
Abstract
The replicative helicase-catalyzed unwinding of the DNA double helix is the initiation of DNA replication. Helicases and primases are functionally related enzymes that have even been expressed as fusion proteins in some organisms and viruses. However, the mechanism underlying DNA unwinding initiation by these helicase-primase fusion enzymes and the functional association between domains have not been elucidated. Herein, we report the cryo-EM structures of mpox virus E5, the founding member of these helicase-primase enzymes, in various enzymatic stages. Notably, E5 forms a head-to-head double hexamer encircling dsDNA, disrupted by the conformational rearrangement of primase domains upon nucleotide incorporation. Five E5-ssDNA-ATP structures further support an ATP cycle-driven non-classical escort model for E5 translocation. Finally, the helicase ___domain is found to enhance the primase function as a DNA scaffold. Together, our data shed light on the E5-mediated DNA unwinding model including dsDNA loading, DNA melting, ssDNA translocation, and provide a reasonable interpretation for evolutionary preservation of helicase-primase fusion from a functional perspective.
Similar content being viewed by others


Introduction
Genomic DNA replication is the central event in the propagation of organisms containing double-stranded (ds) DNA, i.e., bacteria, archaea, eukaryotes, and dsDNA viruses, and is a complex process involving multiple proteins1. Over the years, biologists have made great efforts to understand the initiation of DNA replication, i.e., the process leading to the formation of the replisome, and have carried out in-depth studies in representative systems of bacteria and eukaryotes2,3,4. However, considerable variations have been shown in this process between different biological systems in terms of the completeness, composition, and function of the set of proteins involved, as well as the order of the initiation steps, leaving a large number of unknowns1. Notably, as the most abundant biological entities on Earth, dsDNA viruses show a much greater diversity in the initiation of DNA replication, which needs further investigation5,6, which needs to be further investigated.
It is widely accepted that the replicative helicase plays a central role in the replisome by separating double-stranded DNA (dsDNA) to provide the template for primer synthesis via primase, which is the prerequisite for polymerase-mediated daughter strand extension7. Helicases are complex enzymes consisting of the ATPase motor accompanied by diverse additional domains and can be classified into six superfamilies (SF1-6) depending on the type of ATPase contained8. SF3-6 form homo- or hetero-hexamers that include all the cellular replicative helicases, such as bacterial DnaB (SF4) and archaea/eukaryotic MCM (SF6)9,10, and the major well-studied replicative helicases of dsDNA viruses, i.e., papillomavirus E1 (SF3) and bacteriophage T7 gp4 (SF4)2,11,12,13. In helicases, the ATPase motors hydrolyze ATP and provide the primary driving force for helicase translocation along the single-strand DNA (ssDNA), pushing the replication fork junction (RFJ) forward8,14. Two distinct translocation models have been proposed for SF3 and SF4 helicases: E1, the typical SF3 helicase, adopts a closed ring shape and passes the ssDNA via an escort model in which the ATP-triggered movement of the ssDNA-binding hairpins within helicase protomers drives the DNA passage through the helicase central channel11,13; SF4 helicases adopt a lock-washer shape and translocate along ssDNA via a hand-over-hand model in which ATP drives sequential inter-protomer movement from one end of the lock-washer to the other2,15.
Helicase translocation models mimic the formation of the origin ssDNA immediately after dsDNA melting, providing the basis for recruitment of primase and polymerase and further formation of the replisome. However, the steps to melt dsDNA into ssDNA are even more complicated and are tightly controlled by numerous components beyond the helicase, such as the initiator, loader, accessory proteins, and ATP, with different strategies in different systems1. The unwinding of the dsDNA begins with origin DNA recognition. In this step, the bacterial initiator DnaA or the initiator proteins of the archaeal (Orc) and eukaryotic (multi-subunit origin recognition complex, ORC) systems load onto the dsDNA following DNA sequence specificity (evidenced in prokaryotes and S. cerevisiae) or not (as shown for multicellular organisms), with the process of nucleotide-dependent oligomerization occurring with DnaA and ORC-Cdc61,16,17,18,19. However, the order of the next two steps after initiator binding, i.e., helicase loading and DNA melting, is quite different between bacteria and eukaryotes1. In bacteria, DNA melting occurs upon the initiator binding, providing accessible ssDNA for helicase loading20. Reciprocally, the helicase loaded onto dsDNA in a double-hexameric form in eukaryotes precedes origin melting21. Although the helicase translocation models of dsDNA viruses have been well elucidated in bacteriophage T7 and papillomavirus E1, the players involved and the mechanism underlying the other steps in the initiation of DNA replication for the dsDNA viruses remain a challenge, especially given the diversity of dsDNA viruses.
The direct association of helicase and primase has been widely shown in different replication systems to co-regulate their functions in the replisome22. Helicase is even expressed in fusion with primase in some organisms and viruses6. As illustrated in the resolved architecture of the T7 replisome, the helicase-primase fusion gp4 translocated along the lagging strand and advanced the RFJ via its C-terminal SF4-type helicase, leaving the N-terminal DnaG-type primase downstream to synthesize primers for the lagging strands that have passed from the helicase channel2. Besides gp4, helicase-primase fusions have also been found in other phages, bacteria plasmids, and the Nucleocytoplasmic Large DNA Viruses (NCLDVs), such as NrS-1 encoded by the deep-sea vent epsilonproteobacterial phage23, pRN1 encoded by the Sulfolobus islandicus plasmid24,25, C962R of African Swine Fever Virus26, and D5 of poxviruses (named E5 in mpox virus)27,28. All these proteins contain a C-terminal SF3-type helicase, an N-terminal archaeo-eukaryotic primase (AEP)-type primase and different additional motifs such as the zinc-binding ___domain (ZBD), the helix-bundle ___domain or the PriCT2 ___domain29. While numerous studies have shed light on the translocation movement of these multifunctional enzymes along the leading strand, a property of SF3-type helicases due to the presence of AAA+ (ATPase associated with various activities)-fold ATPases, several intriguing aspects of these systems still remain elusive. Specifically, how these enzymes function in the initiation steps of DNA unwinding, such as DNA loading and dsDNA melting, is yet to be fully understood, providing exciting avenues for further investigation. Moreover, the recently resolved structure of mpox virus E5, the founding member of the helicase-primase fusion enzymes and essential for virus replication29,30, in complex with ssDNA and nucleotides, has revealed a distinct conformation of the primase domains that is associated with the interference of the unwinding function of the helicase domains31. This puzzling functional antagonism of primase to helicase, which appears in evolutionarily retained helicase-primase fusion, presents a valuable example for further elucidation of the evolutionary significance behind the preservation of the architecture of the helicase-primase fusion and ___domain organization.
In this work, we focus on the role of E5 in the initiation of DNA unwinding, and the following key findings are revealed: firstly, the formulation of a nucleotide cycle-driven non-classical escort model for E5 translocation along ssDNA; secondly, the unveiling of a head-to-head double hexamer of E5 for initiating the DNA unwinding; thirdly, the identification of nucleotide incorporation-driven conformational changes in primase serving as the external triggers for E5 from dsDNA capture (double hexamer) to ssDNA translocation; and finally, the elucidation of the functional relevance and cooperative mechanism between the helicase and primase domains in this helicase-primase fusion enzyme.
Results
Biochemical characterization and the apo structure of MPXV E5
The full-length MPXV E5 protein was expressed as a homohexamer and demonstrated helicase activity and primase activity in terms of primer synthesis (Supplementary Fig. 1a–d). In addition, we found that E5 also exhibited DNA-dependent polymerase activity, with a mild substrate preference for NTPs than dNTPs as evidenced by both synthesis rate and a higher maximum of full-length product production in the extension assay (Fig. 1a and Supplementary Fig. 1e–g). Due to this dual function of E5, both in initiating RNA primers and in extending pre-existing primers with dNTPs, we proposed to incorporate E5 into the group of enzymes known as PrimPols (primase-polymerase)32,33. Taken together, MPXV E5 is a multifunctional enzyme, possessing both DNA unwinding function, RNA primer synthesis function, and DNA-dependent polymerase function using either NTPs or dNTPs.

