
Abstract
Electrochemical upcycling of waste polyethylene terephthalate (PET) into biodegradable polyglycolic acid (PGA) is a promising solution to relieve plastic pollution. However, both the low current density and tedious separation process for target glycolic acid (GA) products in a flow electrolysis have hindered industrial-scale applications. Here, we show an interfacial acid-base microenvironment regulation strategy for the efficient oxidation of PET-derived ethylene glycol (EG) into GA using Pd-CoCr2O4 catalysts. Specifically, only a cell voltage of 1.25 V is needed to deliver a current density of ca. 290 mA cm–2. Moreover, a green separation method is developed to obtain high-purity GA (99%). 20 kg of waste PET is employed for the pilot plant test (stack electrolyzer: 324 cm2 × 5), which exhibits 93.0% GA selectivity at 280 mA cm–2 (current: 90.72 A) with a yield rate of 0.32 kg h–1. After polymerization, PGA yield can reach up to 87%, demonstrating the potential of this technique for large-scale PGA production from waste PET.
Similar content being viewed by others


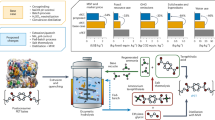
Introduction
Global plastic production reached 400 million tons in 2022 and is projected to increase to exceed 1200 million tons by 20501,2. Given its incredible durability, plastic wastes are predicted to persist in landfills or oceans for centuries to millennia, threatening the environment and biological health3,4,5. Considering the above, researchers are actively investigating the development of biodegradable plastics (e.g., polylactic acid (PLA), poly(butylene adipate-co-terephthalate) (PBAT), and polyglycolide acid (PGA)) as alternatives, which can be efficiently degraded into CO2 and H2O in the natural environment and by microorganisms6. Among them, PGA is an important biopolymer, especially in the medical, oil-gas exploitation, and ecology fields because of its high thermal stability (Tm = 220–230 oC), mechanical strength (115 GPa), high gas-barrier properties, excellent biocompatible and rapid biodegradable properties7,8. However, the large-scale application of PGA is significantly hindered by the high cost (US$10,000 t–1) of the corresponding monomer-glycolic acid (GA)9. The traditional GA production relies on the hydrolysis of chloroacetic acid (CA) in 50% NaOH aqueous solution at 90–130 oC (Fig. 1a), followed by a complex separation and purification process using ion-exchange resins10. In addition to high energy consumption and separation costs, CA, as a highly toxic and corrosive substance, is harmful to the environment and human health. Therefore, it is imperative to develop a cost-effective and sustainable method for GA production.

a Traditional industrial production route using toxic raw materials. b Proposed electrochemical route through upcycling of waste PET.
Recently, our and other research groups demonstrated the electrosynthesis of GA from waste polyethylene terephthalate plastics, driven by renewable electricity under mild conditions11,12,13,14,15,16,17,18,19. This approach is based on the catalyzed hydrolyzation of PET followed by the electrochemical oxidation of EG in alkaline solution. These findings provide an attractive alternative route for GA production (Fig. 1b), which not only eliminates the demand for toxic raw materials but also incentivizes PET recycling20. Specifically, the conversion of waste PET into PGA involves four steps: (i) alkali-catalyzed PET hydrolysis to EG, (ii) the EG oxidation reaction (EGOR) to form glycolate (GA–), coupling with the hydrogen evolution reaction, (iii) the acidification and purification of GA– to GA, and (iv) the polymerization of GA to PGA (Fig. 1b). However, the scale-up synthesis of PGA from PET still suffers from the following issues. 1) the low current density (usually < 120 mA cm–2) in a flow electrolysis system, limiting EG-to-GA– conversion at low space-time yield12,13,14,21. During EGOR in an alkaline medium, the OH– species will be consumed quickly at the anode-electrolyte interface because the diffusion rate of OH– from bulk electrolyte to interface is much slower than the consumption of OH– species at the anode-electrolyte interface, resulting in a local alkali-deficient microenvironment22. In this regard, the EGOR process at the interface will be greatly suppressed, especially under high current densities, due to the fact that the local OH–-deficient discourages activation of the C-H/C-O bond23. However, recently-published studies mainly focus on increasing the number of active sites (e.g., NiMoO4-supported Pd catalysts) and accelerating the generation of *OH active species by incorporation of oxophilic metals (e.g., Co or Ni in CuCo2O4 and Ni(OH)2), but ignore the effect of local OH–-deficient conditions at the electrode surface12,14,16. 2) excessively high-concentration bases are used as an electrolyte to maintain high reaction activity, which requires neutralization before product separation, thus inevitably resulting in significant waste of both bases and acids24. Moreover, the complex reaction pathway and intermediates of EGOR processes generate a diverse and complicated product distribution, necessitating high-cost product separation and purification. Based on the above discussion, the development of efficient catalytic systems and economic separation approaches is highly desirable.
In this work, we comprehensively considered the effects of the number of active sites, local pH changes, and *OH coverage at the electrode surface and designed a Pd-CoCr2O4 cooperative catalyst. Each component of the hybrids fulfills an important specific role: 1) Pd serves as the active sites for EGOR; 2) To inhibit local acidification at the surface of the anode, an interfacial acid-base microenvironment regulation strategy was proposed based on classic hard and soft acid-base theory. Cr2O3 was purposely chosen because Cr3+ is the hardest Lewis acid among transition metals with an extremely low pKa value of 2.0525,26, Cr3+ Lewis acid sites can accelerate the diffusion and enrichment of OH– toward the anode surface; 3) the oxophilic Co is particularly active for the adsorption/activation of OH– to form *OH on Pd surfaces at a relatively low potential. These *OH species not only play a critical role in assisting O-H/C-H bond cleavage of EG but also facilitate the oxidative removal of carbonaceous poisons on the adjacent Pd sites. These effects collectively achieve a top-level activity for EG-to-GA conversion (ca. 290 mA cm–2, 1.25 V, selectivity > 94.5%) and stability (300 h) in a flow cell system. The semi-industrial scale experiments are carried out in a flow stack electrolyzer (324 cm2 × 5) with a GA yield rate of 0.32 kg h–1 and 93.0% selectivity. Moreover, a neutralization-free separation process is designed, achieving a high-purity GA (>99%). Finally, the highly functional PGA (MW = 131198 g/mol, crystallinity = 82.2%) is polymerized through an azeotropic distillation method. Techno-economic analysis demonstrated that the levelling cost of PGA is approximately US$1240.12 t–1, which is close to that of the most widely used polyolefin plastics (ca. US$1000 t–1). In this work, we provide an environmentally friendly and economic strategy for the large-scale PGA synthesis from waste PET.
