
Abstract
Prader-Willi Syndrome (PWS) is caused by the loss of expression of paternally expressed genes in the human 15q11.2-q13 imprinting ___domain. A set of imprinted genes that are active on the paternal but silenced on the maternal chromosome are intricately regulated by a bipartite imprinting center (PWS-IC) located in the PWS imprinting ___domain. We previously discovered that euchromatic histone lysine N-methyltransferase-2 (EHMT2/G9a) inhibitors are capable of un-silencing PWS-associated genes by restoring their expression from the maternal chromosome. Here, in mice lacking the Ehmt2 gene, we document un-silencing of the imprinted Snrpn/Snhg14 gene on the maternal chromosome in the late embryonic and postnatal brain. Using PWS and Angelman syndrome patient derived cells with either paternal or maternal deletion of 15q11.2-q13, we have found that chromatin of maternal PWS-IC is closed and has compact 3D folding confirmation. We further show that a distinct noncoding RNA (TSS4-280118) preferentially transcribed from the upstream of the PWS-IC of maternal chromosome interacts with EHMT2 and forms a heterochromatin complex in CIS on the maternal chromosome. Inactivation of TSS4-280118 by CRISPR/Cas9 editing results in unsilencing of the expression of SNRPN and SNORD116 from the maternal chromosome. Taken together, these findings demonstrate that allele-specific recruitment of EHMT2 is required to maintain the maternal imprints. Our findings provide mechanistic insights and support a model for imprinting maintenance of the PWS imprinted ___domain.
Similar content being viewed by others


Introduction
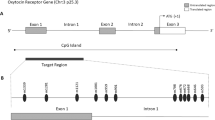
Imprinted genes, unique to mammals and flowering plants, are regulated such that they are expressed from either the maternal or paternal allele but not both1,2. Frequently clustered in the same chromosomal ___domain, imprinted genes are coordinately controlled by an imprinting center (IC) or control region (ICR). Since the discovery of genomic imprinting3,4, investigations have focused on two fundamental questions: how cells recognize the paternal and maternal alleles and how cells maintain allele-specific repression of imprinted genes. The imprinting ___domain of chromosome 15q11.2-q13, implicated in the pathogenesis of both Prader-Willi syndrome (PWS) and Angelman syndrome (AS), has long been an established model for investigating genomic imprinting5,6,7. The 15q11.2-q13 imprinting ___domain contains a PWS imprinting center (PWS-IC), an AS-imprinting center (AS-IC) and two functionally distinct and oppositely imprinted regions8,9,10. Paternally expressed genes SNURF/SNRPN, SNORD116, SNORD115, and SNHG14 are located within a 0.5 Mb region distal to the PWS-IC, and paternally expressed genes NDN, MAGLE2, and MKRN3 are ~1.5 Mb proximal to the PWS-IC. However, how the PWS-IC controls allele-specific expression bidirectionally remains poorly understood.
The genomic organization of the 15q11.2-q13 imprinting ___domain is highly conserved in the chromosome 7C region in mice11, and the general imprinting regulation mechanism is highly similar in humans and rodents. However, at a sequence level, specific regulatory elements are not highly conserved, presenting challenges for mechanistic analyses in animal models. At a structural level, the critical region of PWS-IC has been mapped to a 4.1 kb region that includes the CpG island (CGI) encompassing exon 1 of SNRPN8,9,12. PWS-IC is methylated (5mC) on the maternal chromosome in oocytes but unmethylated on the paternal chromosome in sperm13. Thus, after fertilization, the maternal methylation signature provides an allele-specific methylation imprint that becomes resistant to epigenetic reprogramming14,15. Although the maternal methylation signatures correlate with repressed expression of paternally expressed genes on the maternal chromosome, exactly how the methylated CGI within the PWS-IC silences these genes has remained elusive.
The PWS-IC on the paternal chromosome regulates the expression of paternally expressed genes8,16, and when deleted from the paternal chromosome in germ cells in both human and mice, the unmethylated pattern of the paternal chromosome became methylated pattern that resembles of maternal chromosome, and the paternal gene expression is lost in offspring16,17,18,19. In contrast, the role of PWS-IC on the maternal chromosome and the mechanism underlying silenced expression in the maternal chromosome are unknown. In mouse embryonic stem cells (mESCs) and embryos null for the DNA methyltransferase Dnmt1, methylation of CGI in the PWS-IC on the maternal chromosome was lost20,21. Although transcription of the paternally expressed Snrpn gene on the maternal chromosome remained repressed in Dnmt1-null mESCs20, Snrpn gene expression was unsilenced in Dnmt1-null embryos21. Similarly, treatment with DNA methylation inhibitors in PWS patient-derived cells with a paternal 15q11.2-q13 deletion reduced CGI DNA methylation (5mC) and unsilenced SNRPN expression from the maternal chromosome22,23. In our large-scale small molecule screen in Snrpn-EGFP mouse embryonic fibroblasts, we discovered that inhibitors of the nuclear histone lysine methyltransferase EHMT2, also known as G9a, were capable of unsilencing the imprinted SNRPN and SNHG14 genes from the maternal chromosome, both in human fibroblasts derived from patients with PWS and in a PWS mouse model22. Unexpectedly, EHMT2 inhibitors unsilenced the repressed expression but did not change the DNA methylation of PWS-IC22.
EHMT2 catalyzes mono- and di-methylation of histone H3 lysine 9 (H3K9) or other non-histone proteins24. H3K9me2 is a repressive mark for epigenetic transcriptional silencing, leading to heterochromatin assembly with chromodomain-containing proteins of the HP 1 family24,25,26,27. EHMT2 plays a significant role to maintain imprinted DNA methylation, including at the PWS-IC, in embryonic stem cells; this activity is independent from its catalytic activity responsible for H3K9 methylation28. Recent chromatin structure studies support an important role for EHMT2-mediated H3K9 in the 3D genome organization of chromatin29. Consistently, chromatin loops and topological associated domains (TADs) undergo alterations in EHMT2-deficient mouse ESCs or EHMT2 inhibitor-treated hepatocytes, resulting in derepression of a subset of nuclear envelope-bound genes30,31,32.
Here, we set out to decipher how EHMT2 inhibition instructs unsilencing of PWS imprinted genes. We found that Ehmt2 deficiency in mouse forebrain is sufficient to unsilence the expression of imprinted Snrpn on the maternal chromosome, without any reduction in DNA methylation of PWS-IC. In human PWS and AS patient-derived fibroblasts with paternal or maternal deletion of 15q11.2-q13, respectively, we found that EHMT2 preferentially binds to maternal PWS-IC and enriches H3K9me2, independent of the allele-specific DNA methylation status at the PWS-IC, and that chromatin of the maternal PWS imprinting ___domain is in a closed and compact 3D folding confirmation. We further show that maternal-specific noncoding RNA (ncRNA) TSS4-280118, preferentially transcribed from the maternal chromosome of upstream the PWS-IC interacts with EHMT2 and forms a heterochromatin repressor complex. Inactivation of TSS4-280118 ncRNA by CRISPR/Cas9 editing resulted in unsilencing of the expression of SNRPN and SNROD116 in CIS from the maternal chromosome. These fundamental findings support that EHMT2 plays a central role of maintaining the silenced expression of paternally expressed genes in the maternal chromosome.
