
Abstract
Borylated carbocycles occupy a pivotal position as essential components in synthetic chemistry, drug discovery, and materials science. Herein, we present a photorearrangement that uniquely involves a boron atom enabled by energy transfer catalysis under visible light conditions. The boron functional group could be translocated through energy transfer mechanism and valuable borylated cyclopropane scaffolds could be generated smoothly. Furthermore, we showcase a 1,5-HAT (hydrogen atom transfer)/cyclization reaction, which is also enhanced by energy transfer catalysis excited by visible light. This method enables the synthesis of borylated cyclobutane frameworks. These boron-involved photorearrangement and cyclization reactions represent two techniques for synthesizing highly desirable borylated strained ring structures, which offering avenues for the synthesis of complex organic molecules with medicinal and material science applications.
Similar content being viewed by others

Introduction
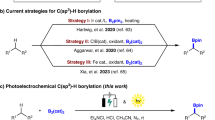
In the investigation of pharmaceutically interesting compounds, strain ring systems, such as cyclopropanes or cyclobutanes, have often been integrated into their structures to modify and enhance their biological activity as well as confer conformational rigidity. These properties can impact the performance and efficacy of novel drugs.1,2,3 Therefore, borylated strain rings are highly versatile and desirable building blocks in the fields of synthetic chemistry, drug discovery, and material science. Their unique properties offer numerous opportunities for further chemical manipulation and transformation.4,5,6,7,8,9,10,11 Synthetic transformations involving boron atoms are extensively researched, encompassing renowned reactions like the Nobel prize-wining Suzuki-Miyaura coupling,12 conjunctive cross-coupling,13 Matteson-type reaction,14 radical based transformations15,16,17,18,19 and many others.20 Molecular rearrangement reactions are among the fundamental transformations in chemistry, enabling the reconstruction of molecules and precise editing of their architectures with high atom and step efficiency.21 This level of precision and efficiency is often difficult to achieve through other synthetic methods.22 In particularly, radical rearrangement has been a popular and mild approach to reconstruct molecule architectures through single electron transfer (SET) mechanism.23,24,25,26,27,28,29 Pioneering by Batey,30 Aggarwal,31,32,33 and Studer,34,35 radical migration involving 1,2-boron shift has been achieved successfully through the SET mechanism (Fig. 1A).36,37,38,39 Remarkably, Studer40 and Song41 have also demonstrated radical translocation involving 1,4-boron shift, which occurs via the SET mechism (Fig. 1B). However, boron-enabled radical rearrangement through energy transfer mechanism is rare but high in demand due to further explosion in new reactivity and potential synthetic application.42,43 The diradical species, resulting from energy transfer catalysis, unveils novel reaction models that incorporate sequential boron migration and radical cyclization, creating useful borylated strain rings, which were difficult to be formed through traditional boron migration approaches via electron transfer mechanisms.

A Radical migration involving 1,2-boron shift via SET. B Radical migration involving 1,4-boron shift via SET. C Boronic Ester Enabled [2 + 2]-Cycloadditions. D Our design-Borylated Carbocycles Synthesis Enabled by Energy Transfer Catalysis.
Recently, energy transfer catalysis has emerged as a valuable synthetic strategy, enabling a range of transformations including σ-bond cleavage, [2 + 2] reaction, isomerization and others.44,45,46,47,48 For example, Bach,49 Yoon,50 Knowles,51 Brown52,53,54 and Molloy55 have pioneered groundbreaking research involving boronic ester enabled [2 + 2]-cycloadditions49,50,51,52,53,55 and photo-ene reactions,54 which have allowed them to construct a diverse array of borylated architectures (Fig. 1C, upper). Masarwa and his colleagues have showcased the utilization of polyborylated alkenes as coupling partners in an elegant set of examples. Through energy transfer catalysis, they have successfully accessed multi-borylated cyclobutanes via the [2 + 2] reaction (Fig. 1C, down).56 Although the reaction types facilitated by energy transfer catalysis are relatively limited, the di-π-methane rearrangement, first discovered by Zimmerman and his team in 1967, presents an alternative synthetic approach.57,58 However, the progress in developing the di-π-methane rearrangement has been somewhat sluggish.59,60,61 Our recent research has shown that the di-π-ethane rearrangement concept can achieve radical translocation of nitrile62 and aromatic ring63,64 functional groups. This advancement indicates that the further development of energy transfer-enabled rearrangements holds the potential to broaden the scope and applicability of this synthetic methodology.
