars representing 95% binomial confidence intervals (Clopper-Pearson method) around the observed proportion (N = 1639 across all trial arms and sites). The denominators for each trial site are shown in Table 1. Colours denote different genotypes. Model predictions from the combined fit are shown by the lines (posterior median) and shaded areas (95% Credible interval), dhps AKA (A437-K540-A581), dhps GKA (437G-K540-A581), dhps GEA (437G-540E-A581), dhps GEG (437G-540E-581G); dhps: dihydropteroate synthase.
SPAS provided a very similar duration of protection to SP against dhps- sulfadoxine-susceptible parasites (dhps AKA) and the dhps GKA mutant, as expected given the short elimination half-life of artesunate of <15 min32. However, SPAS provided significantly longer protection (16.5 days, 95% CrI: 11.2–37.4) against the dhps GEA genotype than did SP alone (10.7 days, 95% CrI: 8.9–21.9). The estimated 30-day protective efficacy of SPAS/SP against new infection in the 12 sites ranged between 15.4% to 98.1%, depending on the ratios of the genotypes present (Supplementary Table 4). The distribution of protective efficacy over time since treatment is shown in Fig. 3 for each genotype. Neither day 0 drug concentrations nor initial parasite density were associated with time to reinfection (Supplementary Note 2), though these were only available from a single trial24,33.

For each set of model parameters across 10,000 iterations, the probability of protection was calculated as a function of time since dose, and the Weibull shape and scale parameters. The solid lines denote the posterior median probability of protection and the shaded areas the 95% credible intervals. Mutations are underlined and shown in bold. Estimated parameters for SPAS were used to obtain these curves, except for the dhps GEG genotype which uses SP-related parameters. The vertical line denotes the estimated mean duration of protection provided by the drug against each genotype. dhps AKA (A437-K540-A581) in panel (a), dhps GKA (437G-K540-A581) in panel (b), dhps GEA (437G-540E-A581) in panel (c), dhps GEG (437G-540E-581G) in panel (d); dhps: dihydropteroate synthase.
We performed validation analysis against two IPTi trials conducted in Mozambique (2002–2004) and Tanzania (2004–2008). Using our model parameters estimated in the main analysis and the frequency of genotypes in the trial sites we predicted the mean duration of protection and the number of reinfections over time that would be expected in the IPTi trials following the first dose (Fig. 4). Accounting for weekly fluctuations in incidence without incorporating any genotype effects, the overall duration of protection offered by SP against clinical malaria was estimated to be 25.0 days (95% CrI: 12.0–41.5) and 10.9 days (95% CrI: 3.8–29.8) in the Mozambique and Tanzania IPTi trials, respectively. Similar estimates for protection were obtained when a time-constant force of infection was assumed across follow-up (Supplementary Table 5). Withholding the control group from the analysis resulted in slightly shorter protection for the trial in Mozambique (20.7 days) and slightly longer protection for the trial in Tanzania (14.9 days). The expected duration of protection against any infection using the estimated genotype-specific parameters in the main analysis, allowing for the frequencies of genotypes in each site, was similar (22.1 days and 13.7 days for the Mozambican and Tanzanian IPTi trials, respectively). The model predictions closely follow the observed proportion of patients reinfected after the first SP dose in both trials (Fig. 4). In the IPTi trial in Mozambique, the model predicts that nearly all infections are dhps GEA over the first 30 days after SP. In the IPTi trial in Tanzania, a small number of new infections were observed between the first and second doses, with similar numbers in the SP and placebo arms, consistent with a short protection conferred by SP.

All panels show the predicted proportion infected over time since the SP dose among those at risk of new infection for the Macete et al. trial20 (a, b) and the Gosling et al. trial12 (c, d). In panels a and c, dots represent the observed proportion of infections, with error bars denoting 95% binomial confidence intervals (Clopper-Pearson method) around the observed proportion. The sample size for plots a and c was 1497 for Mozambique (SP = 747 and placebo = 750) and 639 for Tanzania (SP = 319 and placebo = 320). Solid lines and shaded areas denote the model fit to the data (posterior median) and associated 95% Credible Intervals. Dashed lines show the predicted proportion infected given the estimated protection parameters from the main analysis and estimated frequency of each genotype (56% dhps GEA and 44% dhps AKA in the Mozambique trial21, and 53.6% dhps GEA and 41.1% dhps GEG in Northern Tanzania13). Panels b and d show the predicted proportions of infection with each genotype for each treatment. c dhps: dihydropteroate synthase.
