
Abstract
Macrocyclic peptides represent an attractive drug modality due to their favourable properties and amenability to in vitro evolution techniques such as phage or mRNA display. Although very powerful, these technologies are not without limitations. In this work, we address some of their drawbacks by developing a yeast display-based strategy to generate, screen and characterise structurally diverse disulfide-cyclised peptides. The use of quantitative flow cytometry enables real-time monitoring of the screening of millions of individual macrocyclic peptides, leading to the identification of ligands with good binding properties to five different protein targets. X-ray analysis of a selected ligand in complex with its target reveals optimal shape complementarity and extensive surface interaction, explaining its exquisite affinity and selectivity. The yeast display-based approach described here offers a facile, quantitative and cost-effective alternative to rapidly and efficiently discover and characterise genetically encoded macrocyclic peptide ligands with sufficiently good binding properties against therapeutically relevant targets.
Similar content being viewed by others

Introduction
Macrocyclic peptides are increasingly proving to be a valuable molecular format for drug development1. At present, there are around eighty peptide therapeutics on the global market, of which more than 40 are macrocyclic peptides, with a growing number of drugs approved per year2,3. Macrocyclic peptides have several favourable properties that make them an attractive modality for the development of therapeutic agents4. They can bind to macromolecular targets with high affinity and selectivity. Moreover, they often have good proteolytic stability, and in some cases even display membrane permeability. Furthermore, they usually show a low inherent toxicity or antigenicity. Additionally, macrocyclic peptides can be efficiently produced by chemical synthesis and are easily modified. Their modular structure and the facile access to hundreds of different amino acid building blocks simplify the development of macrocyclic peptide variants with tailored properties. All these qualities position macrocyclic peptides to bridge the gap between small-molecule drugs and larger biologics such as antibodies4.
While numerous macrocyclic peptides continue to originate from the investigation and exploitation of naturally occurring peptides, recent technological advances and major breakthroughs in molecular biology have paved the way for the development of de novo generated macrocyclic peptide ligands with desired qualities5. Indeed, the advent of powerful combinatorial technologies such as phage display6,7, mRNA display8,9, bacteria display10,11, and the split-intein based approach SICLOPPS12 has exponentially accelerated the generation of macrocyclic peptide ligands to diverse protein targets for which no natural peptide ligands have been discovered. All these in vitro directed evolution approaches rely on a physical linkage between the ‘phenotype’ (expressed macrocyclic peptide) and ‘genotype’ (encoding DNA or RNA sequence). By applying such technologies, macrocyclic peptide ligands with desired properties are usually evolved following a similar scheme, comprising the generation of large combinatorial libraries of random genetically encoded macrocyclic peptides, multiple iterative cycles of selection, amplification and diversification. In addition to traditional disulfide-tethered peptides, the recent development of innovative strategies for post-translational chemical and enzymatic modification has greatly expanded the number of macrocyclic peptide formats that can be screened13,14,15,16. These strategies, coupled with the latest technological advances in screening procedures and automatisation, new reagents and tools, and next-generation sequencing (NGS) techniques, have enabled the construction of larger libraries and the isolation of macrocyclic peptide ligands with exquisite binding properties towards increasingly challenging targets17,18,19.
Although these technologies have proven capable of generating and screening large and structurally diverse macrocyclic peptide libraries, thus enabling the rapid identification of ligands against virtually any target, they still rely on procedures difficult to control, which do not allow for the monitoring of the performance of individually selected clones or populations during the high-throughput screening. The success of a selection campaign is typically only seen after several weeks of work, once the isolated macrocyclic peptide molecules are identified. Moreover, the biophysical characterisation of selected ligands often requires chemical synthesis or sub-cloning and recombinant production, followed by purification, all steps which contribute to additional delays and costs15,20,21. Finally, the number of selected macrocyclic peptide ligands that can be synthesised, purified and characterised is typically limited to 10–100 molecules.
Herein, we demonstrate how the use of yeast display technology helps to address the abovementioned concerns. Since its invention, yeast display has proven to be a transformative tool for the directed evolution of multiple immunoglobulin- and non-immunoglobulin-based proteins22,23. This technique was initially validated to enhance the binding affinity of an existing antibody fragment, but later proved highly effective also for isolating de novo proteins with fine-tuned binding affinities and specificities toward a wide range of targets from naïve combinatorial libraries24,25. An advantage of yeast display technology coupled to fluorescence-activated cell sorting (FACS) is that it offers real-time monitoring of the iterative screening of large combinatorial libraries of diverse macrocyclic peptide ligands. By applying flow cytometry, information on the affinity, specificity, stability, and enrichment of individual yeast clones encoding diverse macrocyclic peptide ligands could be monitored in a continuous and quantitative manner through successive rounds of sorting22,26. Moreover, by applying different selection approaches, including equilibrium, kinetic and competition binding, yeast display combined with flow cytometry could allow for the accurate adjustment of selection stringency, enable rapid and fine epitope mapping, and favour quantitative discrimination between single macrocyclic peptide variants directly as cell surface fusions without the need for chemical synthesis or recombinant expression and purification24,25,27.
Yeast display was previously applied for the screening of peptide libraries, but mostly for the engineering of naturally occurring linear and cystine knot peptides28 or for the affinity maturation or binding characterisation of existing molecules29,30. Recently, studies have reported the exploitation of yeast display for the de novo generation of cyclic peptide ligands, such as binders to lysozyme and human interleukin-17 with dissociation constant (KD) values ranging from 300 nM to 3 μM31. Similarly, screening libraries of post-translationally enzymatically modified octapeptides yielded cyclic peptide ligands capable of binding two distinct domains of the yes-associated protein (YAP) with KD values ranging from 700 nM to 1.5 μM32. Although the binding affinities obtained were rather weak, the size of these libraries was small (< 108) and the structural diversity limited to monocyclic peptides of only 7 or 8 amino acids, these studies are the first to demonstrate the ability of yeast display technology to isolate cyclic peptide ligands from naïve combinatorial libraries of linear peptides that have been post-translationally modified using chemical or enzymatic strategies.
In this work, we aim to thoroughly explore the full potential of yeast display for the isolation and characterisation of macrocyclic peptide ligands with fine binding properties. Toward this goal, we generated large and structurally diverse naïve combinatorial libraries encoding macrocyclic peptide ligands with different topologies. To provide a more robust procedure, we turned to disulfide-cyclised peptide ligands because the spontaneous intramolecular oxidation of cysteine residues is substantially easier and more reliable than post-translational cyclisation. In this regard, we foresaw that the eukaryotic expression machinery of yeast cells could support precise disulfide isomerization of cysteine-rich sequences, ultimately enabling the generation of unique disulfide-tethered patterns that would be refractory to other systems22,26,28. A key step in our work is the use of a different yeast surface protein than the commonly applied α-agglutinin Aga1 and Aga2 proteins, where the ligand of interest is usually expressed as a fusion to Aga2, which is itself covalently linked to the membrane-anchored Aga1 via two intermolecular disulfide bonds. In our work, we turned to a cysteine-free glycosylphosphatidylinositol (GPI) cell-surface anchor protein and expressed the disulfide-linked macrocyclic peptide ligands on the surface of yeast cells as amino-terminal fusions with the GPI anchor protein33. We show that by applying quantitative flow cytometry-based selections, yeast-displayed macrocyclic peptides with high structural diversity and fine binding properties against a panel of distinct protein targets can be rapidly identified. The technology appears capable of effectively picking and enriching rare macrocyclic peptides with different motifs and topologies for each target tested, even though the library sizes are comparable or even smaller than those of other in vitro evolution tools. Furthermore, X-ray analysis of a selected ligand in complex with its target reveals optimal shape complementarity, a large interaction surface, constrained peptide backbones and multiple inter- and intra-molecular interactions, explaining its high affinity and exquisite selectivity.
Results
Generation of yeast-encoded macrocyclic peptide libraries with large backbone diversity
We generated large and structurally diverse macrocyclic peptide libraries displayed on the surface of Saccharomyces cerevisiae cells (Fig. 1a). Yeast-displayed macrocyclic peptides were obtained by forming intra-molecular disulfide bridges between cysteine residues present in the genetically encoded peptide sequences. To avoid the formation of undesired inter-molecular disulfide bonds, we appended the cysteine-rich peptide sequences at the N-terminus of a cysteine-free GPI cell-surface anchor that was previously developed to tether small proteins on the surface of yeast cells33.

a Schematic representation of the yeast display macrocyclic peptide (MP) system developed in this work. The cysteine-rich peptide sequence of interest (blue) is expressed as N-terminal fusion of a cysteine-free glycosylphosphatidylinositol cell-surface anchor protein (black line). A linker is placed between the peptide and the hemagglutinin (HA) tag (red). Bridging of one pair of cysteines yields MPs with ‘one ring’ and a unique topology, bridging two pairs of cysteines yields MPs with ‘two rings’ (three different topologies). b Five naïve libraries were generated to include either two or three cysteines (C, blue) at fixed positions and between 7 and 12 random amino acids (X). Naïve libraries were numbered as follows: 1 (CX7C), 2 (CX9C), 3 (CX3CX9C), 4 (CX6CX6C), and 5 (CX9CX3C). The size of each library (unique peptide sequences) is reported and was determined as described in other works22. c Yeast-displayed MP naïve libraries were constructed using degenerated primers that allow all 20 amino acids in the randomised positions (X = ‘NNK’) and homologous recombination-based methods22. Heat map indicating the frequency of cysteines (d) and the ‘TAG’ amber stop codon (‘Z’) (e) in each library. Frequencies were determined by NGS analysis. The intensity of the colour correlates with the frequency of a given cysteine or ‘TAG’ stop codon in the peptide sequence. High and low frequency values are shown in dark and light colours, respectively. f Heat map of experimental frequency of each individual amino acid determined by NGS compared to the theoretical probability expected values for ‘NNK’ codon. Individual amino acids are indicated by a one-letter code. The intensity of the colour correlates with the frequency ratio. Enriched amino acids in naïve libraries are shown in dark blue, whereas depleted amino acids are shown in dark red. Amino acids with an experimental frequency/theoretical probability ratio of one are shown in white. g Frequency of the occurrence and distribution of additional cysteine in library 3. The three constant cysteines are shown in blue, while the fourth random is highlighted in red. Data for d–g are provided as Source data.
We postulated that the use of a cysteine-free GPI anchor protein to display disulfide-tethered macrocyclic peptide ligands on the surface of yeast cells should not only minimise the risk of undesired inter-molecular disulfide bridge formation between the peptide ligand and Aga1 and Aga2 proteins but should also facilitate the use of the system in different yeast strains, ultimately enhancing its flexibility. While the commonly applied galactose-inducible heterodimeric Aga1-Aga2 yeast display system requires the use of engineered yeast strains bearing the Aga1 gene stably integrated into the yeast chromosome, the monomeric cysteine-free GPI anchor protein can be cloned into an episomal plasmid and expressed in any yeast strain. To minimise potential steric hindrance with detection reagents, we placed a flexible glycine-serine (G4S)3 linker between the peptide sequence and the immunofluorescence hemagglutinin (HA) tag (Supplementary Fig. 1). Detection of the HA tag using a fluorescently labelled antibody combined with flow cytometry allowed not only for the rapid quantification of the macrocyclic peptide ligands’ expression level, but also for the normalisation of binding signal to that of the expression level.
We generated peptide libraries preferentially encoding either ‘one ring’ or ‘two rings’ macrocycle topologies with sequence diversity comprised between 3 × 108 and 2 × 109 (Fig. 1b and Supplementary Table 1). We designed yeast-encoded ‘one ring’ macrocyclic peptide libraries of the format CXmC (X = any amino acid, m = 7 or 9), in which all sequences contain predominantly two fixed cysteines (C). Similarly, we created yeast-encoded ‘two rings’ macrocyclic peptide libraries of the format CXmCXnC in which all peptides contain predominantly three constant cysteines spaced by different numbers of random amino acids (X) wherein m = 3, 6, or 9 and n = 9, 6 or 3 (Fig. 1b). The random amino acid positions in all libraries were encoded by ‘NNK’ codons resulting in an ~3% probability of finding a cysteine residue in each of the randomised positions. The theoretical probability of encountering an additional cysteine residue in the ‘two rings’ CXmCXnC macrocyclic peptide libraries was estimated to be ~26% (Supplementary Tables 2–3). We reasoned that a first disulfide bond can be formed between any of the three fixed cysteines, and a second disulfide bridge could be formed between the remaining and the additional cysteine appearing in any of the randomised positions. In total, up to (m + n) × 3 different macrocyclic peptide topologies could be formed, where m = 3, 6 or 9 and n = 9, 6 or 3 (Supplementary Figs. 2–3). Such a large structural diversity should increase the chance of finding ‘two rings’ macrocyclic peptides that have a shape complementary to the surface of protein targets. NGS analysis of the naïve libraries confirmed that the macrocyclic peptides present the expected number of cysteines, amino acid frequency and topological diversity (Fig. 1c–g, Supplementary Figs. 3–7, Supplementary Tables 4–7, and Supplementary data file 1–6).
Screening of yeast-encoded macrocyclic peptides toward a wide range of protein targets
To validate our technology, we screened yeast-encoded macrocyclic peptide libraries towards five highly diverse protein targets (PT), namely aldolase (PT1), streptavidin (PT2), carbapenemase GES-5 (PT3), carbonic anhydrase (PT4) and α-chymotrypsin (PT5; Fig. 2a, b and Supplementary Table 8). These proteins were chosen based on their completely unrelated sequences and structures, and different biochemical properties (Fig. 2c, d and Supplementary Table 9). The use of such distinct proteins was key because it allowed us to evaluate whether the technology would be widely applicable or if it would be instead biased for certain types of protein targets. For instance, the presence of multiple copies (104–105) of a macrocyclic peptide ligand displayed on the surface of a yeast cell could lead to undesired polyvalent interactions that might synergise to enhance the apparent binding affinity. This effect is commonly referred to as ‘avidity’ and it can occur in the presence of a multivalent soluble target that can be recognised by multiple copies of a macrocyclic peptide ligand present on the yeast surface25,27,34. To better comprehend this potential issue, we included two tetrameric proteins (PT1 and PT2) in addition to monomeric ones (PT3–PT5) in our screening campaign (Supplementary Table 9). Furthermore, to enable the comparison of our technology to previous state-of-the-art techniques, we picked two protein targets (PT2 and PT5) for which macrocyclic peptide ligands have already been isolated using well-established in vitro directed evolution tools (Supplementary Fig. 8 and Supplementary discussion). Four out of the five PTs (PT1, PT3, PT4 and PT5) were chemically biotinylated, while streptavidin (PT2) was used in the form of streptavidin-coated magnetic beads (MBs) and fluorescently labelled protein. The purity, degree of monodispersivity and multimeric state of all five PTs were confirmed using size-exclusion chromatography (Supplementary Fig. 9).

a Three-dimensional structure of five different protein targets (PT) used during selection screening: aldolase (PT1, PDB identification code 1QO5), streptavidin (PT2, PDB identification code 7EK8), carbapenemase GES-5 (PT3, PDB identification code 6TS9), carbonic anhydrase (PT4, PDB identification code 1VE9), and α-chymotrypsin (PT5, PDB identification code 6DI9). The overall target secondary structure is shown as cartoon and coloured by secondary structure (α-helices = firebrick, β-sheet = sky-blue, random coil = white). Proteins are ordered according to their molecular weight (MW), from largest (left) to smallest (right). b Heat map showing the amino acid identity among different PTs. The intensity of the colour correlates with the identity percentage. High and low identities are shown in dark blue and white, respectively. c From left to right, bar chart showing the distribution of the MW, solvent accessible surface area (ASA), number of pockets74, and surface/volume ratio of each PT. d Columns graph reporting the percentage of secondary structure content (α-helices = firebrick, β-sheet = sky blue, random coil = light grey) for each PT tested. Data for b–d are provided as Source data.
To increase the likelihood of isolating macrocyclic peptide ligands with desired binding properties against each PT, we applied highly avid MB separations followed by multiple rounds of FACS (Fig. 3a, b). The initial use of MB-based screening allowed the isolation of macrocyclic peptides within a relatively short period of time and in a high-throughput combinatorial manner22. Moreover, we assumed that the multivalency of the yeast display system, combined with the multiple copies of biotinylated PT immobilised on each streptavidin-coated MBs, would facilitate highly avid interactions, thus promoting the isolation of weak macrocyclic peptide ligands otherwise difficult to identify by flow cytometry26,34. In this study, we applied two iterative cycles of MB-based selections for each PT (Fig. 3a, b). Each MB-based selection comprised the growth of yeast cells, expression of the macrocyclic peptide on the yeast surface, binding to the immobilised biotinylated PT, washing, and expansion of the isolated bound yeast cells. In the case of protein targets PT1, PT3, PT4 and PT5, prior to ‘positive selection’, we also performed a ‘negative selection’ to deplete macrocyclic peptides that bound the streptavidin of the streptavidin-coated MBs. In the case of streptavidin (PT2), only the ‘positive selection’ step was performed, in which naïve libraries were directly exposed to the streptavidin-coated MBs. Notably, rather than exposing each naïve library separately to each different PT, we mixed the five libraries together and let the individual PTs pick the most suitable macrocyclic peptide topology.