Results
Ehmt2 deficiency unsilenced the expression of imprinted Snprn gene from the maternal chromosome
Based on our previous EHMT2/G9a inhibitor study22, we hypothesized that expression of imprinted PWS genes from the maternal chromosome is unsilenced upon EHMT2 depletion. Ehmt2 deficiency in germline results in early embryonic lethality before E9.5 day33. Mice with conditional inactivation of Ehmt2 in brains using Nestin-Cre, CamkII-Cre, Drd1-Cre, and Drd2-Cre are viable but have transcriptional alterations and complex behavioral phenotypes34,35. We bred Ehmt2 flox/flox mice (Ehmt2f/f)36 with Nestin-Cre mice to generate a brain-specific Ehmt2 knockout starting embryonic day 11 on the maternal Snrpn-EGFP reporter background (Nestin-Cre+/−; Ehmt2f/f; mSnrpn-EGFP/p+) (Fig. 1a). We then confirmed that EHMT2 protein levels and H3K9me2 levels were reduced in p10 Ehmt2 conditional knockout mouse forebrain tissue (Fig. 1b, c) and that unsilencing of Snrpn-EGFP from the maternal chromosome was readily detected by RT-PCR (Fig. 1d, e). Through ChIP analysis, we validated that H3K9me2 levels on the PWS-IC were significantly decreased in Ehmt2 deficient brains (Fig. 1f and Supplementary Fig. 1a) but that CGI DNA methylation was not affected (Fig. 1g). Through RNA-seq analysis, we documented overexpression of Snurf-Snrpn (a bicistronic transcript) in Ehmt2 deficient brains (Fig. 1h). We noted the increased expression of Ipw/Snhg14 and Snord116 which is the part of extended transcript of Snurf/Snrpn was less consistent, and this is likely due to the low sensitivity of bulk RNA-seq for non-coding RNAs containing low copy repeat. Notably, the expression of other PWS-associated paternally expressed genes of Magel2, Ndn, and Mkrn3 in the distal side of PWS-IC in chromosome 7C region was not consistently upregulated. This observation indicates that silencing of paternally expressed genes at distal and proximal sides is mediated by a different mechanism. We were intrigued about the finding that other paternally expressed genes, such as Cdh15, Galnt6, and Dlk1, were either significantly upregulated (Cdh15) or downregulated (Galnt6 and Dlk1) (Supplementary Fig. 1b). We performed influence analysis related to EHMT2. Only Cdh15 transcript was negatively correlated with Ehmt2 expression (Supplementary Fig. 1c). In line with this finding, ChIP analysis showed less binding affinity of EHMT2 on Cdh15 loci with reduced H3K9me2 (Fig. 1i, j) in Ehmt2-deficient brains at p10. These results suggested that EHMT2 also acts as a repressor in regulating Cdh15 as well as PWS-IC. Meanwhile, there was no change of UBE3A expression in the brain of Ehmt2 deficiency (Supplementary Fig. 1d, e). Furthermore, when conducting gene ontology analysis, we found that genes differentially regulated in the forebrain of Ehmt2-deficient mice were significantly enriched for RNA splicing and DNA repair processes (Supplementary Fig. 1f, g). Taken together, these findings demonstrate that EHMT2 rather than DNA methylation in PWS-IC is associated with silencing of paternally expressed genes Snurf-Snrpn at the proximal side but not Ndn, Magel2, and Mkrn3 at the distal side in the maternal chromosome.

a Schematic figure shows a strategy for generating Ehmt2 cKO mice with maternal Snrpn-EGFP (Exon 3 of Snrpn is fused in-frame with EGFP, Ex3-EGFP). b The level of EHMT2 protein was significantly decreased in forebrains of Ehmt2 cKO (Nestin-Cre + ;f/f;p+/mS-E) mice at p10. c H3K9me2 level was decreased in forebrains of Ehmt2 cKO mice at p10. b, c Data are presented as mean ± SD. (Two-tailed Student’s t test; p = 0.0083 and p = 0.0023, respectively). d RT–PCR analysis detected the expression of Snrpn-EGFP (Ex3-EGFP) in the forebrains of Ehmt2 cKO mice carrying maternal Snrpn-EGFP at p10 (RTase: +/−, with or without reverse transcriptase). e Quantitative RT–qPCR analysis of Snrpn and Snrpn-EGFP mRNA levels in the forebrains at p10. Data are presented as mean ± SEM (Two-tailed Student’s t test; ****p < 0.0001). f ChIP–qPCR quantification of H3K9me2 and H3K9ac in brains of Ehmt2 cKO mice at E18. Enrichment of H3K9me2 was significantly reduced PWS-IC (IC-1 and IC2) of Ehmt2 cKO mice. Data are presented as mean ± SEM (Two-tailed Student’s t test; p value is indicated in graphs). g Comparison of the DNA methylation in PWS-IC between control and Ehmt2 cKO mice at E18 (Two-tailed Student’s t test; ns, not significant). h The genes in human 15q11.2-q13 and its homologous region mouse central chromosome 7. Created in BioRender. Qiao, S. (2025) https://BioRender.com/v2500u0. The paternally expressed genes are highlighted in blue. Heat map of the expression of genes located in the central chromosome 7 between control (Ehmt2f/f;m+/pSnrpn-EGFP) and Ehmt2 cKO (Nestin-Cre+/−;Ehmt2f/f;mSnrpn-EGFP/p+) forebrain at p10 (adj p value < 0.05). i, j ChIP-qPCR quantification of EHMT2 and H3K9me2 on imprinted genes in control (Ehmt2f/f;mSnrpn-EGFP/p+) and Ehmt2 cKO (Nestin-Cre+/−;Ehmt2f/f;mSnrpn-EGFP/p+) forebrain at p10. Data are presented as mean ± SEM (One-tailed Student’s t test; p value is indicated in graphs). Source data are provided as a Source Data file.
EHMT2 binds preferentially to the PWS-IC of maternal chromosome that is independent from DNA methylation
To examine interactions between EHMT2 and the maternal and paternal alleles of the PWS-IC, we performed ChIP analysis in human fibroblasts derived from a patient with PWS (a 6 Mb paternal deletion of 15q11.2-q13) and a patient with AS (a 6 Mb maternal deletion of 15q11.2-q13) (Fig. 2a, b). Compared to the paternal chromosome, we found that EHMT2 binding and H3K9me2 levels were significantly enriched in the PWS-IC of the maternal chromosome (Fig. 2c, d). To investigate whether methylation of CpGs of PWS-IC is a prerequisite for H3K9me2 enrichment, we treated fibroblasts with DNA methylation inhibitor of 5-Aza-2’-deoxycytidine (5-Aza) and confirmed by bisulfite-based PCR that CpG methylation (5mC) on PWS-IC including CGI and exon 1 of the SNRPN gene was reduced (Supplementary Fig. 2a). Because H3K9me2 enrichment in the PWS-IC was not altered despite the reduction of DNA methylation, we conclude that EHMT binding and H3K9me2 enrichment in the PWS-IC on the maternal chromosome does not require CpG methylation (Fig. 2e and Supplementary Fig. 2b).

a Schematic diagram of PWS-associated imprinted ___domain on chromosome 15 (blue allele; paternally expressed genes, gray allele; biallelic expressed gene, BP; break point found in PWS patient with 15q11.2-q13 deletion. Green bar; CGIs). b Schematic diagram of primer binding region (a,b,c within PWS imprinting center. c–e ChIP–qPCR quantification of EHMT2 and H3K9me2 on PWS-IC in human fibroblasts derived from Angelman syndrome (AS) and PWS patients (AS; paternal 15q11.2-q13 deletion, PWS; maternal 15q11.2-q13 deletion, Ctr; Control). H3K9me2 is significantly abundant on PWS-IC of maternal chromosome compared to AS and Control after treatment of 5-Aza as a DNMT1 inhibitor. (One-way ANOVA test followed by Dunnett’s multiple comparison test; Adj p-value is indicated in graph; 50th percentile (median value, line), 25th to 75th percentiles of dataset (box), 5th and 95th percentile (Whiskers)). f Schematic diagram of primer binding site on CGI in PWS-IC for bisulfite genomic sequencing. g Comparison of DNA methylation in AS (paternal CGI), PWS type I deletion, and Uniparental disomy (UPD) (maternal CGI), and a control (gray; unmethylated CpG, black; methylated CpG). h Quantification of DNA methylation level on PWS-IC (Two-way ANOVA test followed by Tukey’s multiple comparison test; ****p < 0.0001). Data are presented as mean ± SEM. i Genome-wide methylation analysis shows that allele-specific DNA methylation is shown in PWS-IC but not other CGIs. Heat map depicts average methylation scores (0; unmethylated CpG, 1; methylated CpG). Source data are provided as a Source Data file.