Trial data on other antimalarial therapies were included in the analysis (Table 2 and Supplementary Fig. 3). Despite SP showing a relatively short protection against dhps GEA, SPAQ showed a substantially longer protection of 42.5 days against this genotype, as expected given the long duration of action of amodiaquine’s main active metabolite, desethylamodiaquine. This estimate is informed by trial data in Malawi, where the day 0 prevalence of Pfcrt 76T, Pfmdr1 86Y and Pfmdr1 1246Y mutations associated with amodiaquine resistance were low (0%, ~10% and 3%, respectively). In this study, there were no reinfections in the SPAQ arm by day 28 and only four reinfections were observed on day 42. In the same study, another SP-combination treatment, SPCQ, also showed a longer protection compared to SP (Table 2). Accounting for heterogeneity in the risk of transmission resulted in similar estimates of mean duration of protection across all drugs (Supplementary Table 7).
Applications in predicting impact of chemoprevention
Using our results on genotype-specific duration of SP protection, an SP protective efficacy prediction tool was developed which can be used to estimate PMC efficacy for any ___location where the frequencies of SP resistance markers are known: https://andriamousa.shinyapps.io/SP_PE_prediction_tool/ (Supplementary Note 3). In Fig. 5, applications of the tool are displayed for three example sites, each representing the main genotype profiles present in West Africa, East Africa and high-resistance pockets in East Africa.

Each insert uses published data on genotype prevalence for three sites in Donga, Benin55, Nord Kivu, DRC56, and Inhambane, Mozambique57. To estimate the 30-day protective efficacy, we used an incidence of malaria of 0.84, 0.15, and 0.29 infections per person per year for Donga, Nord-Kivu, and Inhambane, respectively. The dhps AKA is the sulfadoxine-susceptible genotype. Mutations are underlined and shown in bold. Red markers denote surveys conducted in Africa which reported a prevalence of >50% for dhps A581G and >90% for dhps K540E; dhps AKA (A437-K540-A581), dhps GKA (437G-K540-A581), dhps GEA (437G-540E-A581), dhps GEG (437G-540E-581G); dhps: dihydropteroate synthase, DRC: Democratic Republic of the Congo.
Using our estimated protective efficacy profiles against each genotype, we used previously published estimates of dhps genotype frequencies34 to predict the SP 30-day protective efficacy against any new infection and the median duration of protection (Fig. 6 and Supplementary Fig. 4). Across sub-Saharan Africa, 30-day protective efficacy varied from 59.3% to 91.5%, and the median duration of protection ranged from 17.2 to 37.2 days. We estimated that this protective efficacy corresponded to a mean of 4.7 clinical cases averted per 100 children aged 0 to 2 following a single-dose in areas of moderate-to-high transmission (PRPf2-10 ≥ 10%) (Fig. 7, Supplementary Fig 5). Similar reductions were estimated in the 0 to 5 year age group (Supplementary Fig 5).The highest mean number of clinical cases averted per 100 per dose was estimated for Liberia (8.9), followed by Benin (8.5), Sierra Leone (7.3) and Democratic Republic of the Congo (6.9).

Estimated 30-day protective efficacy (panel a) and median duration of protection (panel b). These are based on the genotype-specific Weibull shape and scale parameters estimated in this analysis and the frequency of each genotype estimated in a previous study for 202034. Protective efficacy in this figure incorporates reinfections (protective efficacy against any episode, rather than first episode). See Supplementary Note 3 for details on how these metrics were estimated. Coordinates are shown in radians.

These are based on prevalence rate estimates from the Malaria Atlas Project in 2–10 year olds (PfPR2-10) and the relationship between PfPR2-10 and incidence in children aged 0 to 2, obtained from the Imperial College malaria transmission model47,48. Incidence following a single SP dose was estimated using the 30-day protective efficacy shown in Fig. 6.
Discussion
Understanding the length of protection conferred by SP against new infections is paramount in informing and shaping effective chemoprevention policies. Our method allows estimation of the length of protection against different parasite genotypes, providing a valuable tool for tailoring preventive strategies in diverse settings. Using reinfection data from therapeutic efficacy studies of SP or SPAS, we estimated a significantly shorter duration of SP protection against parasites carrying more mutations. Protective efficacy was maintained against sulfadoxine-susceptible parasites and those with only the dhps A437G mutation. However, parasites with the dhps GEA or GEG genotypes were associated with shorter durations of protection.
We observed a long duration of protection (56 days) against sulfadoxine-susceptible parasites, consistent with findings from a chemosensitivity study indicating a duration of in vivo inhibitory concentration of >52 days35. Evidence from an IPTi-SPAS trial conducted in a Senegalese setting where parasite genotypes were either dhps AKA (sulfadoxine-susceptible)(67%) or dhps GKA (29%) further supports the findings on the long duration of protection against sulfadoxine-susceptible strains36. In this trial, new infections in the month following the first SPAS dose occurred in 22% of the control cohort and in <2% of the intervention arm.