a General flowchart applied to identify yeast-encoded macrocyclic peptide ligands (MPs) towards five highly diverse PTs. The five yeast-encoded MP libraries (CX7C, CX9C, CX3CX9C, CX6CX6C, and CX9CX3C) were pooled together and then incubated separately with each PT through two rounds of magnetic beads (MB) separation followed by four rounds of FACS-based selection. In the flowchart of MB-based selection, the biotinylated PT immobilised on MB (PT-MB) is represented as a grey circle (PT) surrounded by a dotted ring (MB). In FACS-based selection, the soluble biotinylated PT is represented as a grey circle (PT) and the fluorescently labelled anti-HA antibody (IF) as an orange star. b Schematic representation of the iterative selection pathways applied to isolate yeast-encoded MPs against five different PTs. Two cycles of MB-based screening followed by four cycles of FACS sorts were applied. c Density plots of a representative polyclonal population of yeast cells encoding different MPs against PT5 that has been enriched from 11% to 82% through four FACS sorts (I, II, III and IV). Each dot represents two fluorescent signals of a single yeast cell. The fluorescence intensity on the y-axis is a measure of the amount of PT bound to the surface of a yeast cell (DyLight 650; ‘target binding’) whereas the fluorescence intensity on the x-axis is a measure of the number of MPs expressed on the yeast cell surface (DyLight 488; ‘macrocyclic peptide display’). d Columns graph reporting the geometric mean fluorescence (MFI) measured for the polyclonal population of yeast cells encoding different MPs against PT2 through four cycles of FACS. e Density plots of the enriched polyclonal population of yeast cells encoding different MPs against the respective PT after the fourth and last FACS cycle. f Heat map displaying the ratio between the binding and display MFI of the polyclonal population after each round of FACS. The intensity of the colour correlates with MFI value. High and low intensities are shown in dark blue and white, respectively. Data for c–f are provided as Source data.
We then promoted the enrichment of yeast cells isolated after the MB separations by allowing them to evolve through sequential cycles of equilibrium-based FACS selections (Fig. 3a, b)22. By applying a two-colour labelling scheme, with one fluorescent probe to monitor the expression of macrocyclic peptides and another to monitor the binding of macrocyclic peptides to biotinylated PTs, we could foster the selection and enrichment of yeast-encoded macrocyclic peptides with the desired affinities, specificities, and stabilities (Fig. 3c, d). FACS-based screening of yeast-displayed macrocyclic peptide ligands against protein targets PT1, PT3, PT4, and PT5 involved the labelling of yeast cells with the corresponding biotinylated PT followed by staining with fluorescently labelled streptavidin. In the case of streptavidin (PT2), binding was instead monitored by direct staining with fluorescently labelled streptavidin. We decided to maintain a constant and relatively high concentration of each PT (1 μM) during all rounds of FACS-based selection (Supplementary Fig. 10) in order to identify ligands with a wide range of binding affinities and to compare the structural diversity of ligands obtained against each PT. In the case of protein targets PT1, PT3, PT4, and PT5, we alternated the use of fluorescently labelled streptavidin and neutravidin during FACS to avoid the enrichment of macrocyclic peptide ligands against the detection reagents.
Overall, such evolutionary strategy enabled the isolation and enrichment of yeast-encoded macrocyclic peptide ligands against all five distinct PTs (Fig. 3e, f and Supplementary Fig. 10). Though PTs containing large binding pockets, such as proteases, are apparently easier to target than proteins with featureless surfaces, here we have shown that the chances of identifying macrocyclic peptides against varying surface structures increases if screening is performed in an unbiased manner using large and structurally diverse combinatorial libraries.
Selected yeast-encoded macrocyclic peptides revealed differences in amino-acid sequences and topologies
To uncover the identity of the selected macrocyclic peptide ligands, we applied both Sanger and NGS methodologies. We started with Sanger sequencing because it allowed us to prepare DNA samples from the same yeast cells that were used to characterise the macrocyclic peptide variant as a cell surface fusion, enabling the simultaneous analysis of amino acid composition and binding properties in just a few days. To prepare DNA samples for Sanger sequencing, we plated a fraction of each flow cytometry-sorted population on selective solid media, picked individual colonies, extracted and purified the DNA plasmids present within the yeast cells. Sequence analysis of approximately fifteen unique clones derived from each collected cell population revealed the presence of thirteen unique macrocyclic peptide ligands (MP) with different topologies and varying amino-acid compositions and ring sizes (Fig. 4). Notably, we did not detect cross-contamination among sequences derived from different populations, further proving the technology’s ability to effectively separate and enrich unique binders against different protein targets in parallel. Though rapid and cost-effective, Sanger sequencing allowed for the analysis of a very limited portion of each flow cytometry-sorted population (< 0.0001% of the total collected cells).

a DNA extracted from FACS-enriched yeast cell populations was sequenced using both Sanger and next-generation sequencing (NGS) methodologies. DNA samples for high-throughput sequencing analysis were amplified by PCR, indexed using target-specific barcodes, processed using NovaSeq Illumina NGS technology and the obtained FASTQ files analysed using MATLAB scripts. The amino acid sequences are arranged in groups according to sequence similarities. The amino acids are indicated by a one-letter code. Identical or similar amino acids between different peptide sequences are highlighted in colours (C: grey; E and D: red; G: light green; V and A: intense light green; I and L: dark green; S and T: light orange; Y and F: purple; W: violet; H: indigo; P: light brown; Q and N: light blue; R and K: dark blue; M: yellow). Within a single macrocyclic peptide family, the amino acid sequences were listed starting from the clone with the highest abundance (top) to the one with the lowest (bottom). Only sequences with a percentage of abundance >0.1% are reported. b Heat map visualisation of the type of amino acid residues that were enriched or depleted during selection when compared to naïve libraries. The amino acids are indicated by a one-letter code. The colour intensity correlates with the occurrence of each amino acid, with enriched or depleted residues shown in dark blue and dark red, respectively. c Pie chart visualisation of the different macrocyclic peptide topologies enriched during selection against the five different PTs. Macrocyclic peptides with ‘one ring’ and ‘two rings’ are shown in red and blue, respectively. d Pie chart visualisation of the relative abundance of the most frequently selected macrocyclic peptide topologies across different PTs: CX7C (dark red), CX8C (dark salmon), CX9C (light salmon), CX3CX5CX3C (dark blue), CX2CX6CX3C (blue-grey), CCX2CX5C (light blue), CX3CX4CX3C (pink), CCX4CXC (dark grey), CX6CX5CC (dark green), CX2CX5CC (light grey), CX3CX6CX2C (light green), and others (white). Data for b–d are provided as Source data.
To gain a better insight into the genetic diversity and abundance of target-specific macrocyclic peptide ligands, we applied NGS analysis on the pooled DNA plasmids extracted from flow cytometry-sorted polyclonal yeast cells (Fig. 4a). We implemented sequence data filters to reduce bias originating from the sequencing method and applied sequence correction algorithms to prevent identification of false consensus motifs. Next, we compared the obtained peptide sequence datasets to each other in order to i) identify target-specific peptide binding motifs, ii) uncover the topology and ring size distribution, iii) determine the frequency of each amino acid within each peptide loop, and iv) cluster the most abundant peptides in consensus families (Supplementary Fig. 4, Supplementary Tables 10, 11, and Supplementary data file 7–9). This analysis i) confirmed the presence of all thirteen MPs previously identified by Sanger sequencing, ii) revealed the existence of at least three unique peptide ligand families against four of the five tested PTs, iii) uncovered six new macrocyclic peptide families (MP2.3, MP3.3, MP4.2, MP4.3, MP5.3 and MP5.5), and iv) provided a better view of the sequence diversity and abundance distribution among peptide sequences present within each macrocyclic peptide family (Fig. 4a and Supplementary data file 10). Each family contained several sequences that differed in one or more amino acids. The abundance of single macrocyclic peptide sequences present within each family varied. We cannot exclude the possibility that some low-abundance sequences might be the result of artefacts occurring during NGS preparation and processing rather than peptide ligands encoded by the yeast. However, a dominant family with relative abundance greater than 25% was identified for each PT. Moreover, the amino acid sequences of the isolated macrocyclic peptides differed from target to target (Fig. 4a, b). These differences appeared to persist even at the level of macrocyclic peptides that bound the same PT, although some shared consensus motifs were identified for two targets (Fig. 4a). For example, three of the four macrocyclic peptide families isolated against PT2 contain the ‘HPQ’ motif, a well-known peptide binder of streptavidin (Supplementary Fig. 8, Supplementary discussion, and Supplementary data file 11), though localised within diverse ring sizes and topologies (‘one ring’ and ‘two rings’ macrocyclic peptides; Supplementary Fig. 11). Similarly, three out of the four peptide families selected against PT4 present a conserved ‘GPYY’ motif. Interestingly, this sequence motif has been found within ‘one ring’ macrocyclic peptides with different sizes, including an 8-amino acid loop that was not included in the initial design. Furthermore, sequence analysis allowed us to better appreciate what kind of topologies suited each PT best. While protein targets PT1 and PT4 appear to be favourably recognised by ‘one ring’ macrocyclic structures, ‘two rings’ topologies were preferentially enriched during selections against PT2, PT3 and PT5, although with a different relative abundance distribution (Fig. 4c). In general, ‘one ring’ macrocyclic peptides with a larger ring (9-amino acid ring: CX9C) were more frequent than those with a smaller ring (7-amino acid ring: CX7C). However, a family of ‘one ring’ macrocyclic peptides with a 7-amino acid ring that has a 5-fold higher abundance than those with 9-amino acids was identified solely for PT5 (Fig. 4a, c, d). Interestingly, the sequences of MP4.2, MP4.4, MP5.3, and MP5.4 peptide families were unexpected and are probably the result of either impurities in oligonucleotide synthesis or the introduction of two additional cysteines into the monocyclic peptides. Notably, among the seventy different ‘two rings’ macrocyclic peptide topologies available in the naive libraries (Supplementary data file 1–4, and Supplementary Figs. 6–7), only eight (CX3CX5CX3C, CX2CX6CX3C, CCX2CX5C, CCX4CXC, CX2CX5CC, CX6CX5CC, CX3CX6CX2C and CX3CX4CX3C) were effectively enriched during selection (Fig. 4d). Except for the CX3CX5CX3C topology, which is present in a family of macrocyclic peptides selected against both PT3 and PT5, the other six ‘two rings’ topologies are exclusive to individual protein targets, with CX2CX6CX3C being identified only in a peptide family targeting PT2, CCX2CX5C in a peptide family binding PT4, while CCX4CXC, CX2CX5CC, CX6CX5CC and CX3CX6CX2C topologies were detected only in peptide families enriched against PT5 (Fig. 4d). Notably, no peptide sequences containing three cysteines were isolated even though they were highly represented in the naïve libraries (from 45% to 60% of total clones). This further demonstrates the capability of our tool to generate and enrich exclusively ‘one ring’ and ‘two rings’ macrocyclic peptides.
In summary, sequencing analysis proved the ability of our technology to effectively isolate, finely discriminate and amplify very rare yeast-encoded disulfide-cyclised peptides with different motifs and topologies for each target tested, even starting from combinatorial libraries with comparable or smaller size than those of other in vitro evolution tools.
The majority of selected yeast-encoded macrocyclic peptides exhibit binding affinities in the nanomolar range
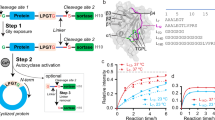
To determine the binding affinities of the isolated macrocyclic peptides, we titrated yeast-displayed macrocyclic peptides into solutions with varying concentrations of the corresponding biotinylated PT (Fig. 5a). This technique allowed for selected macrocyclic peptide ligands to be rapidly characterised directly on the yeast cell surface, eliminating the need for chemical synthesis and purification. The mean fluorescence intensity from binding was normalised to the mean fluorescence intensity from expression and then plotted as a function of PT concentration to obtain an apparent equilibrium dissociation constant (KDapp) of each individually selected clone. We characterised the most abundant macrocyclic peptide of each consensus family (Fig. 5b–f and Supplementary Table 12). The determined KDapp values span 10,000-fold, ranging from 0.2 nM to 1 µM. Three out of the five PTs tested (PT1, PT2 and PT5) yielded macrocyclic peptide ligands with KDapp values below 10 nM (Fig. 5b, c, f, and Supplementary Table 12). For the other two targets examined (PT3 and PT4), the KDapp values were on average higher (0.04 to >1 µM) with the exception being PT3, for which a macrocyclic peptide (MP3.2) with KDapp value of 2.2 nM was also isolated (Fig. 5d, e, and Supplementary Table 12). Notably, the KDapp values obtained for a specific PT were often very close to each other (Fig. 5g). We assume that, rather than the topology of the macrocyclic peptide, it is the propensity of the PT itself that determines the attainable binding affinity.

a Schematic representation of the apparent binding affinity determination using yeast surface titrations. Yeast cells expressing the desired macrocyclic peptide (MP) on the cell surface are incubated with varying concentrations of soluble biotinylated PT. The binding is reported as mean fluorescence intensity, and it is proportional to the amount of PT bound to the macrocyclic peptides expressed on the surface of the yeast cell. Binding isotherms of the most abundant yeast-displayed macrocyclic peptide clones to soluble biotinylated PT1 (b), PT2 (c), PT3 (d), PT4 (e), and PT5 (f) are shown. The apparent equilibrium dissociation constant (KDapp) of each individually selected clone was determined by normalising the mean fluorescence intensity from the binding signal to the mean fluorescence intensity from the display signal (y-axis), as a function of PT concentration (x-axis). The indicated KDapp values are the results of three independent experiments and are presented as mean (dots) ± s.d., standard deviation (bars). g Plot reporting apparent binding affinity values of the twenty characterised yeast-encoded ‘one ring’ (red dots) and ‘two rings’ (blue rhombuses) macrocyclic peptides to soluble biotinylated PT. The indicated values represent the means of three independent experiments. Data for b–g are provided as Source data.
Next, we compared the different macrocyclic peptide sequences selected for each PT and checked whether there was a correlation between the determined binding affinities and the topology or length of the isolated ligands (Fig. 5g and Supplementary Fig. 12). Contrary to our expectation, we often found that macrocyclic peptides with ‘one ring’ topology bound protein targets tighter than those with ‘two rings’. For example, the 9-amino acid ‘one ring’ macrocyclic peptides MP2.2, MP2.3 and MP2.6 appear to bind PT2 with KDapp values 10-fold lower (KDapp = 200–300 pM) or comparable (KDapp = 1.9 nM) to that of the longer 13-amino acid ‘two rings’ macrocyclic peptide MP2.1 (KD = 2.1 nM) (Fig. 5c, g, and Supplementary Fig. 12). Furthermore, the 9-amino acid ‘one ring’ macrocyclic peptide MP4.1 (KDapp = 41 nM) shows 16-fold higher binding affinity for PT4 than the similar length ‘two rings’ macrocyclic peptide MP4.4 (KDapp = 680 nM; Fig. 5e, g, and Supplementary Fig. 12). Likewise, the 7-amino acid ‘one ring’ macrocyclic peptide MP5.1 (KDapp = 1.9 nM) bound 5-fold tighter PT5 than the 13-amino acid ‘two rings’ macrocyclic peptide MP5.5 (KDapp = 9.2 nM; Fig. 5f, g, and Supplementary Fig. 12). Further analyses revealed that macrocyclic peptides with a longer loop often bound more tightly than those with a shorter loop. For example, the 9-amino acid ‘one ring’ macrocyclic peptide MP2.2, which includes the ‘HPQFYVI’ motif, also present in the 7-amino acid ‘one ring’ macrocyclic peptide MP2.5, bound PT2 more tightly (3-fold). Similarly, the 9-amino acid ‘one ring’ peptide MP2.6, which shares the same ‘HPQFYMR’ as the 7-amino acid ‘one ring’ macrocyclic peptide MP2.4, bound PT2 more tightly (6-fold) (Fig. 5c, g, and Supplementary Figs. 12 and 13). A similar correlation was also observed for the ‘one ring’ macrocyclic peptides that bound PT4 and include the ‘GPYY’ motif. The 9-amino acid ‘one ring’ macrocyclic peptide MP4.1 bound PT4 more tightly than MP4.2 (8-amino acid loop) and MP4.3 (7-amino acid loop), by 4- and 9-fold respectively (Fig. 5e, g, and Supplementary Fig. 12). Less often, there were cases where the loop length did not correlate to the binding affinity. For example, the 7-amino acid ‘one ring’ macrocyclic peptide MP5.1 had a 2-fold higher binding affinity to PT5 than the longer 9-amino acid ‘one ring’ macrocyclic peptide MP5.2 (KDapp = 3.7 nM; Fig. 5f, g, and Supplementary Fig. 12). Similarly, the short 7-amino acid MP5.3 (KDapp = 2.4 nM) and 9-amino acid MP5.4 (KDapp = 200 pM) ‘two rings’ macrocyclic peptides bound 3- and 46-fold tighter to PT5 than the longer 13-amino acid bicyclic peptide MP5.5 (KDapp = 9.2 nM), respectively (Fig. 5f, g, and Supplementary Fig. 12).