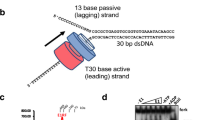
a DNA polymerization assays catalyzed by wild-type (WT) E5 or E5 mutant (DDD/A). The “-” is used to represent the mock group, in which an equal volume of protein buffer was added to the reaction to replace the E5 proteins. The DNA substrate used in the assay is shown diagrammatically at the left side, with the end-labeled FAM is shown as a red star. The experiments were repeated three times independently with similar results, and the representative image is provided. b The schematic representation of E5. The ___domain organization of the helicase ___domain is highlighted. c Overall structure of the hexameric E5, with protomers A–F shown in different colors and distances between protomers marked. d Close-up view of the collar ___domain in E5. Amino acids involved in hydrophobic interactions (upper box) and polar interactions (lower box) are shown as sticks and labeled. e The overall architecture of DNA-bound E5 complex (structure 0). f Close-up view of the primase ___domain. g Local structure of the primase ___domain after 3D focused refinement. The metal ion is depicted as a sphere, and coordinating amino acids in ZBD-F are shown as sticks. The EM density of the metal binding site is highlighted in the inset. h Representative 2D class averages of wild-type (WT) E5 or E5 mutants after addition of different substrates. The pink arrow indicates the conformation of the inter-primase assembly and the green arrow points to the bound DNA.
To capture the apo structure of full-length MPXV E5, the protein sample was then analyzed by single-particle cryo-electron microscopy (cryo-EM) (Supplementary Fig. 2). After 3D reconstruction, the apo E5 structure was refined to an overall resolution of ~3.33 Å (Supplementary Table 1). The clear cryo-EM density was observable for the helicase ___domain, while the N-terminal primase ___domain was absent from the density map, indicating the intrinsic flexibility of the primase ___domain in the apo state. The well-resolved structure revealed four apparent layers in the side view architecture of the E5 helicase (Fig. 1b, c and Supplementary Fig. 1h–j). The N-termini of the D5N motif form a collar-like structure via tight inter-protomer interaction (Supplementary Table 2). Detailed analysis revealed two sets of major interactions, i.e., hydrophobic interactions between F327 of one protomer and L369 and L384 of the other, and a group of polar interactions in which D398 electrostatically bridged R389 and further stabilized by hydrogen bonding to T365 (Fig. 1d). In contrast, no inter-protomer interactions were observed between the AAA+ subunits or the transition motifs that bridge the collar and AAA+s, leaving apparent gaps between the protomers in the AAA+ and transition motif layers (Fig. 1c). The C-terminal winged helix ___domain (WHD) protruded towards the bottom of the AAA+ subunit of the adjacent protomer (Fig. 1c), although only the main chains can be traced due to the relatively low resolution (Supplementary Fig. 2). In contrast to the rigidity of the collar, there is a certain flexibility in the arrangement of the other subunits of the E5 helicase, which is reflected in the slight difference in distance between the six AAA+ subunits as well as in the tapering density from the collar to the WHDs shown in the local density map (Fig. 1c and Supplementary Fig. 2).
The apo structure of E5, obtained by treatment of an endogenous nucleotide incorporated E5 with an ATP hydrolyzing enzyme, has recently been reported (Supplementary Fig. 1k)31. Despite the overall similarity, with a root mean square deviation (r.m.s.d.) of 2.375 Å over 1546 aligned Cα, the structural comparison revealed a more relaxed arrangement of the AAA+ subunits in the present structure. This is clear evidence of a larger mean distance between neighboring protomers (51.93 Å versus 49.20 Å) and a farther average position relative to respective opposite protomers (103.77 Å versus 98.30 Å) (Supplementary Fig. 1l, m). The present structure may suggest a more native apo state of E5, given that it has not been treated with anything before structural study.
Rearrangement of the primase configuration derived by nucleotide incorporation
To further explore the working mechanism of E5, the protein was incubated with the DNA substrate consisting of 30 bp dsDNA and 20 nt 3′-single strand poly-deoxythymidine (dT) in the presence of Mg2+ and ATP, and applied to cryo-EM (Supplementary Figs. 3 and 4). A marked conformational change was observed in the 2D class averages of the cryo-EM images, with the N-terminal primase ___domain appearing a more rigid configuration compared to that in the apo state. The overall DNA-bound E5 complex (structure 0) was resolved at 2.95 Å using 339,072 particles. In the resolved structure, at least ZBDs from two protomers (ZBD-F and ZDB-A) and AEPs from two protomers (AEP-A and AEP-E) were rigid enough to be well modeled and formed a similar configuration arching over the collar-like subdomain as recently reported31,34 (Fig. 1e, f and Supplementary Fig. 4a–c). Besides, four diffuse densities were visible above the D5N subdomains and in an anti-clockwise arrangement with the respective protomers, suggesting the position and flexibility of the remaining four ZBDs (ZBD-B/C/D/E). Likewise, a third AEP (AEP-F) can be identified above the well-resolved parts of the primase domains, as a bulk of density was clearly present there (Supplementary Fig. 4b, c).
Further 3D focused refinement, targeting the primase ___domain, has provided additional details at the interface between the ZBD-F and the AEP-A (Supplementary Fig. 3). Notably, a bulk of extra density was clearly detected sandwiching ZBD-F and AEP-A (Fig. 1g). Although with a relatively low local resolution, this extra density was proposed to be an ATP molecule for the following reasons: First, within this cryo-EM sample of the E5-DNA complex, only DNA substrate, Mg2+, and ATP were additionally introduced, with ATP aligning precisely with this extra density. Second, nucleotide binding is an intrinsic ability of primase for its primer synthesis activity; the latter has been previously validated in E5. The superimposition of the E5 primase and the human PrimPol in its catalytically competent conformation35 reveals that the ATP located within the E5 primases has a similar position to the nucleotides previously elucidated at the elongation site (E-site) or initiation site (I-site) within the AEP catalytic core, with the conserved primase catalytic motifs in E5 also well aligned (Supplementary Fig. 5). In contrast, the ZBD-F of E5 spatially overlapped the DNA template-primer strand in the human PrimPol complex structure (Supplementary Fig. 5b), suggesting that the working conformation of E5 primase is different from that of human PrimPol or that the present local structure of E5 primase cannot represent the primer synthesizing state of E5. Moreover, the EM map shows clear evidence for one metal ion coordinated in a tetrahedral geometry by three cysteine residues (C282, C285 and C314) and a histidine (H290) in the ZBD-F (Fig. 1g). An identical coordination geometry was previously reported in the structure of zinc-binding ___domain of DNA primase from Bacillus stearothermophilus, in which the metal ion is defined as a Zn2+ ion36.
This distinct configuration of the E5 primase domains has previously been proposed to limit the helicase unwinding activity31. To investigate the trigger of this primase configuration, E5 was analyzed via cryo-EM under conditions differing by the presence or not of DNA or nucleotide (Fig. 1h and Supplementary Fig. 6). The 2D class averages revealed that this inter-primase assembly could only be observed upon ATP addition while being independent of the presence of DNA substrate. Given that the ATP molecules are involved in the enzymatic cycles of both primase and the helicase, the E5 inactivating mutants of either primase (mutant DDD/A, explained in Supplementary Fig. 1d) or helicase (mutant KRR/A, explained in Supplementary Fig. 1c) activity were then used to further determine the influences of the individual functional domains in the primase assembly. Remarkably, inactivating mutant of the primase completely disrupted the spatial assembly of primase domains. In contrast, despite the helicase ___domain interacting directly with the primases in this assembly conformation via the D5N subdomain (Fig. 1e, f), its inactivating mutant did not interfere with the primase assembly. Among the three conserved active-site motifs (I, II, and III) of AEP primases37 (Supplementary Fig. 5d), motif III, corresponding to D170 in E5, has been reported to be important for initiating nucleotide incorporation via stabilizing the 2′-OH of the nucleotide at the I-site38. Therefore, we proposed that the E5 DDD/A mutant might abolish ATP incorporation between the ZBD-F and the AEP-A and thus further influence the formation of this spatial configuration of primase domains.
Conformational changes of E5 helicase upon DNA and ATP binding
To further clarify the mechanism of E5 helicase-mediated translocation, a 3D focused classification was further performed on the helicase ___domain. Eight structurally distinct maps were obtained at overall resolutions ranging between ~2.88 Å and ~3.13 Å (Supplementary Fig. 3). Three of these maps, structures 6, 7, and 8, did not present clear density of DNA strand in the hexamer helicase channel and was therefore not taken into further consideration. As for the other five structures with ssDNA binding (structures 1–5), the density map showed consistent features in terms of the local resolution of individual protomers (Supplementary Fig. 4d–h). Within the helicase homohexamer, four protomers are characterized by their stability, especially the two located in the middle with the highest resolution, while the remaining two are more flexible. The density of ssDNA in the helicase channel was consistently leaning towards and in contact with the four stable protomers in all ssDNA-engaged structures. In the context of the overall conformation, the differences between structures 1-5 were only in the relative positions of the two flexible protomers when the primase ___domain was aligned in the same orientation (Supplementary Fig. 4d–h); therefore, structure 1 with the highest local resolution of the helicase ___domain was selected for further structural comparison with the E5 helicase ___domain in the apo state.