In the validation analysis, using IPTi trial data from two studies, one in a setting of dhps AKA (sulfadoxine-susceptible) genotype and the other of dhps GEA genotypes, we were able to replicate the trial results using genotype-specific parameters derived from our primary analysis. This underscores the reliability and generalizability of our findings, providing valuable validation for the application of estimated protection parameters to broader epidemiological contexts. However, the outcome in the TES analysis was patent infection, whereas the IPTi trial outcome was clinical infection. Any small differences between time to parasitaemia and time to clinical symptoms may not be captured between weekly follow-up visits. Furthermore, evidence from a systematic review suggest that efficacy against parasitaemia is not significantly different to efficacy against clinical infection3. However, more evidence is needed to assess whether protective efficacy against any infection is proportional to efficacy against clinical infection, particularly in high-transmission areas with high rates of acquired immunity and highly prevalent asymptomatic parasitaemia.
In malaria-endemic regions characterized by high prevalence of SP-resistance-associated mutations, our study highlights the need for the strategic use of alternate regimens, such as SP plus Amodiaquine (SPAQ). SPAQ exhibits significantly higher chemoprevention efficacy compared to SP alone in areas where dhps GEA and GEG parasites are predominant. The long duration of protection of SPAQ is higher than SP or AQ alone, suggesting a potential boosting effect when co-administered37. To date, there is only one chemoprevention study using SPAQ in an area of saturated prevalence of dhps GEA38. However, in the study by Nuwa et al. there was a near-zero prevalence of mutations in the chloroquine resistance transporter (crt) and multiple drug resistance 1 (mdr 1) genes that are associated with reduced efficacy of amodiaquine, similar to the TES in Malawi included here24. To allow extrapolation to other settings, analysis of SPAQ trial data from settings with a higher prevalence of these mutations is essential for understanding their impact on protective efficacy.
The need to use data from previous TES trials, not originally designed to estimate chemoprevention, adds to the uncertainty around the predicted duration of protection. Where AS was added to SP, we expect no added protection against parasites acquired after the drug dose compared to SP alone, as AS has a short half-life of <1 h32,37. However, based on published TES studies, the model predicted a shorter duration of protection for SP alone compared to SPAS. This is most likely due to misclassification errors whereby recrudescent episodes were scored as new infections following the drug dose in the TES data39. For instance, a parasite present on day 0 may be missed by microscopy, but then be detected during follow-up. In areas of high SP-resistance, this misclassification is more likely to occur in the SP group where more recrudescent infections are expected compared to the SPAS group24,30, in which most day 0 infections will be cleared. Therefore, the duration of protection for SP alone may be underestimated, and trials of SPAS may provide a more reliable estimate of protective efficacy. In future chemoprevention trials, analysing the risk of new infections in recipients who are parasite-negative at day 0 would solve this issue by removing cryptic recrudescent parasites as a source of misclassification.
One of the limitations of this study is the lack of control groups in the trial settings. This means the true underlying transmission rates are not directly measured and assumed to be constant. In the absence of a control group, a long follow-up is needed to isolate drug effects, and is particularly challenging in settings with fluctuating or seasonal transmission which can result in inaccurate estimates of protection31. Estimates for protection against the sulfadoxine-susceptible dhps genotypes were longer than the duration of follow-up in the studies where this genotype was present (42 days) so studies with a longer follow-up would be needed to confirm that. Nevertheless, the duration of SP protection is likely longer than 42 days, as supported by other studies35. Another limitation is that genotype data on the day of reinfection were limited for some of the studies and differences in the specific laboratory methods used by each study may also influence the findings. More data are needed to inform our parameter estimates, particularly on the more resistant dhps GEG genotype. This genotype was only covered by one of the included studies, which had a shorter follow-up of 28 days13. Other possible confounders such as the effect of the effect of age and immunity were not explored. Underdosing in young children is associated with treatment failure40, and was not explored in this analysis. We expect that this effect is the same across studies, except in those using different age groups, where the genotype effects on protection may be harder to distinguish from age effects. However, our accurate prediction of the IPTi trial results despite differences in age groups, suggests that this confounding effect is minimal. Lastly, the analysis of historical studies can give no insight into protection against novel genotypes, such as dhps-431V which has emerged in West and Central Africa since SP was withdrawn as first-line treatment41. Furthermore, there was no individual-level data on the dhfr-164L mutation, though data by WWARN and a published literature review suggest that it is either absent or extremely rare in the countries of the included studies9,42. Quantifying the impact of the dhfr-164L mutation on protective efficacy would improve predictions when extrapolating to areas where these are present.