To validate the KDapp values measured using yeast surface titrations, we determined the binding affinities of some macrocyclic peptides using surface plasmon resonance (SPR). While yeast surface titrations are performed using membrane-anchored macrocyclic peptides against soluble PT, the SPR measurements are executed using soluble macrocyclic peptides against chip-immobilised PTs (Fig. 6a). This opposite orientation would allow us to rule out potential overestimation of binding affinity caused by multivalent binding phenomena which can often occur on yeast when using PTs that form multimers in solution. Toward this goal, three ‘one ring’ (MP1.1, MP2.2, and MP4.2) and two ‘two rings’ (MP3.2 and MP5.4) macrocyclic peptides with the highest binding affinities for their respective PT were produced using solid-phase peptide synthesis, cyclised, purified by reversed-phase high performance liquid chromatography, their molecular mass determined by electrospray ionisation mass spectrometry (Supplementary Figs. 14–16) and their binding kinetics toward immobilised PTs assessed using SPR (Fig. 6b–d, Supplementary Fig. 17, and Supplementary Table 13). The resulting KD values were on average comparable to those measured using yeast surface titrations (Fig. 6f). A large discrepancy (~500-fold) was observed only for the 9-amino acid ‘one ring’ macrocyclic peptide MP1.1 that appears to bind PT1 with a KD value of 600 pM and 326 nM when using yeast surface titration and SPR, respectively (Fig. 6f). Such a difference is not surprising and may reflect the known tetrameric structure of PT1 interacting with multiple yeast-displayed macrocyclic peptides, thus confusing avidity effects with higher affinity.

a Schematic representation of the binding affinity determination using surface plasmon resonance (SPR). The protein target (PT) covalently immobilised on the surface of a chip is incubated with varying concentrations of soluble chemically synthesised macrocyclic peptides (MPs). SPR sensorgram traces for the interaction of soluble macrocyclic peptides MP1.1 (‘one ring’; b), MP3.2 (‘two rings’, three isomers: MP3.2.1, MP3.2.2, and MP3.2.3; c), and MP5.4 (‘two rings’, one isomer: MP5.4.3; d) with the immobilised PT1, PT3 and PT5, respectively. Sensorgram traces are presented with injection and flow fill steps removed and were fitted with the 1:1 binding model. Kinetic constants kon (association constant), koff (dissociation constant) and KD (equilibrium binding affinity) are presented as geometric mean average values. Raw data are shown as solid lines, while fitting curves are shown as dashed lines. e Residual activities of PT5 (α-chymotrypsin) measured at different concentrations of ‘two rings’ macrocyclic peptide isomers MP5.4.1, MP5.4.2, and MP5.4.3. The inhibitory activities of all macrocycle variants towards PT5 protease (α-chymotrypsin) were determined at 25 °C and physiological pH 7.4 using the chromogenic N-Succinyl-Ala-Ala-Pro-Phe p-nitroanilide substrate at a concentration of 100 μM. The Km value of PT5 protease was determined by standard Michaelis–Menten kinetics and used in the calculation of the reported Ki values. The shown values are the means of three independent experiments. Data are presented as means (symbol) ± s.d., standard deviation (bars). f Plot reporting the KD values determined using yeast surface titration (YSD; blue-coloured filled circles) and those measured using SPR (red-coloured filled rhombus). Data for b–f are provided as Source data.
In addition, SPR allowed us to identify which of the three ‘two rings’ macrocyclic peptide isomers potentially attainable from a sequence containing four cysteines were the tightest binders to a given PT. In the case of macrocyclic peptide MP3.2, only the isomer MP3.2.3 bound PT3 with good affinity (KD = 3.2 nM) while no or very weak interactions were detected for the other two isomers MP3.2.2 and MP3.2.1 (Fig. 6c and Supplementary Table 13). Notably, the KD value determined for MP3.2.3 using SPR (KD = 3.2 nM) is similar to that measured using yeast surface titrations (KDapp = 2.2 nM). Similarly, of the three possible isomers for macrocyclic peptide MP5.4, only MP5.4.3 bound PT5 with good affinity (KD = 840 pM; Fig. 6d). To identify which MP5.4 isomer bound tighter to PT5, we employed a complementary approach and determined the inhibitory constant values (Ki) of each chemically synthesised isomer (MP5.4.1, MP5.4.2, and MP5.4.3) by measuring the residual activity of enzyme PT5 using a colorimetric substrate at physiological pH and room temperature. MP5.4.3 showed a Ki value of 360 pM, ~30- and 140-fold better than those for MP5.4.2 (Ki = 11.3 nM) and MP5.4.1 (Ki = 50.8 nM; Fig. 6e and Supplementary Table 14). The good correlation between the determined Ki and KD values further supports the effectiveness of yeast display technology for the rapid and quantitative characterisation of individual macrocyclic peptide variants as cell-surface fusions.
Overall, our data revealed that macrocyclic peptides with fine binding affinities toward a panel of distinct target proteins can be rapidly identified even from low-diversity combinatorial libraries if using quantitative flow cytometry for the continuous and precise monitoring of the directed evolution process.
Selected yeast-encoded macrocyclic peptides exhibit fine binding specificities
To assess the extent of specificity of the selected macrocyclic peptides, we measured the binding of each yeast-displayed macrocyclic peptide ligand directly as cell surface fusions using flow cytometry and a panel of diverse proteins. We initially ruled out potential non-specific polyreactivity by assessing the binding of our macrocyclic peptides toward the same highly diverse PTs used in our screening, which share < 10% sequence identity. No or very weak binding signals were detected for all macrocyclic peptides analysed, confirming their specificity toward the PT for which they were selected (Fig. 7a). Next, we assessed the binding of some selected macrocyclic peptides toward homologue proteins (PHs) that share >30% sequence identity. We initially determined binding specificities of six different yeast-displayed macrocyclic peptides that were selected against PT2 by titrating them into solutions with varying concentrations of two PHs of streptavidin, namely strep-tactin (PH2.1, 98% identity) and neutravidin (PH2.2, 32% identity). No binding was detected for all six macrocyclic peptides MP2.1, MP2.2, MP2.3, MP2.4, MP2.5 and MP2.6 toward the low homology-sharing PH2.2, while a 2– to 12-fold difference in affinity was observed for the highly similar PH2.1 (Fig. 7b, Supplementary Fig. 18, and Supplementary Table 15). Interestingly, while MP2.2, MP2.4, MP2.5 and MP2.6 retained a 4– to 12-fold higher binding affinity for PT2 than PH2.1, MP2.1 and MP2.3 bound PH2.1 ~10- and 2-fold more tightly than PT2, respectively (Fig. 7b, Supplementary Fig. 18, and Supplementary Table 15). Although not a striking difference, it is still surprising to observe that there can be an up to 12-fold variation in binding affinity between proteins that share >95% sequence identity, further highlighting the exquisite specificity of our yeast-encoded macrocyclic peptides. To additionally confirm the specificity of these interactions, we performed competition experiments using biotin, a well-characterised small molecule that binds with exceptionally high affinity (KD = 10 fM) to all three PHs (Fig. 7d and Supplementary Fig. 19a). Exposure of yeast-displayed macrocyclic peptides to solutions of PT2 pre-incubated with a molar excess of biotin resulted in a loss of binding for all six binders tested (Fig. 7e). The ability of biotin to prevent the interaction of our macrocyclic peptides with PT2 also allowed us to unveil the binding site. Similarly, great specificity was observed when we determined the binding affinity of MP3.2, a yeast-displayed ‘two rings’ macrocyclic peptide selected against PT3, a carbapenemase GES-5 from Klebsiella pneumoniae35 that was titrated into solutions with varying concentrations of two other class A β-lactamases, namely the carbapenemase KPC-236 from Klebsiella oxytoca (PH3.1) and the extended-spectrum β-lactamase CTX-M-1537 from Escherichia coli (PH3.2; Fig. 7b, Supplementary Fig. 19b, and Supplementary Table 16). Further kinetic competition studies using a molar excess of avibactam, a non-β-lactam inhibitor of carbapenemase GES-538, revealed that the ‘two rings’ macrocyclic peptide MP3.2 recognises the same catalytic pocket (Fig. 7f). To assess the specificity of MP5.4.3, a ‘two rings’ macrocyclic peptide that inhibits chymotrypsin with an IC50 value of 0.7 nM, we determined its inhibition constants against a group of structurally and functionally related serine proteases, namely human chymotrypsin (PH 5.1), human neutrophil elastase (PH 5.2), elastase from porcine pancreas (PH5.3), human cathepsin G (PH5.4), human kallikrein 3 (PH5.5) and human trypsin (PH5.6). Notably, no inhibition of either PH5.2, PH5.4, PH5.5 and PH5.6 proteases was detected even at the highest concentration tested (300 μM) (Fig. 7c and Supplementary Table 17). Inhibition was observed only for porcine pancreas elastase (PH5.3) and the human ortholog of α-chymotrypsin (PH5.1) for which IC50 values of 274 μM and 108.4 nM were measured, respectively (Fig. 7c and Supplementary Table 17). While PH5.3 is only weakly inhibited, revealing a >100,000-fold selectivity, inhibition of PH5.1 is more pronounced (~150-fold selectivity). Although not impressive, this specificity is still appreciable considering the similarity between the two orthologous enzymes and the fact that no pressure on selectivity was applied during the screening process.

a Heat map of residual binding of all 18 yeast-displayed macrocyclic peptides (MPs) against the five soluble protein targets (PTs) screened. Normalised binding/display signal intensities range from light to dark blue colours, indicating low and high titres, respectively. b Left, binding isotherms of MP2.2 towards streptavidin (PT2, blue), strep-tactin (PH2.1, red), and neutravidin (PH2.2, grey). Right, binding of MP3.2 towards carbapenemase GES-5 (PT3, blue), KPC-2 (PH3.1, red), and CTX-M-15 (PH3.2, grey). Apparent equilibrium binding affinity (KDapp) values are reported. c Residual activities of bovine chymotrypsin (PT5), human chymotrypsin (PH5.1), human neutrophil elastase (PH5.2), elastase from porcine pancreas (PH5.3), human cathepsin G (PH5.4), human kallikrein 3 (PH5.5), and human trypsin (PH5.6) incubated with the ‘two rings’ isomer MP5.4.3. The half maximum inhibitory concentration (IC50) values are reported. d Schematic representation of the competitive binding assay of yeast-encoded MPs for binding to their respective PT in the presence (+) or absence (–) of a well-known PT-binding ligand. The determined residual fluorescence levels are reported in percentage (%). When a known site-specific binding soluble ligand recognises the same PT site of MP, a decrease in fluorescence is expected (red plots; ‘competitive binding’). In the absence of a known site-specific binding soluble ligand (grey plot; ‘no ligand’) or if it binds a PT site other than the one recognised by MP (blue plot; ‘non-competitive binding’), no decrease in fluorescence should be observed. e Competitive binding assay of MP2.1, MP2.2, MP2.3, MP2.4, MP2.5, and MP2.6 for binding to PT2 in the presence or absence of biotin. f Competitive binding assay of MP3.2 for binding to PT3 in the presence or absence of avibactam (AVI). g Competitive binding assay of MP5.1, MP5.2, MP5.3, MP5.4, MP5.5, and MP5.6 for binding to PT5 in the presence or absence of PMSF. All reported values are the means of three independent experiments. Data are presented as mean (dots) ± s.d., standard deviation (bars). n.b., no binding; n.i., no inhibition. Data for a–g are provided as Source data.
To assess whether all six macrocyclic peptides (MP5.1–MP5.6) identified against PT5 bound to the same active site, we conducted binding experiments using a site-specific chemically modified PT5 variant. Exposure of yeast-displayed macrocyclic peptides to solutions of PT5 pre-incubated with phenylmethylsulfonyl fluoride (PMSF), a serine protease inhibitor known to irreversibly sulphonate the Oγ atom of the catalytic serine residue and therefore obliterate the active site of PT5, resulted in a loss of binding for five out of six macrocyclic peptides tested (Fig. 7g). Similar results were obtained when MP5.2 and MP5.4 were exposed to solutions of PT5 pre-incubated with the bulkier aprotinin, a natural cyclic peptide serin protease inhibitor. MP5.2 retained a partial binding (75%) to chymotrypsin, while MP5.4 binding was completely inhibited (Supplementary Fig. 18f and Supplementary Fig. 19c, d).
Taken together, these data indicate that yeast-encoded macrocyclic peptides are highly specific for the protein target they have been selected for. If present, cross-reactivity is often limited to highly similar proteins, while no binding occurs with low identity or unrelated ones. Furthermore, competition experiments not only confirmed the ability of our macrocyclic peptides to bind PTs with good specificity but also highlighted the possibility of using the technology to rapidly ensure recognition of properly folded PTs, as well as unveil the binding site without the need for chemical synthesis and purification.
Yeast-encoded macrocyclic peptide reveals a large contact interface with a protein target
In order to investigate the binding mode of one yeast-encoded macrocyclic peptide to its target, we applied X-ray crystallography and determined the structure of PT5 (bovine α-chymotrypsin) in complex with the ‘two rings’ macrocyclic peptide MP5.4.3 at 2.42 Å maximum resolution (PDB identification code: 9F6H; Fig. 8a and Supplementary Tables 18–19). A single molecule of PT5 in complex with MP5.4.3 is present in the asymmetric unit. The overall structure of PT5 does not show any striking rearrangements of the main backbone (root mean square deviations of the Cα-atoms never exceed 0.5 Å) when compared to its apo form (PDB identification code: 4CHA).

a Molecular surface representation of bovine α-chymotrypsin (PT5; grey) in complex with the ‘two rings’ macrocyclic peptide MP5.4.3 (light blue). A large surface (784 Å2) of PT5 is covered by MP5.4.3. Detailed view of the first (b, top) and second (c, bottom) ring of MP5.4.3 (light blue) bound to PT5 (white) shown in two orientations (180° rotation). The side-chains of the residues are shown as sticks. Only the residues involved in key inter-molecular interactions (black dashed lines) are shown. Amino acid side-chains are shown as sticks and coloured by atom type (carbon: light blue, oxygen: firebrick, nitrogen: deep blue, sulfur: yellow-orange). d Zoomed-in view of the protein complex structure showing the residues of PT5 (white), known to define the primary substrate-binding pocket called S1 binding pocket (S189–S195, S214–C220 and P225–Y228)60,95, surrounding and forming multiple interactions with the side-chain of Trp4 (W4) of MP5.4.3. e Left, columns graph (grey) reporting the total number of polar (both direct and H2O mediated) and non-polar inter-molecular interactions established by PT5 with the ‘two rings’ macrocyclic peptide MP5.4.3. Right, columns graph (grey) reporting the total number of inter-molecular interactions mediated by either side-chain atoms (s) and/or main-chain atoms (m) of MP5.4.3 (top) with either side-chain atoms (s) and/or main-chain atoms (m) of PT5 (bottom). Data for e are provided as Source data.
The electron density of MP5.4.3 is well defined, allowing an unambiguous assignment of side-chain orientations (Supplementary Fig. 20 and Supplementary data file 18). No classical secondary structure elements are found in the macrocyclic peptide. The 63 non-covalent intra-molecular interactions present are mainly mediated by main-chain to main-chain contacts and appear to confer structural rigidity to the macrocyclic peptide (Fig. 8a–c, Supplementary Fig. 20, Supplementary Tables 20–21, and Supplementary data file 17, 18). The ‘two rings’ macrocyclic peptide MP5.4.3 forms an extended structure that fits well into the cleft formed by the active site and the surrounding substrate pockets, covering a total surface of 784 Å2. The high affinity and specificity of the ‘two rings’ peptide can be explained by the large number of polar and non-polar inter-molecular contacts mediated by both catalytic (His57, Asp102, Ser195) and surrounding residues of PT5 (Fig. 8, Supplementary Fig. 20, Supplementary Tables 20–21, and Supplementary data file 17). Both peptide rings of MP5.4.3 interact directly with PT5. The first ring (residues from Cys2 to Cys12) is more rigid and forms a greater number of interactions than the second one (residues from Cys5 to Cys11) and hence contributes more to the overall binding (Supplementary Fig. 20). The residues of MP5.4.3 contacting PT5 through both main- and side-chain hydrogen bond interactions are eight (Ala1, Cys2, Trp4, Cys5, Leu6, Arg7 and Gly14). Most of these interactions are mediated by the main-chain oxygen of Trp4 that establishes three hydrogen bonds with the main-chain nitrogen of Gly193, Asp194 and Ser195 of PT5, with the first and last residue being key in the formation of the oxyanion hole (Fig. 8, Supplementary Fig. 20, Supplementary Table 21, and Supplementary data file 17). Additional important polar interactions are established by the side-chain of Arg7 that forms a hydrogen bond with the main-chain oxygen of Cys58, a salt bridge with the side-chain of Asp64 and a cation-π interaction with the side-chain of Phe41 (Fig. 8, Supplementary Fig. 20, Supplementary Table 21, and Supplementary data file 17). The side-chain of Trp4 occupies the primary specificity S1 hydrophobic pocket of chymotrypsin, as expected for natural substrates of this protease (Fig. 8d, Supplementary Fig. 21, Supplementary Table 21, and Supplementary data file 17). The scissile peptide bond between Trp4 and Cys5 of MP5.4.3 appears to retain a planar and undistorted conformation reminiscent of the natural serine protease inhibitor turkey ovomucoid third ___domain inhibitor (OMTKY3; Supplementary Fig. 22a)39. The carbonyl atom of the peptide bond seems to maintain a favourable geometry and distance (2.8 Å) from the side-chain oxygen (Oγ) of the catalytic Ser195, thus favouring the establishment of a non-covalent contact. Unlike other chymotrypsin-inhibitor complexes, no tetrahedral intermediate adducts are observed (Supplementary Fig. 22b, c)40,41. Interestingly, 37 out of 63 inter-molecular interactions are mediated by peptide main-chain atoms, supporting the key role of the backbone conformation and further explaining the weaker binding of the MP5.4.1 and MP5.4.2 isomers (Fig. 8e, Supplementary Fig. 20, Supplementary Table 21, and Supplementary data file 17). To assess whether our macrocyclic peptide binds chymotrypsin in a substrate-like manner, we performed proteolysis experiments using different enzyme concentrations and incubation times. Chromatographic and mass spectrometric analysis of MP5.4.3 degradation products revealed the formation of a nicked form, as shown by a mass increase of 18 Da corresponding to the hydrolysis of a peptide bond, which occurred primarily at high chymotrypsin concentrations and prolonged incubation times (Supplementary Fig. 23).