For the E5 helicase in this working state, distinct changes in the inter-protomer arrangement were observed, with the four stable protomers, namely A to D, moving to a compact arrangement, while the two flexible protomers (E and F) were still in the apo-like relaxed configuration (Fig. 2a–c). As such, a significant inward and lateral movement of the AAA+ and WHD subunits towards the ssDNA binding channel was observed for the four stable protomers, leading to the transition from the relaxed configuration as shown by the two flexible protomers to the connected compact configurations as shown by the four stable protomers (Fig. 2c, d). The conformational or inter-protomer shift on the collar-like structure, on the other hand, is subtle, suggesting a rigid body. Notably, in the side view alignment, a marked upward elevation of the AAA+ subunits has been observed in the four stable protomers (Fig. 2e, f). This movement results in a tilting of the AAA+ plane in the complex structure respect to that in the apo state. Taken together, the helicase of E5, although belonging to the SF3, underwent more pronounced conformational changes upon substrate incorporation, manifested as an inward-upward-lateral movement between protomers (Fig. 2g).

Top view of the DNA-bound E5 complex (structure 1, a) and the E5 in the apo state (b). The bound DNA is shown in red surface in the DNA binding channel of E5. A red circle marks the collar region. The superimposition of the DNA-bound E5 complex with the E5 apo form, shown in top view (c) and side view (d). The blue arrows highlight the movement direction of the AAA+ domains and the WHD. Protomers A-F in the DNA-bound E5 are multicolored, while the apo E5 form is shown in gray. e, f Side view of E5-ssDNA-ATP complex (structure 1) and apo E5 (f), with the AAA+ domains are highlighted by a blue box and the collar region marked by a red rounded rectangle. The blue arrows highlight the movement direction of the AAA+ domains and the WHD. g The schematic model of the inward-upward-lateral movement between protomers after substrate incorporation of E5. The black dots and the colored dots represent the corresponding subunits of E5 in the apo state and in the DNA-bound state, respectively. The blue arrows highlight the movement of the AAA+ ___domain and the WHD. h Superimposition of protomers (A–F) in E5-ssDNA-ATP complex (structure 1), with the collar subunits fixed. Blue arrows indicate the lateral movement (left) and the inward and upward movement towards the ssDNA binding axis (right) of helicase domains after substrate incorporation. The transition motif is highlighted by a red dashed oval.
Nucleotide cycling and dynamic landscape of E5 helicase conformation
To investigate the correlation between conformational changes of the E5 helicase and substrate incorporation into the AAA+ ATPase motor, ATP identity and occupancy were analyzed for each protomer of structure 1 and structure 3, which possesses the largest difference in flexible protomers position compared to structure 1 among all ssDNA-engaged structures (Supplementary Fig. 4d–h). Consecutive ATP hydrolysis was evidenced by the gradual change of the ATP density in the nucleotide-binding pocket of individual protomer as shown in the EM density maps (Fig. 3). In both structures, intact ATP densities were consistently presented within the three nucleotide-binding pockets (A–B, B–C, C–D) between the four stable protomers, while the D–E contained an ADP and F-A always showed interrupted densities or was empty. In contrast, the nucleotide holding within the nucleotide-binding pocket between the two flexible protomers (E–F) are variable. From structure 1 to structure 3, the β-phosphate density of the nucleotide in the E–F site tapers off and is therefore recognized as a transition state from ADP binding to ADP dissociation. Overall, the analysis of the nucleotide identity and occupancy within the twelve protomers of the two structures revealed a consecutive nucleotide hydrolysis cycle within E5: three ATP stable sites, followed by two ATP hydrolysis sites and one nucleotide turnover site (Fig. 3a). In the ATP hydrolysis sites, two ADPs or one ADP accompanied by another ADP in the to-be-dissociated state (ADP’) were observed. The nucleotide turnover site suggests a thorough dissociation of the ADP and preparation for the next cycle with a new ATP incorporation.

a Schematic representation of the nucleotide cycling. b Top view of the AAA+ domains of E5-ssDNA-ATP complex (structure 1 and structure 3), colored according to the local resolution. c Tables presenting the identity and occupancy of nucleotides in the nucleotide binding pockets of structure 1 and structure 3. All EM densities of nucleotides are shown at the same isosurface threshold within each dataset. Protomer holding ATP or ADP is defined as cis, and clockwise adjacent protomer is defined as trans.
The above observation revealed a connection between the stages of ATP hydrolysis and either the stability or arrangement of the protomers (Fig. 3a). To understand the structural basis for this relevance, we analyzed the detailed interaction network within and around the nucleotide binding pocket at different stages of nucleotide hydrolysis. At various stages of hydrolysis, from ATP to ADP, a similar orientation of the nucleotides was shown within the nucleotide-binding pocket, where the phosphate heads point towards the ssDNA binding axis of the E5 helicase and the nucleobases, together with 5-carbon sugar groups, are completely wrapped by a hydrophobic tunnel of the cis-protomer (Fig. 4a and Supplementary Fig. 7a). The local interactions were further stabilized by H-bonds between D652 and the nucleobase as well as D656 and the 2’-OH group of the 5-carbon sugar (Supplementary Fig. 7b). In particular, extended sequences between the relatively conserved β5 strand and the α6 helix (the name of the secondary structure was previously annotated in the arrangement of the AAA+ ___domain39,40,41) were observed in E5 and formed the base of the nucleotide binding pocket (Fig. 4a and Supplementary Fig. 7c). This motif was persistently traced as two parallel β-strands intervening by a short α-helix in all ATP stable sites and ATP hydrolysis sites, despite gradually decreasing local resolution with nucleotide hydrolysis especially in the ADP’ site, while disordered in the nucleotide turnover site as well as in the E5 apo structure (Supplementary Fig. 7d, e). We therefore defined it as the Nucleotide sensor motif of E5 helicase.

a The structural superimposition of the AAA+ domains between MPXV E5 (in green) and BPV E1 (in gray, PDB Code 2GXA). The Nucleotide sensor motif is highlighted. The surface of the nucleotide binding pockets of MPXV E5 (left) and BPV E1 (right) are shown, with the nucleotides bound inside shown as sticks. b Details of nucleotide-protein interactions (left) and cis-trans interaction (right) at representative ATP stable sites. Key amino acids involved in polar interactions (depicted with yellow dotted lines) are shown as sticks and labeled. c Comparisons of nucleotide binding pockets containing the nucleotide substrate in different hydrolysis states reveals a gradually increasing distance between tran- and cis-protomers. d The conformational difference (black arrow) of the Walker A loop in different nucleotide hydrolysis states. The protomer containing ATP or to-be-dissociated ADP (ADP’) are colored cyan and purple, respectively. Amino acids involved in polar interactions (depicted with yellow dotted lines) are shown as stick and labeled.
In contrast to the consistent interactions surrounding the sugar and nucleobase groups, the interaction network at the phosphate head of the nucleotide undergoes gradual changes corresponding to the nucleotide cycle, which involves alterations in the position and density of the amino acids within the conserved AAA+ ATPase motifs Walker A (P-loop), Walker B and Sensor-1 of the cis-protomer and the arginine finger (R-finger) of the adjacent trans-protomer (Supplementary Fig. 7c). In all ATP stable sites, the Walker A motif (G503-T511) and the Sensor-1 (N605) of the cis-protomer was responsible for most of the interactions with the phosphate groups (Supplementary Table 3), where the α-phosphate was stabilized by the H-bond network with the T507, G508 and T511; the β-phosphate formed H-bonds with A506, T507, G508, K509 and S510; and the γ-phosphate bonded to K509, S510 and N605 (Fig. 4b). The conserved Mg2+ ion was well defined in all ATP stable sites that tightly coordinated with β-, γ-phosphates and the side chain of S510 (Fig. 3 and Supplementary Table 3). Additional contacts around the ATP were provided by the R-fingers (R619 and R620) of the trans-protomer, which bind exclusively to the γ-phosphate (Fig. 4b). Furthermore, three sets of cis-trans interactions around the nucleotide binding pocket further indirectly stabilized the ATP-binding mode: one set involves H-bonds between the main chains of S556 and E557 (the cis Walker B motif) and the side chain of trans-protomer K576; the second set is mediated by the H-bond interaction between T505 of cis Walker A and trans R-finger (R619); the last set includes the salt bridge between cis D560 and trans R612 and the spatially adjacent H-bond between cis Y606 and trans D614 (Fig. 4b). As for the ATP hydrolysis sites where ADP was bound, the density of the Mg2+ ion was absent and a mild decrease in the number of interaction forces between the cis-protomer and the nucleotide was observed, with the Walker A still carried the most interaction forces with the phosphate groups (Fig. 3 and Supplementary Table 3). The trans-protomer, however, significantly lost interactions with both the nucleotide and the cis-protomer. In the ADP’ site where the density of γ-phosphate is absent, the trans R-fingers were no longer tied and appeared flexible in the density map (Supplementary Fig. 7f). Accordingly, the cis-trans interaction mediated by trans R619 and cis T505 was lost and the R-finger containing helix shifted away form that in the ATP stable site (Fig. 4c). Disruption of the cis-trans interaction was also observed between cis Walker B and trans K576, as the helix containing the trans K576 shifted ~3 Å away from the cis-protomer compared to that in the ATP stable site (Fig. 4c). The to-be-dissociated state of ADP (ADP’) removed all interactions related to the phosphate group (Supplementary Table 3), leading to a distinctly upturned and flexible conformation of the Walker A loop (Fig. 4d and Supplementary Fig. 7g). This motif reorientation disrupted the intra-protomer H-bond interactions between T505 and A506 of Walker A and H629, F630 and Y645 of the Nucleotide sensor motif, and may thus be responsible for the latter’s increased flexibility (Fig. 4d). Moreover, the remaining set of cis-trans interactions (the last set as mentioned above) around the nucleotide binding pocket disappeared, and the entire trans AAA+ ___domain presented an obvious shift away from the cis-protomer with further increased inter-protomer distance (Fig. 4c). In the nucleotide turnover site, both the inter-protomer distance and the flexibility of the cis-protomer return to that of the apo structure.