Quantifying the effects of genotype-specific protection is essential for modelling the suitability of SP in chemoprevention. This approach establishes a valuable methodology which can be applied across all epidemiological settings where resistance profiles have been characterised, and can be applied to other treatment regimens including SPAQ and DP, by quantifying the effects of relevant markers such as mdr 1 and crt. This study highlights the need for molecular surveillance data to guide drug selection and roll out of PMC and other chemoprevention strategies. Integrating estimates of genotype-specific protective efficacy with molecular surveillance provides a robust foundation for evidence-based stratification of malaria chemoprevention across a range of transmission settings.
Methods
Data sources
We systematically screened SP and SPAS efficacy trials included in the WWARN Clinical Trials Publication Library43. Eligible studies (1) were conducted in Africa, (2) included SP or SPAS treatment groups, (3) applied polymerase chain reaction (PCR) methods on day 0 and day of failure samples to distinguish reinfection from recrudescence, (4) had a minimum of 42 days follow-up, and (5) collected genotype data on dhps mutations. SPAS trials were included because artesunate (AS) is a short-acting drug that provides no post-treatment prophylaxis32,37. No time of publication or age limits were applied, though studies of pregnant women were excluded due to confounding effects of immunity and multigravidity. Results from a previous analysis31 indicated that a 28-day follow-up in single-arm trials may be insufficient to estimate the interval of protection. A follow-up of ≥42 days allows for more accurate disaggregation of drug protection and underlying transmission effects because the incidence is observed towards the end of follow-up when drug concentration levels are low. However, we did include one study with 28 days follow-up as it was the only one where the highly resistant dhps GEG (437G-540E-581G) genotype was present13.
We requested individual-participant data directly from research groups of identified studies. Data on time to infection with each genotype since the time of drug administration were either obtained from individual-participant data or, where unavailable25, extracted from publications. Individuals were followed up after drug administration and monitored for treatment failure. All studies used PCR genotyping (msp-2, msp-1 or glurp genes) to distinguish new infections from infections present on the day of drug administration (day 0) which had failed to clear or recrudesced (Supplementary Table 1). Recrudescent cases were censored on the day of failure when they received rescue treatment, except when new parasite variants were detected on the same day as the recrudescence. The presence of multiple parasite strains in a single sample in high transmission areas, may result in the majority sensitive genotype being detected but minority resistant infections being missed on day 0. For each individual, the dhps genotypes present in the samples collected on day 0 were compared with those present on the day of failure. Where participants had a mixed infection on both the day of failure and the day of new infection, and where the dhps genotype of the new infection could not be distinguished from the original infection, the genotype of the new infection was considered undetermined, but was still analysed.
Drug concentration data on sulfadoxine and pyrimethamine on day 7 following drug administration were only partially available for one study24,33. For this study, we summarise the mean and median post-treatment drug concentrations on day 0 and day 7, along with initial parasite density by treatment outcome (new infection vs. no new infection during follow-up). In the Supplementary Note 2, we report results from a Cox-regression model on time to new infection accounting for day 0 drug concentrations and parasite density to explore the possibility of confounding effects.
Analysis methods
Trial data were used to estimate the protection against the four genotypes: dhps AKA (sulfadoxine-susceptible), GKA, GEA, GEG. We recently developed a deterministic multi-strain model describing new infection after treatment31 which we used here to quantify SP protective efficacy, building on previous modelling approaches44,45 (Supplementary Note 1). In brief, the probability of being protected by the drug was quantified at each time-step following treatment, by fitting Weibull survival curves to the reinfection data using Hamiltonian Monte Carlo (HMC) methods in RStan46. We fit the model to all data across 12 trial sites simultaneously estimating the site-specific underlying incidence of infection (assumed constant over study follow-up), the site-specific frequency of each genotype in the parasite population, and independent protection curves against each genotypic strain. For studies with more than one treatment group, we fit different Weibull protection curves for each drug. Incidence of infection was assumed to be constant over the 42 days follow-up, due to the challenge of identifying fluctuating transmission effects in the absence of a control group.
30-day protective efficacy against first infection was estimated as the percentage of new infections with each strain prevented by the drug compared to a theoretical control group of no chemoprevention over 30 days. A single-strain model was used in places with limited genotype data and where drug resistance was high (prevalence of resistance genotype on day 0 > 85%), assuming that new infections consisted of resistant parasites. A two-strain model, that incorporates the frequency of each genotype, was used for studies where more than one genotype was present. The time-step used in the model (dt) was 0.5 days. We used relatively uninformative priors for all parameters related to drug protection effects and frequency of genotypes (Supplementary Table 3).