Overall, the crystal structure of the macrocyclic peptide MP5.4.3 in complex with the protease PT5 resembled those of protein complexes with large interfaces of interaction, constrained peptide backbones and multiple directional hydrogen and electrostatic bonds from both loops of the peptide, leading to high-binding affinity and exquisite selectivity. While in the crystal structure the chymotrypsin-bound macrocyclic peptide MP5.4.3 is visualised in its fully intact form, experiments in solution showed that MP5.4.3 can undergo proteolytic cleavage, thus revealing a substrate-like binding mode40.
Yeast display technology can effectively select and enrich macrocyclic peptides with binding affinities comparable to those obtained using other display technologies
We next compared the macrocyclic peptide ligands identified using yeast display to those previously reported in the literature, which bind the same targets and were identified from well-established in vitro directed evolution techniques. A comparison of the KD values of streptavidin-binding macrocyclic peptide ligands reported in this work with those of similar size (< 20 aa) selected using other display technologies and combinatorial naïve libraries revealed comparable or better binding affinities (Fig. 9a, Supplementary discussion, Supplementary Fig. 8, and Supplementary data file 11)42,43,44,45,46,47,48,49,50,51,52. To more fairly compare the different streptavidin-binding cyclic peptide ligands, we plotted the affinity values of the tightest binder as a function of the library size from which it was derived53. Overall, this analysis showed that the highest affinity yeast-encoded streptavidin macrocyclic peptide ligands distribute above the other ligands selected using alternative display technologies (Fig. 9b). However, this comparison does not take into account that, in addition to size, these libraries also differ in the format and number of random residues present in the encoded cyclic peptide ligands. Thus, we attempted to compare only macrocyclic peptide ligands with analogous topology and encoded by libraries of similar size. Among the different macrocyclic peptides binding to streptavidin and chymotrypsin described in this work and available in the literature, the comparable ones are only those encoded by ‘one ring’ libraries of the CX7C format that were isolated using yeast and phage display techniques (Fig. 9c, Supplementary Fig. 8, Supplementary discussion, and Supplementary data file 12, 13)44,46. Distribution analysis of the 7-amino acid ‘one ring’ cyclic peptide ligands of streptavidin and chymotrypsin targets selected using comparable yeast- and phage-encoded CX7C libraries revealed that those isolated using yeast display appear to have better binding properties than those isolated using phage display (Fig. 9c and Supplementary data file 13). Comparison of the Ki values of the macrocyclic peptide inhibitors of chymotrypsin reported in this work with those of naturally evolved protein inhibitors revealed that, despite the smaller size (< 2 kDa), the potency of the yeast-derived macrocyclic peptide inhibitors are similar to those of larger macromolecular inhibitors (2–8 kDa; Fig. 9d)54,55,56,57,58,59,60.

a Plot of the KD values of unique streptavidin-binding macrocyclic peptide ligands isolated using phage display (blue dots), mRNA display (light grey rhombuses), ribosome display (dark grey hexagons), bacteria display (white triangles), and yeast display (this work, red squares). b Plot depicting the most potent cyclic peptide ligand selected against streptavidin and derived from different literature-reported screening campaigns, which are divided based on selection techniques: yeast display (red square), phage display (blue dots), mRNA display (light grey rhombus), ribosome display (dark grey hexagon), and bacteria display (white triangle). The log10(1/KD) of the most potent cyclic peptide ligand is reported on the y-axis while the square root (log10(size of the naïve library)) is reported on the x-axis. c Plot depicting 7-amino acid ‘one ring’ cyclic peptide ligands selected against streptavidin (PT2) and α-chymotrypsin (PT5). Macrocyclic peptide ligands were isolated by screening either phage-encoded (blue dots) or yeast-encoded (red squares) CX7C naïve libraries of similar size. d Plot of the Ki values of best macrocyclic peptide inhibitors (MP) of α-chymotrypsin isolated using phage display (white triangle) and yeast display (this work, red squares). As reference, the Ki values of the natural eglin c from leech Hirudo medicinalis (70 amino acids; Ki = 0.4 pM)54, the Bowman–Birk inhibitor (BBI) from soybean Glycine max (BBI G.m.; 71 amino acids; Ki = 6 nM)55, the BBI from Torresea cearensis seeds (BBI T.c.; 63 amino acids; Ki = 50 nM)56, the Black-eyed pea trypsin and chymotrypsin inhibitor (BTCI) from Vigna unguiculata (83 amino acids; Ki = 120 nM)57, the natural cyclic peptide chymotrypsin inhibitor HECI from the Asian green frog Hylarana erythraea (17 amino acids, H-TVLRGCWTFSFPPKPCI-NH2; Ki = 3.9 μM)58, the PMP-C inhibitor from insect Locusta migratoria (36 amino acids; Ki = 200 pM)59 and the turkey ovomucoid third ___domain (OMTKY3; 56 amino acids; Ki = 20 pM)60 are also plotted (blue dots). Data are provided as Source data.
The numerous streptavidin-binding cyclic peptide ligands reported in the literature also allowed us to evaluate whether similar amino acid motifs (at least >3 identical contiguous residues) were present in our naïve libraries and/or in the selected and enriched yeast-encoded ligands. Comparison of the selected ligands revealed that two of our macrocyclic peptides (MP2.1 and MP2.7) include the 5-amino acid motif ‘HPQGD’, which is also present in four streptavidin-binding cyclic peptide ligands previously isolated using phage display (Supplementary data file 14)47. Although present in both yeast- and phage-selected ligands, the 5-amino acid motif ‘HPQGD’ appeared in different macrocyclic peptide topologies. While in the case of yeast-encoded ligands it was found exclusively within ‘two rings’ macrocyclic peptides, in the case of phage-encoded ligands it occurred mainly within ‘one ring’ cyclic peptide structures. (Supplementary data file 14). The same ‘HPQGD’ motif, as well as shorter versions of it (e.g., ‘HPQG’), were also identified in our naïve libraries, although at very low frequencies (Supplementary data file 15). Even though NGS data cover < 3% of the entire size of our naïve libraries, these findings further highlight the ability of our approach to select and enrich even very rare clones (frequency < 0.001%).
We then examined whether similar amino acid motifs were present within macrocyclic peptide ligands with analogous topology and encoded by similarly sized naïve libraries. However, among the streptavidin-binding cyclic peptide ligands selected using different display technologies, no molecule containing a motif with at least >3 identical contiguous residues located within a similar topology was identified. Conversely, we observed that some 4-amino acid motifs (e.g., ‘ERER’, ‘RDGN’, ‘SVKL’, ‘QPRV’) located within streptavidin-binding cyclic peptide ligands isolated from phage-encoded CX7C naïve libraries were also present in yeast-encoded CX7C naïve libraries (Supplementary data file 16). The reason why yeast-encoded macrocyclic peptides containing these motifs, although present in the original naïve library, were not isolated and enriched during the selection steps could depend on many factors that are difficult to ascertain based on so few data and sequences. Perhaps, one explanation lies in the ability of the yeast display technology itself, which, when combined with flow cytometry sorting, allows for the precise control of the selection process and quantitative discrimination of clones with different binding affinities during the screening process. Indeed, all yeast-encoded macrocyclic peptide ligands selected against streptavidin described in this work have a binding affinity 100- to 1000-fold better than the cyclic peptides containing these motifs isolated using phage display (Supplementary data file 13). Therefore, we hypothesise that, although present in the original naïve libraries, yeast cells encoding macrocyclic peptide ligands containing these motifs might not have been selected because they bind streptavidin weaker than those with the ‘HPQ’ motif; therefore, they did not fit into the FACS gate that was designed to collect and enrich yeast cells displaying high-affinity binders.
Although these analyses seem to indicate that yeast display performs similarly to, and in some cases better than, other display technologies, the available data may yet be too small to draw conclusions. Further studies using multiple and more similar library designs, closer library sizes, equivalent experimental and stringency selection conditions, different protein targets, and multiple display systems in parallel are needed to better compare performance across different technologies.
Discussion
In summary, we have shown that yeast display, an in vitro selection technology that has proven transformative for the discovery and engineering of multiple antibodies and diverse protein scaffolds, can also be applied to generate macrocyclic peptide ligands with desired binding properties. In this proof-of-concept study, we showed that large naïve combinatorial libraries encoding sequence and structurally diverse disulfide-tethered macrocyclic peptide ligands can be rapidly generated and effectively expressed on the surface of yeast cells. The size of the generated libraries (~109) is at the higher end of those previously reported using yeast display, is comparable or slightly smaller than the largest phage display libraries, and is still orders of magnitude smaller than those attainable by mRNA display. The small size of the libraries has often been considered an Achilles’ heel of yeast display technology. However, while this drawback is central when engineering large proteins such as antibodies (>100 amino acids), it might be less critical for short peptide sequences (< 10 amino acids) where the diversity to be covered is smaller.
Next, we demonstrated the robustness and simplicity of our approach by rapidly isolating high-binding affinity disulfide-tethered macrocyclic peptide ligands against five highly diverse protein targets. By combining MB separations with multiple rounds of FACS, we successfully identified macrocyclic peptide ligands against all five protein targets. Isolated macrocyclic peptide ligands showed different amino acid sequences, topologies and ring size distribution.
We demonstrated the ability of yeast display technology to enable the rapid and efficient characterisation of isolated macrocyclic peptide ligands directly on the cell surface, eliminating the need for costly and time-consuming chemical synthesis and purification. All identified macrocyclic peptide ligands showed binding affinities lower than 1 µM. Interestingly, for four out of the five protein targets screened, we isolated at least one macrocyclic peptide ligand with a binding affinity below 10 nM. Importantly, yeast-encoded macrocyclic peptides showed high specificity for the protein target they were selected for. These exquisite binding properties are remarkable considering that macrocyclic peptide ligands were rapidly isolated starting from relatively small naïve combinatorial libraries without the need for affinity maturation. Interestingly, there appears to be no correlation between the binding affinities determined and the topology or size of the isolated macrocyclic peptide ligands. Contrary to expectations, we found that ‘one ring’ macrocyclic peptides can bind a protein target tightly than those with ‘two rings’. We speculate that, rather than the topology and the size of the macrocyclic peptide ligand, it is the structure of the target protein itself that determines the achievable binding affinity. This supports the importance of using multiple unbiased library designs to increase the chances of identifying macrocyclic peptide ligands with desired binding properties. Interestingly, by performing kinetic competition experiments in the presence of well-characterised soluble ligands with known binding affinities, we could also rapidly unveil the binding site of macrocyclic peptide ligands directly on the yeast cell surface, without the need for time-consuming and painstaking downstream synthesis and purification processes.
The use of sequence and structurally distinct protein targets allowed us to better evaluate the capabilities of the technology as well as to address remaining limitations. For instance, aware of the possible ‘avidity’ issues that could arise when using yeast display technology in the presence of multivalent soluble targets25,27,34, we included multimeric protein targets in the screening and determined the binding affinities of the selected macrocyclic peptide ligands using opposite orientations and complementary techniques. For the tetrameric protein streptavidin, we observed no difference between the binding affinity values measured by yeast surface titrations (cell-anchored macrocyclic peptide against soluble protein target) and those determined using surface plasmon resonance (chip-anchored protein target against soluble macrocyclic peptide ligand), whereas for the tetrameric protein aldolase, we observed a ~500-fold discrepancy. One possible explanation for such a difference between aldolase and streptavidin is that the extent of ‘avidity’ effect depends not only on the number of identical binding sites present on the protein target to be detected, but also on the size of the protein itself and thus the distance and the density of the epitope that must be concomitantly recognised by different copies of macrocyclic peptide ligands displayed on the surface of yeast cells. Nevertheless, the binding affinity values measured for tetrameric protein aldolase using surface plasmon resonance are still in the nanomolar range. Moreover, we cannot exclude the possibility that avidity may actually prove advantageous when working with otherwise undetectable weak macrocyclic peptide ligands, especially during the first rounds of screening, when each member of the library is represented in small numbers. If necessary, multivalent binding can be overcome during FACS sorts by applying kinetic strategies in the presence of a large excess of a competitive unlabelled protein target, which can compete in a concentration-dependent manner24. All these strategies, which have been previously developed and optimised for the engineering of different protein scaffolds and have made yeast display technology a versatile and highly successful tool, can now be rapidly recapitulated for the engineering of macrocyclic peptide ligands.
Another noteworthy aspect is that while only one species is expressed for ‘one ring’ macrocyclic peptides, up to three different isomers can be expressed for ‘two rings’ macrocyclic peptides. Whether a yeast cell is able to preferentially express one or a mixture of these possible isomers is still unknown and certainly a subject of further investigation. However, while this information becomes fundamental when planning to recombinantly produce the macrocyclic peptide, either alone or fused to other proteins, it is less relevant during screening and discovery. In fact, it is known that a single yeast cell can express about 105 copies of a ligand on its surface23, so even if only 10% (~104 copies) of the displayed ‘two rings’ macrocyclic peptides represent the ‘active’ form, such an isomer would still be selected for and enriched during FACS if this isomer has a good binding affinity, especially when using high concentrations of a protein target or if the target is multimeric. However, if the ‘active’ isomer is displayed at a much lower relative level, then that isomer may escape selection because the binding intensity normalised to the total expression intensity, which includes the large number of ‘inactive’ isomers, may fall below the FACS gate diagonal.
Though the aim of this work was solely to demonstrate the ability of yeast display to efficiently generate and isolate high-affinity and specificity disulfide-bound macrocyclic peptide ligands from large, topologically diverse combinatorial libraries, yeast display can also be combined with other in vitro directed evolution tools to further harness the unique advantages of each, thus enabling previously unexplored applications. These include the evaluation and improvement of binding and stability properties and rapid and fine epitope mapping. Moreover, based on the enormous success that yeast display technology has had in rapidly screening and assessing the biophysical properties of hundreds of computationally designed proteins28,61,62, we envisage the tool to also be suitable for effectively characterising in silico developed genetically encoded macrocyclic peptide ligands. The binding affinities of the macrocyclic peptide ligands identified in this work could be further improved by affinity maturation. Such a process involves the design of macrocyclic peptide libraries based on one or more of the selected clones and the application of equilibrium, kinetic and/or competitive binding approaches28.
Although the design of the herein described yeast-encoded disulfide-tethered macrocyclic peptides restricts their immediate application to extracellular protein targets, this does not preclude that their binding affinity, selectivity, stability, and membrane permeability could be further enhanced and tailored by applying various biocompatible post-translational modifications, medicinal chemistry and rational design strategies63,64,65,66,67. To this end, it is worth emphasising that post-translational cyclisation of linear peptides displayed on the surface of yeast cells has already been successfully applied. Indeed, yeast-displayed cyclic peptide ligands of lysozyme and interleukin-17 have been identified by screening libraries encoding linear peptides bearing two lysine residues that were cross-linked intramolecularly using the primary amine-reactive disuccinimidyl glutarate31. Similarly, cyclic peptide ligands of the YAP protein were isolated by screening linear peptides that were ‘head-to-side chain’ cyclised on the surface of yeast cells using the enzyme transglutaminase32. Although beyond the scope of the present work, we speculate that our cysteine-rich peptide libraries could be compatible with similar chemical conjugation strategies based on thiol-reactive agents, giving rise to even larger molecular repertoires68.
Moreover, the modular structure of peptides and the commercial availability of hundreds of amino acid building blocks simplify the rapid development of macrocyclic peptides with desired properties. For instance, the incorporation of cysteine-reactive chemical linkers, N-methylated amino acids, D-amino acids, non-proteinogenic amino acids, N-terminal capping/acetylation, deamination, systematic truncations, extensions of N- or C-termini, N-to-C-terminal cyclisation and amide bond mimetics are chemical modifications that have often proved key to transforming disulfide-cyclised macrocyclic peptides into potent analogues with better drug-like properties3,69,70.
Although many challenges remain, the yeast display–based approach described here has the potential to enable the facile and cost-effective development of target-tailored, disulfide-tethered macrocyclic peptides with the desired binding properties. These peptides can then be readily used either as chemical molecules or as gene fusion products. In the future, it will be important to prove the efficacy and broad applicability of this technology to generate genetically encoded macrocyclic ligands against challenging protein targets and demonstrate their therapeutic uses in vivo.
Methods
Bioinformatic analysis of protein targets
The biochemical and biophysical properties of each protein target (PT; Supplementary table 8) were determined using different bioinformatic tools. The volume (Å3) of each PT was assessed using MOLEonline web interface71. A probe radius of 5 Å and an interior threshold of 1.1 Å were applied. The solvent accessible surface area (ASA) of each PT was calculated using PISA software and a spherical probe of 1.5 Å radius72. The surface charge distribution of each PT was calculated using PyMOL73. The cavities volume and surface representation of each PT were determined using VEGAZZ software setting a minimum and maximum sphere radius of 3 and 6 Å, respectively74. The composition of secondary structure of each PT was determined using PDBsum database75. The three-dimensional structure of each PT was generated and rendered using PyMOL73. Molecular weight (MW), isoelectric point (pI), and protein extinction coefficient (ε) were determined using ProtParam tool76. The properties were determined using the three-dimensional structures of PTs in the absence of bound ligands (apo form; Supplementary table 9).