We also investigated conformational changes between individual protomers during the nucleotide cycle via structural superimposition of protomers holding the nucleotide in different hydrolysis states, with the collar subunits fixed (Fig. 2h). Notably, a progressive movement of the AAA+ and WHD subunits within protomers towards the ssDNA binding axis was observed, reminiscent of the hanging leg raise movement, with protomer A exhibiting the most contraction and protomer F showing stretching. A sideview presentation also showed a progressive lateral movement of the cis-protomer in the direction of the trans-protomer. These two types of movements, identified in different views, together represent the inward-upward-lateral rearrangement of the protomers after substrate bound described in the above section. This pronounced ___domain rearrangement within protomers started at the end of the transition motif, where an extended linker loop bridges D5N and the AAA+s, providing plasticity for the downstream subunits (Fig. 2h). Moreover, the ssDNA-binding hairpins were well defined in the four rigid protomers and presented a spiral staircase-like arrangement corresponding to that delineated in the escort translocation model11,13, with the one in protomer A, the first step in nucleotide cycling, located highest and protomer D lowest (Supplementary Fig. 8a). The interactions between ssDNA and E5 helicase are mediated by the residues R585 and F588, with mainly one nucleotide bound by each protomer (Supplementary Fig. 8b). For the two flexible protomers, ambiguous density was observed for their hairpins, indicating weak or absent interaction with ssDNA (Supplementary Fig. 8c, d).
Based on all the observations described above, we propose a non-classical escort translocation model for E5. In this model, in addition to the conformational changes within the individual protomers (the common feature of the escort model defined by E1), a nucleotide cycle-triggered inter-protomer movement is also present, albeit to a mild extent compared to that in the hand-over-hand translocation model (Supplementary Movie 1).
Head-to-head E5 double hexameric assembly for dsDNA capture
To further investigate the role of E5 in helicase-mediated DNA unwinding steps prior to ATP-triggered translocation along the ssDNA, such as origin loading and dsDNA melting, the dsDNA duplex of 60 bp was incubated with E5 in the absence of ATP, and the samples was then analyzed by cryo-EM. To our surprise, the 2D average revealed that nearly half of the E5 particles formed a distinct head-to-head double hexamer via their primase regions after dsDNA addition (Supplementary Fig. 6). The flexibility of this configuration is evident as the docking angle between two hexamers varies from nearly linear to splayed (Fig. 5a and Supplementary Movie 2), with the maximum tilted angle observed being about ~65° in the 2D classification. Moreover, certain particles showed a twisted stacking pattern between two hexamers (Fig. 5a), reminiscent of the double hexamer arrangement of MCM in the initial origin melting process previously described9.

a The representative 2D class averages (left) and schematic models (right) showing structural flexibility and intermolecular arrangements of the E5 double hexamer. b Unsharpened EM density map of E5 double hexamer. The density map displays the local resolution distribution, the diffuse densities of AEPs (black arrows), and the density of the bound dsDNA. The latter is highlighted in the inset, presented in the cut-off view, and indicated with a red dashed box along with a red arrow. c, d Sharpened EM density map of the E5 double hexamer. The ZBDs and the helicase are colored orange and green, respectively. The anti-clockwise arrangement of ZBDs with respect to the corresponding helicase domains is highlighted by black dashed lines. e Cut-off view of the dsDNA binding channel reveals a positively charged inner wall suitable for DNA accommodation. f Model of dsDNA incorporation into the double hexamer. The length and position of the dsDNA is determined from the unsharpened EM density map. B-form DNA was used for the generation of this model. The precise status of the DNA duplex incorporated in the double hexamer remains to be clarified. g Representative 2D class averages of wild-type (WT) E5 and E5 mutant (DDD/A) under varying substrate conditions. “-” indicates substrate absence, and “+” indicates substrate presence. h DNA polymerization assays catalyzed by wild-type E5 (WT) or E5 mutants (DDD/A, KRR/A, RF/A, and ΔHelicase). The “-” is used to represent the mock group, in which an equal volume of protein buffer was added to the reaction to replace the E5 proteins. The experiments were repeated three times independently with similar results, and the representative image is provided.
To gain more information on the interface of the E5 double hexamer, particles with double hexameric configuration were selected for further 3D reconstruction. After rounds of 3D classification and refinements, the EM map of the double hexamer was obtained at an overall resolution of ~3.31 Å (Supplementary Fig. 9). Specifically, one hexamer exhibit a higher resolution of 3–5 Å, while the other hexamer demonstrated a lower resolution due to inherent flexibility (Fig. 5b and Supplementary Movie 3). The key structural features of the two entire helicases and half of the primase domains, the ZBD subunits, of both hexamers are well modeled (Fig. 5c–f). Specifically, all ZBDs stand upright upon the collar regions, slightly anti-clockwise aligned with the corresponding helicase domains, and wrap around to form an extended channel (Fig. 5d). In this conformation, the basic amino acids of the ZBDs, such as K286, K315, and K317, provide a positively charged inner wall of the channel thus make it suitable for DNA accommodation (Fig. 5e). Although the density of the dsDNA was weak, it can be traced in the unsharpened EM density map located in the central of the ZBD channel and extending to the collar, indicating dsDNA capture by this double hexamer structure (Fig. 5b, f). Moreover, the ring-like diffuse density visible wrapped outside the interface of the double hexamers in the unsharpened EM density map suggests a flexible arrangement of the AEPs around the ZBDs channel (Fig. 5b).
This head-to-head double hexameric organization of the helicases has previously been well delineated for the eukaryotic MCM helicase, which assembles at the chromatin origin as an inactive double hexamer and then melts the dsDNA after recruitment of a set of firing factors and ATP-triggered DNA base pair breakage42. A comparable head-to-head double hexameric architecture at the origin sequence of genome duplication was observed in Simian Virus 40 (SV40) formed by the viral helicase, large T-Antigen (T-Ag)43,44. Therefore, the E5 double hexamers loaded on the dsDNA may mimic the very beginning of poxvirus genome replication. The flexibility of double hexamer assembly may provide the internal factors to trigger the disruption of the double hexamer interface and expose duplex DNA for further melting (Supplementary Movies 2 and 3). Considering the similar assembly of E5 as for the MCM in the origin binding, we asked whether the ATP would also trigger the subsequent DNA melting in the E5-mediated DNA unwinding. To address this question, ATP was added to the E5-dsDNA complex and the sample was then analyzed by cryo-EM. Remarkably, the addition of ATP completely abolished the double hexameric assembly of E5, with the primase ___domain returning to the ATP-harboring configuration described above (Figs. 5g and 6). In contrast, although the E5 DDD/A mutant can also form double hexamers in the presence of the dsDNA substrate, the double hexameric configuration in this case was insensitive to the ATP (Fig. 5g). This suggests that the ATP recruitment by the primase ___domain and the accompanying formation of primase assembly disrupt the head-to-head double hexameric organization of E5 and result in the release of the dsDNA. Collectively, these results suggest that nucleotide recruitment by the primase ___domain acts as at least one of the external triggers for E5 to switch from dsDNA capture (double hexamer) to translocation along ssDNA, although the mechanism for one DNA strand ejection is unknown.