Results
Catalyst preparation and characterization
The Pd-CoCr2O4 catalysts were prepared by a three-step process (“Methods” section and Supplementary Fig. 1). In the first step, Co-Cr hydroxide was grown on Ni foam (NF) by a hydrothermal method, followed by annealing to obtain the CoCr2O4. Finally, Pd-CoCr2O4 was prepared through an impregnation method, in which the precursor was impregnated in a H2PdCl4 solution. The spinel-structure CoCr2O4 and cubic Pd were verified by X-ray diffraction (XRD) (Fig. 2a). The chemical composition of Pd-CoCr2O4 was further determined by Fourier transform infrared (FTIR) and Raman spectroscopy (Supplementary Fig. 2)27,28,29,30. Inductively coupled plasma optical emission spectrometry (ICP-OES) analysis confirms that the Co/Cr ratio of 1:2 and the content of Pd is 30.6 wt% in Pd-CoCr2O4 (Supplementary Table 1). Scanning electron microscopy (SEM) images display that CoCr2O4 nanosheets are uniformly and densely deposited onto NF (Supplementary Fig. 3a, b). Transmission electron microscopy (TEM) image reveals the smooth surface of CoCr2O4 nanosheets (Supplementary Fig. 3c). The high-resolution TEM (HRTEM) images of Pd-CoCr2O4 show the clear lattice fringes of CoCr2O4 (311) and Pd (111) (Fig. 2b, c). Energy-dispersive spectroscopy (EDS) spectrum of Pd-CoCr2O4 confirms a Pd ratio of 26.1% (Supplementary Fig. 4), roughly consistent with the ICP-OES result. Pd is uniformly dispersed on the nanosheets in the form of nanoparticles, which can be distinguished in elemental mapping images (Supplementary Fig. 5). X–ray photoelectron spectroscopy (XPS) was used to characterize the chemical states and the electronic interactions between Pd and CoCr2O4 (Figs. 2d–f, and Supplementary Fig. 6). As for pure Pd, the major peaks at 335.4 eV and 340.7 eV can be attributed to the metallic Pd0 (Fig. 2d). After the introduction of CoCr2O4, the binding energies of Pd positively shift by 0.1 eV, demonstrating the occurrence of charge transfer from Pd to CoCr2O4. Conversely, the peaks of Pd-CoCr2O4 in Co 2p and Cr 2p spectra shift toward lower binding energy (Fig. 2d–f). The high-resolution Co 2p spectrum of CoCr2O4 possesses four pairs of deconvoluted peaks, suggesting the presence of multiple Co species with different chemical states. The broad (non-symmetrical) peak shape of Cr3+ can be fitted to the fine structure of multiple splitting31,32 This electronic interaction between CoCr2O4 and Pd is further proved by the charge density difference and Bader charge calculations (Supplementary Fig. 7 and Supplementary Note 1). Pdδ+ in an electron-deficient state can more readily adsorb OH– since 0.68 electrons are transferred from Pd to CoCr2O433,34. Thus, the introduction of CoCr2O4 is expected to increase the surface density of OH– species around the Pd active sites.

a XRD patterns of CoCr2O4 and Pd-CoCr2O4. b, c HRTEM images of Pd-CoCr2O4 with different resolutions. High-resolution XPS spectra of d Pd 3 d, (e) Co 2p, and f Cr 2p. g NH3-TPD of Pd-CoCr2O4, Pd-Cr2O3, Pd-Co3O4, and Pd. h CV curves obtained at a scan rate of 90 mV s–1 for Pd-CoCr2O4, Pd-Cr2O3, Pd-Co3O4, and Pd in 1 M KOH. i Relationship between current density and scan rate. j PZCs of Pd-CoCr2O4, Pd-Cr2O3, Pd-Co3O4, and Pd in 0 (100 mM KCl), 20, 40, 60, 80, 100 mmol KOH solutions. k Bode plots of Pd-CoCr2O4 in 1 M KOH under different potentials. l Plots of Cφ vs. potential for Pd-CoCr2O4, Pd-Cr2O3, Pd-Co3O4, and Pd in 1 M KOH.
The effective OH– adsorption (OH–ads) process of Pd-CoCr2O4 was first supported by strong surface acidity (NH3 desorption peak, ca. 390 °C) in the ammonia temperature-programmed desorption (NH3-TPD) curve (Fig. 2g)35. Cr3+ sites are recognized as the strong Lewis acid sites by comparing with other curves36. Theoretical support for the enrichment of OH– in the local microenvironment was provided by density functional theory (DFT) simulations (details provided in the Methods and Supplementary Fig. 8). The adsorption energy (Eads) of OH– on the surface Co2+ sites flanking the Pd cluster (Pd-Co sites) of Pd-CoCr2O4 (–2.29 eV) is greater than those of controls, demonstrating that the introduction of CoCr2O4 favors *OH near the Pd sites (Supplementary Fig. 9). To further explore the structure-function relationships of Lewis acid sites, the OH– adsorption-desorption curves of samples were first studied by cyclic voltammetry (CV) measurements. As shown in Fig. 2h, Pd-CoCr2O4 exhibits an obvious OH– oxidation adsorption peak at a lower potential (0.36 V vs. RHE) than Pd-Cr2O3 (0.43 V vs. RHE), Pd-Co3O4 (0.47 V vs. RHE), and Pd (0.48 V vs. RHE). Notably, the diffusion coefficient of OH– (DOH) can be calculated from the relationships between the scan rate (v1/2) and OH–ads peak current density (Figs. 2i and Supplementary Fig. 10). The Pd-CoCr2O4 sample displayed the highest DOH, (10.05 N2), higher than that of Pd-Cr2O3 (4.80 N2), Pd-Co3O4 (3.69 N2), and Pd (2.19 N2)36. Recognizing if specific adsorption takes place on a surface can be reflected directly using the potential of zero charge (PZC), which is the potential at which there is no excess charge on the electrode surface. The minimal differential capacitance curve in a dilute electrolyte could be used to calculate PZC37. According to the Esin-Markov effect, the PZCs of samples shift negatively with the increase of OH– concentration, showing a specific adsorption of OH– on the surface (Fig. 2j and Supplementary Fig. 11)36. Most interestingly, a more effective OH– adsorption on the inner Helmholtz plane (IHP) is shown by a more noticeable negative shift in PZC of Pd-CoCr2O4 upon the addition of OH–, as compared to controls. The OH–ads capacity was further determined by in-situ electrochemical impedance spectroscopy (EIS) measurements in 1 M KOH. Among them, the characteristic peak that shifts from 100 Hz to 101 Hz with the potential increase is associated with the adsorption of OH–38,39. The OH–ads peak in the Bode plots of Pd-CoCr2O4 is located at a higher frequency and lower phase angle in comparison to controls, indicating faster OH–ads kinetics (Figs. 2k and Supplementary Fig. 12, 13). Then we fitted the EIS data based on the circuit model to quantify *OH adsorption (Supplementary Fig. 14)38. As shown in Fig. 2l and Supplementary Tables 2-5, the Cφ of Pd-CoCr2O4 is higher than those of controls in a wide potential range, identifying the extensive surface coverages of *OH at the junction of the electrode and the IHP. In summary, benefiting from the hard Lewis acid sites, the OH–-rich local microenvironment is formed near the electrode interface, which made the Pd-CoCr2O4 exhibit high catalytic performance12,36.