We further investigated the methylation status of CpG dinucleotides in the 15q11.2-q13 imprinting ___domain in PWS patient fibroblasts with a paternal deletion or maternal uniparental disomy (UPD) and in AS patients with a maternal deletion (Fig. 2f, g). Using bisulfite genomic sequencing, we confirmed that maternal allele-specific DNA methylation for the CGI was associated with the PWS-IC and noted that, relative to the distal region (CpG 3’), CG dinucleotides in the proximal CGI (CpG 1’) region were more methylated in paternal chromosomes (Fig. 2h). We then used methylome arrays to extend the methylation analysis to the remaining 15q11.2-q13 regions. This analysis confirmed allelic methylation of CGI of SNRPN but unexpectedly, did not reveal consistent allele-specific methylation of CGIs associated with other paternally expressed genes, such as previously reported for MAGEL2, NDN, and MKRN3 using traditional low throughput methods37,38,39,40,41 (Fig. 2i). The discrepancy may reflect the inter-individual variability of methylation status or different resolution of method used for DNA methylation analyses.
Allele-specific chromatin accessibility is associated with PWS imprinting ___domain but not affected by EHMT2 inhibition
Next, to examine chromatin accessibility in the PWS-associated region, we performed ATAC-seq using PWS and AS fibroblasts with a paternal or maternal deletion. This analysis revealed that regulatory regions, including the CGI in the PWS-associated region on maternal chromosome (PWS), were in a closed state compared to the paternal chromosome (AS) and normal control (Ctr) (Fig. 3a). The peaks of chromatin accessibility in the CGIs were similar between control and AS, suggesting that the paternal region was in an open state to maintain active transcription of PWS-associated genes. The regulatory UBE3A regions that are bi-allelically expressed in fibroblasts were in an open chromatin state in both maternal and paternal chromosomes (Fig. 3a). We noted a significant peak of chromatin accessibility at the upstream region of SNRPN (50 kb from u1B) that was associated with the paternal chromosome (64 CpGs, marked with * in Fig. 3a). ChIP analysis revealed that this region was more enriched with H3K9me2 on the maternal chromosome than the paternal chromosome (Fig. 3b).

a Genome viewer screenshot of ATAC-seq analysis illustrating a closed chromatin state of maternal imprinting domains (PWS) and an open chromatin state of paternal imprinting domains (AS). The control (Ctr) also shows an open chromatin state. b ChIP–qPCR quantification of H3K9me2 on the upstream region of SNRPN gene (§, ATAC-seq peak) in human fibroblasts derived from Angelman syndrome (AS) and PWS patients with a 15q11.2-q13 deletion (One-way ANOVA test followed by Dunnett’s multiple comparison test; Adj p-value is indicated in graph; 50th percentile (median value, line), 25th to 75th percentiles of dataset (box), 5th and 95th percentile (Whiskers). c Genome viewer screenshot illustrating maternal imprinting domains (PWS) remain to be a closed chromatin after treatment of MS1262 (EHMT2 inhibitor) or 5-Aza (DNMT1 inhibitor). d Genome viewer screenshot demonstrating Ehmt2 catalytic mutant (CM) or Ehmt2/Ehmt1 double knockout (DKO) not contributing to open chromatin status on PWS-IC in mouse embryonic stem cells (ESC). Source data are provided as a Source Data file.
To determine whether EHMT2 or DNMT1 inhibitors change the chromatin states of the PWS imprinting ___domain on the maternal chromosome, we treated PWS fibroblasts with a paternal deletion with MS126242,43, a recently developed EHMT2 inhibitor, or 5-Aza, a known DNMT1 inhibitor. We confirmed that both MS1262 and 5-Aza treatments led to the reactivation of the SNPRN gene from the repressed maternal chromosome. Through the ATAC-seq analysis, we did not reveal that these treatments resulted in a more open chromatin state of PWS imprinting ___domain despite the observed unsilencing of paternally expressed gene from the maternal chromosome (Fig. 3c), and despite Principal Component Analysis (PCA) supporting that treatment with DNMT1and EMHT2 inhibitors resulted in significant gene expression differences in PWS fibroblasts (Supplementary Fig. 3a-3b). In line with this finding, increased chromatin accessibility in PWS-IC was not observed in Ehmt2 catalytic mutant (CM) or Ehmt2/Ehmt1 double knockout (DKO) mouse ESCs (Fig. 3d). In contrast, the gain of chromatin accessibility along with overexpression of Snurf-Snrpn was observed in mouse Dnmt1/3a/3b triple KO (TKO) ESCs (Fig. 3d). The paternally expressed genes of Dlk1, Cdh15, and Galnt6 did not show distinguishable chromatin state changes in Ehmt2/1 double knockout (DKO) or Dnmt1/3a/3b TKO mouse ESCs (Supplementary Fig. 3c–e) even though their expression was upregulated. These results indicate that chromatin accessibility change may not be essential to enable gene expression of repressed imprinted PWS alleles and that, instead, other regulatory factors inducing spatiotemporal chromatin architecture may be considered, as suggested in recent work44,45,46.
Chromatin organization of PWS imprinting ___domain shows allele-specific chromatin conformation
Next, to determine physical chromatin interactions in maternal or paternal chromosome, we performed Hi-C analysis of human fibroblasts derived from patients with a large 15q.11-q13 deletion on the paternal chromosome (PWS) or maternal chromosome (AS). Each Hi-C contact matrix was aligned with reference tracks of CTCF and histone marks contributing to the overall chromatin structure (Fig. 4a). In line with a previous report47, there are no strong TAD boundaries in the PWS-critical region, including the PWS-IC (Fig. 4b and Supplementary Fig. 4a), consistent with the scarcity of CTCF binding in this region that is in contrast with other imprinting domains such as H19-IGF2 in human chromosome 11p15 region47 and Peg13-Kcnk9 in mouse chromosome 15 region48. (Fig. 4a). We observed maternal and paternal specific chromatin loops in imprinting loci, with significantly more loops in paternal than maternal loci (Fig. 4c). Biallelic loci showed more loops and open chromatin peaks than imprinted loci (Fig. 4c). Interestingly, imprinted genes located >1 Mb upstream from the PWS-IC showed strong CTCF peaks with a paternal-specific loop (Supplementary Fig. 4b). Taken together, these results indicate an allele-specific chromatin conformation of PWS imprinting ___domain and suggest a role of chromatin conformation in regulating the imprinting maintenance to silence gene expression on the maternal chromosome.

a This Juicebox screenshot visualizing a region of 1.7 Mb locus (chr15:24,500,000-26,200,000, hg38) at 5-Kb resolution. (Normalization; SCALE). b Wash U Epigenome Browser snapshot showing CTCF (control) Hi-C track of a 5.5 Mb region of human chromosome 15q11.2-q13 at 500 bp resolution normalized using SCALE. The triangle shapes in the Hi-C track depict chromatin domains in human fibroblasts derived from PWS and AS patients (The color scale of the heatmap; higher contact counts corresponding to a darker color). c Genome viewer screenshot illustrating 1D representation of the DNA fragment that forms the loop in PWS-associated imprinted domains of maternal or paternal chromosome, partially matched with ATAC-seq peaks. (UBE3A; a paternal and neuron cell type specific imprinted gene in brain).