Inspired by these examples and our keen interest in energy transfer-enabled photorearrangement involving boron atoms, we are intrigued to investigate whether a π-bond could potentially be substituted with a σ-bond. This question arises from our desire to explore novel reaction pathways that involve boron-containing compounds. With this concept in mind, we have designed a substrate that incorporates a carbon-boron σ-bond and a carbon-carbon π-bond, separated by a single sp3 carbon (analogous to methane). A photorearrangement involving this type of substrate could aptly be termed “π, σ-methane rearrangement”. This approach holds promise for exploring photorearrangement mediated by energy transfer processes involving boron atoms. As depicted in Fig. 1D, we envision that the borylated substrate could potentially be activated under visible light conditions with the aid of suitable energy transfer catalysis to form a diradical intermediate. This activation would enable the terminal primary radical I to interact with the boronic ester, leading to the cleavage of the carbon-boron σ-bond and subsequently achieving 1,4-boron migration.40,41 Alternatively, when the borylated substrate possesses a relatively weak C—H bond, the diradical intermediate III could still be generated through an energy transfer mechanism. In this case, the primary radical species of intermediate III may undergo 1,5-hydrogen atom transfer rather than attacking the boronic ester. This alternative pathway efficiently forms the 1,4-diradical intermediate IV. The successful implementation of this energy transfer driven, π, σ-methane reaction and HAT/cyclization represents two complementary approaches in the evolving fields of energy transfer catalysis and the synthesis of polyborylated strained rings.4,10,65
Results and discussion
Reaction design and optimization
Bearing the idea in mind, we synthesized the substrate 1a through a simple SN2 reaction and then subjected it to a basic set of conditions (Fig. 2, entry 1: 2 mol% Ir(ppy)3, acetonitrile as solvent, and illumination with 450 nm LEDs). However, the outcome was disappointing, as only the starting material 1a was recovered. Even when we attempted using a carbazole-based photocatalyst (4CzIPN) in a second experiment (entry 2), it failed to yield the desired results. Fortunately, when we employed Ir-F as the photocatalyst (entry 3), we were able to achieve the formation of the desired product 3 with an isolated yield of 87% and 1.8:1 dr. Although there was minimal diastereoselectivity observed, the diastereomeric mixtures of 3 could be readily separated via column chromatography. Furthermore, both Ir-Me and thioxanthone were suitable photocatalysts for the current π, σ-methane rearrangement process (entries 4 & 5). Control experiments conducted further emphasized that both light and a photocatalyst are necessary for the success of the current catalytic system (entries 6–7). Notably, we also conducted a screening of various organic solvents, and the results indicated that this boron-involved π, σ-methane rearrangement exhibited good tolerance to a wide range of organic solvents. Consequently, the desired product 3 was obtained in yields ranging from good to excellent (entries 8–15). Interestingly, the diastereoisomeric ratio of product 3 underwent a slight improvement, reaching approximately a 3:1 ratio, when trifluoromethylbenzene was utilized as the organic solvent (entry 11). Entries 1 through 5 demonstrate that the yield of 3 is contingent on the triplet energy of the photocatalyst, rather than its reduction potentials. This suggests that the ongoing π, σ-methane rearrangement is probably facilitated by a triplet-triplet energy transfer (TTEnT) mechanism.

Reaction condition: 1a (0.2 mmol), photocatalyst (2 mol%), and MeCN (2 mL) at room temperature under 4 × 30 W 450 nm LEDs for 12 h under N2; then the solvent was removed and the mixture was treated with aq. NaOH (2 M, 2 mL, 10 equiv.), aq. H2O2 (30%, 1 mL), THF (1 mL, 0.1 M). The product of two isomers could be separated through column chromatography. a 3 mol% 4CzIPN is used. b 10 mol% thioxanthone and 405 nm LEDs are used. c isolated yield.
Substrate scope
Utilizing the optimized conditions, we started to explore the versatility of this energy transfer enabled π, σ-methane rearrangement (Fig. 3). Our findings suggest that the scope of this photorearrangement is indeed general when considering mono-boron-containing substrates. Our initial findings are encouraging, as various substituted aromatic rings proved to be tolerable, resulting in good to excellent yields. The two isomers of desired boron products could not be separated in most examples (except 2w and 2ae). Thus, we decided to oxidize the mixture, and the corresponding alcohols could be obtained efficiently. Specifically, fluoro (4–6), chloro (7), methyl (8), phenyl (9), trifluoromethoxy (10), trifluoromethyl (11), ketone (12), ester (13 & 17), naphthyl (14), difluoro (15 & 16), trifluoro (18) substituents all performed well. Notably, this π, σ-methane rearrangement is also compatible with complex motifs, such as menthol (19), ibuprofen (20), and β-rhodinal (21). In addition, we screened different substitutions next to the boronic ester under the optimized conditions. Again, a range of aromatic substituents, including fluoro (22–24), chloro (25), methyl (26), isopropyl (27), tertbutyl (28), phenyl (29), trifluoromethoxy (30), naphthyl (31), di-substituted aromatic ring (32) were all tolerated in this newly developed catalytic system. X-ray analysis further confirmed the structure of product 29 (CCDC 2367147). Expanding our investigation, we also looked at di-substituted substrates. The desired cyclopropane products containing two all-carbon quaternary centers (33–35) could be obtained with moderate yields efficiently. The desired product 2ah could not be produced efficiently when only one aliphatic group was present in the substrate. However, when the substrate contained two aliphatic groups, the desired product 2ai was formed efficiently, suggesting that a tertiary radical intermediate is important for promoting this photorearrangement. Additionally, the alkene motif must be a styrene to lower the triplet energy of the substrate. Consequently, the desired product 2aj could not be formed. We also synthesized the substrate 1ak in an attempt to produce a five-membered ring product, but unfortunately, the desired product 2ak was not formed efficiently under standard conditions. Therefore, we discovered that the equilibration of radical species via a 1,4-boron shift results in the formation of thermodynamically favored benzylic radicals (3–32) and tertiary radicals (33–35, 2ai). However, this process does not favor the formation of secondary radicals (2ah).