Electrochemical performance for EGOR
To confirm the EGOR performance of the as-prepared catalysts, linear sweep voltammetry (LSV) curves were first evaluated. The optimized Pd (the true active sites in EGOR) dosage was determined to be 1 mg cm–2 (Supplementary Fig. 15). Pd-CoCr2O4 exhibited the highest current density (1.06 A cm–2) and the lowest onset potential (0.56 V vs. RHE) compared with the other controls (Fig. 3a). The catalytic activity advantage conferred by the OH–-rich microenvironment was also observed in solutions with low OH– concentrations. However, since KOH serves not only as electrolyte but also as essential reactants in the EGOR, the system operating under low OH– concentration conditions fails to achieve sufficiently high current densities (Supplementary Fig. 16). Considering the difference in electrochemical active surface area (ECSA) calculated from the reduction process of Pd in Fig. 2h, Pd-CoCr2O4 still exhibited the highest intrinsic activity (Supplementary Fig. 17). The catalytic activity advantage is also supported by the Bode plots measured by Operando EIS. The inflection point potential (0.55 V vs. RHE) in the high-frequency region of Pd-CoCr2O4 suggests that EGOR occurs at this potential (Fig. 3b)40. In contrast, the inflection point potentials of Pd-Cr2O3, Pd-Co3O4, and Pd appear at 0.6 V, 0.65 V, and 0.65 V vs. RHE, respectively, representing higher onset potentials (Supplementary Fig. 18). Moreover, in the potential range of the EGOR (0.6-0.8 V vs. RHE), the phase angle value of Pd-CoCr2O4 is lower than those of controls, which also confirms faster EGOR kinetics. An identical conclusion can be obtained from relatively Nyquist plots. The results of fitting circuit show that Pd-CoCr2O4 has the smallest charge transfer resistance (40.67 Ω) compared with Pd-Cr2O3 (65.4 Ω), Pd-Co3O4 (84.2 Ω), and Pd (123.2 Ω) in EGOR (Fig. 3c and Supplementary Fig. 19).

a LSV curves without iR correction obtained at a scan rate of 10 mV s–1 of Pd-CoCr2O4, Pd-Cr2O3, Pd-Co3O4, and Pd in 1 M KOH with 1 M EG. b Bode plots of Pd-CoCr2O4 in 1 M KOH with 1 M EG. c Nyquist plots of Pd-CoCr2O4, Pd-Cr2O3, Pd-Co3O4, and Pd in 1 M KOH with 1 M EG. d I-t curves of Pd-CoCr2O4, Pd-Cr2O3, Pd-Co3O4, and Pd in 1 M KOH with 1 M EG under 0.8 V vs. RHE. e CO stripping experiments of Pd-CoCr2O4, Pd-Cr2O3, Pd-Co3O4, and Pd in 1 M KOH. f 1H NMR spectra of electrolyte before and after long-term electrolysis in H-cell system at 0.8 V vs. RHE. Inset: 13C NMR spectrum of electrolyte after long-term electrolysis in H-cell system at 0.8 V vs. RHE. g The concentrations of EG and GA– during long-term electrolysis in H-cell system at 0.8 V vs. RHE. h Schematic diagram of electrostatic interactions at the interface. i OCP changes of Pd-CoCr2O4, Pd-Cr2O3, Pd-Co3O4, and Pd in 1 M KOH before and after 60 mmol GA was injected.
Keeping high GA space-time yield in a flow cell system is also dependent on catalyst stability and product selectivity, both of which are facilitated by the OH–-rich local microenvironment12,36. At the beginning, the current density of all the samples decreased rapidly as the charge current from the oxidation process of the Pd active sites disappeared in the stability test (Fig. 3d)41. After operating for 3600 s, Pd-CoCr2O4 retained 55% of the initial current density, higher than Pd-Cr2O3, Pd-Co3O4, and Pd. The stability advantage of Pd-CoCr2O4 was consistently observed over a wide potential window (Supplementary Fig. 20). Meanwhile, no new vibration modes of Pd-CoCr2O4 were found on the surface after long-term electrolysis in Raman and FTIR spectra, indicating that the surface composition of Pd-CoCr2O4 was maintained (Supplementary Fig. 21, 22). No new oxidation state and obvious crystal structure changes were observed (Supplementary Fig. 23–25). Meanwhile, ECSA has not changed significantly (Supplementary Fig. 26). When Pd-Cr2O3 was utilized as anode, the electrolyte turned light yellow over time due to the rapid leaching of CrO42− ions, which was confirmed through ICP-OES (Supplementary Fig. 27). In contrast, the leaching amount of CrO42− ions in the electrolyte using Pd-CoCr2O4 as the anode is extremely limited in the same time. These results reveal the weak OH– capacity of Pd-Cr2O3 caused by Eskolaite structural collapse can be avoided by introducing Co2+ to construct spinel skeleton structure (Supplementary Fig. 28). The high-spin divalent Co2+ cations mostly occupy the tetrahedral sites, while the low-spin trivalent Cr3+ cations are fixed in the octahedral sites42,43. This rigid geometry avoids excessive oxidation of Cr3+ cations. In addition, CO stripping curves were measured (Fig. 3e). Pd-CoCr2O4 exhibits the lowest CO oxidation potential peak (0.71 V vs. RHE) and an obvious OH–-assisted CO oxidation peak (0.4 V vs. RHE), indicating superior COads oxidation activity44. On the one hand, the abundant adsorbed-OH– contributes to the rapid CO oxidation removal and further improves the anti-CO poisoning ability. On the other hand, the downshift of d-band center revealed by the valence band spectra (–4.07 eV for Pd-CoCr2O4 vs. –3.31 eV for Pd, Supplementary Fig. 29) implies relatively weak adsorption of COads, which is beneficial for anti-CO poisoning.
After a long-term electrolysis, GA– is the main product with a small fraction of formate (FA), oxalate (OA) and carbonate (CA) (Fig. 3f). NMR spectra with various charge consumption show the dynamic process of EGOR (Fig. 3g and Supplementary Fig. 30). As electrolysis proceeds, the desired product GA– grows linearly with continued EG consumption. Finally, Pd-CoCr2O4 displays a GA– selectivity of 94.6% when the EG conversion is 95.7%. In contrast, Pd exhibited a maximum selectivity of only 46% at a low conversion (Supplementary Fig. 31). This high product selectivity of Pd-CoCr2O4 can be attributed to a weaker adsorption of GA– in the IHP of Pd-CoCr2O4 than that of Pd caused by electrostatic repulsion between OH– and GA– (Fig. 3h)45. The difference of adsorption strength was measured by open circuit potential (OCP), which reflected absorbates in the IHP40. Upon the addition of 60 mmol GA, a smaller decrease in OCP for Pd-CoCr2O4 was observed than that of controls, suggesting favorable desorption of GA– on Pd-CoCr2O4 (Fig. 3i)40,46. In contrast, when 60 mmol KOH was injected into 0.1 M KCl, the OCP of Pd-CoCr2O4 exhibited the biggest decline, implying stronger OH– adsorption in IHP (Supplementary Fig. 32). The GA–-deficient IHP directly leads to the weak electrooxidation ability of Pd-CoCr2O4 to GA– (Supplementary Fig. 33), avoiding excessive oxidation of GA–. Therefore, the introduction of Lewis acid sites is highly effective in obtaining high GA– selectivity.
Electrochemical in situ FTIR measurements on Pd-CoCr2O4 were performed in order to more clearly examine the intermediates and comprehend the reaction mechanism during EGOR. As shown in Supplementary Fig. 34, the peaks in 1230 cm–1 ~ 1600 cm–1 corresponding to glycolate. A distinct vibration peak at 1640 cm–1 can be assigned to ν(C = O) of adsorbed 2-hydroxyacety (*OC-CH2OH) and interfacial water, which is consistent with previous research47. Based on these results, a plausible conversion route of EG to GA– was proposed (Supplementary Fig. 35). One EG molecule is first adsorbed on Pd-CoCr2O4, and then deprotonated and oxidized to glycolaldehyde intermediate, which is finally oxidized to glycolate. Note that a small fraction of FA, OA, and CA was obtained by further oxidation of glycolate along with C–C cleavage or over-oxidation (as shown in Fig. 3f).