Chromatin tracing reveals allele-specific 3D folding organization of PWS imprinting domains in the human fibroblasts
To further characterize the chromatin 3D folding architecture, we applied a chromatin tracing method based on multiplexed DNA fluorescence in situ hybridization (FISH)49,50 in the human PWS and AS fibroblasts with a paternal or maternal deletion. We partitioned the critical PWS imprinting ___domain into 42 consecutive 50-kb segments, spanning the 2.1-Mb genomic region from genome coordinates of chr15:23,500,000 to 256,500,000 (hg38), and labeled each segment with 500 unique primary oligonucleotide probes (Fig. 5a). Then, through sequential hybridization, we generated matrices of median spatial distance between each pair of targeted segments for the PWS-associated region on paternal or maternal chromosomes (Fig. 5b, c). To validate the chromatin traces, we compared distances with the corresponding contact frequencies from our Hi-C analysis (Fig. 5d, e). We found that the inter-loci median spatial distances of paternal or maternal region of interest were highly correlated with the Hi-C contact frequencies, with correlation coefficients of −0.7473 and −0.8222 for paternal and maternal region, respectively (Fig. 5f). To compare chromatin compaction between maternal and paternal regions, we calculated the log2-fold change of median spatial distances between PWS and AS (Fig. 5g). The traced region, which is largely maternally silenced, is overall more compactly folded (smaller distances) in PWS cells than in AS cells, whereas a sub-region containing PWRN4-1 was more compacted in AS cells than in PWS cells. To investigate whether EHMT2 maintains the chromatin conformation of the imprinting ___domain in the PWS critical region (chr15:24,950 kb-25,300 kb, Hyb #26-#32), we treated PWS fibroblasts with a paternal deletion with the EHMT2 inhibitor (Fig. 5h). When we calculated the log2 fold-change of the median spatial distance between PWS treated with EHMT2 inhibitor and control (Fig. 5i), we did not detect a significant systematic change in 2.1 Mb PWS imprinting ___domain. However, it should be noted that the interpretation is limited by the 50 kb genomic resolution for the probe design, and the micro-change of chromatin structure may be missed.

a (i) Schematic illustration of the chromatin tracing strategy. (ii) Representative images showing foci corresponding to maternal/paternal chromosome, with representative chromatin trace, respectively. b, c Median spatial distance matrix of the traced genomic region (42 consecutive 50-kb loci) in human fibroblasts. d, e Hi-C contact frequency matrix of the same genomic region as in (b, c). f Comparison of median inter-loci spatial distance from chromatin tracing with contact frequency measured by Hi-C. g Log2 fold change of inter-loci distance of PWS versus AS. h Median spatial distance matrix of the traced genomic region (42 consecutive 50-kb loci) in PWS fibroblasts treated with EHMT2 inhibitor. i Log2 fold change of inter-loci distance of PWS fibroblasts treated with EHMT2 inhibitor versus control PWS. Source data are provided as a Source Data file.
Maternal noncoding RNAs upstream of PWS-IC recruit EHMT2 and interact with PWS-IC on the maternal chromosome
Our findings that the DNA methylation at PWS-IC is not essential for maternal allele-specific EHMT2 binding in fibroblasts suggest an alternative mechanism to be tested. There are many non-coding RNAs between PWS-IC and NDN from published literature and in silico analysis. We then hypothesized that EHMT2 is recruited to PWS-IC by non-coding RNAs to maintain silencing on the PWS imprinting ___domain on the maternal chromosome. We focused on the non-coding RNAs that are extended from 5′ UTR of SNRPN/SNHG14 transcripts and also associated with CpG islands that suggest a transcription start sites. Previous reports delineated three transcript start sites (TSS) of SNRPN in humans that include canonical site from exon 1 of SNRPN (TSS1, which is paternal-specific), upstream u1A (TSS3), and u1B (TSS5) (Fig. 6a)51. The same structure of exon 1 of Snrpn, U1, and U2 is also described in mice. U1 and U2 are known as oocyte-specific11. Four CGIs (40, 37, 18, 77) are mapped within this region, of which the CGI-77 overlaps with PWS-IC and TSS1, whereas TSS2 and TSS3 are not associated with any CGIs. Our silico analysis revealed the presence of PWRN1 and non-coding RNA (ncRNA) of ENSG00000280118 (280118) upstream of TSS1(Supplementary Fig. 5a). PWRN1 is a previously reported non-coding RNA that displays an isoform and paternal-specific in fetal brain but biallelic expression in kidney and testis52. 280118 is a transcript (3899 bp in hg38) with 3 exons that does not overlap with any exon of PWRN1 and 5′ untranslated exons of coding SNRPN transcripts. The exact size of 280118 transcript could not be determined at this time. Using bulk RNA-seq, we examined allele-specific expression of transcripts upstream of TSS1 in PWS and AS fibroblasts with a paternal or maternal deletion of 15q11.2-13. We found several transcript peaks that overlap with CGI and were either predominantly paternal, maternal, or biallelically expressed (Fig. 6a). As expected, non-imprinted gene loci were transcribed from both paternal and maternal chromosomes in fibroblasts (Supplementary Fig. 6). The in silico analysis predicted possible TSSs associated with these non-coding RNA transcript peaks. We delineate these upstream non-coding transcripts as TSS2-TSS5 as diagramed (Fig.6a). The TSS4 peak associated with transcript ENSG00000280118 (TSS4-280118) and CGI-40. The expression of TSS4-280118 ncRNA is predominantly maternal-specific by chromatin-associated RNA-seq (chrRNA-seq) (Fig. 6b)53. Through RNA immunoprecipitation (RIP) or RNA fluorescence in situ hybridization (FISH) with a EHMT2 antibody, we were able to detect binding between these maternal or paternal ncRNA TSS4-280118 and EMHT2 (Fig. 6c, d) as well as co-localization of EHMT2 and ncRNA TSS4-280118 (Fig. 6e). We also captured EHMT2 binding to the genomic region of TSS4-280118 by ChIP-qPCR, but without any change in H3K9me2 level (Fig. 6f). These results suggested that TSS4-280118 may recruit EHMT2 and was the focus for the further study. We next examined whether EHMT2 inhibition affected the interaction of EHMT2 with ncRNAs in cultured fibroblasts. After EHMT2 inhibitor treatment, which unsilenced expression of SNRPN from the maternal chromosome43, the interaction between ncRNA and EHMT2 was significantly reduced (Fig. 6g). To examine the causal relationship between ncRNA and imprinting maintenance, we designed the sgRNAs to specially inactivate the ncRNA 280118 by CRISPR/Cas9 method (Fig. 6h). Co-transduction of lenti-Cas9 and two sgRNAs targeting TSS4-280118 was performed in PWS (maternal chr15q11.2-q13) fibroblasts. AS fibroblasts (paternal chr15q11.2-q13) were used as a positive control, and dCas9 with sgRNAs was used as a negative control. As a result, inactivation of ncRNA TSS4-280118 resulted in unsilencing of the expression of SNRPN and SNORD116 from the maternal chromosome (Fig. 6i). These results support that TSS4-280118 and also the binding of ncRNA and EHMT2 are important for the silencing of SNRPN gene on the maternal chromosome. EHMT2 is reported to interact with other chromatin regulators such as SUV39H1, heterochromatin proteins of (HP)-alpha (HP1α), and β-actin25,54,55,56. We performed co-immunoprecipitation analysis that confirmed the interaction of EHMT2 with SUV39H1, HP1α, and β-actin (Fig. 6j). Accordingly, EHMT2 protein complex with SUV39H1, HP1α, and β-actin was significantly more enriched in the maternal imprinted ___domain of 15q11.2-q13 compared to the paternal chromosome of the same region (Fig. 6k). Taken together, our analyses suggest that maternal ncRNA TSS4-280118 transcribed from the maternal and upstream region of PWS-IC recruit EHMT2 and form a heterochromatin repressor complex to PWS-IC that instructs silencing of imprinted genes distal to PWS-IC in the maternal chromosome.