Purification and chemical biotinylation of protein targets
Protein targets (PT) rabbit aldolase (PT1, GE-Healthcare, Chicago, IL, USA), Klebsiella pneumoniae carbapenemase GES-5 (PT3, recombinantly produced)35, Streptomyces avidinii streptavidin (PT2, Thermo Fisher Scientific, Dreieich, Germany), bovine carbonic anhydrase (PT4, Fluka, Darmstadt, Germany) and bovine α-chymotrypsin (PT5, Fluka, Darmstadt, Germany) were dissolved in 1X PBS pH 7.4 and further purified by size-exclusion chromatography using either a Superdex 75 16/600 GL or a Superdex 200 16/600 GL column (Cytiva, Marlborough, MA, USA) equilibrated with buffer 1X PBS pH 7.4 and connected to an ÄKTA pure 25 M system (Cytiva, Marlborough, MA, USA). The fractions containing the expected monodisperse PT were pulled and further concentrated to a final concentration of 10 µM using 10,000 Da MWCO Amicon ultrafiltration devices (Merck, Nottingham, UK) at 4000 × g and 4 °C on a Heraeus Multifuge X1R centrifuge (Thermo Fisher Scientific, Dreieich, Germany). Reactive EZ-link sulfo-NHS-LC-biotin (Thermo Fisher Scientific, Dreieich, Germany) was dissolved in 1X PBS pH 7.4 to obtain a final concentration of 10 mM. Protein-biotin conjugates PT1, PT3, PT4 and PT5 were prepared by incubating each PT at a concentration of 10 µM in 1X PBS pH 7.4 with 10-fold molar excess of EZ-link sulfo-NHS-LC-biotin (100 µM) for 1 hr at room temperature. The reaction was quenched with 1 M Tris-HCl. Excess of unreacted or hydrolysed biotinylation reagent was removed by gel filtration using a HiPrep 26/10 desalting column (Cytiva, Marlborough, MA, USA) equilibrated with buffer 1X PBS pH 7.4 and connected to an ÄKTA pure 25 M system (Cytiva, Marlborough, MA, USA). The fractions containing the expected monodisperse PT were pulled and concentrated to a final concentration ranging from 10 μM to 50 μM using 10,000 Da MWCO Amicon ultrafiltration devices (Merck, Nottingham, UK) at 4000 × g and 4 °C on a Heraeus Multifuge X1R centrifuge (Thermo Fisher Scientific, Dreieich, Germany). Final PT concentrations were determined using a BioPhotometer D30 UV spectrophotometer (Eppendorf, Hamburg, Germany). Purified PTs were flash-frozen in liquid nitrogen and stored at −80 °C. Protein target Streptomyces avidinii streptavidin (PT2) was not chemically biotinylated since streptavidin-coated MBs and fluorescently labelled streptavidin are commercially available (Thermo Fisher Scientific, Dreieich, Germany). The monodisperse state of concentrated biotinylated PT1, PT3, PT4, PT5 and fluorescently labelled PT2 was confirmed by size-exclusion chromatography using a Superdex 200 10/300 GL column (Cytiva, Marlborough, MA, USA) equilibrated with buffer 1X PBS pH 7.4 and connected to an ÄKTA pure 25 M system (Cytiva, Marlborough, MA, USA). Purified biotinylated PT1, PT3, PT4, PT5 and fluorescently labelled PT2 were eluted as a single peak at elution volumes (Ve) that correspond to apparent molecular masses of 160 kDa for aldolase (PT1, tetramer), 50 kDa for streptavidin (PT2, tetramer) and ~25–30 kDa for monomeric carbapenemase GES-5 (PT3), carbonic anhydrase (PT4) and α-chymotrypsin (PT5).
Generation of the yeast-encoded macrocyclic peptide naïve libraries
The yeast-encoded macrocyclic peptide naïve libraries were constructed using homologous recombination-based methods22. Macrocyclic peptides were displayed on the surface of yeast as N-terminal fusion of the cysteine-free GPI cell-surface anchor protein33. The yeast display vector was based on a modified version of the pCT-CON backbone22, here renamed pCT-GPI. This vector contains a DNA sequence encoding for a secretory leader sequence, a NheI restriction site, a sequence encoding for a long and flexible glycine-serine spacer (G4S)3, a sequence encoding for the HA tag (YPYDVPDYA) followed by a sequence encoding for a GPI cell-surface anchor protein which has been modified to include a silent BamHI restriction site. The synthetic genes were codon optimised for expression in Saccharomyces cerevisiae cells and obtained from Integrated DNA Technologies (Coralville, IA, USA). Yeast-encoded macrocyclic peptide naïve libraries were created by inserting the DNA sequences encoding the random peptide sequences ACXmCSG (X = any amino acid, m = 7 or 9) and ACXmCXnCSG (X = any amino acid, m = 3, 6 or 9 and n = 9, 6 or 3), the flexible (G4S)3 linker, the HA tag and a N-terminal portion of the GPI protein into the pCT-GPI vector. The insert was created by appending the DNA encoding the random macrocyclic peptide sequences to the N-terminus of the (G4S)3–HA–GPI encoding gene in a PCR reaction (30 cycles) using the forward degenerate primers F1, F2, F3, F4 and F5 and the universal reverse primer R (Supplementary Table 22). The oligonucleotides were obtained from Integrated DNA Technologies (Coralville, Iowa, IA, USA). The pCT-GPI vector was used as DNA template. The PCR amplifications were performed in a reaction volume of 50 μL containing the oligonucleotides (500 nM each), dNTP mix (250 μM), DNA template (1 ng), 10X DreamTaq buffer (1X), DreamTaq DNA polymerase (1.25 U, Thermo Fisher Scientific, Dreieich, Germany) and H2O mQ. The pCT-GPI vector was double digested using NheI-HF and BamHI-HF restriction enzymes (New England Biolabs, Woburn, MA, USA). The linearised pCT-GPI vector and the PCR products were further purified using ethanol precipitation and concentrated to 1 µg/µL, combined together in a 1:50 molar vector to insert ratio, and electroporated into freshly prepared RJY100 competent cells, where the full constructs are reassembled via homologous recombination22,77. Transformed yeast cultures were recovered and expanded in SD-CAA media. Small portions of transformed cells were serially diluted and titrated on SD-CAA plates to assess the final library sizes22. Library quality and diversity were further assessed by NGS.
Determination of the probability of encountering additional cysteine residues within macrocyclic peptide sequences encoded by ‘NNK’ codons
The theoretical probability to encounter additional cysteine residues within a given peptide sequence encoded by ‘NNK’ codons (where N = G/A/T/C and K = G/T) was calculated as follows. Considering that all the outcomes of ‘NNK’ codon are equally likely and since only 1 of the 32 possible codons is allocated to a cysteine residue (‘TGT’), the probability that ‘NNK’ codon yields a cysteine residue is P = 1/32 = 0.03125 or 3.125%. The probability of occurrence of a number k of cysteines within a sequence of n positions is defined as P(k) and described by the binomial distribution: \(P\left(k\right)=\left(\frac{n}{k}\right){P}^{k}{(1-P)}^{n-k}\), where n is the number of residues, each encoded by a ‘NNK’ codon, k is the number of cysteine residues appearing in the peptide sequence, P is the probability that a ‘NNK’ codon yields a cysteine residue (0.03125), \(\left(\frac{n}{k}\right)=\frac{n!}{k!\left(n-k\right)!}\) represents the binomial coefficient (the number of ways to choose the k positions of cysteine residues from n positions), \({P}^{k}\) indicates the probability of a cysteine appearing k times in the sequence and \({\left(1-P\right)}^{n-k}\) indicates the probability of non-cysteine residues appearing in the remaining n–k positions, that is equal to \({(0.96875)}^{n-k}\). For ‘one ring’ macrocyclic peptide naïve libraries CX7C (1) and CX9C (2), the number (n) of residues X encoded by ‘NNK’ codons is n = 7 and n = 9, respectively. For ‘two rings’ libraries CX3CX9C (3), CX6CX6C (4), and CX9CX3C (5), the number (n) of residues X encoded by ‘NNK’ codons is n = 12. The number of additional cysteine residues (k) appearing in the peptide sequence of n residues X is equal to 2 for two additional cysteines (k = 2; +2 cys), 3 for three additional cysteines (k = 3; + 3 cys) and 4 for four additional cysteines (k = 4; + 4 cys).
Sample preparation and analysis of NGS data
Transformed yeast cultures were grown to mid-log phase (OD600 = 2–3) in fresh SD-CAA media at 30 °C with shacking (250 rpm) and the DNA plasmid pools extracted using Zymoprep Yeast Plasmid Miniprep II kit following manufactureʼs instructions (Zymo Research, Irvine, CA, USA)22. Amplicons for NGS were generated by performing two consecutive PCR reactions using yeast-extracted DNA plasmids as template. For each yeast-encoded macrocyclic peptide library a specific 10 nucleotide barcode and an Illumina sequencing tag were used (Supplementary data file 7). The oligonucleotide primers were obtained from Integrated DNA Technologies (Coralville, IA, USA). The first PCR was performed in a reaction volume of 30 µL containing 5X HF buffer (1X), DMSO (30% v/v), dNTPs (200 μM), NGS-F1 oligonucleotide (500 nM), NGS-R1 oligonucleotide (500 nM), Phusion high-fidelity DNA polymerase (New England Biolabs, Woburn, MA, USA), and yeast-extracted DNA plasmids (10 μL). The PCR amplicons (280 bp) were purified using the Clean and Concentrator PCR purification kit (Zymo Research, Irvine, CA, USA) and their quality verified on a 1.5% w/v agarose gel. The second PCR was performed in a reaction volume of 50 µL containing 5X HF buffer (1X), DMSO (30% v/v), dNTPs (200 μM), NGS-F2-X barcode oligonucleotide (500 nM, where X is a letter from A to I) and NGS-R2 oligonucleotide (500 nM), Phusion high-fidelity DNA polymerase (New England Biolabs, Woburn, MA, USA), and initial purified PCR products (10 ng). The PCR products were analysed on a 1% w/v agarose, gel-extracted and purified using Qiagen Gel Extraction Kit (Qiagen, Hilden, Germany). The final DNA concentration and quality were assessed using a BioPhotometer D30 UV spectrophotometer (Eppendorf, Hamburg, Germany). The amplicons were sequenced using a NovaSeq6000 (IGA Technology Services, Udine, Italy), yielding paired-end reads of 150 bp. Raw data were processed using MATLAB scripts that have been adapted to the need of this study78. The applied command lines and the codes are reported in Supplementary information and Supplementary Table 10.
Selection of yeast-encoded macrocyclic peptides
Selection of yeast-encoded macrocyclic peptide ligands toward each protein target (PT) was performed using an amount of yeast cells at least 10-fold larger than i) the initial estimated naïve library size or ii) the number of cells isolated from the previous round of either MBs or flow cytometry cell sorting (FACS). Each single naïve yeast-encoded macrocyclic peptide library was grown separately in SD-CAA media at 30 °C with shacking (250 rpm) and surface expression of macrocyclic peptides induced in SG-CAA media for 16 hrs at 20 °C with shacking (250 rpm). Before ‘positive selection’, separately expanded and induced yeast populations were mixed ensuring to cover at least 10-fold the diversity of each library and subjected to two sequential cycles of ‘negative selection’ using streptavidin-coated Dynabeads biotin binder MBs (Thermo Fisher Scientific, Dreieich, Germany). A 10-fold diversity mixed library depleted of streptavidin-coated MBs binders was next screened against highly diverse biotinylated PT1, PT3, PT4 and PT5 captured on MBs. For protein target streptavidin (PT2) the ‘negative selection’ process was not applied, and the 10-fold diversity naïve yeast-encoded macrocyclic peptide libraries were exposed directly to streptavidin-coated MBs. Overall, two iterative cycles of MB-based ‘positive selections’ followed by four cycles of FACS were applied for each PT. Each ‘positive selection’ cycle comprises the growth of yeast cells, the expression of the macrocyclic peptide on the surface of yeast cells, binding to PT, washing, and expansion of the isolated bound yeast cells. For each cycle of MB-based ‘positive selection’ against PT1, PT3, PT4 and PT5, streptavidin-depleted yeast cells displaying macrocyclic peptide ligands (2 × 109 cells/mL) were washed twice with ice-cold PBSA buffer (1X PBS pH 7.4 supplemented with 0.1% w/v bovine serum albumin fraction V), incubated at 4 °C for 1 hr with 50 pmol biotinylated PT1, PT3, PT4 and PT5 immobilised on 4 × 106 streptavidin-coated MBs, washed three times using ice-cold PBSA buffer, cells rescued with 5 mL of fresh SD-CAA medium and grown for 16 hrs at 30 °C with shacking (250 rpm). In the case of MB-based selection against PT2, yeast cells displaying naïve undepleted macrocyclic peptide ligands (2 × 109 cells/mL) were washed twice with ice-cold PBSA buffer, incubated directly with 4 × 106 streptavidin-coated MBs for 1 hr at 4 °C, washed three times using ice-cold PBSA buffer, cells rescued with 5 mL of fresh SD-CAA medium and grown for 16 hrs at 30 °C with shacking (250 rpm). The use of highly avid reagents such as streptavidin-coated MBs (either ‘nude’ as in the case of PT2 or saturated with biotinylated proteins as in the case of PT1, PT3, PT4 and PT5) increases the likelihood of isolating low-affinity macrocyclic peptide ligands from the naïve library by exploiting the multivalent interaction between yeast cells and the PT pre-loaded on MBs. For FACS-based ‘positive selection’, yeast cells displaying macrocyclic peptide ligands were isolated using a two-colour labelling scheme based on fluorescent-conjugated detection reagents for expression of macrocyclic peptides on the surface of yeast cells (anti-HA epitope tag) and binding of the same macrocyclic peptides to PTs (anti-biotin) at recommended dilutions (Supplementary Table 23). In the case of protein targets PT1, PT3, PT4 and PT5, binding of yeast-displayed macrocyclic peptide ligands to each PT was determined by labelling yeast cells with corresponding biotinylated PT proteins (1 μM) followed by staining with the secondary reagent neutravidin or streptavidin conjugated to a fluorophore. In the case of PT2, binding of the yeast-displayed macrocyclic peptide ligands was assessed by labelling yeast cells directly with fluorescently labelled streptavidin-DyLight 650 (1 μM). In the case of protein targets PT1, PT3, PT4, and PT5, the fluorescently labelled secondary detection reagents streptavidin-DyLight 650 and neutravidin-DyLight 650 were alternated to avoid enrichment of potential streptavidin or neutravidin binders during FACS-based selections (Supplementary Table 23). Such alternation was not applied during the FACS-based ‘positive selection’ against streptavidin (PT2) where solely streptavidin-DyLight 650 was used. Sorting was performed on BD FACSAriaIII sorter instrument (BD Life Sciences, Franklin Lakes, NJ, USA) and data evaluated using FlowJo v.10.0.7 software (BD Life Sciences, Franklin Lakes, NJ, USA). After four cycles of iterative FACS-based selections, the DNA plasmid was extracted from the collected polyclonal yeast cells and sequenced.
Sequencing analysis of selected yeast-encoded macrocyclic peptide ligands
The identity of the selected macrocyclic peptide ligands was revealed by both Sanger and NGS methodologies. For Sanger sequencing, a fraction of each polyclonal yeast cell population collected after the fourth round of FACS sorting was spread and grown on selective SD-CAA solid agar media at 30 °C. Single colonies were further picked and grown to mid-log phase (OD600 = 2–3) in fresh SD-CAA media at 30 °C with shacking (250 rpm). The DNA plasmid present within the yeast cells was extracted using Zymoprep Yeast Plasmid Miniprep II kit following manufactureʼs instructions (Zymo Research, Irvine, CA, USA)22. Purified yeast-extracted DNA plasmid was then used to transform competent Escherichia coli TOP10 cells. Bacteria-extracted DNA plasmids were analysed by Sanger sequencing (BMR Genomics, Padova, Italy) using the following oligonucleotide: 5’-GACTTGGAAGGTGACTTCG-3’. For NGS analysis (IGA Technology Services, Udine, Italy), a fraction of polyclonal yeast cell populations collected after the fourth round of FACS sort was grown to mid-log phase (OD600 = 2–3) in fresh SD-CAA media at 30 °C with shaking (250 rpm) and the DNA plasmid pools extracted using Zymoprep Yeast Plasmid Miniprep II kit following manufacture instructions (Zymo Research, Irvine, CA, USA). Purified yeast-extracted DNA plasmids were then used as templates to generate amplicons through two consecutive PCR reactions. For each selected yeast-encoded macrocyclic peptide population a specific ten-nucleotide barcode and an Illumina sequencing tag were used. The amplicons were sequenced using a NovaSeq6000 (IGA Technology Services, Udine, Italy), yielding paired-end reads of 150 bp. Raw data were processed using command lines, and the codes are reported in Supplementary information.