In the absence of a control group, without a reasonably informative prior for malaria incidence, the risk of infection is difficult to distinguish from the protective effect of the drug. Hence, priors used for malaria incidence in each site were semi-informative and were based on predictions using the Imperial College model of malaria transmission47,48 calibrated to reported prevalence of parasitaemia where available26,30. If unavailable, we calibrated the model to predicted prevalence from the Malaria Atlas Project49,50 specific to the year and place of the survey13,24,25,26,27,28,29(Supplementary Table 3 and Supplementary Fig 6). We ran 10,000 model iterations and four chains (5000 burn-in iterations per chain). Convergence of all MCMC chains was assessed by visually assessing the posterior distributions and traceplots, and using a threshold of <1.05 for the Gelman-Rubin’s convergence diagnostic \((\hat{R})\) and >1000 for the effective sample size (ESS) and effective tail distribution (Tail-ESS) per chain51 (Supplementary Table 6). As a sensitivity analysis, we explored the effect of including heterogeneity in risk of transmission in the main analysis. To account for variation in risk between individuals, three risk groups were used, partitioned using Gaussian quadrature (Supplementary Table 7).
If the studies included additional drug arms other than SP/SPAS, these were also used in the model fitting to provide information on the underlying incidence of infection and genotype frequencies (Supplementary Fig. 3). Studies were analysed together, by fitting site-specific background malaria incidence and underlying genotype frequencies, and a pooled estimate of SP protection against each genotype (by drug arm). Using the parameter estimates derived from this analysis, we developed a simple web-based interactive tool to predict the chemoprevention efficacy of SP alone for areas of varying resistance and endemicity profiles (Supplementary Note 3).
A recently published study estimated the frequency of dhps haplotypes across sub-Saharan Africa, using a Bayesian spatiotemporal model and molecular surveillance data from WWARN34. Using these estimates (Supplementary Fig. 4) we apply protective efficacy parameters obtained from the current analysis to predict SP chemoprevention impact across sub-Saharan Africa in 2020 (Fig. 6).
We used prevalence estimates in children aged 2 to 10 for the year 2020 obtained from the Malaria Atlas Project at 5 km × 5 km resolution49. The relationship between parasite prevalence in 2 to 10 year olds and incidence in 0 to 2 year olds, obtained from an agent-based malaria transmission model47,48, was then used to infer incidence in each 5 km × 5 km pixel across Africa. The 30-day protective efficacy was then applied to these incidence metrics to estimate the number of clinical cases aged 0 to 2 averted per population after a single SP dose. The same process was repeated with incidence in 0 to 5 year olds to compare impact in these two age groups. To calculate mean clinical cases averted per dose for each country in areas of moderate to high transmission, we weighted the values by the population in each 5 km × 5 km, using 2020 WorldPop population data52.
Validation
Placebo-controlled IPTi-SP trial data12,20 were used for validation of our results, and were obtained from trial investigators or digitized from published Kaplan-Meier survival curves for both treatment and control arms after the first dose of SP in the trial and before any further doses. Where no dhps genotype data were available from the IPTi trial participants, we used data on the frequency of SP-resistance markers in that area and year from other sources. In the case of the IPTi trial in Mozambique20, the genotype frequency information was obtained from samples collected from symptomatic malaria cases in the placebo group of the IPTi trial21, representing population frequencies in the absence of drug selection. In the case of the IPTi trial in Tanzania, frequency estimates were obtained from samples collected in a health facility within 30 km of the IPTi implementing district, and during the same time as the IPTi trial (in 2006)13. Using both control and treatment arms, we estimated weekly clinical incidence and a Weibull survival curve for protective efficacy using RStan as above. We then predicted new infections given the estimated incidence and local frequencies of resistance genotypes13,21 and compared the predicted duration of SP protection with that estimated in the observed IPTi data used for validation.
As an additional sensitivity analysis we repeated the validation analysis with a time-constant force of infection and compared the estimated incidence rate in the IPTi trial by including and excluding the placebo-controlled arm. We further compared the estimated duration of protection with that obtained from the analysis using time-varying incidence, and that expected using the protection parameters from the main analysis of the TES trials.
All analyses and visualisations were performed in R version 4.0.3. All maps presented use national boundaries obtained from the global administrative areas database (GADM, version 4.1).
Ethics statement
All studies included in this analysis have been published. Informed consent was obtained from participants or their parents or guardians. All individual studies were approved by both institutional ethics committees and local ethics review committees. This secondary analysis of trial data has been approved by the London School of Hygiene and Tropical Medicine ethics committee (reference: 29340).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available with no restrictions in https://github.com/AndriaMousa/SP-resistance-protective-efficacy-code. The repository containing the data has been archived, with accession number: 10.5281/zenodo.1498881953.
Code availability
The analysis code that supports the findings of this study is available with no restrictions in https://github.com/AndriaMousa/SP-resistance-protective-efficacy-code. The repository containing the code has been archived, with accession number: 10.5281/zenodo.1498881953.
References
World Health Organization. World Malaria Report 2023. (Geneva, 2023).
World Health Organization. WHO Guidelines for Malaria. (Geneva, 2022).