a,b Model of the formation of the double hexameric E5 on the dsDNA. The red question marks indicate that the mechanism by which the two copies of E5 are loaded onto the dsDNA ends in the correct orientation and translocated along the dsDNA until they meet and initiate DNA melting is still unknown. c The inherent flexibility of the double hexameric conformation of E5, which may provide the internal triggers for the initiation of E5-mediated DNA melting. d ATP serves as an external trigger, inducing reorientation of the primase ___domain, triggering disruption of the E5 double hexamer and eventually leading to dissociation of E5 from dsDNA, although the mechanism underlying single-strand ejection requires further investigation. ATP binding in the primase and helicase domains is depicted as a red oval and two encircled blue S respectively. e The nucleotide cycling triggered non-classical escort translocation model of E5. The top view of the helicase ___domain of structures 1-5 was displayed with the spatial orientation of primase domains aligned. f The proposed model of primer synthesis and unwinding by E5 during DNA replication, with the red arrows representing the direction of E5 translocation along the leading strand and the red question marks indicating primer synthesis on the lagging strands.
Functional relevance of the helicase and primase domains of E5
Given the direct involvement of primase in DNA unwinding procedure shown above, we wondered whether the helicase also contributes to primase activity. To this end, a truncated mutation containing only the E5 primase ___domain (residues 1–323, namely ΔHelicase) was expressed and used for the evaluation of primase activity in the primer extension assay (Supplementary Fig. 1b). As shown in Fig. 5h, a significant decrease in the substrate utilization and 30-nt product synthesis of ΔHelicase mutant compared to wild-type E5 was observed during extension assay, indicating an important role of the helicase ___domain in supporting primase function. To attribute this influence more precisely to the different function of the helicase ___domain, inactivating mutations of E5 for either NTPase activity (KRR/A) or ssDNA binding activity (a double alanine mutant of residues R585 and F588A, RF/A) were used to evaluate the respective contribution in primer activity. The primer extension assay revealed that the KRR/A mutant possesses a comparable or even enhanced activity to the wild-type E5, while the RF/A mutant shows a pronounced reduction in primer extension to a similar extent as the ΔHelicase mutant. This result suggests that it is the ssDNA binding, rather than the DNA translocation ability, of the helicase ___domain that may provide a docking station for the DNA template and further supports primase activity.
Discussion
Replicative helicase-catalyzed unwinding of the DNA double helix is the starting event of the DNA replication, and the underlying mechanism has therefore long been of central interest to molecular biologists. In this study, we examined the working mechanism of the mpox virus helicase-primase E5, the founding member of a special class of replicative helicases in which the primase and helicase domains are fused together and expressed as a single polypeptide, delineating the E5-mediated DNA unwinding initiation model in terms of dsDNA loading, DNA melting and ssDNA translocation (Fig. 6).
The double hexameric architecture of E5 loaded on the dsDNA suggests that the MPXV may adopt a similar strategy to initiate DNA replication as its cellular host in terms of the timing of helicase loading relative to origin melting, albeit in a simplified form. This initiation strategy of DNA replication was also observed in SV40, suggesting that it might be widely used by viral helicases among DNA viruses. In light of the random design of the dsDNA sequence being used in the cryo-EM assay, and the formation of a double hexamer immediately following the mixing of the purified E5 protein with dsDNA, it can be inferred that the origin binding by E5 is quite simple, requiring neither strict origin sequence specificity nor the involvement of other proteins as an initiator and/or loader. In other words, E5 combines origin recognition and helicase loading into a single step that requires no ATP as a motive force. Given the rigid body of the collar region, it can be concluded that the dsDNA loading for E5 did not include an open ring state, and therefore, it might be loaded onto the viral genome from the hairpin ends of the genome. However, the mechanism by which the two copies of E5 are loaded onto the dsDNA ends in the correct orientation and translocated along the dsDNA until their meeting is unknown (Fig. 6a, b).
After loading onto dsDNA as an inactive double hexamer, the eukaryotic helicase MCM is activated by the recruitment of a set of firing factors to form the holo-helicase, CMG, which leads to a distinct splayed configuration of the double hexamer with three base pairs untwisted inside the helicases4,42,45. These ATP-induced changes in MCMs were thought to provide the motive power for starting DNA double helix melting. The further extension of dsDNA unwinding was proposed to follow a DNA shearing model, as evidenced by both MCM and T-Ag43,44. The inherent flexibility observed in the double hexameric conformation of E5 suggested the deficiency of a strict regulatory mechanism in E5-mediated DNA melting initiation (Fig. 6c), comparable to that of MCM. Since viral genome replication does not have the same strict requirement to respond to the cell cycle phase as its host, this loose regulation is somewhat reasonable for the virus. More details of the interaction between dsDNA and E5, the role of the AEP subunit in E5 dodecamer-mediated DNA unwinding, in particular whether and how base pair breakage occurs within E5 and whether E5 adopted the DNA shearing mechanism of duplex unwinding as MCM and T-Ag, need to be investigated further.
Before translocation along the ssDNA, the double hexameric architecture of E5 should be disrupted, with one strand of the DNA duplex released from the helicase. Although the precise mechanism of single strand ejection remains unknown, our results provide evidence that the conformational reorientation of the primase domains induced by ATP incorporation completely disrupts the head-to-head E5 dodecamer and leads to dissociating of E5 from the dsDNA. In addition to this role as an external trigger for DNA melting by E5, this ATP-harboring configuration of the primase domains has previously been observed to affect the unwinding function of E5 by partially obstructing the DNA channel at the collar ___domain31,34. Given that dsDNA has a larger radius than ssDNA, this steric hindrance is more likely to limit dsDNA entry through the E5 collar. We therefore propose two plausible roles for this distinct primase conformation: first, it disrupts the local environment for dsDNA capture, leading to double hexamer departure; and second, it acts as a roadblock against dsDNA entry into the helicase channel, promoting single strand ejection (Fig. 6d).
After origin melting, the E5 further unwinds the double strand by translocating along the ssDNA in a non-classical escort translocation model, in which the nucleotide cycling controls both the intra-protomer reorientation and the inter-protomer movement of the AAA+s and WHDs of E5 helicase (Fig. 6e). The consecutive nucleotide cycling by hexameric AAA+ ATPase has also been well delineated in the RuvAB-Holliday junction branch migration model46,47. Since the density of nucleotides was observed to change gradually even when they were defined with the same identity, we suggest that the commonly observed difference in the ratio of ATP to ADP within the AAA+ ATPase structures simply represents various snapshots in nucleotide cycling and is influenced by the substrate used (ATP or ATPγS) or the hydrolysis rate of the ATPases. The number of ATP consumed in each step and the step size of the helicase translocation along the ssDNA are determined by the number of ATP turnover sites and the number of nucleotides captured by each ssDNA binding hairpin, respectively. For E5, one ATP was consumed to move the RFJ one nucleotide forward, similar to other SF3 helicases.
Although the quality of the EM density makes it difficult to define the 5’- or 3’-termini of the ssDNA bound in the E5 helicase channel, the top-to-bottom arrangement of the ssDNA binding hairpins from protomer A to protomer D, together with the direction of the nucleotide cycling, reveals that the latest protomer (protomer A) in the nucleotide cycling possess ssDNA binding hairpin closest to the N-terminal primase ___domain (Supplementary Fig. 8a). This means that the ATPase-motored translocation in E5 is towards its primase ___domain. As a result, when E5 translates along the leading strand, the primase domains are located upstream of the helicase motor, close to the RFJ (Fig. 6e, f). In this model, the ATP-driven primase configuration and the E5 collar are rigid to provide the non-specific wedge for dsDNA unwinding at the RFJ. By comparing E5 with the architecture of the T7 replisome2, which is also a helicase-primase fusion, we found the intriguing fact that despite different translocation directions, either along the leading or lagging strand, their respective ___domain organization strategies leads to a consistent outcome, i.e., the primases located close to the lagging strand persistently to ensure parallel synthesis of Okazaki fragments and a similar rate of synthesis of the leading and lagging strands. Despite being proposed to act as an origin-binding ___domain in helicases containing the D5/E5-like helicase fold6,48, we fail to identify any roles for WHD in either origin binding or DNA strand separation. Instead, we observed that it may contribute to the stabilization of the D5/E5-like helicase-specific nucleotide sensor motif during nucleotide cycling, which may be one of the reasons for its evolutionary preservation in the D5/E5-like helicase.
Although the primase and helicase are highly correlated during the initiation of DNA replication, fusion of these two proteins into a single polypeptide is not a prerequisite. Our results indicate that both the primase and helicase domains of the E5 contribute to each other’s functions. This unique cooperative mechanism may be the primary reason for the development or maintenance of this helicase-primase fusion enzyme during the evolutionary process. While our study provides a clue to the mechanism of DNA replication initiation mediated by helicase-primase fusion enzymes, the precise role of each functional domains as well as the overall processing are still await a high-resolution structure involving the replication fork.