Generation of unique yeast-encoded macrocyclic peptide ligands identified using NGS
While binding properties of the thirteen most abundant macrocyclic peptide ligands identified using Sanger sequencing (MP1.1, MP2.1, MP2.2, MP3.1, MP3.2, MP4.1, MP4.4, MP5.1, MP5.2, MP5.4, MP5.5, MP5.6 and MP5.7) could be characterised directly as cell-surface fusions starting from the same single picked colonies of sorted yeast cells, those identified by NGS analysis (MP2.3, MP2.4, MP2.5, MP2.6, MP3.3, MP4.2, MP4.3, MP5.3 and MP5.5) required additional cloning steps. DNA constructs encoding NGS-derived macrocyclic peptide ligands fused to the N-terminus of the GPI cell-surface anchor were generated using homologous recombination-based methods. Briefly, DNA inserts encoding the macrocyclic peptide sequences were appended to the N-terminus of the (G4S)3–HA–GPI encoding gene in a PCR reaction (30 cycles) using the pCT-GPI vector as template, forward and reverse oligonucleotides (Supplementary data file 10) and the Phusion high-fidelity DNA polymerase (Thermo Fisher Scientific, Dreieich, Germany). The oligonucleotides were obtained from Integrated DNA Technologies (Coralville, IA, USA). The pCT-GPI vector was double digested using NheI-HF and BamHI-HF restriction enzymes (New England Biolabs, Woburn, MA, USA). The linearised vector and the PCR products were further purified using ethanol precipitation, concentrated to 100 ng/µL, combined using DNA assembly methods (1:15 vector to insert molar ratio)79 and transformed into Escherichia coli TOP10 competent cells. All DNA constructs encoding a single macrocyclic peptide ligand were verified by Sanger sequencing and further used to transform new competent RJY100 cells using Frozen-EZ Yeast Transformation II Kit (Zymo Research, Irvine, CA, USA).
Determination of equilibrium binding affinities using yeast surface display titrations
The apparent equilibrium dissociation constant (KDapp) of individually selected macrocyclic peptide ligands towards each single PT was determined using yeast surface display titrations22. Individual yeast colonies were inoculated into 5 mL SD-CAA cultures, grown to mid-log phase (OD600 = 2–5) in SD-CAA media at 30 °C with shaking (250 rpm). Cells were induced in SG-CAA media for 16 hrs at 20 °C with shaking (250 rpm). The binding assays were conducted in 96-well conical V-bottom plates (Corning, Tewksbury, MA, USA) containing 2 × 105 induced yeast cells per well. Yeast cells displaying macrocyclic peptide ligands were incubated with varying concentrations of soluble biotinylated PT1, PT3, PT4 and PT5 (ranging from 100 pM to 3 μM) overnight at 4 °C with gentle shaking (150 rpm). After PT incubation, yeast cells were pelleted (2200 × g for 3 min at 4 °C) and washed twice with 200 μL ice-cold PBSA buffer. Labelling with secondary reagents such as streptavidin or neutravidin conjugated to DyLight dye was performed at recommended dilutions (Supplementary Table 23). The KDapp values of selected macrocyclic peptide ligands towards PT2 were determined using a similar approach where yeast cells displaying macrocyclic peptide ligands were incubated with varying concentrations of soluble streptavidin conjugated to DyLight dye (streptavidin-DyLight 650). The level of cell surface expression of each macrocyclic peptide ligand was estimated by incubating 2 × 105 induced yeast cells with mouse anti-HA epitope tag (1:1000) antibody (Thermo Fischer Scientific, Dreieich, Germany). After incubation, yeast cells were pelleted (2200 × g for 3 min at 4 °C) and washed twice with 200 μL ice-cold PBSA buffer. Labelling with secondary goat anti-mouse antibody conjugated to DyLight 488 was performed at recommended dilutions (Supplementary Table 23) and incubated for 30 min at 4 °C. After the incubation, cells were pelleted (2200 × g for 3 min at 4 °C) and washed twice with 200 μL ice-cold PBSA buffer. The 96-well plates were run on a Attune NxT (Thermo Fischer Scientific, Dreieich, Germany) and data analysed using FlowJo v.10.0.7 software (BD Life Sciences, Franklin Lakes, NJ, USA). To ensure that the difference in binding was not due to variations in the number of copies of macrocyclic peptides expressed on the surface of yeast cells, the geometric mean fluorescence intensity (MFIBIND) from the binding signal was normalised to the geometric mean fluorescence intensity (MFIDISP) from the display signal. The normalised (binding/display = MFIBIND/ MFIDISP) geometric mean fluorescence intensity as a function of PT concentration was used to determine the KDapp values for all clones of interest. The KDapp values were determined by fitting a one-site-specific binding curve on GraphPad Prism v8.2.1 (GraphPad Software Inc., San Diego, CA, USA). Reported values are the results of three independent experiments (n = 3) and are presented as mean (dots) ± s.d., standard deviation (bars).
Chemical synthesis of macrocyclic peptides
Linear peptides containing both cysteines protected with Trt(trityl), a free amine at the N-terminus and an amide at the C-terminus, were chemically synthesised by standard Fmoc (9-fluorenylmethoxycarbonyl) solid-phase peptide synthesis (SPPS)80. Fmoc-protected amino acids, Fmoc-rink amide MBHA resin (100–200 mesh, loading 0.4–0.9 mmol/g resin, 0.01 mmol scale), N,N-dimethylformamide (DMF) and anisole were purchased from Novabiochem (Darmstadt, Germany). Acetic anhydride, acetonitrile (ACN), formic acid, trifluoroacetic acid (TFA), octanedithiol (ODT), diethyl ether, dichloromethane (DCM), triisopropylsilane (TIS), piperidine, N-methylmorpholine (NMM), dimethyl sulfoxide (DMSO) and 2,6-Lutidine were purchased from Merck (Darmstadt, Germany). Thioanisole and 1,2-ethanedithiol (EDT) were purchased from FlukaChemie GmbH (Buchs, Switzerland). N-methylpirrolidone (NMP) was purchased from VWR (Pennsylvania, PA, USA). O-Benzotriazole-N,N,N’,N’-tetramethyl-uroniumhexafluoro-phosphate (HBTU) and Hexafluorophosphate Azabenzotriazole Tetramethyl Uronium (HATU) were purchased from ChemPep (Wellington, FL, USA). All chemicals were used as received without further purification. Peptides were synthetised using either a ResPepSLi or a MultiPepRSi automated peptide synthesiser (Intavis Bioanalytical Instruments, Köln, Germany). The deprotection step was carried out twice using a 20% v/v solution of Piperidine in DMF for 5 min. The amino acid coupling was carried out twice (60 min × 2) for each Fmoc-amino acid (2.15 eq., 0.18 M solution in DMF) using HATU/NMM coupling mixture (2 eq. 0.17 M/4.7 eq. 0.4 M/4% v/v solution in DMF). The capping step was performed once using a solution of 5% v/v acetic anhydride and 6% v/v 2,6-Lutidine in DMF. Washes in between were performed using DMF (600 µL × 2). At the end of the synthetic process, washes were performed twice with DCM (600 µL × 2). The final peptides were deprotected and cleaved from the resin under reducing conditions using a TFA/H2O/thionalisole/anisole/ODT mixture (90/2.5/2.5/2.5/2.5% v/v) for 3 hrs at room temperature with shaking (300 rpm). The resin was removed by vacuum filtration and the peptides were precipitated with cold diethyl ether (50 mL) and centrifugation at 4000 × g for 5 min at 4 ◦C either on a Heraeus Multifuge X1R centrifuge (Thermo Fisher Scientific, Waltham, MA, USA) or a Sigma 4-16KS centrifuge (Merck, Darmstadt, Germany) under inert atmosphere. The precipitated linear peptides were washed twice with cold diethyl ether (35 mL × 2) and cyclised. Macrocyclic peptides with ‘one ring’ were generated by dissolving crude linear peptides in 10% v/v DMSO and 90% v/v aqueous buffer (20 mM NH4HCO3, pH 8.0) at a final concentration of 500 µM. The reaction mixture was incubated at room temperature for 48 hrs with shaking (150 rpm). Macrocyclic peptides with ‘two rings’ were generated using two orthogonal cysteine protecting groups. Linear peptides containing one pair of cysteines protected with Mmt (Monomethoxytrityl) and the second pair with Dpm (1,2-Diphenylmaleyl), a free amine at the N-terminus and an amide at the C-terminus were chemically synthetised by standard Fmoc SPPS as described above. Deprotection of Mmt groups was conducted on resin under mild reducing conditions using DCM/TFA/TIS (93/2/5 % v/v) mixture with shaking (300 rpm) for 10 min at room temperature. The entire procedure was repeated five times. The on-resin Mmt-deprotected linear peptide was sequentially washed with 100% v/v DCM (3 mL × 3), 100% v/v MeOH (3 mL × 3), 100% v/v DCM (3 mL × 3) and 100% v/v DMF (3 mL × 3). The on-resin ‘one ring’ macrocyclic peptides (first disulfide bridge formation) were obtained by adding N-chlorosuccinimide (NCS, 2 eq. 0.04 M in DMF) with shaking (300 rpm) for 15 min at room temperature. The reaction solution was removed by vacuum filtration, and the ‘one ring’ macrocyclic peptides washed three times with 100% v/v DMF (3 mL × 3) and 100% v/v DCM (3 mL × 3). ‘One ring’ macrocyclic peptides were further fully deprotected and cleaved from the resin under reducing conditions using TFA/H2O/TIS (95/2.5/2.5% v/v) mixture for 3 hrs at room temperature with shaking (300 rpm). The resin was removed by vacuum filtration and the ‘one ring’ macrocyclic peptides precipitated with cold diethyl ether (50 mL) and by centrifugation at 4000 × g for 5 min at 4 °C on a Heraeus Multifuge X1R centrifuge (Thermo Fisher Scientific, Waltham, MA, USA) or Sigma 4-16KS centrifuge (Merck, Darmstadt, Germany) under inert atmosphere. The precipitated ‘one ring’ macrocyclic peptides were washed twice with cold diethyl ether (35 mL × 2) and the second cyclisation performed. ‘Two rings’ macrocyclic peptides (second disulfide bridge formation) were generated by dissolving ‘one ring’ macrocyclic peptides in 10% v/v DMSO and 90% v/v aqueous buffer (20 mM NH4HCO3, pH 8.0) at final concentration of 500 µM. The reaction mixture was incubated at room temperature for 48 hrs with shacking (150 rpm). Finally, the ‘two rings’ macrocyclic peptides were dissolved in H2O:ACN (1:1), freeze-dried and lyophilised on a LIO-5PDGT (5Pascal, Milan, Italy).
Purification and characterisation of macrocyclic peptides
The macrocyclic peptides were purified by semi-preparative reversed-phase high-performance liquid chromatography (RP-HPLC) using a C18 SymmetryPrep functionalized silica column (7 μm, 19 mm × 150 mm, Waters, Millford, MA, USA) connected to a Waters Delta Prep LC 4000 System equipped with a Waters 2489 dual λ absorbance detector, a Waters 600 pump and a PrepLC Controller (Waters, Millford, MA, USA)81. A flow rate of 4 mL/min and a linear gradient (30% to 70% in 25 min) with a mobile phase composed of eluent A (99.9% v/v H2O, 0.1% v/v TFA) and eluent B (99.9% v/v ACN and 0.1% v/v TFA) was applied. The purified peptides were freeze-dried. The purity and molecular mass of each macrocyclic peptide (linear, ‘one ring’ and ‘two rings’ forms) were determined by electrospray ionisation mass spectrometry (ESI–MS) performed either on a single quadrupole liquid chromatograph mass spectrometer LCMS-2020 (Shimadzu, Kyoto, Japan) or on an InfinityLab LC/MSD mass spectrometer coupled to a 1260 Infinity II LC system (Agilent Technologies, Santa Clara, CA, USA). Both systems operated with the standard ESI source and in the positive ionisation mode. Peptides were dissolved in DMSO:ACN:H2O (1:50:50) solution at a final concentration of 50 µM and run at flow rate of 1 mL/min with a linear gradient (10%–100% v/v) of solvent B over 5 min (solvent A: 99.95% v/v H2O, 0.05% v/v formic acid; solvent B: 99.95% v/v ACN, 0.05% v/v formic acid). The reversed-phase HPLC column was a Nucleosil 100-5 C18 (5 μm, 125 mm × 4 mm; Macherey-Nagel, Dueren, Germany). Data were acquired, processed, and analysed using the Shimadzu LabSolutions software (Kyoto, Japan) and MestReNova v.12.0.1-20560 (Mestrelab Research, Santiago de Compostela, Spain). Each macrocyclic peptide was analysed once (n = 1). Concentrations of macrocyclic peptides were determined using a BioPhotometer D30 UV spectrophotometer (Eppendorf, Hamburg, Germany).
Determination of equilibrium binding affinities using surface plasmon resonance
The equilibrium binding constant (KD), association rate constant (‘on rate’, kon) and dissociation rate constant (‘off rate’, koff) for the interaction of chemically synthesised macrocyclic peptides with PTs were determined by surface plasmon resonance (SPR) using a Biacore 8 K+ instrument (BIAcore Inc., Piscataway, NJ, USA). Protein targets PT1 (aldolase), PT2 (streptavidin), and PT5 (α-chymotrypsin) were diluted in 10 mM sodium acetate (NaAc) pH 5.2 at 200 µg/mL, 100 µg/mL and 30 µg/mL final concentration, respectively. Protein target PT3 (carbapenemase GES-5 from Klebsiella pneumoniae) and PT4 (carbonic anhydrase) were instead diluted in 10 mM NaAc pH 4.5 at 10 µg/mL final concentration. All PTs were captured on a series S sensor chip CM5 (Cytiva, Marlborough, US) using amine coupling in 1X PBS pH 7.4 and 0.005% v/v Tween-20. The coupling was performed at 25 °C under a continuous flow rate of 10 µL/min till a protein immobilisation level of approximately 1000 resonance units (RUs) per flow cell was reached. The coupling involved four steps: i) injection of 70 µL of 0.4 M EDC for 420 s, ii) injection of 70 µL of 0.1 M NHS for 420 s, iii) injection of 70 µL of PT for 420 s and iv) injection of 83 µL of 1 M ethanolamine-HCl pH 8.5 for 500 s. Binding experiments with multiple two-fold dilutions of each macrocyclic peptide were performed in running buffer (1X PBS pH 7.4 supplemented with 0.005% v/v Tween-20 and 2% v/v DMSO) at 25 °C and a continuous flow rate of 30 µL/min. Serial 20–50 µL injections of two-fold dilutions of the macrocyclic peptides were applied through the sensor surface with a continuous flow rate of 10 µL/min for a time period ranging from 120–320 s and with a dissociation time of 600 s. The bound macrocyclic peptides were allowed to dissociate for 120 s before further analysis was performed. A solvent correction of four points (3.0%, 2.5%, 2.0%, 1.5% v/v DMSO in 1X PBS pH 7.4 supplemented with 0.005% v/v Tween-20) was performed before and after each analysis. In all experiments, an untreated flow cell without PT protein was used as a reference to correct binding response for bulk refractive index changes and non-specific binding. The association (kon) and dissociation (koff) rate constants were determined by global fitting mode. Data were fitted to a Langmuir binding model assuming stoichiometric (1:1) interactions by using Biacore 8 K Insight Evaluation software (Piscataway, NJ, USA). The equilibrium binding constant values were calculated as the ratio of koff to kon (KD = koff/kon). The KD was also determined by plotting binding responses in the steady-state region of the sensorgram (Req, 5 s before injection ends with a window of 5 s) versus the analyte peptide concentration (C). Each macrocyclic peptide was examined by SPR once (n = 1), and data were analysed using Biacore 8 K Evaluation Insight software with the predefined evaluation methods LMW multi-cycle or LMW single-cycle kinetics.
Determination of inhibitory activity of macrocyclic peptides
The inhibitory activity of macrocyclic peptides MP5.1, MP5.4.1, MP5.4.2 and MP5.4.3 was assessed by monitoring the residual activity of the enzyme bovine α-chymotrypsin (PT5) in the presence of a chromogenic substrate and different concentrations of each macrocyclic peptide. The activity assay was performed by incubating 0.5 nM PT5 with 100 μM chromogenic substrate N-Succinyl-Ala-Ala-Pro-Phe p-nitroanilide (100 μM; Merck, Darmstadt, Germany) and two-fold macrocyclic peptide dilutions ranging from 0.0005 to 2000 nM. All reagents were diluted in 10 mM Tris-Cl, pH 7.4, 150 mM NaCl, 10 mM MgCl2, 1 mM CaCl2, 1% w/v BSA and 0.1% v/v Triton-X100 buffer. The final reaction volume was 150 μL. The measurements were performed on a Tecan Infinite 200 PRO microplate reader (Tecan Trading AG, Switzerland) using a standard flat-bottom 96-well plate (Thermo Fisher Scientific, Dreieich, Germany). The enzymatic reactions were performed at 25 °C for 1 hr, under shaking with an absorbance wavelength of 410 nm. The initial velocities were monitored as changes in absorbance intensity. The sigmoidal curves were fitted to the data using the following non-linear regression equation for the inhibitory dose-response curves with variable slope (1):
where x is the peptide concentration, y is the residual percentage of protease activity and p is the hill slope. Half-maximum inhibitory concentration (IC50) values were determined using GraphPad Prism v8.2.1 software (GraphPad Software Inc., San Diego, CA, USA). The apparent inhibitory constants Kiapp values were subsequently calculated using the Cheng-Prusoff Eq. (2):
where Km (114 μM) is the Michaelis constant for the hydrolysis of substrate N-Succinyl-Ala-Ala-Pro-Phe p-nitroanilide catalysed by PT5, which has been determined by the standard Michaelis–Menten equation. Values were obtained using either OriginPro 8 G software (OriginLab Corporation, Northampton, MA, USA) or GraphPad Prism v8.2.1 software (GraphPad Software Inc., San Diego, CA, USA). Reported values are the results of three independent experiments (n = 3) and are presented as mean (dots) ± s.d., standard deviation (bars).