Scale-up synthesis of PET-into-PGA with continuous electrosynthesis
To explore the catalytic performance in continuous electrosynthesis processes, a Pd-CoCr2O4 catalytic electrode was used as the anode, and Ni foam was used as the cathode to assemble a membrane electrode flow cell (Pd-CoCr2O4//NF, 10 × 10 cm2) (Fig. 4a). Due to the large consumption of strong alkaline substances in the hydrolysis and electrosynthesis processes, less expensive NaOH (US$300 t–1) was chosen to replace KOH (US$700 t–1) as the OH– source, and this change did not result in a significant change in catalytic activity (Supplementary Fig. 36). In order to obtain high hydrolysis efficiency of PET and high space-time yield, the hydrolysate containing 2 M EG and 5 M NaOH was determined (Supplementary Figs. 37, 38). The qualitative and quantitative analysis of additives in waste PET is based on methanol transesterification according to GB/T 14190, revealing compositional profiles of diethylene glycol (DEG, 0.13 wt%), Fe (<0.01 wt‰), TiO2 particulates (0.12 wt%), and pigments (0.85 wt%) (detailed in the “Methods” section, Supplementary Fig. 39, 40, and Supplementary Table 7). Fe, TiO2 powders, and pigment in hydrolysate can be removed from hydrolysate during the pretreatment by simple filtration and activated carbon adsorption. Trace amounts of DEG and the possible presence of diglycolate will not affect the electrolysis process and product purity (Supplementary Figs. 41, 42). The electrolyzer polarization curves for overall water splitting and electrolysis of purified hydrolysate in a flow cell system were measured as a reference for optimizing production parameters (Fig. 4b). When feeding purified hydrolysate, ca. 770 mV decrease of cell voltage for obtaining the same current density was detected, suggesting that electrolysis of EG was an energy-efficient process. To validate the persistence of the OH–-rich microenvironment strategy within 5 M NaOH-containing hydrolyzate, the OH⁻ diffusion rates and electrocatalytic activity were systematically quantified (Supplementary Fig. 43). These multimodal analyzes demonstrate that in purified hydrolysate, the Pd-CoCr2O4 can still improve the alkali-microenvironment on the electrode surface by promoting OH– diffusion to the electrode surface, thereby improving the catalytic performance. As a key indicator in continuous flow electrosynthesis, the space-time yield is strongly correlated to flow rate and cell voltage. A flow rate design that maximizes selectivity and utilization is critical in optimizing the overall electrosynthesis system efficiency48,49. The optimal flow rate (3 mL min–1) was determined, considering that further increase of flow rate can not improve the space-time yield (Fig. 4c). The highest GA– selectivity (94.5 %) and space-time yield (0.195 g h–1 cm–2) were achieved at a cell voltage of 1.25 V and decreased rapidly when the cell voltage is too high (Fig. 4d). After obtaining the optimal production conditions, continuous flow electrolysis up to 300 h was performed. In the initial stage, the current density increased and finally stabilized at ca. 290 mA cm–2 when the system temperature rose from 25 °C to 50 °C (Fig. 4e). Building upon the results of the stability test characterization (Supplementary Figs. 21-26), it was clearly observed that the catalyst remained unchanged in terms of its active surface area, surface structure, bulk structure, and composition after electrolysis. Consequently, the improved current density cannot be accounted for by changes in these factors mentioned above. It is credible that the enhanced performance can be attributed to the increase in the reaction system temperature, which is induced by the electrothermal effect. The results of the variable-temperature experiments and another electrolysis experiment carried out again, shown in Supplementary Fig. 44, effectively corroborate this point. The long-term stability is benefited from the timely removal of the generated product during dynamic electrolysis in a flow electrolyzer50,51. Impressively, this highly efficient production capacity surpasses many recently reported about GA electrosynthesis in flow cell systems (Supplementary Table 6)12,13,14,15,16,52,53. The selectivity and high space-time yield attained in the flow cell system provide opportunities for refining the entire scale-up upcycling of PET to PGA.

a Schematic diagram of the membrane electrode flow cell for the continuous electrosynthesis of GA– from EG coupling with H2 generation. b LSV curves of flow cell system with/without EG under high flow rate (10 mL min–1). c The selectivity and yield of GA– in flow cell under different flow rate at a cell voltage of 1.25 V. d The selectivity and conversion of GA– on Pd-CoCr2O4 across a wide cell voltage range from 0.95 to 1.45 V in flow cell. e Stability test of the Pd-CoCr2O4//NF system in membrane electrode flow cell at a cell voltage of 1.25 V.
Based on the above results, we use a continuous flow stack electrolyzer with multiple units (electrolysis area = 324 cm2 × 5) connected in series for semi-industrial electrosynthesis simulation. A schematic of the scale-up synthetic routes of PGA from PET, including four major processes, is developed and shown in Fig. 5. Photos of the waste PET and equipment required are shown in Supplementary Fig. 45. To be specific, first, 20 kg of waste PET (polyester fibers) was cleaned, fragmented, and then hydrolyzed using NaOH aqueous solution in a stirred reactor. After treated with activated carbon, the solid in the hydrolysate was filtered. The resulting powder was dissolved, filtered, and acidified to obtain terephthalic acid (> 99%, Supplementary Fig. 46). The obtained hydrolysate filtrate 1 was adjusted to a fixed concentration (2 M EG, 5 M NaOH, Supplementary Fig. 47) and then pumped into the flow stack electrolyzer using a peristaltic pump set at a flow rate of 50 mL min–1. Under the conditions of a stack cell voltage of 6.25 V and a current of 90.72 A (electrolytic power = 567 W), continuous flow electrosynthesis was conducted to achieve 75% single-pass conversion of EG in outflow anolyte 2 (Supplementary Fig. 48). The outflow anolyte 2 contained the unconsumed NaOH and EG in the electrosynthesis reaction, along with the main product C2H3O3Na (selectivity 93%), the by-products Na2C2O4 (selectivity 2.5%), Na2CO3 (selectivity 2.8%), and HCOONa (selectivity 1.4%). Subsequently, vacuum distillation and cyclic extraction with EtOH solution containing methanol and acetonitrile were developed, in which the main product, C2H3O3Na, not dissolve while EG and NaOH dissolve. These processes not only complete the separation of the main product C2H3O3Na, but also achieve residual recycling of EG and NaOH to prevent the acidification process of superfluous alkali and EG waste. Altered concentrations of recovered EG and NaOH were reintroduced into the flow stack electrolyzer to achieve the reuse of recycled raw materials. After that, the by-products Na2C2O4 and HCOOH were separated by filtration and vacuum distillation, respectively, in the acidification process of GA–. Finally, a low-temperature cooling crystallization method in a mixed solution of acetone and ethyl acetate was developed for the purification of GA. The solubility of GA in mixed solution is 490 g L–1 at 60 °C and 32 g L–1 at –10 °C, respectively, leading to the precipitation of the largest amount of GA crystals with high purity through the cooling crystallization process. NMR data for each step of qualitative and quantitative analysis and detailed descriptions of the synthesis of GA from PET are included in Supplementary Figs. 47–54 and Note 2. The above technique achieves 99% recovery of NaOH and EG, 91% recovery of the main product, GA (purity > 99%), significantly reducing the production costs without any waste discharge.