Methods
Protein expression and purification
The cDNA of full-length MPXV E5 (GenBank: YP_010377099.1) and variants, including DDD/A (D70/D72/D170A) mutant, KRR/A (K509/R619/R620A) mutant, and RF/A (R585/F588A) mutant were fused to an N-terminal 6 × His tag, cloned into the pFastbacTM1 (Invitrogen) vector and expressed using the Bac-to-Bac baculovirus expression system, as previously described49,50,51. In brief, the plasmid was transformed into DH10Bac competent cells (Biomed, catalog no. BC112-01) and the recombinant bacmid was then used to transfect the Sf9 cells (Invitrogen, catalog no. B825-01). For protein expression, the recombinant baculovirus stock (P3) was harvest and used to infect the High FiveTM cells (Invitrogen, catalog no. B855-02). Cells were harvested by centrifugation 48 h post infection and ultrasonicated in buffer A containing 20 mM Tris-Hcl (pH 7.4), 500 mM NaCl, 10% glycerol and 1 mM protease inhibitor cocktail (MCE, catalog no. HY-K0010). The cell debris was then centrifuged for 1 h at 91,000 × g at 8 °C. After centrifugation, the supernatant was further filtered with a 0.22 μm filter and purified by affinity chromatography using a HisTrap column (GE Healthcare), followed by size-exclusion chromatography using a Superose 6 Increase 10/300 GL column (GE Healthcare). Purified proteins were stored in protein buffer B containing 20 mM Tris-HCl, pH 7.4, 300 mM NaCl, 10% glycerol.
For the expression of the E5 ΔHelicase mutant, the cDNA encoding E5 residues 1–323 was fused to an N-terminal 6 × His tag and cloned into the NdeI and XhoI sites of the pET-28a vector. The expressing plasmid was transformed into the Escherichia coli (E. coli) strain BL21 competent cells, which were then induced with 1 mM IPTG (isopropyl--D-thiogalactopyranoside) and cultured overnight at 16 °C. Cells were collected, resuspended in the buffer A and lysed by sonication. After centrifugation, the supernatant containing the soluble E5 ΔHelicase protein was further filtered with a 0.22 μm filter and purified by affinity chromatography using a HisTrap column (GE Healthcare), followed by size-exclusion chromatography using a Superdex 200 column (GE Healthcare). Purified proteins were stored in protein buffer B containing 20 mM Tris-HCl, pH 7.4, 300 mM NaCl, 10% glycerol.
Primase assays
Primase reactions were carried out as previously described52. The reaction mixtures (10 μL) consist of 20 mM Tris-HCl (pH 7.4), 60 mM NaCl, 6 mM MnCl2, 10 mM DTT, 100 μg/mL BSA, 1 mM ATP, 250 μM UTP, 25 μM GTP, 250 μM CTP, 0.0066 μM [α-32P]GTP (3000 Ci/mmol) (PE, catalog no. NEG006H250UC), 1 μg (0.57 pmol) ϕX174 phage DNA (NEB, catalog no. N3023S), and 600 ng (100 nmol) wild-type or mutant E5 proteins. Reactions were incubated at 37 °C for 1 h and then stopped by heating at 90 °C for 10 min. The products were treated with 5 units of calf intestinal alkaline phosphatase (NEB, catalog no. M0525S) at 37 °C for 0.5 h. The reactions were denatured by adding 10 μL of termination buffer (95% deionized formamide, 20 mM EDTA, plus bromophenol blue and xylene cyanol) followed by heating at 90 °C for 10 min. The reaction products were then analyzed on a denaturing gel containing 18% polyacrylamide and 7 M urea and visualized using Typhoon FLA 7000 (GE Healthcare). The oligonucleotide size markers were chemically synthesized and end-labeled with 32P using polynucleotide kinase (Thermo Fisher Scientific, catalog no. EK0031) and [γ-32P] ATP (3000 Ci/mmol) (PE, catalog no.NEG502A100UC).
In vitro DNA polymerization assays
The template strand (5′-GGCTAAACCGCTGTTATCTTCCTGATTCGG-3′) and the 5′FAM-labeled primer strand (5′-CCGAATCAGGAAGAT-3′), both of which were synthesized by Sangon Biotech (Shanghai) Co., Ltd., were then mixed with a molar ratio of 1:1 in DEPC water. The mixtures were heated at 95 °C for 5 min, followed by slow cooling to room temperature. The annealed dsDNA substrate (1 μM) was then mixed with 100 nM wild-type, E5 mutants and ΔHelicase proteins in buffer composed of 20 mM Tris-HCl (PH 7.4), 60 mM NaCl, 6 mM MnCl2, 2.5 mM NTPs/dNTPs and 10 mM DTT, 100 μg/mL BSA. Reaction mixtures were incubated at 37 °C for 1 h before the addition of formamide loading buffer (90% formamide, 20 mM EDTA, 0.05% bromophenol blue, and 0.05% xylene blue). After boiling at 95 °C for 5 min, the samples were loaded onto pre-warmed 12.5% urea gels and run for 1.5 h. The gel was visualized using Fusion FX (Vilber). Reaction products were monitored over a time course and quenched at various time points (as indicated in Supplementary Fig. 1 and legends). All quantification was performed using ImageJ. Data were plotted and analyzed using GraphPad Prism 6.
DNA unwinding assay
The 60 nt DNA strand (Top strand, 5′-T(18)CAGTCCGAAGCGCATCCCGTT TGACCAT(15)-3′) and the 12 nt 5’-FAM-labeled DNA strand (Bottom strand, 5′-FAM-CGGGATGCGCTT-3′) (synthesized by Sangon Biotech (Shanghai) Co., Ltd.) were dissolved in DEPC water and mixed with a molar ratio of 1:1. The mixture was heated at 95 °C for 5 min, followed by slow cooling to room temperature. The annealed DNAs (200 nM) were then mixed with E5 wild-type or mutant proteins (the concentrations of the proteins were labeled on the gel) in the buffer composed of 20 mM Tris (pH 7.4), 60 mM NaCl, 7.5 mM MgCl2, 2 mM dCTP and 1 mM DTT. To prevent the re-annealing of the unwound DNA single strands, 2 μM of unlabeled bottom strand was also contained in the reaction system. Reaction mixtures were incubated at 33 °C for 30 min and then digested with 5 mg/mL Proteinase K (Thermo Fisher Scientific, catalog no. EO491) at room temperature for 20 min, followed by the addition of loading buffer (10% glycerol, 20 mM EDTA, 0.5% SDS and 0.2% bromphenol blue) to stop the reaction. Samples were loaded onto 10% TBE polyacrylamide gel for electrophoresis and the gel was visualized using Fusion FX (Vilber).
Cryo-EM sample preparation and data collection
The DNA duplex used for the DNA-bound E5 complex was obtained by annealing equimolar amounts of a 50 nt DNA strand (5′-CCGAATCAGGAAGATAACAGCGGTTTAGCCT(20)-3′) and a 30 nt DNA strand (5′-GGCTAAACCGCTGTTATCTTCCTGATTCGG-3′) (Sangon Biotech (Shanghai) Co., Ltd.). For the MPXV E5 in the apo state, the protein was diluted to 0.6 mg/mL in buffer B. For the MPXV DNA-bound E5 complex, the E5 protein was mixed with the above DNA duplex in a 1:1 molar ratio and incubated for 30 min on ice. ATP was then added to the sample to a final concentration of 1 mM and incubated for 5 min at room temperature before being applied to grids. For the E5 double hexamer, the E5 protein was mixed with dsDNA (The Top strand 5′-CCGAATCAGGAAGATAACAGCGGTTTAGCCT(30)-3′) and the bottom strand 5′-A(30)GGCTAAACCGCTGTTATCTTCCTGATTCGG-3′) (synthesized by Sangon Biotech (Shanghai) Co., Ltd.) at molar ratio 1:2 in buffer B and incubated on the ice for 30 min before being applied to grids.
Each aliquot of 4 μL of the protein samples was loaded onto the glow-discharged QUANTIFOIL® R 1.2/1.3 holey carbon grids with 2 nm continuous carbon on top. The grids were blotted at 4 °C and 100% humidity for 0.5 s, and then flash-frozen in liquid ethane using the FEI Vitrobot Mark IV (Thermo Fisher Scientific). Cryo-EM data were collected on Titan Krios G3i electron microscopes operated at 300 kV and equipped with a Gatan K3 direct electron detector. All datasets were automatically recorded using EPU in super resolution mode and defocus values ranged from −1.3 μm to −2.3 μm.
Image processing
The detailed data processing workflow is summarized in Supplementary Figs 2, Supplementary 3 and Supplementary 9. All the raw dose-fractionated images stacks were 2 × binned, aligned, dose-weighted and summed using MotionCor253. The contrast transfer function estimation, particle picking and extraction, 2D classification, ab-initio model generation, 3D refinements were performed in cryoSPARC v4.2.054.