Determination of binding specificity of selected macrocyclic peptides
To assess the specificity of macrocyclic peptides, their equilibrium dissociation constants and/or inhibitory activities towards proteins with different extents of sequence and structural identity were evaluated. The apparent equilibrium dissociation constants (KDapp) were determined using yeast surface display titrations combined with flow cytometry. The binding assays were conducted in 96-well conical V-bottom plates (Corning, Tewksbury, MA, USA) containing 2 × 105 induced yeast cells per well. Yeast cells displaying macrocyclic peptides (MP2.1, MP2.2, MP2.3, MP2.4, MP2.5 and MP2.6) selected against streptavidin-DyLight 650 (PT2) were incubated with varying concentrations of soluble strep-tactin-DyLight 650 (PH2.1; concentrations ranging from 0.1 to 300 nM) and neutravidin-DyLight 650 (PH2.2; concentrations ranging from 30 to 300 nM) for 1 hr at 4 °C with shaking (150 rpm). Similarly, yeast cells displaying macrocyclic peptide (MP3.2) selected against the carbapenemase GES-5 from Klebsiella pneumoniae (PT3) were incubated with varying concentrations of soluble biotinylated carbapenemase KPC-2 from Klebsiella oxytoca (PH3.1; concentrations ranging from 100 to 1000 nM) and the extended-spectrum β-lactamase CTX-M-15 from Escherichia coli (PH3.2; concentrations ranging from 100 to 1000 nM) for 1 hr at room temperature with shaking (150 rpm). After primary incubation, cells were pelleted (2200 × g for 3 min at 4 °C) and washed twice with 200 μL of ice-cold PBSA buffer. In the case of biotinylated PTs, secondary labelling was performed with neutravidin-DyLight 650 at the recommended dilution (Supplementary Table 23) and incubated for 30 min at 4 °C. After the secondary incubation, cells were pelleted (2200 × g for 3 min at 4 °C) and washed twice with 200 μL ice-cold PBSA buffer. The 96-well plates were run on a Attune NxT (Thermo Fischer Scientific, Dreieich, Germany) and data analysed using FlowJo v.10.0.7 software (BD Life Sciences, Franklin Lakes, NJ, USA). To ensure that the difference in binding was not due to variations in the number of copies of macrocyclic peptides expressed on the surface of yeast cells, the mean fluorescence intensity (MFIBIND) from binding signal was normalised to the mean fluorescence intensity (MFIDISP) from display signal. The normalised (binding/display = MFIBIND/MFIDISP) geometric mean fluorescence intensity as a function of PT concentration was used to determine the KDapp values for all clones of interest. The KDapp values were determined by fitting a one-site-specific binding curve on GraphPad Prism v.8.2.1 software (GraphPad Software Inc., San Diego, CA, USA). Reported values are the results of three independent experiments (n = 3) and are presented as mean (dots) ± s.d., standard deviation (bars). The inhibitory activities were assessed by monitoring the residual activity of different enzymes in the presence of either chromogenic or fluorogenic substrates and different concentrations of each macrocyclic peptide. Residual activities were measured in 150 μL volume of buffer containing 10 mM Tris-Cl, pH 7.4, 150 mM NaCl, 10 mM MgCl2, 1 mM CaCl2, 0.1% w/v BSA, 0.01% v/v Triton-X100 and 5% v/v DMSO. Final concentrations of serine proteases were: 0.5 nM bovine α-chymotrypsin (PT5), 3 nM human chymotrypsin (PH5.1; IHUCHYLY from Molecular Innovations, Novi, MI, USA), 5 nM human neutrophil elastase (PH5.2; IHUELASD from Molecular Innovations, Novi, MI, USA), 3 nM elastase from porcine pancreas (PH5. 3; Merck, Darmstadt, Germany), 3 nM human cathepsin G (PH5.4; Merck, Darmstadt, Germany), 7.5 nM human kallikrein 3 (PH5.5; hKLK3 from MedChemExpress, Monmouth Junction, NJ, USA) and 6 nM human trypsin (PH5.1; hTRYP from Molecular Innovations, Novi, MI, USA). Human kallikrein 3 was purchased as inactive proenzyme (pro-hKLK3) and further activated by incubating 3 µM pro-hKLK3 with 300 nM thermolysin from Geobacillus stearothermophilus (Merck, Darmstadt, Germany) at 37 °C for 5 min according to the manufacturer’s instructions. Three-fold dilutions of macrocyclic peptide inhibitor MP5.4.3 were prepared ranging from 0.1 to 300 µM for all the proteases. For PT5, an additional two-fold dilutions of MP5.4.3 inhibitor, were prepared ranging from 0.3 pM to 100 nM. The inhibitory activities of macrocyclic peptide inhibitor MP5.4.3 against bovine α-chymotrypsin (PT5), human chymotrypsin (PH5.1) and human cathepsin G (PH5.4) were assessed using chromogenic substrate N-Succinyl-Ala-Ala-Pro-Phe p-nitroanilide (Merck, Darmstadt, Germany) at a final concentration of 100 μM. For elastase from porcine pancreas (PH5.3) and human neutrophil elastase (PH5.2), we used chromogenic substrate N-Methoxysuccinyl-Ala-Ala-Pro-Val p-nitroanilide (Merck, Darmstadt, Germany) at a final concentration of 100 μM. For human kallikrein 3 (PH5.5) we used chromogenic substrate N-Methoxysuccinyl-Arg-Pro-Tyr p-nitroanilide (Merck, Darmstadt, Germany) at final concentration of 1 mM. For human trypsin (PH5.6) we used the fluorogenic substrate Z-Gly-Gly-Arg-AMC (Bachem, Bubendorf, Switzerland) at final concentration of 50 μM. The initial velocities were monitored as changes in absorbance or fluorescence intensity on a Tecan microplate reader (Tecan infinite 200 pro, Tecan Trading AG, Switzerland) using either standard flat bottom 96-well plate or black microfluor 96-well plate Nunc MicroWell (Thermo Fisher Scientific, Dreieich, Germany). In the case of PT5, PH5.1 and PH5.4, the enzymatic reactions were performed at 25 °C for 1 hr, under shaking with an absorbance wavelength of 410 nm. In the case of PH5.2 and PH5.3, the enzymatic reactions were performed at 25 °C for 1 hr, under shaking with an absorbance wavelength of 400 nm. In the case of PH5.5, the enzymatic reactions were performed at 25 °C for 1 hr, under shaking with an excitation wavelength of 355 nm and an emission recording at 460 nm. The sigmoidal curves were fitted to the data using Eq. (1). Half-maximum inhibitory concentration (IC50) values were derived from the fitted curves from GraphPad Prism v.8.2.1 software (GraphPad Software Inc., San Diego, CA, USA). Reported values are the results of three independent experiments (n = 3) and are presented as mean (dots) ± s.d., standard deviation (bars).
Competitive binding assay on yeast cells
A competitive flow cytometry-based binding assay was performed to rapidly identify the binding site of some selected yeast-encoded macrocyclic peptides. The binding assays were conducted in 96-well conical V-bottom plates (Corning, Tewksbury, MA, USA) containing 2 × 105 induced yeast cells per well. Yeast cells displaying the desired macrocyclic peptide were incubated for 1 hr at 4 °C with a concentration of soluble protein target (PT) that was 10-fold higher than the measured KDapp values. Yeast cells were incubated with either PT alone or PT pre-complexed with at least 100-fold molar excess of a well-known site-specific PT-binding soluble ligand under gentle shacking. Competitive binding assay of yeast-encoded macrocyclic peptides MP2.1, MP2.2, MP2.3, MP2.4, MP2.5 and MP2.6 for binding to streptavidin (PT2) were conducted in the presence or absence of biotin (Merck, Darmstadt, Germany). Competitive binding assay of yeast-encoded macrocyclic peptide MP3.2 for binding to carbapenemase GES-5 from Klebsiella pneumoniae (PT3) was conducted in the presence or absence of the non-β-lactam β-lactamase inhibitor avibactam (Merck, Darmstadt, Germany). Competitive binding assay of yeast-encoded macrocyclic peptides MP5.1, MP5.2, MP5.3, MP5.4, MP5.5 and MP5.6 for binding to bovine α-chymotrypsin (PT5) was conducted in the presence or absence of PMSF (Merck, Darmstadt, Germany) and aprotinin (Cytiva, Marlborough, USA). After the incubation, cells were pelleted (2200 × g for 3 min at 4 °C) and washed twice with 200 μL ice-cold PBSA buffer. Cells were labelled with neutravidin-DyLight 650 at the recommended dilution (Supplementary Table 23) and incubated for 30 min at 4 °C. After the incubation, cells were pelleted (2200 × g for 3 min at 4 °C) and washed twice with 200 μL ice-cold PBSA buffer. The 96-well plates were run on a Attune NxT (Thermo Fischer Scientific, Dreieich, Germany) and data analysed using FlowJo v.10.0.7 software (BD Life Sciences, Franklin Lakes, NJ USA). The binding (MFI) values were normalised to the value obtained in the absence of soluble ligand providing us with a percentage, ranging from 0 to 100%, that corresponds to the residual binding observed upon incubation with the known site-specific soluble ligand. Reported values are the results of three independent experiments (n = 3) and are presented as mean (dots) ± s.d., standard deviation (bars).
Crystallisation and structure determination of a protein target in complex with a macrocyclic peptide
Lyophilised α-chymotrypsin from bovine pancreas (UniProt ID: P00766) was purchased from Fluka (Burlington, MA, USA), dissolved in 50 mM MES, pH 6.0 and further purified by size-exclusion chromatography using a HiLoad 26/600 Superdex 200 prep grade column (Cytiva, Marlborough, MA, USA) pre-equilibrated with the same buffer and connected to an ÄKTA pure 25 M system (Cytiva, Marlborough, MA, USA). The fractions containing the monodisperse protein were pooled and further concentrated by ultrafiltration using 10,000 Da MWCO Amicon ultrafiltration devices (Merck Life Science, Darmstadt, Germany) at 4000 × g and 4 °C on a Heraeus Multifuge X1R centrifuge (Thermo Fisher Scientific, Waltham, MA, USA) to a final value of 25 mg/mL (1 mM). The protein concentration was determined using a BioPhotometer D30 UV spectrophotometer (Eppendorf, Hamburg, Germany). Crystallisation trials of α-chymotrypsin in complex with macrocyclic peptide MP5.4.3 were carried out in SWISSCI MRC 96-well crystallisation plates (Hampton Research, Aliso Viejo, CA, US) using the isothermal (293 K) sitting-drop vapour diffusion method and the PACT premier, LMB and Morpheus II screening kits (Molecular Dimensions Ltd., Sheffield, UK). Formation of protein-peptide complex was induced by incubating α-chymotrypsin (1 mM) with the macrocyclic peptide MP5.4.3 (1.5 mM, 5% v/v DMSO) for 2 hrs at 25 °C. Droplets of 0.6 μL volume (0.3 μL of protein-peptide complex in the presence of 5% v/v DMSO and 0.3 μL of reservoir solution) were dispensed by an Oryx 8 crystallisation robot (Douglas Instruments Ltd., Berkshire, UK) and equilibrated against 70 μL of reservoir solution. Best crystals were obtained in 48–72 hrs using the following precipitant agent: 70% v/v MPD, 0.1 M HEPES, pH 7.5. For X-ray data collection, crystals were soaked in a solution of 20% v/v ethylene glycol in the precipitant buffer, mounted on LithoLoops (Molecular Dimensions Ltd, Suffolk, UK) and flash-frozen in liquid nitrogen. X-ray diffraction data were collected at the ID30-A beamline of the European Synchrotron Radiation Facility (ESRF, Grenoble, France). Crystals belong to the P61 space group. The asymmetric unit contains 1 α-chymotrypsin molecule and a solvent content of 56% of the crystal volume. Reflections were indexed and integrated by the GrenADeS automated processing pipeline82, merged and scaled by AIMLESS83 in the CCP4i2 crystallographic suite84. Phases were determined by molecular replacement with Molrep85 using the PDB entry 4CHA as a template86, via the CCP4i2 interface84. Refinement of the protein model was carried out manually by Coot87 and automatically by Refmac588 and PDB_REDO89. The peptide ligand was parametrised by eLBOW90 via the PHENIX interface91. The presence of macrocyclic peptide MP5.4.3 in α-chymotrypsin active site was confirmed by omit map at 2.5 Å. Since the first cycles of refinement, the electron density corresponding to the bound macrocyclic peptide MP5.4.3 was clearly visible in the electron density map, and the peptide model was built in a well-defined, unique conformation. The final model of the complex contains 1778 protein atoms, 97 macrocyclic peptide atoms and 41 water molecules. The final crystallographic R factor is 0.213 (Rfree 0.28). Geometrical parameters of the model are as expected or better for this resolution. Buried surface calculations were performed using PDBePISA72,92. Intra-molecular and inter-molecular interactions were analysed by LIGPLOT + 93 and PLIP94. All figures were made with PyMOL73. The structure of α-chymotrypsin in complex with macrocyclic peptide MP5.4.3 has been deposited in the Protein Data Bank (PDB) under identification code 9F6H.
Proteolytic stability of a macrocyclic peptide
The stability of the macrocyclic peptide MP5.4.3 was assessed by incubating the peptide (final concentration of 30 μM) with bovine α-chymotrypsin (PT5) at a final concentration of 3 nM, 30 nM and 300 nM for 32 hrs at 37 °C and the resulting species analysed by chromatography and mass spectrometry. All reagents were diluted in 1X PBS pH 7.4 buffer. The final reaction volume of each mixture was 300 μL. The mixtures were incubated at 37 °C on a water bath (VWR, Pennsylvania, PA, USA). After 0, 4, 8, 16 and 32 hrs, 50 μL samples were taken, mixed with 50 μL of filter-sterilised H2O containing 0.1% v/v TFA, and stored at −20 °C until analysis. The samples (100 μL each) were characterised by LC-MS using a 1260 Infinity II LC system coupled to an InfinityLab LC/MSD mass spectrometer (Agilent Technologies, Santa Clara, CA, USA). The system operated with the standard ESI source and in the positive ionisation mode. The reversed-phase HPLC column was a Xterra™ MS C18 (3.5 μm, 150 mm × 4.6 mm, Waters, Millford, MA, USA). The linear gradient elution was 10 − 100% v/v solvent B over 35 min at a flow rate of 1 mL/min (A: 95% v/v H2O, 5% v/v ACN, and 0.1% v/v TFA; B: 95% v/v ACN, 5% v/v H2O, and 0.1% v/v TFA). Data were acquired, processed, and analysed using MestReNova v.12.0.1-20560 (Mestrelab Research, Santiago de Compostela, Spain). Each sample was analysed once (n = 1).
Statistics and reproducibility
Statistical analyses were conducted using FlowJo v10.0.7 software (BD Biosciences), Biacore Insight Evaluation software (Cytiva), GraphPad Prism v8.2.1 software (Dotmatics) and Excel Office 365 (Microsoft), employing predefined evaluation protocols. Data are reported as mean ± standard deviation (s.d.), unless otherwise specified. Details regarding the number of biological replicates (n) and the statistical tests applied can be found in the corresponding figure legends. Each experiment was independently repeated at least three times with reproducible outcomes. While no statistical methods were used to predetermine sample size, the chosen sample sizes are consistent with those commonly used in similar studies. The experiments were not randomised, and the investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
NGS raw and analysed data, mass spectrometry raw data, X-ray crystallography data and MATLAB scripts generated and applied in this study have been deposited in the Code Ocean open data repository database under accession code 18-535681-2 [https://codeocean.com/signup/nature?token=15583644fba64b518288224ac852a35d]. Additional flow cytometry raw and analysed data, inhibitory activity assay data, NGS analysed data, X-ray crystallography analysed data and surface plasmon resonance raw data generated in this study are provided in the Supplementary Information and Source data. X-ray structure: PDB code 9F6H. NGS raw data: BioProject NCBI accession code PRJNA1262878. Source data are provided with this paper.
Code availability
MATLAB code is available in the Code Ocean open data repository database [https://codeocean.com/signup/nature?token=15583644fba64b518288224ac852a35d].
References
Ji, X., Nielsen, A. L. & Heinis, C. Cyclic peptides for drug development. Angew. Chem. Int. Ed. 63, e202308251 (2024).
Muttenthaler, M., King, G. F., Adams, D. J. & Alewood, P. F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 20, 309–325 (2021).
Fetse, J., Kandel, S., Mamani, U.-F. & Cheng, K. Recent advances in the development of therapeutic peptides. Trends Pharmacol. Sci. 44, 425–441 (2023).
Zhang, H. & Chen, S. Cyclic peptide drugs approved in the last two decades (2001-2021). RSC Chem. Biol. 3, 18–31 (2022).
Li, X., Craven, T. W. & Levine, P. M. Cyclic peptide screening methods for preclinical drug discovery. J. Med. Chem. 65, 11913–11926 (2022).
Smith, G. P. & Petrenko, V. A. Phage Display. Chem. Rev. 97, 391–410 (1997).
Deyle, K., Kong, X.-D. & Heinis, C. Phage Selection of Cyclic Peptides for Application in Research and Drug Development. Acc. Chem. Res. 50, 1866–1874 (2017).
Kamalinia, G., Grindel, B. J., Takahashi, T. T., Millward, S. W. & Roberts, R. W. Directing evolution of novel ligands by mRNA display. Chem. Soc. Rev. 50, 9055–9103 (2021).
Peacock, H. & Suga, H. Discovery of de novo macrocyclic peptides by messenger RNA display. Trends Pharmacol. Sci. 42, 385–397 (2021).
Lee, S. Y., Choi, J. H. & Xu, Z. Microbial cell-surface display. Trends Biotechnol. 21, 45–52 (2003).
Palei, S., Jose, J. & Mootz, H. D. Preparation of bacterial cell-surface displayed semisynthetic cyclic peptides. Methods Mol. Biol. 2371, 193–214 (2022).