a Genome browser screenshot illustrating distribution of chrRNA-seq and bulk RNA-seq reads of ncRNA transcripts upstream of PWS-C in human fibroblasts derived from PWS and AS patients. The transcription start site close to the codding exon 1 of SNRPN is defined as a canonical transcription start site 1(TSS1). u1A (TSS3) and u1B (TSS5) are two oocyte specific transcript start sites previously reported51. TSS2 and TSS4 that correspond to CGI-18 and CGI-40 are identified from this study. TSS4 is preferentially maternal, as shown the expression peak in significantly higher in PWS chrRNA than in AS chrRNA. b Genome browser screenshot showing chrRNA-seq reads TSS4 of ncRNA of 280118 in human fibroblasts derived from PWS and AS patients. c Native RNA-IP (RIP) with EHMT2 antibody showed the interaction between EMHT2 and maternal ncRNAs of using the primers derived from 280118. Data are presented as mean ± SEM (One-way ANOVA test followed by Dunnett’s multiple comparison test; Adj p-value is indicated in graph). d qPCR analysis following CLIP with EHMT2 antibody in human fibroblasts. Data are presented as mean ± SEM (Two-tailed student’s t test; *p = 0.0136). e Representative images showing foci corresponding to EHMT2 and ncRNA280118, respectively. (Scale bar 5 μM, colocalized foci are indicated by white arrowhead). f Allele-specific ChIP-qPCR with EHMT2 and H3K9me2 in genomic loci associated with ncRNA TSS4-280118 in human fibroblasts. f, g Data are presented as mean ± SEM (One-way ANOVA test followed by Dunnett’s multiple comparison test; Adj p-value is indicated in graph). g RIP-qPCR with EHMT2 antibody after treatment with EHMT2 inhibitor (MS1262) in human fibroblasts. h Schematic diagrams illustrating gRNA binding sites (top) and strategy for co-transduction of Lenti-Cas9, Lenti-dCas9, and Lenti-gRNA (bottom). Created in BioRender. Qiao, S. (2025) https://BioRender.com/i66o883. i Quantitative RT-qPCR analysis of TSS4-280118, SNRPN, and SNORD116 transcript levels in human fibroblasts with three replications. Data are presented as mean ± SEM (One-way ANOVA test followed by Dunnett’s multiple comparison test; Adj p value is indicated in graph). j Representative blots showing components of EHMT2 repressive complex following immunoprecipitation (IP) of lysates from human fibroblasts with Ab against EHMT2. k Quantification of co-IP result. Protein levels were normalized to inputs. EHMT2 repressive complexes were more abundant in PWS fibroblasts compared to AS fibroblasts or Ctr. Source data are provided as a Source Data file. Data are presented as mean ± SD (One-way ANOVA test followed by Dunnett’s multiple comparison test; Adj p value is indicated in graph). Source data are provided as a Source Data file.
Discussion
Here, we deploy comprehensive molecular and high-resolution chromatin analyses using PWS and AS patient-derived fibroblasts with paternal or maternal deletion, respectively, to significantly advance our understanding of mechanisms underlying imprinting regulation in 15q11.2-q13 region. Firstly, we show that EHMT2-mediated H3K9me2 but not DNA methylation on the maternal PWS-IC is essential for maintaining the silencing of paternally expressed genes in the maternal chromosome. Secondly, distinct mechanisms operate to silence the expression of paternally expressed genes proximal and distal to the bipartite PWS-IC in the maternal chromosome. It is known that the microdeletion of PWS-IC on the paternal chromosome results in loss of the expression of paternally expressed genes of both distal and proximal sites, both in humans and mice16,17,18,19. In contrast, the deletion of PWS-IC in the maternal chromosome does not alter the expression of the paternally expressed genes in the maternal chromosome. These results indicate that the DNA regulatory element of PWS-IC in the maternal chromosome is not directly implicated in silencing of imprinted genes in the maternal chromosome. A prevailing hypothesis is that maternal allele-specific DNA methylation in the PWS-IC silences expression of paternally expressed genes on the maternal chromosome because of unsilencing of SNRPN gene expression in vitro by DNA methylation inhibitors8. However, to our knowledge, unsilencing of SNRPN gene by DNA methylation inhibitors from the maternal chromosome has not been described in vivo. Loss of methylation of PWS-IC is observed in both mouse Dnmt1−/− ESC and embryos. However, expression of Snrpn is mono-allelic in ESC but biallelic in embryos of Dnmt1−/−21,55. In contrast, expression of Snrpn is biallelic in Ehmt2−/− ESC and embryos20,28. Interestingly, methylation of PWS-IC in the maternal chromosome is lost in ESC but intact in embryos of Ehmt2-/-20. These results indicate that both DNMT1 and EHMT2 contribute to the methylation of PWS-IC in ESC but only DNMT1 is involved in the embryo. These are consistent with other reports that DNMT1 and EHMT2 interact directly57,58. This is also in line with the finding that different epigenetic machinery proteins are implicated in the epigenomic reprogramming of demethylation during early development59. Consistent with these findings, we reported that EHMT2 inhibitors unsilence the paternally expressed SNRPN/SNHG14 from the imprinted genes on the maternal chromosome without changing the DNA methylation of PWS-IC22. Here, we showed that conditional inactivation of Ehmt2 in the embryonic mouse brain reduces H3K9me2 levels without any changes in DNA methylation of the PWS-IC and unsilenced the expression of maternally imprinted Snprn. Unexpectedly, the expression of paternally imprinted genes Magel2, Ndn, and Mkrn3 was not affected. These findings support that EHMT2 and H3K9me2 play a critical role in maintaining the silenced expression of PWS-associated genes proximal to PWS-IC in the maternal allele in humans. Surprisingly, our findings indicate that a distinct mechanism controls the silenced expression of paternally expressed genes in the distal side of PWS-IC in the maternal chromosome in mice. Because of the lacking the chromatin loops in the proximal region between MKRN3 and PWS-IC, we hypothesize that chromatin 3D structure is the mechanism to the silencing of MAGEL2, NDN, and MKRN3 on the maternal chromosome and warranted for the further investigation.
Unique to this study, we were able to determine allele-specific chromatin accessibility, looping, and 3D chromatin folding of PWS imprinting ___domain by employing the high-resolution epigenome and chromatin profiling technologies in human PWS and AS cell models with a paternal or maternal deletion, respectively. The paternal allele is associated with a more open chromatin state and more chromatin loops, while the maternal allele represents a more closed chromatin state and less chromatin loops. Notably, there is a scarcity of CTCF binding in the imprinted ___domain of 15q11.2-q13 compared to the 11p15.5 imprinted region associated with imprinted genes of H19 and IGF247. Through application of the developed high-resolution chromatin tracing method, we were able to uncover an allele-specific 3D-folding organization of the PWS critical region with a 50 kb genomic resolution.
We found that EHMT2 and H3K9me2 were enriched in the maternal PWS-IC. This enrichment was not affected upon inhibition of DNA methylation, suggesting that maternal DNA methylation is not required to maintain EHMT2 binding to the PWS-IC. The transcript structures upstream and downstream of coding SNRPN exons are complex and have not been fully delineated. Numerous noncoding RNA transcripts, including microRNAs and snoRNAs, are found both sides of PWS-IC in humans and mice. The exact structure and functions of these transcripts remain to be fully delineated. The tissue and transcript-specific imprinted and non-imprinted expression pattern for these long non-coding RNAs has been described in refs. 60,61. Our in silico analysis identified ENSG00000280118 and many other transcripts in an extended region between PWS-IC and NDN. We targeted ENSG00000280118 as a transcript in the extended region of 5′ UTR of the SNRPN gene and also associated with a CpG island (CGI-40), suggesting a transcriptional starting site. Additionally, RNA-IP experiments using EHMT2 antibody confirmed the interaction of EHMT2 and ENSG00000280118 or other non-coding RNAs (ENSG00000286110, SNRPN transcript including u1B or u1A) in the region (Supplementary Fig. 7). More importantly, we discovered that the TSS4-280118 is preferentially expressed from the maternal chromosome. The exons of TSS4-280118 do not overlap with other adjacent transcripts that are predominantly paternal or biallelic. These results indicate that TSS4-280118 transcript is distinct from the previously described paternally expressed transcripts upstream of SNRPN. Careful review also indicate that the TSS4-280118 is also distinct from the oocyte-specific u1A (TSS3) and u1B (TSS5) previously reported in human51. Our RNA immunoprecipitation and co-localization analysis for TSS4-280118 ncRNA and EHMT protein further supports that TSS4-280118, which is preferentially expressed from the maternal chromosome, plays a role in EHMT2 recruitment in CIS to the maternal PWS-IC. EMHT2 forms a local repressor or heterochromatin chromatin complex with SUV39H1 and HP1α. The inactivation of TSS4-280118 ncRNA resulted in unsilencing of the expression of SNRPN/SNHG14 from the maternal chromosome. These results together support a causal role of TSS4-280118 ncRNA in silencing of PWS-associated genes of SNRPN and SNORD116 on the maternal chromosome. It is noted that a similar mechanism has been described for the maternally expressed Igf2r gene: the paternally expressed antisense noncoding Air RNA mediates silencing of Ig2r expression in cis by recruiting EHMT2 to chromatin2,26,62,63. DNA elements within its locus are not required for Air RNA to silence distant imprinted genes. The same is observed for PWS-IC: deletion of PWS-IC DNA in the maternal chromosome does not affect the imprinted expression of paternally expressed genes19.