The complete process of scale-up upcycling of waste PET plastics to biodegradable PGA plastics includes four steps: GA electrolysis, GA separation, GA crystallization and PGA polymerization. The parameters and prices of all the equipment used in the schematic are shown in Supplementary Table 13.
The obtained GA crystals were subjected to bulk polycondensations via a solvent-free method to obtain high-molecular-weight PGA. Approximately 5.3 kg of PGA was obtained (Supplementary Fig. 55) with a final yield of 87% (the yield of each step in the whole process is shown in Supplementary Table 8). The stretching band related to carbonyl ester at 1740 cm–1 and C-O-C bend at 1145 cm–1 in the FTIR spectrum of the synthesized PGA, suggesting evidence of structure for the synthesized PGA from GA monomer (Supplementary Fig. 56)54. The two crystallites observed at 22° and 28° in XRD are the main crystal forms in PGA (Supplementary Fig. 57)55. The high molecular weight (Mn = 131198) and low polydispersity (pD = 1.14) of the synthesized PGA were measured by gel permeation chromatography (GPC) analysis (Supplementary Fig. 58). The high crystallinity (82.2%) of the product means that it has enough gas barrier properties for bioplastics packaging (Supplementary Fig. 59)56,57. The synthesized PGA possesses similar mechanical properties (3.3% vs. 3.6% elongation at break) and higher degradation ability (74.3% vs. 51.4% weight percent loss in 40 °C PBS solution after 28 days) to commercial PGA (Supplementary Figs. 60, 61), which proves the commercial application potential of this synthetic route.
Techno-economic analysis and environmental impact assessment
To evaluate the feasibility of implementing this technology for the upcycling of PGA from waste PET, we applied a detailed calculation model (Supplementary Note 3, Supplementary Fig. 62, and Supplementary Tables 8–15) for techno-economic analysis. For a reference single cell operating at 280 mA/cm2 and 1.25 V, a plant would require an electrode area of about 857.1 m2, yielding a production capacity of roughly 10,000 tons of PGA annually. The estimated costs per ton of PGA produced were US$3,460.15 (79.2%) for raw and auxiliary materials, US$598.06 (14.3%) for electricity, US$57.46 (1.3%) for catalyst preparation, and US$226.32 (5.2%) for capital (Fig. 6a). Considering byproduct sales (TPA, Na2SO4, and H2), the levelized cost for PGA could decrease to 1363.85 US$/tPGA, making it competitive with mainstream bioplastics and nearing polyolefin plastics (Fig. 6b). The cost of waste PET is expected to be further reduced as the high profits allow more PET to avoid being simply discarded and subsequently provide yet more positive feedback.

a Production cost of PGA synthesis from waste PET. b Costs of mainstream polyolefin and bioplastics versus cost of PGA synthesized from waste PET. Detailed data and data sources see Supplementary Table 15. c CO2 emissions from different PET recycling methods. Detailed data and data sources see Supplementary Table 14. d Energy efficiency of different GA synthesis methods. Abbreviations: GN glycolonitrile, FA formaldehyde, MG methyl glycolate.
Environmental impact assessment (EIA) of the electrosynthesis of PGA from waste PET is crucial for its promotion. Compared with mechanical recyling and traditional chemical recycling of PET58, electrochemical recycling of PET exhibits the lowest CO2 emissions (841 kg-CO2/tPET) (Fig. 6c and Supplementary Table 14). A metric that is poised to further diminish as the proportion of renewable energy in the power grid escalates. In contrast to traditional GA production technologies59, the GA electrosynthesis route exhibits superior energy efficiency (87%) and are non-toxic in raw materials (Fig. 6d). In essence, the green characteristic could be caused by the following three reasons: (1) the extensive utilization of electrical energy has eliminated the requirement for high-temperature and high-pressure equipment reliant on steam. (2) the high space-time yield of GA in the flow cell system is realized by introducing hard Lewis acid sites. (3) A reasonable purification process reduces unnecessary consumption of raw materials, products, and energy.
Discussion
In this investigation, we formulated an integrated process for scale-up upcycling of waste PET to PGA. Pd-CoCr2O4 was established to achieve high electrosynthesis activity (290 mA cm–2 at a cell voltage of 1.25 V) and long-term stability (>300 h), which are benefiting from the OH–-rich local microenvironment. After a non-neutralization separation process, a high space-time yield (0.195 g h–1 cm–2) and low cost (1363.85 US$/tPGA) were realized simultaneously. An EIA based on kilogram-scale PGA synthesis shows that the process is green and eco-friendly. This advancement in the field of plastic pollution prevention set the stage for wiping out waste plastics through reducing PGA costs and promoting PET upcycling.
Methods
Materials
All materials were of analytical grade and used as received without further purification. Nickle foam (NF) and Nafion N117 were obtained from SCI Materials Hub. Cr(acac)3, Co(acac)2, Pd/C, Palladium chloride (PdCl2) were obtained from Energy chemical. Hydrochloric acid (HCl), potassium hydroxide (KOH), sodium hydroxide (NaOH), ethylene glycol (EG), glycolic acid (GA), and D2O were obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). All chemicals were of analytical grade and used without further purification.
Synthesis of CoCr-Pre
The CoCr-Pre grown on NF were prepared by a facile solvothermal method. A piece of commercial NF (2.5 cm × 4 cm) was sequentially washed with acetone, diluted HCl (1 M), and deionized (DI) water using ultrasound treatment. In a typical synthesis, 0.5 mmol Cr(acac)3 and 0.25 mmol Co(acac)2 were mixed in 30 mL ethanol under magnetic stirring for 30 min at 60 °C. Then the solution was added 6 mL of 0.4 M urea ethanol solution under magnetic stirring for 10 min. The obtained mixture was transferred into 50 mL Teflon-lined autoclave, and then the pre-treated NF was immersed into the solution. The Teflon-lined autoclave was sealed, heated to 150 °C for 12 h, and then cooled down to room temperature. Afterward, the samples were removed and washed with water and ethanol to neutral, then dried at room temperature. The resulting sample is denoted as CoCr-Pre.
Synthesis of Pd-CoCr2O4
The as-prepared CoCr-Pre (1 cm × 4 cm) was transformed into the corresponding CoCr2O4 phases by annealing at 400 °C for 2 h in a muffle furnace under an air atmosphere. Then the CoCr2O4 was immersed in 4 ml 10 mM H2PdCl4 aqueous solution and stationed until the solution became colorless. Finally, the prepared Pd-CoCr2O4 sample was rinsed with pure water and ethanol and then dried in an oven at 60 °C.
Synthesis of Pd, Pd-Co3O4, and Pd-Cr2O3
The samples of Pd-Co3O4 and Pd-Cr2O3 were obtained only by changing the solution of solvothermal method. 36 mL ethanol solution containing 0.75 mmol Co(acac)2 and 2.4 mmol urea was heated to 150 °C for 12 h in 50 mL Teflon-lined autoclave for preparing Co-precursor on nickel foam. 36 mL ethanol solution containing 0.75 mmol Cr(acac)3 and 2.4 mmol urea was heated to 150 °C for 12 h in 50 mL Teflon-lined autoclave for preparing Cr-precursor on nickel foam. After rinsing with pure water and ethanol, the precursors were annealed at 400 °C for 2 h in a muffle furnace under an air atmosphere to form an oxide phase. The as-prepared samples and pure nickel foam were immersed in 4 mL 10 mM H2PdCl4 aqueous solution and stationed until the solution became colorless.