Esu, E. B., Oringanje, C. & Meremikwu, M. M. Intermittent preventive treatment for malaria in infants. Cochrane Database Syst. Rev. 7, Cd011525 (2021).
Okell, L. C., Griffin, J. T. & Roper, C. Mapping sulphadoxine-pyrimethamine-resistant Plasmodium falciparum malaria in infected humans and in parasite populations in Africa. Sci. Rep. 7, 7389 (2017).
Plowe, C. V. Malaria chemoprevention and drug resistance: a review of the literature and policy implications. Malar. J. 21, 104 (2022).
Amimo, F. et al. Plasmodium falciparum resistance to sulfadoxine-pyrimethamine in Africa: a systematic analysis of national trends. BMJ Glob. Health 5, e003217 (2020).
Picot, S. et al. A systematic review and meta-analysis of evidence for correlation between molecular markers of parasite resistance and treatment outcome in falciparum malaria. Malar. J. 8, 89 (2009).
Gregson, A. & Plowe, C. V. Mechanisms of resistance of malaria parasites to antifolates. Pharm. Rev. 57, 117–145 (2005).
Infectious Diseases Data Observatory (IDDO). SP Molecular Surveyor. Infectious Diseases Data Observatory. (InteractiveResource). https://www.wwarn.org/tracking-resistance/sp-molecular-surveyor (2015).
Naidoo, I. & Roper, C. Mapping ‘partially resistant’, ‘fully resistant’, and ‘super resistant’ malaria. Trends Parasitol. 29, 505–515 (2013).
Flegg, J. A. et al. Spatiotemporal spread of Plasmodium falciparum mutations for resistance to sulfadoxine-pyrimethamine across Africa, 1990-2020. PLoS Comput. Biol. 18, e1010317 (2022).
Gosling, R. D. et al. Protective efficacy and safety of three antimalarial regimens for intermittent preventive treatment for malaria in infants: a randomised, double-blind, placebo-controlled trial. Lancet 374, 1521–1532 (2009).
Gesase, S. et al. High resistance of Plasmodium falciparum to sulphadoxine/pyrimethamine in northern Tanzania and the emergence of dhps resistance mutation at Codon 581. PLoS ONE 4, e4569 (2009).
Griffin, J. T. et al. Protective efficacy of intermittent preventive treatment of malaria in infants (IPTi) using sulfadoxine-pyrimethamine and parasite resistance. PLoS ONE 5, e12618 (2010).
Cairns, M. et al. Duration of protection against clinical malaria provided by three regimens of intermittent preventive treatment in tanzanian infants. PLoS ONE 5, e9467 (2010).
Gutman, J. et al. The A581G mutation in the gene encoding plasmodium falciparum dihydropteroate synthetase reduces the effectiveness of sulfadoxine-pyrimethamine preventive therapy in Malawian pregnant women. J. Infect. Dis. 211, 1997–2005 (2015).
Chico, R. M. et al. Influence of malaria transmission intensity and the 581G mutation on the efficacy of intermittent preventive treatment in pregnancy: systematic review and meta-analysis. Trop. Med. Int. Health 20, 1621–1633 (2015).
Hansson, H. et al. Reduced birth weight caused by sextuple drug-resistant Plasmodium falciparum Infection in early second trimester. J. Infect. Dis. 224, 1605–1613 (2021).
Sandison, T. G. et al. Protective efficacy of co-trimoxazole prophylaxis against malaria in HIV exposed children in rural Uganda: a randomised clinical trial. Br. Med. J. 342, d1617 (2011).
Macete, E. et al. Intermittent preventive treatment for malaria control administered at the time of routine vaccinations in Mozambican infants: a randomized, placebo-controlled trial. J. Infect. Dis. 194, 276–285 (2006).
Mayor, A. et al. Molecular markers of resistance to sulfadoxine-pyrimethamine during intermittent preventive treatment for malaria in Mozambican infants. J. Infect. Dis. 197, 1737–1742 (2008).
Chandramohan, D. et al. Cluster randomised trial of intermittent preventive treatment for malaria in infants in area of high, seasonal transmission in Ghana. Br. Med. J. 331, 727–733 (2005).
Cairns, M. et al. Duration of protection against malaria and anaemia provided by intermittent preventive treatment in infants in Navrongo, Ghana. PLoS ONE 3, e2227 (2008).
Bell, D. J. et al. Sulfadoxine-pyrimethamine-based combinations for malaria: a randomised blinded trial to compare efficacy, safety and selection of resistance in Malawi. PLoS ONE 3, e1578 (2008).
Nahum, A. et al. Extended high efficacy of the combination sulphadoxine-pyrimethamine with artesunate in children with uncomplicated falciparum malaria on the Benin coast, West Africa. Malar. J. 8, 37 (2009).