For the case of MPXV E5 in the apo state, a whole dataset of 11,653 micrographs was subjected to template picking, particles extraction (4 × binned, box size 480) and 2D classification. A clean dataset with 894,596 particles from good 2D classes were selected for the initial models generation and hetero-refinement and we chose the top three dominant volumes and particles to perform another two rounds of hetero-refinements. Finally, the predominant class containing a subset of 197,759 good particles. These particles were re-extracted (unbinned, box size 480) and subjected to three rounds of 2D classifications to remove the junk particles. After running one round of non-uniform refinement, we got a reconstruction at 3.33 Å resolution as determined by the Fourier shell correlation (FSC) 0.143 cut-off value. The map was further sharpened by DeepEMhancer55.
For the case of MPXV DNA-bound E5 complex, a total of 11,635 movie stacks were collected for this dataset. The particles were picked out using cryoSPARC blob-pick procedure from 3897 micrographs, and then these particles were subjected to 2D classification and hetero-refinement. One round of homogeneous refinement was performed in order to select good particles in different views for Topaz training and generated the Topaz model. The Topaz56 procedure was then applied to select particles against entire dataset. The 2,448,532 initial particles were picked and extracted with the box size of 480 pixels from 11,635 micrographs. After the extensive 2D classification, ~1,596,469 good particles were selected to generate the initial models and heterogeneous refinement and resulted to four distinct volumes. Next, two parallel methods was used to deal with the current data: (1) Two dominant classes containing 44.1% and 31.9% of total 1,213,562 particles were identified, which displayed clear features of secondary structural elements. These particles were subjected to homogeneous refinement and the cryoSPARC 3D Variability and clustering (8 clusters) to clearly display the intermediate states of the dynamic process. All the 8 classes could be found clearly-enough defined density for actual model building and we chose 6 of them to run the non-uniform refinement and got six reconstructions at 2.99 Å, 2.88 Å, 2.95 Å, 3.12 Å, 3.08 Å, 3.13 Å resolution as determined by the FSC 0.143 cut-off value. The map at 2.88 Å and 3.12 Å resolution were used to build models for the further structural analysis. (2) One dominant class containing 44.1% of the total 703,715 particles, was used to perform another hetero-refinement. The predominant class contains a subset of 400,496 good particles. These particles were re-extracted (unbinned, box size 480) and subjected to three rounds of 2D classifications to remove the junk particles. After running one round of non-uniform refinement, we got a reconstruction at 2.95 Å resolution as determined by the FSC 0.143 cut-off value. The map was sharpened by DeepEMhancer55.
To improve the local resolution of the E5 primase and Zn binding ___domain, subtract the signal of the primase with the adjacent Zn binding ___domain from the input particle images and perform cryoSPARC local refinement on the associated ___location, which yielded a final density map at 2.95 Å resolution estimated by the gold-standard FSC cut-off value of 0.143. The final map was sharpened by DeepEMhancer55, which can clearly reveal the distinct residues density.
For the MPXV E5 double hexamer, the dataset was processed in a similar way to that for the E5 in the apo state. A total of 1,701,512 particles were selected from 34,683 micrographs for 2D classification, hetero-refinement which resulted in two distinct volumes. One class that contains 46.0% of total particles with the characteristics of double-hexamer assembly conformation was identified. These particles were subjected to re-extraction and proceeded a new round of ab-initio reconstruction and hetero-refinement and resulted in two distinct volumes. The double-hexamer-like volume with the corresponding particles was subjected to non-uniform refinement in cryoSPARC v.4.2.0, which yielded a final density map at 3.31 Å resolution estimated by the gold-standard FSC cut-off value of 0.143. The final map was sharpened by DeepEMhancer55.
We then use two parallel ways to deal with the current data: in order to show the conformation variabilities of the volume, this class of particles were used to procced the cryoSPARC 3DVA with the filter-resolution set to 7 Å and the number of modes set to 4 indicated that there were total 4 components could be built through this job. Following is the 3DVA display under the “simple mode” would output a simple linear “movie” of volumes along each component and the number of frames set to 10. However, we chose 2 of the 4 components frame series to show the possible conformation changes using Chimera57 shown in Supplementary Movies 2 and 3.
Model building and structure refinement
The structure of a single protomer of E5 was initially predicated by AlphaFold258, and then was rigidly docked into the density map using Chimera57 with further manual adjustment. The DNA strands were manually built. The initial coordinates were refined in real space using PHENIX59 with Ramachandran restraints and secondary structural restraints applied. The model was further corrected to improve the local fit using COOT60 and refined for several rounds, aided by stereochemical quality assessment using MolProbity61. Each residue was manually checked with the chemical properties taken into consideration during model building. Statistics associated with data collection, 3D reconstruction and model building were summarized in Supplementary Table 1. Figures were generated using Chimera, ChimeraX 1.757,62 and PyMOL.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data needed to interpret and verify the conclusions in the paper are presented in the paper. The data that has been utilized in the generation of the figures and charts presented in this study is available in the Source Data file provided alongside this paper. The cryo-EM maps have been deposited in the Electron Microscopy Data Bank (EMDB) under accession codes EMD-60676 (Cryo-EM structure of MPXV E5 in the apo state), EMD-60684 (The Cryo-EM structure of MPXV E5 head-to-head double hexamer conformation), EMD-60677 (The Cryo-EM structure of MPXV E5 in complex with ssDNA), EMD-60678 (The Cryo-EM structure of MPXV E5 in complex with ssDNA focused on primase and Zn binding ___domain), EMD-60679 (The Cryo-EM structure of MPXV E5 in complex with ssDNA in intermediate state 1), EMD-60680 (The Cryo-EM structure of MPXV E5 in complex with ssDNA in intermediate state 3), EMD-60681 (The Cryo-EM map of MPXV E5 in complex with ssDNA in intermediate state 2), EMD-60682 (The Cryo-EM map of MPXV E5 in complex with ssDNA in intermediate state 4), EMD-60683 (The Cryo-EM map of MPXV E5 in complex with ssDNA in intermediate state 5). The atomic structure coordinates were deposited in the RCSB Protein Data Bank (PDB) under the accession codes 9ILY (Cryo-EM structure of MPXV E5 in the apo state), 9IM3 (The Cryo-EM structure of MPXV E5 head-to-head double hexamer conformation), 9ILZ (The Cryo-EM structure of MPXV E5 in complex with ssDNA), 9IM0 (The Cryo-EM structure of MPXV E5 in complex with ssDNA focused on primase and Zn binding ___domain), 9IM1 (The Cryo-EM structure of MPXV E5 in complex with ssDNA in intermediate state 1) and 9IM2 (The Cryo-EM structure of MPXV E5 in complex with ssDNA in intermediate state 3). Other structures for analysis, including 8HWC, 8HWA, 8XJ6, 5L2X, 2GXA were obtained from the PDB. All the accession codes listed here are also shown in Supplementary Table 1. Source data are provided with this paper.
References
Bleichert, F., Botchan, M. R. & Berger, J. M. Mechanisms for initiating cellular DNA replication. Science 355, 6327 (2017).
Gao, Y. et al. Structures and operating principles of the replisome. Science 363, 6429 (2019).
Miller, T. C. R. et al. Mechanism of head-to-head MCM double-hexamer formation revealed by cryo-EM. Nature 575, 704–710 (2019).
Lewis, J. S. et al. Mechanism of replication origin melting nucleated by CMG helicase assembly. Nature 606, 1007–1014 (2022).
Stenlund, A. Initiation of DNA replication: lessons from viral initiator proteins. Nat. Rev. Mol. Cell. Biol. 4, 777–785 (2003).
Kazlauskas, D., Krupovic, M. & Venclovas, C. The logic of DNA replication in double-stranded DNA viruses: insights from global analysis of viral genomes. Nucleic Acids Res. 44, 4551–4564 (2016).
Brosh, R. M. Jr. & Matson, S. W. History of DNA helicases. Genes 11, 255 (2020).
Singleton, M. R., Dillingham, M. S. & Wigley, D. B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 76, 23–50 (2007).
Li, N. et al. Structure of the eukaryotic MCM complex at 3.8 Å. Nature 524, 186–191 (2015).
Arias-Palomo, E. et al. Physical basis for the Loading of a bacterial replicative helicase onto DNA. Mol. Cell 74, 173–184.e4 (2019).
Enemark, E. J. & Joshua-Tor, L. Mechanism of DNA translocation in a replicative hexameric helicase. Nature 442, 270–275 (2006).
Chaban, Y. et al. Structural basis for DNA strand separation by a hexameric replicative helicase. Nucleic Acids Res. 43, 8551–8563 (2015).
Javed, A., Major, B., Stead, J. A., Sanders, C. M. & Orlova, E. V. Unwinding of a DNA replication fork by a hexameric viral helicase. Nat. Commun. 12, 5535 (2021).