Synthesis of Pd-CoCr2O4 used in flow electrolyzer
In order to synthesize large-area catalysts, nickel foam (0.5 mm × 18 cm × 18 cm) was folded in half and placed in a large-volume Teflon-lined autoclave (Φ180 mm × 140 mm, purchased from Xi’an Yibeier Instrument Equipment Co., Ltd.). Subsequently, 2.6 L ethanol solution containing 36 mmol Cr(acac)3, 18 mmol Co(acac)2, and 170 mmol urea was then added and heated to 120 °C for 8 h. After cooling to room temperature, the nickel foam loaded with the precursor was removed and rinsed with water and ethanol. After annealing at 400 °C for 2 h, the impregnation process was carried out in a surface dish (Φ260 mm). The nickel foam loaded with CoCr2O4 was immersed in 150 mL aqueous solution containing 3.05 mmol H2PdCl4 until the solution was colorless with mechanical stirring.
Material characterizations
The morphologies of samples were characterized by scanning electron microscopy (SEM, TESCAN MIRA LMS) with an accelerating voltage of 15 kV and transmission electron microscopy (TEM, JEOL-2100F) with an accelerating voltage of 200 kV. X-ray diffraction (XRD) was performed via a 18KW/D/max2550VB/PC (40 KV, 200 mA) with Cu Kα1 radiation at a scan rate of 2° min−1. Raman spectroscopy was carried out by a Renishaw inVia Reflex Raman microscope under an excitation of 532 nm laser light. XPS analysis was carried out on a Thermo Scientific ESCALAB 250XI spectroscopy equipped with an Al Kα radiation, and all binding energies were standardized at a binding energy of 284.6 eV for C 1 s. The Avantage 5.52 software is chosen and the fitting algorithm is Powell. The context of peak fitting is based on Gauss-Lorentz product functions (GLP). The exact chemical compositions of Pd-CoCr2O4 and electrolyte were determined by ICP-OES utilizing an Optima 4300 DV spectrometer (Perkin Elmer). Ammonia temperature programmed desorption (NH3-TPD) profiles were recorded on a Micromeritics AutoChem II 2920 instrument.
Electrochemical measurements
All electrochemical measurements were performed on an electrochemical workstation (CHI 760E, CH Instruments, Inc.). Error bars correspond to the standard deviation of three measurements. Pd-CoCr2O4 was applied as the working electrode (0.5 cm2), platinum sheet (1 cm2) as the counter electrode, and Hg/HgO electrode as the reference electrode, respectively. The reference electrode was converted by Eq. (1)
The KOH or NaOH aqueous solutions were freshly prepared each time to avoid the change of electrolyte composition caused by long-term storage. LSV and CV experiments were determined at a scan rate of 10 mV s–1 in alkaline electrolyte (1 M KOH) with and without 1 M EG. ECSA was estimated from the CV curve (Fig. 3h) in 1 M KOH with Eq. (2) 60
Operando EIS measurements were performed over a frequency range from 105 to 0.1 Hz with a perturbation voltage of 5 mV.
OCP measurements: First, the working electrode was placed in 1 M KOH electrolytes for OCP test. After 70 s, GA was injected to 60 mM. OCP test continued for 1800 s.
Two-electrode flow electrolyzer measurements
Electrochemical two-electrode flow electrolyzer measurements were performed using a DC regulated power supply workstation (KPS-3010DU). The electrolyzer is a two-electrode divided cell, and the anode and cathode are separated by a proton exchange membrane (PEM, Nafion N117). PEM was pretreated by soaking in 0.5 M H2SO4 and 30% H2O2 for 30 min. Nickle foam (area: 10 cm × 10 cm) and Pd-CoCr2O4 (area: 10 cm × 10 cm) were used as the cathode and anode, respectively. 5 M NaOH and 5 M NaOH + 2 M EG were used as catholyte and anolyte, respectively. The cell voltage for the anolyte flow rate optimization experiment was selected as 1.25 V. The catholyte was circulated in the flow reactor by using a peristaltic pump at a flow rate of 10 mL min–1. In the cell voltage optimization experiment, the anolyte was pumped into the flow reactor at a flow rate of 3 mL min–1 without circulation. Reaction results are the averages of three experiments conducted in parallel.
Flow stack electrolyzer was made up of five cells separated by bipolar plate. Nickle foam (area: 18 cm × 18 cm) and Pd-CoCr2O4 (area: 18 cm × 18 cm) were used as the cathode and anode, respectively. Electrolyte (5 M NaOH + 2 M EG) was pumped into the flow stack electrolyzer at a total flow rate of 50 mL/min without circulation.
Computational details
Simulations were conducted using the Vienna ab initio simulation package (VASP) with the projector augmented wave (PAW) method61,62. The Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional within the generalized gradient approximation (GGA) scheme was applied to account for electron exchange correlation effects63. The Hubbard-U (U-J) parameters used were 3.32 eV for Co and 3.70 eV for Cr. A plane-wave cutoff energy of 500 eV was employed for valence electrons64. In the Brillouin zone, Gamma k-points were sampled with a 3 × 3 × 1 mesh for slabs with or without Pd cluster. To avoid interlayer interactions, a 20 Å vacuum layer was included in the slab model. Half of the atomic layers in the slab were fixed while the remainder were allowed to relax (the slab referred to the substrate support, i.e., excluding the Pd atoms on the surface). Geometrical structures were optimized until the convergence criteria for forces and energies reached values below 0.05 eV/Å and 1.0-5 eV, respectively. The Bader charge population calculation was incorporated to assess the changes in charge during the interaction between ions and the Pd-CoCr2O4 surface65.
The adsorption energy (Eads) was calculated using Eq. 3. Here, Etotal represented the total energy with and without the adsorbate, Esubstrate was the energy of the substrate, and Eadsorbate denoted the energy of the adsorbate.
Detection of additives in PET
According to the national standards of the People’s Republic of China (GB/T 14189−2015), chemical additives in PET polyester fiber may include diethylene glycol (DEG), Fe powders, TiO2 powders, and pigment. According to the method of methanol transesterification in GB/T 14190, 10 g PET was subjected to methanolysis by filtration to obtain a filtrate and a residue. 0.5 g of activated carbon was used to decolorize the filtrate, followed by filtration and weighing. The difference in the quality of activated carbon before and after adsorption is considered to be the quality of the adsorbed pigment. The content of DEG in the decolorized filtrate was detected by gas chromatography (GC). The GC column was used FFAP. The detector is a flame ionization detector (FID). ICP were used to determine Fe and TiO2 powders additives in residue. The residue was dissolved by HF and HCl, and then the solution was diluted to 1 L.
Polymer characterization
Tensile tests were carried out using a universal material testing machine CMT4204 (Shenzhen SANS, China). The strain rate was 5 mm/min for tensile test.
The hydrolysis degradation test was carried out at 40 °C in PBS solution (pH = 7). Tensile test specimens were submerged in PBS solution, and after hydrolysis, specimens with varying hydrolysis durations (0, 10, 20, and 30 days) were removed and vacuum-dried for 48 h at 60 °C. After that, pictures were taken and the weights of specimens were tested.
Gel permeation chromatography (GPC) was performed to measure the molecular weight using PL GPC 50. HFIP was used to conduct the analysis at 30 °C with a steady flow rate of 0.6 mL/min. Before measurement, samples were filtered using a 0.45 um membrane after 24 h dissolution in HFIP.