Our analysis did not reveal that EHMT2 inhibitor significantly enhances chromatin accessibility or changes the chromatin folding organization of the PWS imprinting ___domain using a global profiling method of ATAC-seq and targeted chromatin tracing method with 50 kb genomic resolution for 3D chromatin conformation. This is in contrast to the increased chromatin accessibility detected in a previous study using quantitative PCR of genomic DNA following in situ nuclease digestion22. One of the caveats is that the resolution of our probe design of 50 kb genomic distance for the chromatin tracing is not optimal, and the change in chromatin accessibility after EHMT2 inhibitor treatment is at a micro-scale and can only be detected by epigenetic profiling methods with higher resolution. On the other hand, we discovered that EHMT2 inhibitor treatment in human fibroblasts significantly reduced the binding of maternal non-coding RNAs and EHMT2. The conformational change in EHMT2 is expected to lead to less formation of heterochromatin complex of EHMT2 with HP1α and SUV39H1 in the PWS-IC and unsilenced the expression of SNPRN from the maternal chromosome.
In summary, our study provides evidence supporting a model of the mechanism of regulation of the PWS imprinted ___domain (Fig. 7). The model highlights the key discoveries from this study as well as incorporates existing knowledge in the literature. First, the DNA methylation of PWS-IC serves in the establishment of imprinting but is not essential for imprinting maintenance. Second, our findings indicate a different mechanism operates to silence expression of paternally expressed genes distal or proximal to the PWS-IC on the maternal chromosome. This is in contrast with reports that PWS-IC likely employs the same mechanism to control the active expression of paternally expressed genes on the paternal chromosome at both distal and proximal sites. Third, we show that maternal non-coding RNA of TSS4-280118 upstream of PWS-IC interacts with and recruits EHMT2 to PWS-IC, forms a heterochromatin repressor complex, and silences the expression of SNRPN/SNHG14 on the maternal chromosome. Thus, EMHT2 at PWS-IC plays a critical role in silencing expression on the maternal chromosome. Our findings have significantly advanced understanding of one of the most investigated imprinting domains in humans to date. Future study in mouse model is warranted to see whether the same mechanism is conserved in rodents in vivo.

Blue allele: maternal imprinted gene, green allele: maternally imprinted noncoding RNA, NPAP1: monoallelic (paternal) expression in fetal brain, biallelic expression in adult testis and brain, UBE3A: neuronal cell type specific paternal imprinted gene, green ncRNA; maternal specific transcription, black ncRNA; biallelic transcription. Created in BioRender. Qiao, S. (2025) https://BioRender.com/u82b098.
Methods
Human fibroblast cell culture
We obtained human fibroblasts from patients with PWS, AS, and Ctr from the Baylor College of Medicine cell repository, Columbia University, and Kansas University Hospital. We handled all fibroblast cells derived from patients according to an Institutional Review Board (IRB) protocol approved by Yale University (#2000026262). We maintained human fibroblast cells in minimum essential medium alpha media (Gibco 12571-063) supplemented with 10% FBS (Gibco 10082-147), 1% l-glutamine (Gibco 25030-081), 100 units/ml penicillin, and 100 μg/ml streptomycin (Gibco 15240-062) at 37 °C and 5% CO2 as previously described in ref. 22.
Animals
We handled all animals according to an Institutional Animal Care and Use Committee (IACUC) protocol approved by Yale University (#2022-20271). Snrpn-EGFP mice64 were previously described. We obtained Ehmt2f/+36 mice from University of British Columbia and Nestin-Cre+ and C57BL/6 J mice from the Jackson Laboratory. We used male and female mice in all studies. All mice were maintained under 12 h light/12 h darkness cycles with constant conditions of temperature (22 °C) and humidity (40%-60%). Neonatal mice at p10 were euthanized and their forebrains were collected for biochemical experiments.
CRISPR-Cas9 mediated genome editing for ENSG00000280118 knock out
In this study, following vectors were used, lentiCas9-Blast (Plasmid #52962, Addgene); lentiGuide-Puro (Plasmid #52963, Addgene); pHAGE-TO-dCas9-3XGFP (Plasmid #64107, Addgene)65,66; pMD2.G (Plasmid #12259, Addgene); psPAX2 (Plasmid #12260, Addgene).
To pick guide sequence, we used CRISPick67 or CRISPR HDR Design tool (http://www.idtdna.com). For cloning, we followed the Target Sequence Cloning protocol provided by Zhang lab68 (Supplementary Table 1). Lenti-virus was produced by co-transfection of lneti-Cas9, lentiGuide-puro, or pHAGE-TO-dCas9-3XGFP with the compatible packaging plasmids, pMD2.G and psPAX2, into Lenti-X 293T cells (632180, TAKARA) using Lipofectamine 3000 (L3000-008, Invitrogen) following the manufacturer’s instructions. We filtered the entire media by passing it through 0.22-micron syringe-driven filters (SLGV033N, Millipore), then added this media with protamine sulfate (8ug/mL; P3369, Sigma-Aldrich) to fibroblasts derived from PWS. After selecting the cells with puromycin (3 mg/ml; A11138-03, Gibco) for 48 h, we harvested the cells and isolated genomic DNA to validate of knockout cells69. The designed primers for PCR and Sanger sequencing are described in Supplementary Table 2.
Co-immunoprecipitation
We washed human fibroblast cells cultured in a 10 cm dish with PBS and collected 5–6 million in 1.5 mL tube. After centrifugation at 4 °C for 5 min at 211 × g, we resuspended cell pellets by pipetting in 0.2 mL of non-SDS lysis buffer (150 mM NaCl, 50 mM Tris-HCl pH 7.4, 10% Glycerol, 1% Triton X-100) containing 1x protease/phosphatase inhibitor cocktail (Cell Signaling). Cells were incubated on ice for 30 min and resuspended by pipetting every 10 min. After centrifugation at 4 °C for 30 min at 15,800 × g, collected supernatants were quantified using Pierce™ BCA Protein Assay Kit (Thermo Fisher). The lysates (500 μg) were immunoprecipitated with 2 μg antibody (EHMT2, Invitrogen) or mouse IgG (Millipore) overnight at 4 °C with rotation. 30 μL of protein G-agarose beads (Roche) was added for 2 h and then washed with HMTG buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.1% Triton X-100, and 10% glycerol) containing 1x protease and phosphatase inhibitor three times at 4 °C for 5 min with rotation. The proteins were eluted in 2x Laemmli buffer (Bio-rad) by boiling at 98 °C for 5 min, and analyzed by immunoblotting.