Reaction condition: 1a-1ak (0.2 mmol), Ir–F (2 mol %), and PhCF3 (2 mL, 0.1 M) at room temperature under 4 × 30 W 450 nm LEDs with a cooling fan for 12 h under N2; then the solvent was removed, and the mixture was treated with aq. NaOH (2 M, 1 mL, 10 equiv.), aq. H2O2 (30%, 1 mL), THF (2 mL), RT, 2 h. The product of two isomers could be separated through column chromatography in most cases. a 4 mol% Ir-F photocatalyst was used. b 4 mol% Ir-F photocatalyst and MeCN (2 mL, 0.1 M) were used. c alternative oxidation condition: NaBO3·4H2O (2.0 equiv.), THF:H2O (2:1, 3 mL), RT, 2 h.
Continuing our investigation, we examined the substrate scope of various diboron-containing substrates (Fig. 4, upper). The desired borylated cyclopropane products were efficiently obtained using 4 mol% Ir-F and acetonitrile as the solvent. We first tested a series of substituted aromatics, including fluoro (41–43), chloro (44), methyl (45), naphthyl (46), trifluoromethoxy (47), trifluoromethyl (48), ester (49), multi-fluoro-containing substrates (50-53). All of these substrates resulted in the synthesis of the desired borylated cyclopropane scaffolds in good to high yields. X-ray analysis further confirmed the structure of product 47 (CCDC 2367148). Interestingly, when the substrate containing relatively weak C—H bond, such benzylic, allylic, or propylic C—H positions, were examined, a sequential 1,5-hydrogen atom transfer (HAT)/cyclization process was observed instead of the rearrangement reaction. This alternative pathway led to the formation of a variety of borylated cyclobutane scaffolds (Fig. 4, down). Different aromatics substituents, including hydrogen (54), trifluoromethoxy (55), trifluoromethyl (56), flouro (57), chloro (58), nitrile (59), ester (60), and alkyne (61), alkene (62) functional groups, were all tolerated in this catalytic system, resulting in good yields. X-ray crystallographic analysis provided further confirmation of the structure of product 61' (CCDC 2367149). Despite observing moderate to little diastereoselectivity, diastereomeric mixtures were readily separated through the use of column chromatography, with the exception of products 61 and 62.

Reaction condition: 1al-1bh (0.2 mmol), Ir–F (4 mol %), and MeCN (2 mL, 0.1 M) at room temperature under 4 × 30 W 450 nm blue LEDs with a cooling fan for 24 or 36 h under N2. In most cases, the product of two isomers could be separated through column chromatography in most cases. a 36 h. b 2 mol% Ir-F photocatalyst and PhCF3 (2 mL, 0.1 M) were used, 12 h. c 2 mol% Ir-F photocatalyst and PhCF3 (2 mL, 0.1 M) were used, 9 h.
Synthetic application and mechanistic studies
After surveying the substrate scope of the photorearrangement enabled by energy transfer catalysis, we then explored the stability and synthetic application of these two newly developed rearrangement and cyclization methods. Firstly, large-scale level experiments were performed, and the desired borylated cyclopropane motif 2a could be obtained in 1.1 gram with an 82% isolated yield (Fig. 5A, left). Meanwhile, the borylated cyclobutane framework 54 could also be obtained in 0.8 grams with a 40% isolated yield (Fig. 5A, right). The continuous-flow experiment was also performed, and the desired borylated cyclopropane product 40 could also be obtained in 1.8 grams with a 60% isolated yield (Fig. 5B). The borylated strain ring product could also be manipulated easily (Fig. 5C). Di-boronic ester product 40' could be converted to vinyl containing product 63 and furan containing product 64 respectively using the corresponding organometallic reagents. The product 40 could also be transferred to the similar products 66 and 67. Additionally, the primary boronic ester could be oxidized to hydroxyl functional group 65. Furthermore, the tertiary boronic ester could react with different organometallic reagents to convert to phenyl or furan fictional groups (68 & 69) and also could undergo homologation to form the primary boronic ester 70. Interestingly, the four-membered cyclobutane motif 54 could be converted to ketone 71 and lactone 72 under different oxidation conditions. The cyclobutane motif 54' could also be transferred to lactone 72, mono-borated product 73, which could be further converted to alkene 74 and alcohol 75 smoothly. We then initiated mechanistic investigations to gain some insight into these two types of synthetic transformations. Firstly, radical inhibitor (TEMPO) and triplet energy quenching reagent were added to the standard condition respectively, we could not obtain the desired product, indicating that the π, σ-methane rearrangement may go through an energy transfer mechanism (Fig. 5D, upper). Traditional radical initiation experiments were also investigated in the current catalytic system, the desired product could not be formed, showing that an electron transfer mechanism is unlikely (Fig. 5D, down). Light on-off experiments clearly show that visible light is necessary for this photochemical rearrangement to occur (Fig. 5E). We then synthesized the deuterated substrate 1az-D2 and subjected it to our standard conditions. We observed the deuterium migration step, and the four-membered product 76 was obtained smoothly, clearly demonstrating that the hydrogen atom transfer (HAT) step occurred (Fig. 5F).66,67 The three-membered ring product 77 could also be formed as a by-product, suggesting that when the deuterium migration step was slower, the boron migration step took place. UV-vis experiment (Fig. 5G) and stern-volmer quenching experiments (Fig. 5H) showed that only the photocatalyst could absorb the visible light and the photo-excited Ir-F could only be quenched by the starting material 1a. The quantum yield of the π, σ-methane rearrangement was measured (ϕ = 0.0061), indicating that the current reaction is more likely a photocatalytic reaction. We also employed electron paramagnetic resonance (EPR) to investigate the π, σ-methane rearrangement of the boron functional group. Upon addition of the spin trap N-tert-butyl-α-phenylnitrone (PBN) to the reaction, a radical signal was observed (Fig. 5I). Analysis of the EPR data revealed that PBN captured a carbon-centered radical, indicating that the reaction proceeds via a radical mechanism.