Under a nitrogen environment, a differential scanning calorimetry (DSC) analyzer Q2000 (TA Instruments, USA) was used to measure thermal performance. For the nonisothermal crystallization experiment, 5 mg of sample was first heated from 40 °C to 260 °C at 30 °C/min and held for 5 min, then cooled to 25 °C at 10 °C/min, and finally heated again to 260 °C at 10 °C/min (second heating scan).
Product analysis
The liquid products can be analyzed qualitatively and quantitatively by using 1H and 13C nuclear magnetic resonance (NMR) spectroscopy on a 600 MHz/AVANCE 400 (Bruker). In a standard NMR tube, 300 μL of post-electrolysis liquid, 285 μL of D2O, and 15 μL of internal standard dimethyl sulfoxide (DMSO) were added. The amount of GA and EG in electrolyte was calculated by comparing the integrations of the -CH2- of GA (δ = 3.84 ppm), -CH2- of EG (δ = 3.56 ppm), and -CH3 of DMSO (δ = 2.62 ppm) based on the following equation:
where IGA is the integral of GA peak; N is the numbers of proton corresponding to GA (2H), EG (4H), and DMSO peak (6H); M is the molar mass of GA or DMSO.
The EG conversion (%) and the selectivity of the GA were calculated using Eq. (5) and (6), respectively.
Data availability
All data supporting the conclusions given in this paper are available within the article and its supplementary files. Any additional data related to this paper may be requested from the corresponding authors. The raw data have been deposited in Figshare https://doi.org/10.6084/m9.figshare.26170351. Source data are provided with this paper.
References
Ali, W., Jeong, H., Lee, J.-S., Zinck, P. & Souissi, S. Biodegradable microplastics interaction with pollutants and their potential toxicity for aquatic biota: a review. Environ. Chem. Lett. 22, 1185–1220 (2024).
Wang, X. et al. Recent progress of waste plastic upcycling based on multifunctional zeolite catalysts. Chem. Synth. 4, 28 (2024).
Borrelle, S. B. et al. Predicted growth in plastic waste exceeds efforts to mitigate plastic pollution. Science 369, 1515–1518 (2020).
Garcia, J. M. & Robertson, M. L. Chemical advances are increasing the proportion of polymer waste that can be recycled. Science 358, 870–872 (2017).
Sheridan, E. A. et al. Plastic pollution fosters more microbial growth in lakes than natural organic matter. Nat. Commun. 13, 4175 (2022).
Kwon, D. Three ways to solve the plastics pollution crisis. Nature 616, 234–237 (2023).
Jem, K. J. & Tan, B. The development and challenges of poly (lactic acid) and poly (glycolic acid). Adv. Ind. Eng. Polym. Res. 3, 60–70 (2020).
Yamane, K., Sato, H., Ichikawa, Y., Sunagawa, K. & Shigaki, Y. Development of an industrial production technology for high-molecular-weight polyglycolic acid. Polym. J. (Tokyo, Jpn.) 46, 769–775 (2014).
Göktürk, E., Pemba, A. G. & Miller, S. A. Polyglycolic acid from the direct polymerization of renewable C1 feedstocks. Polym. Chem. 6, 3918–3925 (2015).
Cao, X. M., Zhang, K., Wang, Y. & He, P. Boosting the production of glycolic acid from formaldehyde carbonylation via the bifunctional PdO/ZSM-5 catalyst. Ind. Eng. Chem. Res. 62, 17671–17680 (2023).
Shi, R. et al. Electrocatalytic reforming of waste plastics into high value-added chemicals and hydrogen fuel. Chem. Commun. 57, 12595–12598 (2021).
Liu, F. et al. Concerted and selective electrooxidation of polyethylene-terephthalate-derived alcohol to glycolic acid at an industry-level current density over a Pd-Ni(OH)2 catalyst. Angew. Chem. Int. Ed. Engl. 62, e202300094 (2023).
Yan, Y. F. et al. Electrocatalytic upcycling of biomass and plastic wastes to biodegradable polymer monomers and hydrogen fuel at high current densities. J. Am. Chem. Soc. 145, 6144–6155 (2023).
Shi, K., Si, D., Teng, X., Chen, L. & Shi, J. Pd/NiMoO4/NF electrocatalysts for the efficient and ultra-stable synthesis and electrolyte-assisted extraction of glycolate. Nat. Commun. 15, 2899 (2024).
Du, M. et al. Electrochemical production of glycolate fuelled by polyethylene terephthalate plastics with improved techno-economics. Small 19, e2303693 (2023).
Liu, K. et al. Energy-saving hydrogen production by seawater splitting coupled with PET plastic upcycling. Adv. Energy. Mater. 14, 2304065 (2024).
Bhattacharjee, S. et al. Photoelectrochemical CO2-to-fuel conversion with simultaneous plastic reforming. Nat. Synth. 2, 182–192 (2023).
Lin Y. et al. Inspiration of bimetallic peroxide for controllable electrooxidizing ethylene glycol through modulating surficial intermediates. Adv. Funct. Mater., 34, 2404594 (2024).
Chen, J. L. et al. Unveiling synergy of strain and ligand effects in metallic aerogel for electrocatalytic polyethylene terephthalate upcycling. Proc. Natl. Acad. Sci. USA. 121, e2318853121 (2024).
Nairobi. Turning Off The Tap. How The World Can End Plastic Pollution And Create A Circular Economy. United Nations Environment Programme (2023).
Wang, Y. et al. Selective electro-reforming of waste polyethylene terephthalate-derived ethylene glycol into C2 chemicals with long-term stability. Green Chem. 25, 5872–5877 (2023).
Guo, J. X. et al. Direct seawater electrolysis by adjusting the local reaction environment of a catalyst. Nat. Energy 8, 264–272 (2023).
Yang, Y. C. & Mu, T. C. Electrochemical oxidation of biomass derived 5-hydroxymethylfurfural (HMF): pathway, mechanism, catalysts and coupling reactions. Green Chem. 23, 4228–4254 (2021).
Nabil, S. K. et al. Acid–base chemistry and the economic implication of electrocatalytic carboxylate production in alkaline electrolytes. Nat. Catal. 7, 330–337 (2024).
Magaz, G. E., Rodenas, L. G., Morando, P. J. & Blesa, M. A. Electrokinetic behaviour and interaction with oxalic acid of different hydrous chromium(III) oxides. Croat. Chem. Acta 71, 917–927 (1998).
Shu, Y. L. et al. Hollow echinus-like PdCuCo alloy for superior efficient catalysis of ethanol. ACS Appl. Mater. Interfaces 10, 4743–4749 (2018).
Mączka, M., Ptak, M., Kurnatowska, M. & Hanuza, J. Synthesis, phonon and optical properties of nanosized CoCr2O4. Mater. Chem. Phys. 138, 682–688 (2013).
Gomes, A. S. O., Yaghini, N., Martinelli, A. & Ahlberg, E. A. Micro-Raman spectroscopic study of Cr(OH)3 and Cr2O3 nanoparticles obtained by the hydrothermal method. J. Raman Spectrosc. 48, 1256–1263 (2017).
Zhong, J. et al. Toluene oxidation process and proper mechanism over Co3O4 nanotubes: Investigation through in-situ DRIFTS combined with PTR-TOF-MS and quasi in-situ XPS. Chem. Eng. J. 397, 125375 (2020).
Al-Mamun, M. et al. Strongly coupled CoCr2O4/carbon nanosheets as high performance electrocatalysts for oxygen evolution reaction. Small 12, 2866–2871 (2016).