O’Donnell, M. E. & Li, H. The ring-shaped hexameric helicases that function at DNA replication forks. Nat. Struct. Mol. Biol. 25, 122–130 (2018).
Itsathitphaisarn, O. et al. The hexameric helicase DnaB adopts a nonplanar conformation during translocation. Cell 151, 267–277 (2012).
Remus, D., Beall, E. L. & Botchan, M. R. DNA topology, not DNA sequence, is a critical determinant for drosophila ORC-DNA binding. EMBO J. 23, 897–907 (2004).
Robinson, N. P. et al. Identification of two origins of replication in the single chromosome of the archaeon sulfolobus solfataricus. Cell 116, 25–38 (2004).
Broach, J. R. et al. Localization and sequence analysis of yeast origins of DNA replication. Cold Spring Harb. Symp. Quant. Biol. 47, 1165–1173 (1983).
Fuller, R. S., Funnell, B. E. & Kornberg, A. The DNA A protein complex with the E. coli chromosomal replication origin (oriC) and other DNA sites. Cell 38, 889–900 (1984).
Duderstadt, K. E., Chuang, K. & Berger, J. M. DNA stretching by bacterial initiators promotes replication origin opening. Nature 478, 209–213 (2011).
Bell, S. P. & Kaguni, J. M. Helicase loading at chromosomal origins of replication. Cold Spring Harb. Perspect. Biol. 5, 6 (2013).
Corn, J. E. & Berger, J. M. Regulation of bacterial priming and daughter strand synthesis through helicase-primase interactions. Nucleic Acids Res. 34, 4082–4088 (2006).
Chen, X. et al. Structural studies reveal a ring-shaped architecture of deep-sea vent phage NrS-1 polymerase. Nucleic Acids Res. 48, 3343–3355 (2020).
Lipps, G. et al. Structure of a bifunctional DNA primase-polymerase. Nat. Struct. Mol. Biol. 11, 157–162 (2004).
Beck, K., Vannini, A., Cramer, P. & Lipps, G. The archaeo-eukaryotic primase of plasmid pRN1 requires a helix bundle ___domain for faithful primer synthesis. Nucleic Acids Res. 38, 6707–6718 (2010).
Shao, Z. et al. Structures and implications of the C962R protein of african swine fever virus. Nucleic Acids Res. 51, 9475–9490 (2023).
Hutin, S. et al. Domain Organization of vaccinia virus helicase-primase D5. J. Virol. 90, 4604–4613 (2016).
Tan, W. & Gao, G. F. Neglected zoonotic monkeypox in Africa but now back in the spotlight worldwide. China CDC Wkly. 4, 847 (2022).
Iyer, L. M., Koonin, E. V., Leipe, D. D. & Aravind, L. Origin and evolution of the archaeo-eukaryotic primase superfamily and related palm-___domain proteins: structural insights and new members. Nucleic Acids Res. 33, 3875–3896 (2005).
Boyle, K. A., Arps, L. & Traktman, P. Biochemical and genetic analysis of the vaccinia virus D5 protein: multimerization-dependent ATPase activity is required to support viral DNA replication. J. Virol. 81, 844–859 (2007).
Li, Y., Zhu, J., Guo, Y. & Yan, R. Structural insight into the assembly and working mechanism of helicase-primase D5 from Mpox virus. Nat. Struct. Mol. Biol. 31, 68–81 (2024).
Lipps, G., Rother, S., Hart, C. & Krauss, G. A novel type of replicative enzyme harbouring ATPase, primase and DNA polymerase activity. Embo. J. 22, 2516–2525 (2003).
Martinez-Jimenez, M. I. et al. The Zn-finger ___domain of human primpol is required to stabilize the initiating nucleotide during DNA priming. Nucleic Acids Res. 46, 4138–4151 (2018).
Zhang, W. et al. Structural and functional insights into the helicase protein E5 of mpox virus. Cell Discov. 10, 67 (2024).
Rechkoblit, O. et al. Structure and mechanism of human primpol, a DNA polymerase with primase activity. Sci. Adv. 2, e1601317 (2016).
Pan, H. & Wigley, D. B. Structure of the zinc-binding ___domain of Bacillus stearothermophilus DNA primase. Structure 8, 231–239 (2000).
Guilliam, T. A., Keen, B. A., Brissett, N. C. & Doherty, A. J. Primase-polymerases are a functionally diverse superfamily of replication and repair enzymes. Nucleic Acids Res. 43, 6651–6664 (2015).
Li, A. W. H. et al. Molecular basis for the initiation of DNA primer synthesis. Nature 605, 767–773 (2022).
Wendler, P., Ciniawsky, S., Kock, M. & Kube, S. Structure and function of the AAA+ nucleotide binding pocket. Biochim. Biophys. Acta 1823, 2–14 (2012).
Iyer, L. M., Leipe, D. D., Koonin, E. V. & Aravind, L. Evolutionary history and higher order classification of AAA+ ATPases. J. Struct. Biol. 146, 11–31 (2004).
Erzberger, J. P. & Berger, J. M. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct. 35, 93–114 (2006).
Douglas, M. E., Ali, F. A., Costa, A. & Diffley, J. F. X. The mechanism of eukaryotic CMG helicase activation. Nature 555, 265–268 (2018).
Cuesta, I. et al. Conformational rearrangements of SV40 large T antigen during early replication events. J. Mol. Biol. 397, 1276–1286 (2010).
Langston, L. D., Yuan, Z., Georgescu, R., Li, H. & O’Donnell, M. E. SV40 T-antigen uses a DNA shearing mechanism to initiate origin unwinding. Proc. Natl Acad. Sci. USA 119, e2216240119 (2022).
Yeeles, J. T. et al. Regulated eukaryotic DNA replication origin firing with purified proteins. Nature 519, 431–435 (2015).
Wald, J. et al. Mechanism of AAA+ ATPase-mediated RuvAB-holliday junction branch migration. Nature 609, 630–639 (2022).
Rish, A. D. et al. Molecular mechanisms of holliday junction branch migration catalyzed by an asymmetric RuvB hexamer. Nat. Commun. 14, 3549 (2023).
Yeo, H. J. et al. Phage P4 origin-binding ___domain structure reveals a mechanism for regulation of DNA-binding activity by homo- and heterodimerization of winged helix proteins. Mol. Microbiol. 43, 855–867 (2002).
Wang, H. et al. Ebola viral glycoprotein bound to its endosomal receptor Niemann-Pick C1. Cell 164, 258–268 (2016).
Peng et al. Structure of monkeypox virus DNA polymerase holoenzyme. Science 379, 100–105 (2023).
Xie, Y. et al. Structural basis of DNA replication fidelity of the Mpox virus. Proc. Natl Acad. Sci. USA 122, e2411686122 (2025).
De Silva, F. S., Lewis, W., Berglund, P., Koonin, E. V. & Moss, B. Poxvirus DNA primase. Proc. Natl Acad. Sci. USA 104, 18724–18729 (2007).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. Cryosparc: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Sanchez-Garcia, R. et al. DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Commun. Biol. 4, 874 (2021).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Jumper, J. et al. Highly accurate protein structure prediction with alphafold. Nature 596, 583–589 (2021).
Adams, P. D. et al. PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D. Biol. Crystallogr. 66, 213–221 (2010).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of coot. Acta Crystallogr. Sect. D., Biol. Crystallogr. 66, 486–501 (2010).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D. Biol. Crystallogr. 66, 12–21 (2010).
Meng, E. C. et al. UCSF ChimeraX: tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Acknowledgements
We express our gratitude to the entire team at the Cryo-EM Center, Shanxi Academy of Advanced Research and Innovation, especially Ms. Xiaomei Li, for their invaluable technical support during the cryo-EM data collection. Additionally, we extend our thanks to Lei Song from the Institute of Microbiology, CAS, for providing technical assistance in handling radioactive products. This project is supported by the following funds: the National Key Research and Development Program of China (grant 2025YFC2311800, to H.W.), National Natural Science Foundation of China (NSFC, grant 32270157, to H.W.; grant 82241076, to Y.S.; grant 92169208, to J.Q.); The Fundamental Research Funds for the Central Universities, Peking University (grant 7100604396, to H.W.).
Author information
Authors and Affiliations
Contributions
H.W. and G.F.G. designed the experiments. Y.C., P.H., Q.P., R.L., H.L., B.Y., Y.Z. and L.K. performed the investigations and assays. H.W., Y.S., K.M., and J.Q. analyzed the data. H.W., G.F.G., Y.C., Q.P. and P.H. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cheng, Y., Han, P., Peng, Q. et al. Assembly and breakage of head-to-head double hexamer reveals mpox virus E5-catalyzed DNA unwinding initiation. Nat Commun 16, 5176 (2025). https://doi.org/10.1038/s41467-025-60539-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60539-1