A Large-scale experiment. B The continuous-flow experiment. C Synthetic application. D Radical inhibitor & initiation experiments. E Light on-off experiments. F Deuteriuim labeling experiment. G UV-Vis experiment. H Stern-Volmer quenching experiment. I EPR experiement. a Vinylmagnesium bromide (4.0 equiv.), THF (0.1 M), RT, 0.5 h, then I2 (4.0 equiv.), − 78 oC, 1 h, then NaOMe (8.0 equiv.), − 78 oC, 1 h; b Furan-2-yllithium (6.0 equiv.), THF (0.05 M), − 78 °C, 1 h, then NBS (6.0 equiv.), − 78 °C, 2 h; c NaBO3•4H2O (5.5 equiv.), THF (0.1 M), H2O (0.1 M), RT, 2 h; d Furan-2-yllithium (3.3 equiv.), THF (0.1 M), -78 °C, then NBS (3.0 equiv.), -78 °C, 2 h; e (3-methoxyphenyl)lithium (4.0 equiv.), THF (0.1 M), − 78 °C, 1 h, then NBS (4.0 equiv.), − 78 °C, 1 h; f Furan-2-yllithium (4.0 equiv.), THF (0.05 M), − 78 °C, 1 h, then NBS (4.0 equiv.), − 78 °C, 2 h; g ICH2Cl (5.0 equiv.), n-BuLi (4.0 equiv.), THF (0.1 M), − 78 °C, then RT, 1 h; h NaBO3•4H2O (2.0 equiv.), THF (0.1 M), H2O (0.1 M), RT, 4 h; i 0.1 mmol scale: 30% H2O2 0.5 mL, 2 M NaOH 0.56 mL, RT, THF (0.1 M), 4 h; j tBuOH (1.0 equiv.), THF (0.5 M), rt, 5 min, then tBuONa (3.0 equiv.), 0 °C to rt, 2 h; k Vinylmagnesium bromide (4.0 equiv.), THF (0.1 M), 0 °C, then RT, 30 min, then I2 (4.0 equiv.), − 78 °C, 20 min, then NaOMe (8.0 equiv.), 0 °C, 30 min, then RT, 3 h; l NaBO3•4H2O (2.0 equiv.), THF (0.05 M), H2O (0.1 M), 0 °C to rt, 3 h.
Computational studies
Density functional theory (DFT) computations with the DFT method were performed to understand further the π, σ-methane rearrangement (Fig. 6A) and 1,5-HAT (hydrogen atom transfer)/cyclization reaction (Fig. 6B). As shown in Fig. 6A, the adiabatic triplet π, π* reactant 1al* is 52.5 kcal/mol radiative to the ground state of 1al. The transition state TS_HAT (17.6 kcal/mol) was obtained, leading to intermediate INT_HAT (-1.3 kcal/mol), which is a 1,4-radical. TS_HAT is 2.8 kcal/mol higher in energy than TS1, indicating that the HAT step is disfavored. This intermediate INT-1al subsequently undergoes intersystem-crossing and collapses of the singlet biradical to the product 40. Furthermore, we performed the DFT calculation of sequential 1,5-HAT/cyclization reaction (Fig. 6B). The transition state TS_HAT (7.4 kcal/mol) was obtained, leading to intermediate INT_HAT (− 12.8 kcal/mol), which is a 1,4-radical. TS_HAT is 4.7 kcal/mol lower in energy than TS-1, indicating that the HAT step is much favored in the substrate of 1az. After intersystem-crossing, the singlet biradical S1az is formed smoothly and converted to the final product 54 after radical coupling.

A DFT calculation of π, σ-methane rearrangement; B DFT calculation of 1,5-HAT (hydrogen atom transfer)/cyclization reaction.