Duan, Y. et al. Anodic oxidation enabled cation leaching for promoting surface reconstruction in water oxidation. Angew. Chem. Int. Ed. Engl. 60, 7418–7425 (2021).
Biesinger, M. C. et al. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and. Ni. Appl. Surf. Sci. 257, 2717–2730 (2011).
Li, Z. et al. Alcohols electrooxidation coupled with H2 production at high current densities promoted by a cooperative catalyst. Nat. Commun. 13, 147 (2022).
Li M. R. et al. Engineering MXene surface via oxygen functionalization and Au nanoparticle deposition for enhanced electrocatalytic hydrogen evolution reaction. Small Methods, 4, 2401569 (2024).
Liu, J.-D., Zhang, T.-T., Jia, A.-P., Luo, M.-F. & Lu, J.-Q. The effect of microstructural properties of CoCr2O4 spinel oxides on catalytic combustion of dichloromethane. Appl. Surf. Sci. 369, 58–66 (2016).
Huang B., Yan J., Li Z., Chen L., Shi J. Anode-electrolyte interfacial acidity regulation enhances electrocatalytic performances of alcohol oxidations. Angew. Chem. Int. Ed. Engl. 63, e202409419 (2024).
Liu, C., Zhang, G., Zhang, W., Gu, Z. & Zhu, G. Specifically adsorbed ferrous ions modulate interfacial affinity for high-rate ammonia electrosynthesis from nitrate in neutral media. Proc. Natl. Acad. Sci. USA. 120, e2209979120 (2023).
Ge, R. X. et al. Selective electrooxidation of biomass-derived alcohols to aldehydes in a neutral medium: promoted water dissociation over a nickel-oxide-supported ruthenium single-atom catalyst. Angew. Chem. Int. Ed. Engl. 61, e202200211 (2022).
Chen, J. L., Jiang, M. M., Zhang, F. Z., Wang, L. & Yang, J. P. Interstitial boron atoms in pd aerogel selectively switch the pathway for glycolic acid synthesis from waste plastics. Adv. Mater. 36, 2401867 (2024).
Zhou, P. et al. Heterogeneous-interface-enhanced adsorption of organic and hydroxyl for biomass electrooxidation. Adv. Mater. 34, e2204089 (2022).
Zhang, Q., Weng, J. & Xu, J. Palladium phosphide/black phosphorus heterostructures with enhanced ethanol oxidation activity and stability. J. Phys. Chem. C 125, 18717–18724 (2021).
Wang, Y., Jia, A.-P., Luo, M.-F. & Lu, J.-Q. Highly active spinel type CoCr2O4 catalysts for dichloromethane oxidation. Appl. Catal. B: Environ 165, 477–486 (2015).
Sun, Y. M. et al. Navigating surface reconstruction of spinel oxides for electrochemical water oxidation. Nat. Commun. 14, 2467 (2023).
Huang, W. et al. Highly active and durable methanol oxidation electrocatalyst based on the synergy of platinum-nickel hydroxide-graphene. Nat. Commun. 6, 10035 (2015).
Yan, C. et al. Regulating the inner Helmholtz plane for stable solid electrolyte interphase on lithium metal anodes. J. Am. Chem. Soc. 141, 9422–9429 (2019).
Lu, Y. X. et al. Tuning the selective adsorption site of biomass on Co3O4 by Ir single atoms for electrosynthesis. Adv. Mater. 33, 2007056 (2021).
Wang, H. et al. Electrocatalysis of ethylene glycol oxidation on bare and Bi-modified Pd concave nanocubes in alkaline solution: an interfacial infrared spectroscopic investigation. ACS Catal. 7, 2033–2041 (2017).
Yao, Y. X., Lei, J. F., Shi, Y., Ai, F. & Lu, Y. C. Assessment methods and performance metrics for redox flow batteries. Nat. Energy 6, 582–588 (2021).
Ke, X. Y. et al. Rechargeable redox flow batteries: flow fields, stacks and design considerations. Chem. Soc. Rev. 47, 8721–8743 (2018).
Qi, J. et al. Energy-saving and product-oriented hydrogen peroxide electrosynthesis enabled by electrochemistry pairing and product engineering. Nat. Commun. 14, 6263 (2023).
Atobe, M., Tateno, H. & Matsumura, Y. Applications of flow microreactors in electrosynthetic processes. Chem. Rev. 118, 4541–4572 (2017).
Du, M. et al. Tandem integration of biological and electrochemical catalysis for efficient polyester upcycling under ambient conditions. Nano Lett. 24, 9768–9775 (2024).
Bhattacharjee, S. et al. Reforming of soluble biomass and plastic derived waste using a bias-free Cu30Pd70|perovskite|Pt photoelectrochemical device. Adv. Funct. Mater. 32, 2109313 (2021).
Sanko, V., Sahin, I., Aydemir Sezer, U. & Sezer, S. A versatile method for the synthesis of poly(glycolic acid): high solubility and tunable molecular weights. Polym. J. 51, 637–647 (2019).
Schwarz, K. & Epple, M. A detailed characterization of polyglycolide prepared by solid-state polycondensation reaction. Macromol. Chem. Phys. 200, 2221–2229 (1999).
Nakafuku, C. & Yoshimura, H. Melting parameters of poly(glycolic acid). Polymer 45, 3583–3585 (2004).
Sangroniz, A., Zhu, J. B., Etxeberria, A., Chen, E. Y. & Sardon, H. Modulating the crystallinity of a circular plastic towards packaging material with outstanding barrier properties. Macromol. Rapid Commun. 43, e2200008 (2022).
Chaudhari, U. S. et al. Systems analysis approach to polyethylene terephthalate and olefin plastics supply chains in the circular economy: a review of data sets and models. ACS Sustainable Chem. Eng. 9, 7403–7421 (2021).
Zhou, X. et al. Glycolic acid production from ethylene glycol via sustainable biomass energy: integrated conceptual process design and comparative techno-economic–society–environment analysis. ACS Sustainable Chem. Eng. 9, 10948–10962 (2021).
Chen, L. et al. Improved ethanol electrooxidation performance by shortening Pd-Ni active site distance in Pd-Ni-P nanocatalysts. Nat. Commun. 8, 14136 (2017).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Efthimiopoulos, I. et al. Pressure-induced transition in the multiferroic CoCr2O4 spinel. Phys. Rev. B 92, 064108 (2015).
Tang, W., Sanville, E. & Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys.: Condens.Matter 21, 084204 (2009).
Acknowledgements
Y.C. thank for their financial support the National Key Research and Development Program of China (2022YFB3803600), the Natural Science Foundation of China (22421005), the CAS-Croucher Funding Scheme for Joint Laboratories. R.S. thank for the Natural Science Foundation of China (52370153). F.Liu thank for the Natural Science Foundation of China (22309193).
Author information
Authors and Affiliations
Contributions
Y.C. and R.S. led the project. Y.W. designed and performed the experiments. R.S. and F.Liu contributed considerably to the revision of the manuscript. J.C. and E.C.M.T. carried out DFT calculations. All authors discussed the results and contributed to the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Y., Liu, F., Chen, J. et al. Scale-up upcycling of waste polyethylene terephthalate plastics to biodegradable polyglycolic acid plastics. Nat Commun 16, 4440 (2025). https://doi.org/10.1038/s41467-025-59667-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59667-5