Chromatin immunoprecipitation
We used a Chromatin immunoprecipitation (ChIP) assay kit (Millipore) following the manufacturer’s instructions. Human fibroblast cells or mice forebrains were fixed in 1% formaldehyde for 10 min at 37 °C, followed by two washes in cold PBS (Thermo Scientific). Cells were scraped in PBS containing 1 × protease/phosphatase inhibitor cocktail and resuspended in 0.2 mL SDS lysis buffer. Cells were incubated on ice for 20 min prior to lysing using a Bioruptor (Diagenode) for 12 cycles (10 s on and 50 s off) or 20 cycles (30 s on and 30 s off), followed by centrifugation at 4 °C for 10 min at 18,407 × g. The sonicated cell supernatant was diluted in ChIP dilution buffer with protease inhibitor, 75 μL of Protein G Agarose (50% Slury) was added, after which the samples were incubated for 30 min at 4 °C with agitation. After brief centrifugation, the supernatant was collected and incubated with immunoprecipitating antibody (EHMT2, Invitrogen; H3K9me2, Abcam) overnight at 4 °C with rotation. Protein G Agarose was added for one hour and then washed with low salt, high salt, LiCl, and TE buffer for 5 min with rotation. To elute the precipitate, 250 μL of elution buffer was added and rotated at room temperature. After collecting the supernatant, the elution step was repeated. 20 μL 5 M NaCl was added to combined elutes for reverse crosslinks at 65 °C for four hours, followed by added 10 μL of 0.5 M EDTA, 20 μL 1 M Tris-HCl, and 2 μL of 10 mg/mL Proteinase K at 45 °C for one hour. DNA was recovered by phenol/chloroform (Sigma) extraction and precipitated by 40 μL 3 M sodium acetate, 95% ethanol, and 20 μg glycogen. Pellets were washed with 70% ethanol and resuspended in double-distilled water for qPCR reaction.
RNA immunoprecipitation
We used a modified version of RNA immunoprecipitation protocol described by Raab et al.70. Human fibroblast cells were fixed in 0.3% methanol-free formaldehyde for 30 min at 4 °C, followed by quenching with 125 mM glycine for 5 min at room temperature. After three PBS washes, cells were collected in PBS containing 1 mM PMSF (phenylmethyl sulfonyl fluoride). After centrifugation, cell pellets were resuspended in 0.5 mL radioimmunoprecipitation assay (RIPA) buffer containing 0.5 mM Dithiothreitol (DTT, Thermo Scientific), 1 × protease inhibitors (Thermo Scientific) and 2.5 μL RNAsin (Promega), followed by incubated on ice for 10 min prior to lysing using a Bioruptor (Diagenode) for four cycles of 5 s on and 55 s off. After centrifugation at 4 °C for 10 min at 18,000 × g, the supernatant was collected for incubation overnight at 4 °C with antibody-conjugated beads. Protein G magnetic beads (NEB) were pre-conjugated with EHMT2 monoclonal antibody (Invitrogen, A8620A) for 2 h at 4 °C. Next day, beads were washed consecutively with fRIP buffer (25 mM Tris-HCl pH 7.5, 5 mM EDTA, 0.5% Ipegal CA-630, 150 mM KCl), followed by three times in ChIP buffer (50 mM Tris-HCl pH 7.5, 140 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS), one time in fRIP buffer for 5 min at 4 °C. After final wash, beads were resuspended in 3x reverse crosslinking buffer (3x PBS, 6% N-lauroyl sarcosine, 30 mM EDTA, 5 mM DTT). Eluted samples were collected to new tube and incubated with 20 μL proteinase K (Roche) and 1 μL RNAsin for one hour at 42 °C, 1 h at 55 °C, and 30 min at 65 °C. RNA was extracted using Direct-Zol RNA miniprep kit (Zymo Research), including the on-column DNase digestion. RNA was eluted in 12 μL double-distilled water and used for qPCR reaction and library preparation.
Cross-linking and immunoprecipitation (CLIP)
For direct RNA-protein cross-linking, we irradiated ultraviolet 0.8 J/cm2 using Spectrolinker XL-1500 for human fibroblasts. The cells were floated with cold PBS and dispersed in 150 mm dishes before irradiation, following the previous study (PMID: 33989516). And then, we followed the manufacturer’s protocol for immunoprecipitation (Pierce™ Crosslink IP Kit, Cat# 26147, Thermo Scientific™).
Bisulfite conversion sequencing
This experiment was performed using a EpiTect Bisulfite kit (Qiagen) following manufacturer’s instructions. In brief, isolated genomic DNA from human fibroblast or E18 mouse brain was treated with bisulfite, and then 200 ng of input DNA was used for PCR amplification. We subcloned PCR products into pGEM-T easy vector (Promega), and we sequenced an average of 15 clones. We analyzed DNA-sequencing results using BISMA web-based analysis platform (http://services.ibc.uni-stuttgart.de/BDPC/BISMA/) with a setting for individual clones with <80% bisulfite conversion and <80% sequence identity to be excluded in the analysis. The primers that we used in this study are listed here. Human CpG 1′ (forward, 5′-ATTGTAATAGTGTTGTGGGGTTTTAGGG-3′; reverse, 5′-CCCAAA CTATCTCTTAAAAAAAACCACC-3′), Human CpG 2′ (forward, 5′-TTTAAGTTTTTAGGATTTGGAG TATTGA-3′; reverse, 5′-AAACTACAATCACCCTAATATACCCAC-3′), Human CpG 3′ (forward, 5′-GGTGGGTATATTAGGGTGAT TGTAGTTT-3′; reverse, 5′- CCTAATCCACTACCATAACCTCCTC -3′), and Mouse PWS-IC (forward, 5′- AATTTGTGTGATGTTTGTAATTATTTGG-3′; reverse, 5′- ATAAAATACACTTTCACTACTAAAATCC -3′’).
Western blotting
We performed western blotting as previously described in ref. 71. Whole cell lysates from human fibroblasts and mouse forebrains were prepared using 1x lysis buffer (Cell Signaling) containing 1x protease/phosphatase inhibitor (Cell Signaling). Histones were extracted using Core Histone Isolation kit (Sigma) following manufacturer’s instructions. Samples were quantified by BCA assay (Thermo Fisher) and boiled in 4x Laemlli buffer (Bio-Rad) at 98 °C for 5 min before loading in 4-20% precast gel (Bio-Rad). The primary antibodies used to detect proteins are given in Supplementary Table 3. Antibodies used in this study have been tested and validated for specificity by the manufacturers.
RT-PCR and qPCR
RNA was extracted from mouse forebrain of p11 or human fibroblasts using Trizol reagent (Sigma). cDNA synthesis was performed using a Reverse Transcription System kit (Promega). For quantitative real-time PCR (qPCR), PCR was performed on a CFX96 TouchTM Real-Time PCR Detection System (Bio-Rad). The primers used to amplify cDNAs are given in Supplementary Table 4. CT values for each sample were obtained using CFX Manager Software version 3.0 (Bio-Rad).
Chromatin-associated RNA (chrRNA) fractionation
We performed chrRNA using a protocol described in Sledziowska et al53. Human fibroblasts were treated with 1 ml of TrypLE(Gibco) per well in a 6-well plate. The cells were incubated until they detached, at which point 2 ml of DPBS was added per well. The cells were collected and centrifuged at 200 × g for 5 min. The supernatant was removed and the cells were resuspended in the 200 μl of cytoplasmic lysis Buffer (0.15% NP-40, 10 mM Tris–HCl pH 7, 150 mM NaCl, 50U RiboLock). After incubating samples for 5 min on ice, they were layered on 500 μl of sucrose buffer (10 mM Tris–HCl pH 7, 150 mM NaCl, 25% Sucrose, 50U RiboLock). Nuclei were collected by centrifugation of 16,000 × g for 10 min at 4 °C. The supernatant containing cytoplasmic fraction was then removed, and nuclei were washed with nuclei wash buffer (1X PBS supplemented 0.1% TritonX-100, 1 mM EDTA, 50U RiboLock) at 1200 g for 1 min at 4 °C. Supernatant was discarded and the nuclei were resuspended in 200 μl of glycerol buffer (20 mM Tris HCl pH 8, 75 mM NaCl, 0.5 mM EDTA, 50% glycerol, 0.85 mM DTT, 50U RiboLock). 200 μl of nuclei lysis buffer (1% NP-40, 20 mM HEPES pH7.5, 300 mM NaCl, 1 M Urea, 0.2 mM EDTA, 1 mM DTT, 50U RiboLock) was added, following 2 min of vortexing by pulsed. After centrifuged at 18,500 × g for 2 min at 4 °C, the pellet containing chrRNA was resuspended in 200 μl of PBS supplemented with 50U RiboLock. Following resuspension, RNA was extracted using Trizol reagent for library prep.