In summary, we have demonstrated the π, σ-methane rearrangement, and boron-involved HAT/cyclization reaction, facilitated by energy transfer catalysis. These two approaches enable efficient access to a diverse library of borylated cyclopropane and cyclobutane architectures. The key to the success of these synthetic transformations lies in the combination of suitable energy transfer catalysis and substrate design. The mild catalytic platform permits a broad substrate scope and good functional group toleration, providing practical and versatile access to various borylated strain rings. We believe that the newly developed energy transfer-enabled photorearrangement represents a significant addition to the field of energy transfer catalysis-enabled synthetic chemistry. Furthermore, both the synthetic approaches to borylated strained rings and their potential applications are expected to inspire synthetic and medicinal chemists in academia and industry alike.
Methods
General method A for the synthesis of products 3–34
An oven-dried 8-mL vial charged with a Teflon® septum was used. The vial was then charged with corresponding 1a-1af (0.2 mmol) and Ir-F (2 mol%). After degassed by vacuum-nitrogen purging, the vial was transferred to the glovebox, and the solvent (PhCF3, 2 mL, 0.1 M) was added. The vial was finally sealed with a poly-tetrafluoroethylene-lined cap tightly and stirred under irradiation with 4 × 30 W blue LEDs (λ = 450 nm, at approximately 3 cm away from the light source) at room temperature. Upon complete conversion, the mixture was passed through a short silica gel column. After the solvent was removed under reduced pressure, the residue was used directly for the next step without further purification. The residue prepared as shown above was added to a 20 mL vial equipped with a Teflon-coated magnetic stir bar. After adding 2 mL THF, the solution was cooled to 0 °C. 2 M NaOH (1.0 mL) was injected, followed by the addition of 30% H2O2 (1.0 mL) dropwise. The reaction mixture was stirred at rt for 2 h, diluted with brine, and extracted with EtOAc (15 × 3 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered. After the solvent was removed under reduced pressure, the residue was purified by flash column chromatography on silica gel to afford analytically pure products 3–34.
General method B for the synthesis of product 35
An oven-dried 8 mL vial charged with a Teflon® septum was used. The vial was then charged with corresponding 1ag (0.2 mmol) and Ir-F (2 mol%). After degassed by vacuum-nitrogen purging, the vial was transferred to the glovebox, and the solvent (PhCF3, 2 mL, 0.1 M) was added. The vial was finally sealed with a poly-tetrafluoroethylene-lined cap tightly and stirred under irradiation with 4 × 30 W blue LEDs (λ = 450 nm, at approximately 3 cm away from the light source) at room temperature. Upon complete conversion, the mixture was passed through a short silica gel column. After the solvent was removed under reduced pressure, the residue was used directly for the next step without further purification. After adding THF/H2O (3 mL, 2:1), the solution was cooled to 0 °C followed by the addition of sodium perborate tetrahydrate (40.7 mg, 0.4 mmol). The reaction mixture was stirred at rt for 2 h, diluted with brine, and extracted with EtOAc (15 × 3 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered. After the solvent was removed under reduced pressure, the residue was purified by flash column chromatography on silica gel to afford analytically pure products 35.
General method C for the synthesis of products 2w, 2ae and 2ai
An oven-dried 8-mL vial charged with a Teflon® septum was used. The vial was then charged with corresponding 1w or 1ae, or 1ai (0.2 mmol) and Ir-F (2 mol%). After degassed by vacuum-nitrogen purging, the vial was transferred to the glovebox, and the solvent (PhCF3, 2 mL, 0.1 M) was added. The vial was finally sealed with a poly-tetrafluoroethylene-lined cap tightly and stirred under irradiation with 4 × 30 W blue LEDs (λ = 450 nm, at approximately 3 cm away from the light source) at room temperature. Upon complete conversion, the mixture was passed through a short silica gel column and concentrated. The residue was purified by flash column chromatography on silica gel to afford the products (2w, 2ae, or 2ai).
General method D for the synthesis of products 40–62
An oven-dried 8 mL vial charged with a Teflon® septum was used. The vial was then charged with corresponding 1al-1bh (0.2 mmol) and Ir-F (4 mol%). After degassed by vacuum-nitrogen purging, the vial was transferred to the glovebox, and the solvent (MeCN, 2 mL, 0.1 M) was added. The vial was finally sealed with a poly-tetrafluoroethylene-lined cap tightly and stirred under irradiation with 4 × 30 W blue LEDs (λ = 450 nm, at approximately 3 cm away from the light source) at room temperature. Upon complete conversion, the mixture was transferred to a 20 mL vial and concentrated under reduced pressure. The crude residue was purified through column chromatography on silica gel to provide the products 40–62.
Data availability
Materials and methods, detailed optimization studies, experimental procedures, mechanistic studies, NMR spectra and computational data are available in the Supplementary Information and from the corresponding authors upon request. CCDC 2367147-2367149 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing [email protected], or by contacting The Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EZ, U.K.; fax: + 44 1223 336033. Source data are provided in this paper.
References
Taylor, R. D., Maccoss, M. & Lawson, A. D. G. Rings in drugs. J. Med. Chem. 57, 5845–5859 (2014).