Barnes, K. I. et al. Sulfadoxine-pyrimethamine pharmacokinetics in malaria: pediatric dosing implications. Clin. Pharmacol. Therap. 80, 582–596 (2006).
Mabuza, A. et al. Therapeutic efficacy of sulfadoxine-pyrimethamine for Plasmodium falciparum malaria. S. Afr. Med. J. 95, 346–349 (2005).
Barnes, K. I. et al. Increased gametocytemia after treatment: an early parasitological indicator of emerging sulfadoxine-pyrimethamine resistance in falciparum malaria. J. Infect. Dis. 197, 1605–1613 (2008).
Bredenkamp, B. L. F., Sharp, B. L. & Barnes, K. I. Failure of sulphadoxine-pyrimethamine in treating Plasmodium falciparum malaria in KwaZulu-Natal. SAMJ 91, 970–972 (2001).
Allen, E. N. et al. Efficacy of sulphadoxine-pyrimethamine with or without artesunate for the treatment of uncomplicated Plasmodium falciparum malaria in southern Mozambique: a randomized controlled trial. Malar. J. 8, 141 (2009).
Mousa, A. et al. Measuring protective efficacy and quantifying the impact of drug resistance: a novel malaria chemoprevention trial design and methodology. PLoS Med. 21, e1004376 (2024).
Morris, C. A. et al. Review of the clinical pharmacokinetics of artesunate and its active metabolite dihydroartemisinin following intravenous, intramuscular, oral or rectal administration. Malar. J. 10, 263 (2011).
Bell, D. J. et al. Measurement of adherence, drug concentrations and the effectiveness of artemether-lumefantrine, chlorproguanil-dapsone or sulphadoxine-pyrimethamine in the treatment of uncomplicated malaria in Malawi. Malar. J. 8, 204 (2009).
Foo, Y. S. & Flegg, J. A. A spatio-temporal model of multi-marker antimalarial resistance. J. R. Soc. Interface 21, 20230570 (2024).
Watkins, W. M., Mberu, E. K., Winstanley, P. A. & Plowe, C. V. The efficacy of antifolate antimalarial combinations in Africa: a predictive model based on pharmacodynamic and pharmacokinetic analyses. Parasitol. Today 13, 459–464 (1997).
Cissé, B. et al. Seasonal intermittent preventive treatment with artesunate and sulfadoxine-pyrimethamine for prevention of malaria in Senegalese children: a randomised, placebo-controlled, double-blind trial. Lancet 367, 659–667 (2006).
Sokhna, C. et al. A trial of the efficacy, safety and impact on drug resistance of four drug regimens for seasonal intermittent preventive treatment for malaria in Senegalese children. PLoS ONE 3, e1471 (2008).
Nuwa, A. et al. A non-randomized controlled trial to assess the protective effect of SMC in the context of high parasite resistance in Uganda. Malar. J. 22, 63 (2023).
Juliano, J. J., Gadalla, N., Sutherland, C. J. & Meshnick, S. R. The perils of PCR: can we accurately ‘correct’ antimalarial trials? Trends Parasitol. 26, 119–124 (2010).
Barnes, K. I., Watkins, W. M. & White, N. J. Antimalarial dosing regimens and drug resistance. Trends Parasitol. 24, 127–134 (2008).
Adegbola, A. J. et al. A snapshot of the prevalence of dihydropteroate synthase-431V mutation and other sulfadoxine-pyrimethamine resistance markers in Plasmodium falciparum isolates in Nigeria. Malar. J. 22, 71 (2023).
Lynch, C. et al. Emergence of a dhfr mutation conferring high-level drug resistance in Plasmodium falciparum populations from southwest Uganda. J. Infect. Dis. 197, 1598–1604 (2008).
Takata, J. et al. The World Wide Antimalarial Resistance Network Clinical Trials Publication Library: a live, open-access database of plasmodium treatment efficacy trials. Am. J. Trop. Med. Hyg. 103, 359–368 (2020).
Okell, L. C. et al. Contrasting benefits of different artemisinin combination therapies as first-line malaria treatments using model-based cost-effectiveness analysis. Nat. Commun. 5, 5606 (2014).
Bretscher, M. T. et al. The duration of chemoprophylaxis against malaria after treatment with artesunate-amodiaquine and artemether-lumefantrine and the effects of pfmdr1 86Y and pfcrt 76T: a meta-analysis of individual patient data. BMC Med. 18, 47 (2020).
Stan Development Team RStan: the R interface to Stan. https://mc-stan.org/ (2024).
Griffin, J. T. et al. Reducing Plasmodium falciparum malaria transmission in Africa: a model-based evaluation of intervention strategies. PLOS Med. 7, e1000324 (2010).
Griffin, J. T., Ferguson, N. M. & Ghani, A. C. Estimates of the changing age-burden of Plasmodium falciparum malaria disease in sub-Saharan Africa. Nat. Commun. 5, 3136 (2014).