Tavassoli, A. SICLOPPS cyclic peptide libraries in drug discovery. Curr. Opin. Chem. Biol. 38, 30–35 (2017).
Passioura, T., Katoh, T., Goto, Y. & Suga, H. Selection-based discovery of druglike macrocyclic peptides. Annu. Rev. Biochem. 83, 727–752 (2014).
Sharma, K., Sharma, K. K., Sharma, A. & Jain, R. Peptide-based drug discovery: current status and recent advances. Drug Discov. Today 28, 103464 (2023).
Jaroszewicz, W., Morcinek-Orłowska, J., Pierzynowska, K., Gaffke, L. & Wȩgrzyn, G. Phage display and other peptide display technologies. FEMS Microbiol. Rev. 46, 1–25 (2022).
Dotter, H., Boll, M., Eder, M. & Eder, A. C. Library and post-translational modifications of peptide-based display systems. Biotechnol. Adv. 47, 107699 (2021).
Smith, T. P. et al. Identification and engineering of potent cyclic peptides with selective or promiscuous binding through biochemical profiling and bioinformatic data analysis. RSC Chem. Biol. 5, 12–18 (2023).
Stellwagen, S. D. et al. The next generation of biopanning: next gen sequencing improves analysis of bacterial display libraries. BMC Biotechnol. 19, 100 (2019).
Sloth, A. B., Bakhshinejad, B., Stavnsbjerg, C., Rossing, M. & Kjaer, A. Depth of Sequencing Plays a Determining Role in the Characterization of Phage Display Peptide Libraries by NGS. Int. J. Mol. Sci. 24, 5396 (2023).
Vodnik, M., Zager, U., Strukelj, B. & Lunder, M. Phage display: Selecting straws instead of a needle from a haystack. Molecules 16, 790–817 (2011).
Wang, H. & Liu, R. Advantages of mRNA display selections over other selection techniques for investigation of protein-protein interactions. Expert Rev. Proteom. 8, 335–346 (2011).
Angelini, A. et al. Protein engineering and selection using yeast surface. Methods Mol. Biol. 1319, 3–36 (2015).
Boder, E. T. & Wittrup, K. D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 15, 553–557 (1997).
Cherf, G. M. & Cochran, J. R. Applications of yeast surface display for protein engineering. Methods Mol. Biol. 1319, 155–175 (2015).
Lopez-Morales, J. et al. Protein engineering and high-throughput screening by yeast surface display: survey of current methods. Small Sci. 3, 2300095 (2023).
Teymennet-Ramírez, K. V., Martínez-Morales, F. & Trejo-Hernández, M. R. Yeast surface display system: strategies for improvement and biotechnological applications. Front. Bioeng. Biotechnol. 9, 1–10 (2022).
Lopez-Morales, J., Vanella, R., Kovacevic, G., Santos, M. S. & Nash, M. A. Titrating Avidity of Yeast-Displayed Proteins Using a Transcriptional Regulator. ACS Synth. Biol. 12, 419–431 (2023).
Linciano, S., Pluda, S., Bacchin, A. & Angelini, A. Molecular evolution of peptides by yeast surface display technology. Medchemcomm 10, 1569–1580 (2019).
van Rosmalen, M. et al. Affinity maturation of a cyclic peptide handle for therapeutic antibodies using deep mutational scanning. J. Biol. Chem. 292, 1477–1489 (2017).
Ishii, J. et al. Cell wall trapping of autocrine peptides for human G-protein-coupled receptors on the yeast cell surface. PLoS ONE 7, e37136 (2012).
Bacon, K. et al. Isolation of chemically cyclized peptide binders using yeast surface display. ACS Comb. Sci. 22, 519–532 (2020).
Bowen, J. et al. Screening of yeast display libraries of enzymatically treated peptides to discover macrocyclic peptide ligands. Int. J. Mol. Sci. 22, 1634 (2021).
McMahon, C. et al. Yeast surface display platform for rapid discovery of conformationally selective nanobodies. Nat. Struct. Mol. Biol. 25, 289–296 (2018).
Erlendsson, S. & Teilum, K. Binding revisited—avidity in cellular function and signaling. Front. Mol. Biosci. 7, 1–13 (2021).
Cendron, L. et al. X-ray Crystallography deciphers the activity of broad-spectrum boronic acid β-lactamase inhibitors. ACS Med. Chem. Lett. 10, 650–655 (2019).
Celenza, G. et al. Phenylboronic acid derivatives as validated leads active in clinical strains overexpressing KPC-2: a step against bacterial resistance. ChemMedChem 13, 713–724 (2018).
Santucci, M. et al. Computational and biological profile of boronic acids for the detection of bacterial serine- and metallo-β-lactamases. Sci. Rep. 7, 17716 (2017).
Gill, C. M., Oliver, A., Fraile-Ribot, P. A. & Nicolau, D. P. In vivo translational assessment of the GES genotype on the killing profile of ceftazidime, ceftazidime/avibactam and meropenem against Pseudomonas aeruginosa. J. Antimicrob. Chemother. 77, 2803–2808 (2022).
Fujinaga, M. et al. Crystal and molecular structures of the complex of α-chymotrypsin with its inhibitor Turkey ovomucoid third ___domain at 1.8 Å resolution. J. Mol. Biol. 195, 397–418 (1987).
Singh, N. et al. Detection of native peptides as potent inhibitors of enzymes. Crystal structure of the complex formed between treated bovine alpha-chymotrypsin and an autocatalytically produced fragment, IIe-Val-Asn-Gly-Glu-Glu-Ala-Val-Pro-Gly-Ser-Trp-Pro-Trp, at 2.2 ang. FEBS J. 272, 562–572 (2005).
Tulinsky, A. & Blevins, R. A. Structure of a tetrahedral transition state complex of alpha-chymotrypsin dimer at 1.8-A resolution. J. Biol. Chem. 262, 7737–7743 (1987).
Oppewal, T. R., Jansen, I. D., Hekelaar, J. & Mayer, C. A strategy to select macrocyclic peptides featuring asymmetric molecular scaffolds as cyclization units by phage display. J. Am. Chem. Soc. 144, 3644–3652 (2022).
Bellotto, S., Chen, S., Rentero Rebollo, I., Wegner, H. A. & Heinis, C. Phage selection of photoswitchable peptide ligands. J. Am. Chem. Soc. 136, 5880–5883 (2014).
Krook, M., Lindbladh, C., Eriksen, J. A. & Mosbach, K. Selection of a cyclic nonapeptide inhibitor to α-chymotrypsin using a phage display peptide library. Mol. Divers. 3, 149–159 (1997).
Giebel, L. B. et al. Screening of cyclic peptide phage libraries identifies ligands that bind streptavidin with high affinities. Biochemistry 34, 15430–15435 (1995).
Jafari, M. R. et al. Discovery of light-responsive ligands through screening of a light-responsive genetically encoded library. ACS Chem. Biol. 9, 443–450 (2014).
Owens, A. E., Iannuzzelli, J. A., Gu, Y. & Fasan, R. MOrPH-PhD: an integrated phage display platform for the discovery of functional genetically encoded peptide macrocycles. ACS Cent. Sci. 6, 368–381 (2020).
Brown, L. et al. Proximity-driven site-specific cyclization of phage-displayed peptides. Nat. Commun. 15, 7308 (2024).
Hacker, D. E., Hoinka, J., Iqbal, E. S., Przytycka, T. M. & Hartman, M. C. T. Highly constrained bicyclic scaffolds for the discovery of protease-stable peptides via mRNA display. ACS Chem. Biol. 12, 795–804 (2017).
Lamla, T. & Erdmann, V. A. Searching sequence space for high-affinity binding peptides using ribosome display. J. Mol. Biol. 329, 381–388 (2003).
Bessette, P. H., Rice, J. J. & Daugherty, P. S. Rapid isolation of high-affinity protein binding peptides using bacterial display. Protein Eng. Des. Sel. 17, 731–739 (2004).
Koivunen, E., Wang, B. & Ruoslahti, E. Isolation of a highly specific ligand for the alpha 5 beta 1 integrin from a phage display library. J. Cell Biol. 124, 373–380 (1994).
Tanaka, M. M., Sisson, S. A. & King, G. C. High affinity extremes in combinatorial libraries and repertoires. J. Theor. Biol. 261, 260–265 (2009).
Faller, B. & Bieth, J. G. Kinetics of the interaction of chymotrypsin with eglin c. Biochem. J. 280, 27–32 (1991).
McBride, J. D. & Leatherbrrow, R. J. Synthetic peptide mimics of the Bowman-Birk inhibitor protein. Curr. Med. Chem. 8, 909–917 (2001).
Tanaka, A. S. et al. Purification and Primary Structure Determination of a Bowman-Birk Trypsin Inhibitor from Torresea cearensis Seeds. 378, 273–282 (1997).
Barbosa, J. A. R. G. et al. Crystal Structure of the Bowman-Birk Inhibitor from Vigna unguiculata Seeds in Complex with β-Trypsin at 1.55 Å Resolution and Its Structural Properties in Association with Proteinases. Biophys. J. 92, 1638–1650 (2007).
Zhang, L. et al. A Bowman-Birk type chymotrypsin inhibitor peptide from the amphibian, Hylarana erythraea. Sci. Rep. 8, 5851 (2018).
Kellenberger, C. et al. Serine protease inhibition by insect peptides containing a cysteine knot and a triple-stranded beta-sheet. J. Biol. Chem. 270, 25514–25519 (1995).
Scheidig, A. J., Hynes, T. R., Pelletier, L. A., Wells, J. A. & Kossiakoff, A. A. Crystal structures of bovine chymotrypsin and trypsin complexed to the inhibitor ___domain of alzheimer’s amyloid β-protein precursor (APPI) and basic pancreatic trypsin inhibitor (BPTI): engineering of inhibitors with altered specificities. Protein Sci. 6, 1806–1824 (1997).
Pan, X. & Kortemme, T. Recent advances in de novo protein design: principles, methods, and applications. J. Biol. Chem. 296, 100558 (2021).
Listov, D., Goverde, C. A., Correia, B. E. & Fleishman, S. J. Opportunities and challenges in design and optimization of protein function. Nat. Rev. Mol. Cell Biol. 25, 639–653 (2024).
Mcconnell, A. & Hackel, B. J. Protein engineering via sequence-performance mapping. Cell Syst. 16, 656–666 (2023).
Yu, T., Boob, A. G., Singh, N., Su, Y. & Zhao, H. In vitro continuous protein evolution empowered by machine learning and automation. Cell Syst. 16, 633–644 (2023).
Jenson, J. M. et al. Peptide design by optimization on a dataparameterized protein interaction landscape. Proc. Natl. Acad. Sci. USA 115, E10342–E10351 (2018).
Freschlin, C. R., Fahlberg, S. A. & Romero, P. A. Machine learning to navigate fitness landscapes for protein engineering. Curr. Opin. Biotechnol. 75, 102713 (2022).
He, J., Ghosh, P. & Nitsche, C. Biocompatible strategies for peptide macrocyclisation. Chem. Sci. 15, 2300–2322 (2024).
Kale, S. S. et al. Cyclization of peptides with two chemical bridges affords large scaffold diversities. Nat. Chem. 10, 715–723 (2018).
Ma, J., Yan, L., Yang, J., He, Y. & Wu, L. Effect of modification strategies on the biological activity of peptides/proteins. ChemBioChem 25, e202300481 (2024).
Jimmidi, R. Synthesis and applications of peptides and peptidomimetics in drug discovery. Eur. J. Org. Chem. 26, e202300028 (2023).
Pravda, L. et al. MOLEonline: a web-based tool for analyzing channels, tunnels and pores (2018 update). Nucleic Acids Res 46, W368–W373 (2018).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007).
The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC (2015).
Pedretti, A., Mazzolari, A., Gervasoni, S., Fumagalli, L. & Vistoli, G. The VEGA suite of programs: an versatile platform for cheminformatics and drug design projects. Bioinformatics 37, 1174–1175 (2021).
Laskowski, R. A., Jabłońska, J., Pravda, L., Vařeková, R. S. & Thornton, J. M. PDBsum: Structural summaries of PDB entries. Protein Sci. 27, 129–134 (2018).
Gasteiger, E. et al. Protein Identification and Analysis Tools on the ExPASy Server BT - The Proteomics Protocols Handbook. Proteomics Protoc. Handb. 571–607 (2005).
Van Deventer, J. A., Kelly, R. L., Rajan, S., Wittrup, K. D. & Sidhu, S. S. A switchable yeast display/secretion system. Protein Eng. Des. Sel. 28, 317–3325 (2015).
Rebollo, I. R., Sabisz, M., Baeriswyl, V. & Heinis, C. Identification of target-binding peptide motifs by high-throughput sequencing of phage-selected peptides. Nucleic Acids Res. 42, e169 (2014).
Angelini, A. et al. Directed evolution of broadly crossreactive chemokine-blocking antibodies efficacious in arthritis. Nat. Commun. 9, 1461 (2018).
Mazzocato, Y. et al. A novel genetically-encoded bicyclic peptide inhibitor of human urokinase-type plasminogen activator with better cross-reactivity toward the murine orthologue. Bioorg. Med. Chem. 95, 117499 (2023).
Chinellato, M. et al. Folding of Class IIa HDAC Derived Peptides into α-helices Upon Binding to Myocyte Enhancer Factor-2 in Complex with DNA. J. Mol. Biol. 436, 168541 (2024).
Monaco, S. et al. Automatic processing of macromolecular crystallography X-ray diffraction data at the ESRF. J. Appl. Crystallogr. 46, 804–810 (2013).
Evans, P. R. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D. Biol. Crystallogr. 67, 282–292 (2011).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D. Biol. Crystallogr. 67, 235–242 (2011).
Vagin, A. & Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D. Biol. Crystallogr. 66, 22–25 (2010).
Tsukada, H. & Blow, D. M. Structure of α-chymotrypsin refined at 1.68 Å resolution. J. Mol. Biol. 184, 703–711 (1985).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D. Biol. Crystallogr. 67, 355–367 (2011).
Joosten, R. P., Long, F., Murshudov, G. N. & Perrakis, A. The PDB_REDO server for macromolecular structure model optimization. IUCrJ 1, 213–220 (2014).
Moriarty, N. W., Grosse-Kunstleve, R. W. & Adams, P. D. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D. Biol. Crystallogr. 65, 1074–1080 (2009).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. Sect. D., Struct. Biol. 75, 861–877 (2019).
Krissinel, E. Crystal contacts as Nature’ s Docking Solutions. J. Comput. Chem. 31, 133–143 (2009).
Laskowski, R. A. & Swindells, M. B. LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786 (2011).
Adasme, M. F. et al. PLIP 2021: expanding the scope of the protein-ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 49, W530–W534 (2021).
Perona, J. J. & Craik, C. S. Evolutionary Divergence of Substrate Specificity within the Chymotrypsin-like Serine Protease Fold *. J. Biol. Chem. 272, 29987–29990 (1997).
Acknowledgements
The authors would like to thank Dr. Giuseppe Borsato, Dr. Alessandro Bonetto, Mr. Giacomo Bettin and Ms. Linda Trevisan for technical assistance with peptide purification and mass spectrometry analysis. We thank Dr. Federica Giummolè for support with statistical analysis. We are grateful to Mr. Federico Cusinato of the Department of Pharmaceutical and Pharmacological Sciences at the University of Padova for technical assistance during FACS experiments. We thank Dr. Kelvin Lau at the protein production and structure core facility at EPFL for technical assistance during the surface plasmon resonance experiments. The authors would like to thank Dr. Gordon Leonard and the staff of ID30-A-3 beamline of the European Synchrotron Radiation Facility (ESRF, Grenoble, France) for assistance with crystal testing and data collection. We are grateful to all the group members for helpful discussions and for critical reading of this manuscript. This material is based upon work supported by the National Recovery and Resilience Plan and the European Union—Next Generation EU, Mission 4, Component 2, CUP B93D21010860004, as part of the research activities of the ‘National Centre for Gene Therapy and Drugs based on RNA Technology’, Spoke 8 ‘Platforms for RNA/DNA delivery’, under the project ALLIANCE (to S.L., Z.R., M.S., A.S., and A.A.).
Author information
Authors and Affiliations
Contributions
S.L. and A.A. conceived the study; S.L. generated the libraries; S.L. and Y.M. performed the screening, bioinformatic analyses and determined the binding affinities and specificities; Z.R. performed competition studies; S.L., Y.M., E.W., and Y.X. synthesised and purified macrocyclic peptides; S.L., L.F.S., and Y.K. performed surface plasmon resonance experiments; F.V. and L.C. solved and analysed the X-ray structure; M.S. supervised bioinformatic analyses; S.C., A.S., and C.H. supervised chemical synthesis, modification and purification. All authors analysed the data, discussed the results, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
S.L., Y.M., Z.R., and A.A. declare that they are co-founders of Arzanya S.r.l. and have submitted a patent application that has been filed in the Italian Patent and Trademark Office on behalf of the Ca’ Foscari University of Venice pertaining to the generation of the yeast display macrocyclic peptides naive libraries of this work (international publication number WO/2025/083029 entitled ‘Generation of disulfide-tethered macrocyclic peptide libraries displayed on the surface of yeast cells’). The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ratmir Derda, Xu-Dong Kong and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Linciano, S., Mazzocato, Y., Romanyuk, Z. et al. Screening macrocyclic peptide libraries by yeast display allows control of selection process and affinity ranking. Nat Commun 16, 5367 (2025). https://doi.org/10.1038/s41467-025-60907-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60907-x