Ebner, C. & Carreira, E. M. Cyclopropanation strategies in recent total syntheses. Chem. Rev. 117, 11651–11679 (2017).
Poplata, S., Tröster, A., Zou, Y.-Q. & Bach, T. Recent advances in the synthesis of cyclobutanes by olefin [2 + 2] photocycloaddition reactions. Chem. Rev. 116, 9748–9815 (2016).
Augustin, A. U., Di Silvio, S. & Marek, I. Borylated cyclopropanes as spring-loaded entities: Access to vicinal tertiary and quaternary carbon stereocenters in acyclic systems. J. Am. Chem. Soc. 144, 16298–16302 (2022).
Fawcett, A., Biberger, T. & Aggarwal, V. K. Carbopalladation of C–C σ-bonds enabled by strained boronate complexes. Nat. Chem. 11, 117–122 (2019).
Nóvoa, L., Trulli, L., Parra, A. & Tortosa, M. Stereoselective diboration of spirocyclobutenes: A platform for the synthesis of spirocycles with orthogonal exit vectors. Angew. Chem. Int. Ed. 60, 11763–11768 (2021).
Nguyen, T. V. T., Bossonnet, A., Wodrich, M. D. & Waser, J. Photocatalyzed [2σ+2σ] and [2σ+2π] cycloadditions for the synthesis of bicyclo[3.1.1]heptanes and 5- or 6-membered carbocycles. J. Am. Chem. Soc. 145, 25411–25421 (2023).
Vyas, H. et al. Generation and application of homoallylic α,α‐diboryl radicals via diboron‐promoted ring‐opening of vinyl cyclopropanes: cis‐diastereoselective borylative cycloaddition. Chemistry 30, e202303175 (2024).
Shen, H.-C. et al. Iridium-catalyzed asymmetric difunctionalization of C–C σ-bonds enabled by ring-strained boronate complexes. J. Am. Chem. Soc. 145, 16508–16516 (2023).
Hanania, N., Nassir, M., Eghbarieh, N. & Masarwa, A. A stereodivergent approach to the synthesis of gem ‐Diborylcyclopropanes. Chemistry 28, e202202748 (2022).
Salvado, O., Dominguez-Molano, P. & Fernández, E. Stereoselective cyclopropanation of 1,1-diborylalkenes via palladium-catalyzed (Trimethylsilyl)diazomethane insertion. Org. Lett. 24, 4949–4953 (2022).
Miyaura, N. & Suzuki, A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 95, 2457–2483 (1995).
Zhang, L. et al. Catalytic conjunctive cross-coupling enabled by metal-induced metallate rearrangement. Science 351, 70–74 (2016).
Sharma, H. A., Essman, J. Z. & Jacobsen, E. N. Enantioselective catalytic 1,2-boronate rearrangements. Science 374, 752–757 (2021).
Kischkewitz, M., Okamoto, K., Mück-Lichtenfeld, C. & Studer, A. Radical-polar crossover reactions of vinylboron ate complexes. Science 355, 936–938 (2017).
Silvi, M. & Aggarwal, V. K. Radical addition to strained σ-bonds enables the stereocontrolled synthesis of cyclobutyl boronic esters. J. Am. Chem. Soc. 141, 9511–9515 (2019).
Zhao, B. et al. An olefinic 1,2-boryl-migration enabled by radical addition: Construction of gem-bis(boryl)alkanes. Angew. Chem. Int. Ed. 58, 9448–9452 (2019).
Tappin, N. D. C., Gn-Gi-lux, M. & Renaud, P. Radical-triggered three-component coupling reaction of alkenylboronates, α-halocarbonyl compounds, and organolithium reagents: The inverse ylid mechanism. Chem. A Eur. J. 24, 11498–11502 (2018).
Quiclet-Sire, B. & Zard, S. Z. Radical instability in aid of efficiency: A powerful route to highly functional MIDA boronates. J. Am. Chem. Soc. 137, 6762–6765 (2015).
Hall, D.G. Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials. (Wiley, 2011).
Trost, B. The atom economy–a search for synthetic efficiency. Science 254, 1471–1477 (1991).
Rojas, C.M. Molecular Rearrangements in Organic Synthesis. (John Wiley & Sons, Inc, 2015).
Li, W., Xu, W., Xie, J., Yu, S. & Zhu, C. Distal radical migration strategy: an emerging synthetic means. Chem. Soc. Rev. 47, 654–667 (2018).
Wu, X., Ma, Z., Feng, T. & Zhu, C. Radical-mediated rearrangements: Past, present, and future. Chem. Soc. Rev. 50, 11577–11613 (2021).
Wu, X. & Zhu, C. Radical-mediated remote functional group migration. Acc. Chem. Res. 53, 1620–1636 (2020).
Studer, A. & Bossart, M. Radical aryl migration reactions. Tetrahedron 57, 9649–9667 (2001).
Holden, C. M. & Greaney, M. F. Modern aspects of the smiles rearrangement. Chem. A Eur. J. 23, 8992–9008 (2017).
Chen, Z. M., Zhang, X. M. & Tu, Y. Q. Radical aryl migration reactions and synthetic applications. Chem. Soc. Rev. 44, 5220–5245 (2015).