Weiss, D. J. et al. Mapping the global prevalence, incidence, and mortality of Plasmodium falciparum, 2000-17: a spatial and temporal modelling study. Lancet 394, 322–331 (2019).
Pfeffer, D. A. et al. malariaAtlas: an R interface to global malariometric data hosted by the Malaria Atlas Project. Malar. J. 17, 352 (2018).
Vehtari, A., Gelman, A., Simpson, D., Carpenter, B. & Bürkner, P.-C. Rank-normalization, folding, and localization: an improved R ̂ for assessing convergence of MCMC (with discussion). Bayesian Anal. 16, 667–718 (2021).
Tatem, A. J. WorldPop, open data for spatial demography. Sci. Data 4, 170004 (2017).
Mousa, A. SP-resistance-protective-efficacy-code. https://doi.org/10.5281/zenodo.14988819 (2025).
Roper, C. et al. Antifolate antimalarial resistance in southeast Africa: a population-based analysis. Lancet 361, 1174–1181 (2003).
Svigel, S. S. et al. Low prevalence of highly sulfadoxine‐resistant dihydropteroate synthase alleles in Plasmodium falciparum isolates in Benin. Malar. J. 20, 72 (2021).
Guémas, E. et al. Evolution and spread of Plasmodium falciparum mutations associated with resistance to sulfadoxine-pyrimethamine in central Africa: a cross-sectional study. Lancet Microbe 4, e983–e993 (2023).
da Silva, C. et al. Targeted and whole-genome sequencing reveal a north-south divide in P. falciparum drug resistance markers and genetic structure in Mozambique. Commun. Biol. 6, 619 (2023).
Acknowledgements
A.M., C.R., R.G., R.M.C., C.J.S., K.B.B., A.C.P., M.A., E.F.H. and H.H. were funded by Unitaid as part of the Plus Project (www.unitaid.org, grant number: 101150IC). G.C.D. and L.O. acknowledge funding from the UK Royal Society and from the MRC Centre for Global Infectious Disease Analysis (reference MR/R015600/1), jointly funded by the UK Medical Research Council (MRC) and the UK Foreign, Commonwealth & Development Office (FCDO), under the MRC/FCDO Concordat agreement and is also part of the EDCTP2 programme supported by the European Union. KIB acknowledges funding from the WHO TDR and The Global Fund to Fight AIDS, Tuberculosis and Malaria (GFATM). The funders had no role in the study design, data collection, analysis, interpretation, decision to publish, or preparation of the manuscript. We would like to acknowledge Ilona Carneiro for help in interpreting IPTi trial data in Tanzania. We would also like to acknowledge WWARN Oxford for the curation of data related to the therapeutic efficacy studies conducted in Mozambique and South Africa.
Author information
Authors and Affiliations
Contributions
A.M., C.R., L.C.O., C.J.S., R.M.C. and R.G. contributed to the conceptualisation of the study. A.M., C.R. and L.C.O. contributed to study design, and C.R. and L.C.O. supervised the work. A.M., C.R., L.C.O., G.C.D. and individual study data contributors had access to the data and have contributed to verifying underlying data and analyses. AM performed analyses reported in the manuscript, and GCD and HAT contributed to the code and methodology. D.J.B., U.D.A., R.G., A.N., K.I.B., J.R., L.W., J.A.F. and Y.S.F. contributed to data acquisition, data curation and cleaning and helped interpretation of the trial data used in this paper. A.M., G.C.D., H.A.T., D.J.B., U.D.A., R.G., A.N., K.I.B., J.R., L.W., Y.S.F., J.A.F., E.F.H., H.H., A.C.P., K.B.B., M.A., R.M.C., C.J.S., L.C.O., and C.R. assisted in the interpretation of data and results. AM wrote the first draft of the paper. A.M., G.C.D., H.A.T., D.J.B., U.D.A., R.G., K.I.B., J.R., L.W., Y.S.F., J.A.F., E.F.H., H.H., A.C.P., K.B.B., M.A., R.M.C., C.J.S., L.C.O., and C.R. reviewed and edited the paper for important intellectual content and approved the final paper for publication.
Corresponding author
Ethics declarations
Competing interests
All authors declare no competing interests as defined by Nature Portfolio, or other interests that might be perceived to influence the results and/or discussion reported in this paper.
Peer review
Peer review information
Nature Communications thanks Ewan Cameron, Sunil Parikh and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mousa, A., Cuomo-Dannenburg, G., Thompson, H.A. et al. Impact of dhps mutations on sulfadoxine-pyrimethamine protective efficacy and implications for malaria chemoprevention. Nat Commun 16, 4268 (2025). https://doi.org/10.1038/s41467-025-58326-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58326-z