Sivaguru, P., Wang, Z., Zanoni, G. & Bi, X. Cleavage of carbon-carbon bonds by radical reactions. Chem. Soc. Rev. 48, 2615–2656 (2019).
Batey, R. A. & Smil, D. V. The first boron-tethered radical cyclizations and intramolecular homolytic substitutions at boron. Angew. Chem. Int. Ed. 38, 1798–1800 (1999).
Kaiser, D., Noble, A., Fasano, V. & Aggarwal, V. K. 1,2-Boron shifts of β-boryl radicals generated from bis-boronic esters using photoredox catalysis. J. Am. Chem. Soc. 141, 14104–14109 (2019).
Wang, H., Wu, J., Noble, A. & Aggarwal, V. K. Selective coupling of 1,2‐bis‐boronic esters at the more substituted site through visible‐light activation of electron donor–acceptor complexes. Angew. Chem. Int. Ed. 61, e202202061 (2022).
Wang, H., Han, W., Noble, A. & Aggarwal, V. K. Dual nickel/photoredox‐catalyzed site‐selective cross‐coupling of 1,2‐bis‐boronic esters enabled by 1,2‐boron shifts. Angew. Chem. Int. Ed. 61, e202207988 (2022).
Jana, K., Bhunia, A. & Studer, A. Radical 1,3-difunctionalization of allylboronic esters with concomitant 1,2-boron shift. Chem 6, 512–522 (2020).
Jana, K. & Studer, A. Allylboronic esters as acceptors in radical addition, boron 1,2-migration, and trapping cascades. Org. Lett. 24, 1100–1104 (2022).
Jiang, X. et al. 1,2‐Boryl migration enables efficient access to versatile functionalized boronates. Eur. J. Org. Chem. 2022, e202101463 (2022).
Tao, X., Ni, S., Kong, L., Wang, Y. & Pan, Y. Radical boron migration of allylboronic esters. Chem. Sci. 13, 1946–1950 (2022).
Kong, D. et al. Photocatalyzed regioselective hydrosilylation for the divergent synthesis of geminal and vicinal borosilanes. Nat. Commun. 14, 2525 (2023).
Paulus, F. et al. Three-component photochemical 1,2,5-trifunctionalizations of alkenes toward densely functionalized lynchpins. J. Am. Chem. Soc. 145, 23814–23823 (2023).
Wang, D., Mück-Lichtenfeld, C. & Studer, A. 1, n-Bisborylalkanes via radical boron migration. J. Am. Chem. Soc. 142, 9119–9123 (2020).
Li, C. et al. Remote boryl and alkenyl radical migration of olefin-bearing aryl bromides. CCS Chem. 7, 279–292 (2025).
Molloy, J. J. et al. Boron-enabled geometric isomerization of alkenes via selective energy-transfer catalysis. Science 369, 302–306 (2020).
Marotta, A. et al. Direct light‐enabled access to α‐boryl radicals: Application in the stereodivergent Synthesis of Allyl Boronic Esters. Angew. Chem. Int. Ed. 62, e202307540 (2023).
Dutta, S., Erchinger, J. E., Strieth-Kalthoff, F., Kleinmans, R. & Glorius, F. Energy transfer photocatalysis: exciting modes of reactivity. Chem. Soc. Rev. 53, 1068–1089 (2024).
Zhou, Q., Zou, Y., Lu, L. & Xiao, W. Visible‐light‐induced organic photochemical reactions through energy‐transfer pathways. Angew. Chem. Int. Ed. 58, 1586–1604 (2019).
Großkopf, J., Kratz, T., Rigotti, T. & Bach, T. Enantioselective photochemical reactions Enabled by Triplet Energy Transfer. Chem. Rev. 122, 1626–1653 (2022).
Zhu, M., Zhang, X., Zheng, C. & You, S.-L. Energy-transfer-enabled dearomative cycloaddition reactions of indoles/pyrroles via excited-state aromatics. Acc. Chem. Res. 55, 2510–2525 (2022).
Neveselý, T., Wienhold, M., Molloy, J. J. & Gilmour, R. Advances in the E → Z isomerization of alkenes using small molecule photocatalysts. Chem. Rev. 122, 2650–2694 (2022).
Coote, S. C. & Bach, T. Enantioselective intermolecular [2+2] photocycloadditions of isoquinolone mediated by a chiral hydrogen-bonding template. J. Am. Chem. Soc. 135, 14948–14951 (2013).
Scholz, S. O. et al. Construction of complex cyclobutane building blocks by photosensitized [2 + 2] cycloaddition of vinyl boronate esters. Org. Lett. 23, 3496–3501 (2021).
Murray, P. R. D. et al. Intermolecular crossed [2 + 2] cycloaddition promoted by visible-light triplet photosensitization: Expedient access to polysubstituted 2-oxaspiro[3.3]heptanes. J. Am. Chem. Soc. 143, 4055–4063 (2021).
Liu, Y. et al. Photosensitized [2+2]‐cycloadditions of alkenylboronates and alkenes. Angew. Chem. Int. Ed. 61, e202200725 (2022).
Liu, Y., Ni, D. & Brown, M. K. Boronic ester enabled [2 + 2]-cycloadditions by temporary coordination: synthesis of artochamin J and Piperarborenine B. J. Am. Chem. Soc. 144, 18790–18796 (2022).
Posz, J. M. et al. Synthesis of borylated carbocycles by [2 + 2]-cycloadditions and photo-ene reactions. J. Am. Chem. Soc. 146, 10142–10149 (2024).
Fang, H. et al. Energy transfer (EnT) catalysis of non‐symmetrical borylated dienes: Origin of reaction selectivity in competing EnT processes. Angew. Chem. Int. Ed. 64, e202418651 (2025).
Hanania, N., Eghbarieh, N. & Masarwa, A. PolyBorylated alkenes as energy‐transfer reactive groups: Access to multi‐borylated cyclobutanes combined with hydrogen atom transfer event. Angew. Chem. Int. Ed. 63, e202405898 (2024).
Zimmerman, H. E., Binkley, R. W., Givens, R. S. & Sherwin, M. A. Mechanistic organic photochemistry. XXIV. The mechanism of the conversion of barrelene to semibullvalene. A general photochemical process. J. Am. Chem. Soc. 89, 3932–3933 (1967).
Zimmerman, H. E. & Samuelson, G. E. Mechanistic and exploratory organic photochemistry. XXVIII. Methylene analogs of cyclohexenones. J. Am. Chem. Soc. 89, 5971–5973 (1967).
Hixson, S. S., Mariano, P. S. & Zimmerman, H. E. Di-.pi.-methane and oxa-di-.pi.-methane rearrangements. Chem. Rev. 73, 531–551 (1973).
Zimmerman, H. E. & Armesto, D. Synthetic aspects of the Di-π-methane rearrangement. Chem. Rev. 96, 3065–3112 (1996).
Riguet, E. & Hoffmann, N. Diπ-methane, Oxa-diπ-methane, and Aza-diπ-methane Photoisomerization. Comprehensive Organic Synthesis. 5, (Elsevier Ltd., 2014).
Zheng, Y., Dong, Q.-X., Wen, S.-Y., Ran, H. & Huang, H.-M. Di-π-ethane rearrangement of cyano groups via energy-transfer catalysis. J. Am. Chem. Soc. 146, 18210–18217 (2024).
Wen, S.-Y., Chen, J.-J., Zheng, Y., Han, J.-X. & Huang, H.-M. Energy‐transfer enabled 1,4‐aryl migration. Angew. Chem. Int. Ed. 64, e202415495 (2025).
Wen, S.-Y., Chen, J.-J., Zheng, Y. & Huang, H.-M. Energy-transfer-enabled rearrangement involving pyridines. CCS Chem. https://doi.org/10.31635/ccschem.024.202405071 (2025).
Kubota, K., Yamamoto, E. & Ito, H. Copper(I)-catalyzed borylative exo -cyclization of alkenyl halides containing unactivated double bond. J. Am. Chem. Soc. 135, 2635–2640 (2013).
Xiong, Y., Großkopf, J., Jandl, C. & Bach, T. Visible light‐mediated dearomative hydrogen atom abstraction/ cyclization cascade of indoles. Angew. Chem. Int. Ed. 61, e202200555 (2022).
Liu, J., Hao, T., Qian, L., Shi, M. & Wei, Y. Construction of benzocyclobutenes enabled by visible‐light‐induced triplet biradical atom transfer of olefins. Angew. Chem. Int. Ed. 61, e202204515 (2022).
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (No. 22201179 & 22471168 to H.-M.H.), China Postdoctoral Science Foundation (No. 2023M742366 to Y. Z.), Shanghai Postdoctoral Excellence Program (No. 223525 to Y.Z.), Double First-Class Initiative Fund of ShanghaiTech University (Y.Z.). We thank the HPC Platform of ShanghaiTech University for computing time. We sincerely thank Dr. Song Yu (The University of Manchester) for the great support on our computational study. We also thank Prof. Hong Yi (Wuhan University), Prof. Pengxin Liu, Fan Wu, Prof. Chaodan Pu, Prof. Jiajun Yan, Zhuo Zhao, Shu-Ya Wen and Jun-Jie Chen (all in ShanghaiTech University) for help with insightful discussion, EPR experiments, mechanistic study, X-ray analysis, and photochemical flow experiments.
Author information
Authors and Affiliations
Contributions
H.-M.H. directed the project; H.-M.H. and Y.Z. designed the experiments; S.-S.C. and Z.-X.X. performed all the experiments and analyzed all the data. H.-M.H. and Y.Z. performed the DFT calculations and drafted the DFT part. H.-M.H. wrote the manuscript with contributions from all authors. S.-S.C. and Y.Z. contributed equally.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Nature Communications thanks Ahmad Masarwa, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, SS., Zheng, Y., Xing, ZX. et al. Borylated strain rings synthesis via photorearrangements enabled by energy transfer catalysis. Nat Commun 16, 3724 (2025). https://doi.org/10.1038/s41467-025-58353-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58353-w