RNA-seq
All procedures were conducted in the Yale Center for Genome Analysis.
RNA Seq Quality Control: total RNA quality was determined by estimating the A260/A280 and A260/A230 ratios by nanodrop. RNA integrity was determined by running an Agilent Bioanalyzer gel, which measures the ratio of the ribosomal peaks. For library prep, we used samples with RIN values of 5 or greater.
RNA Seq Library Prep: for mouse forebrain samples, using the Kapa RNA HyperPrep Kit with RiboErase (KR1351), rRNA is depleted starting from 25 to 1000 ng of total RNA by hybridization of rRNA to complementary DNA oligonucleotides, followed by treatment with RNase H and DNase to remove rRNA duplexed to DNA. Samples are then fragmented using heat and magnesium. 1st strand synthesis is performed using random priming. 2nd strand synthesis incorporates dUTPs into the 2nd strand cDNA. Adapters are then ligated, and the library is amplified. Strands marked with dUTPs are not amplified, allowing for strand-specific sequencing. Indexed libraries that meet appropriate cut-offs for both quantity and quality are quantified by qRT-PCR using a commercially available kit (KAPA Biosystems) and insert size distribution determined with the LabChip GX or Agilent Bioanalyzer. Samples with a yield of ≥0.5 ng/μl are used for sequencing. For human fibroblast samples, using the SMARTer Stranded Total RNA-Seq Kit v3- Pico Input Mammalian from Takara and a normalized RNA input between 250 pg and 10 ng, the RNA is first fragmented prior to first-strand cDNA synthesis. Next, indexing and PCR1 occurs. After a bead clean-up, ribosomal cDNA is depleted by ZapR v3 in the presence of mammalian-specific R-probes. Next, fragments that are not cleaved in the depletion step are enriched in a second PCR before a final bead clean-up is performed. Indexed libraries that meet appropriate cut-offs for both quantity and quality are quantified by qRT-PCR using a commercially available kit (KAPA Biosystems) and insert size distribution determined with the LabChip GX or Agilent Bioanalyzer. Samples with a yield of ≥0.5 ng/μl are used for sequencing.
Flow Cell Preparation and Sequencing: sample concentrations are normalized to 1.2 nM and loaded onto an Illumina NovaSeq flow cell at a concentration that yields 25 million (human fibroblast RNA) or 50 million (mouse forebrain RNA) passing filter clusters per sample. Samples are sequenced using 100 bp paired-end sequencing on an Illumina NovaSeq according to Illumina protocols. The 10 bp unique dual index is read during additional sequencing reads that automatically follow the completion of read 1. Data generated during sequencing runs are simultaneously transferred to the YCGA high-performance computing cluster. A positive control (prepared bacteriophage Phi X library) provided by Illumina is spiked into every lane at a concentration of 0.3% to monitor sequencing quality in real time.
Data Analysis: Signal intensities are converted to individual base calls during a run using the system’s Real Time Analysis (RTA) software. Base calls are transferred from the machine’s dedicated personal computer to the Yale High Performance Computing cluster via a 1-Gigabit network mount for downstream analysis. Primary analysis—sample de-multiplexing and alignment to the mouse genome—is performed using Illumina’s CASAVA 1.8.2 software suite. The sample error rate is less than 2%, and the distribution of reads per sample in a lane is within reasonable tolerance.
RNA-seq data analysis and Gene ontology analysis
Data were utilized in a server provided by Research Support Center for Bio-Bigdata Analysis and Utilization of Biological Resources in Soonchunhyang University, Korea. Reads were mapped to the mouse genome (mm10) using STAR software72. DESeq273 software was applied to the counts of protein-coding genes to estimate the fold-change between the samples from mice that Ehmt2f/f;pS-E/m+ versus Nestin-cre+/−;Ehmt2f/f;mS-E/p+. Analyses in gene set enrichment analysis (v4.1.0) were performed on normalized counts generated in DESeq2 to analyze whether published gene sets were significantly enriched in either the Ehmt2f/f;pS-E/m+ or Nestin-cre+/−;Ehmt2f/f;mS-E/p+74. The analysis used c5.all.v.7.4.symbols.gmt (gene ontology) gene set databases with default parameters. A false discovery rate (FDR) of less than 25% was used as a cut-off for a gene set to be significantly enriched. Gene expression levels were converted into heatmaps and colors quantitatively correspond to fold-changes. For analysis of chrRNA-seq and bulk RNA-seq, reads were mapped to the human genome (hg38) by Burrows-Wheeler Aligner (v0.7.12). The alignment BAM files were converted to bigWig file format and visualized in the UCSC Genome Browser (https://genome.ucsc.edu/)75.
ATAC-seq
ATAC-seq libraries were constructed with 5 × 104 human fibroblast cells following Omni-ATAC protocol (Illumina FC-121-1031)76. The libraries were sequenced on Illumina Nextseq 500 (paired-end run, 42 bp): sequenced reads were trimmed with adaptor sequences (cutadapt v1.9.1)77 and mapped to the human genome (GRCh38/hg38) by Bowtie2 (v2.3.4.1)78. Mitochondrial and duplicated reads were removed by SAMtools (v1.9)79 and Picard (v2.9.0, https://broadinstitute.github.io/picard/), respectively. Peaks were found by MACS2 (v.2.1.1)80 and visualized by deepTools (v3.1.1)81. Motif enrichment analysis of ATAC-seq peaks was done by HOMER (v4.10)82. P values for motif enrichment were calculated using cumulative binomial distribution.
DNA methylation array
The Infinium MethylationEPIC Kit (Illumina) was used to measure DNA methylation profiles from the eight human fibroblast lines. Genomic DNA was extracted using DNeasy blood and tissue kit (Qiagen) following the manufacturer’s instruction. DNA input of 500 ng was preprocessed for bisulfite conversion, and DNA methylation profiling was conducted at Yale Center for Genome Analysis as previously described in ref. 83. GenomeStudio software (Illumina) was used for Methylation EPIC data analysis.
Hi-C and bioinformatics analysis
Hi-C data was generated using the Arima-HiC kit (Arima Genomics, A510008), according to the manufacturer's protocols, and analyzed at Yale Center for Genome Analysis. Briefly, human fibroblasts grown on 100 mm diameter dishes were collected and resuspended in media for crosslinking with formaldehyde (the final concentration; 2%). After incubation at room temperature for 10 min, stop solution 1 was added and incubated at room temperature for 5 min. We then followed the instruction described in User Guide (Document# A160134 v01) to purify DNA for library preparation. Before proceeding to library preparation, we did shallow sequencing for quality determination following the Arima-HiC QC Quality Control protocol (Supplementary Table 5). For studying 3D genome conformation, we obtained 700 million read-pairs per sample. HiC library was sequenced in paired-end mode (2 × 150 bp read length) with NovaSeq (Illumina) and mapped to human genome hg38. For analysis, we used Juicer, providing a pipeline from processing raw Fastq reads to high-order analysis, including contact ___domain and chromatin loops84. To visualize.hic.file, we used Juicebox85, WashU Epigenome Browser86, and IGV87.
Probe design and synthesis for chromatin tracing
To design DNA FISH probes for chromatin tracing, the genomic regions of interest (Chr15: 23,500,000–25,650,000, hg38) were each divided into 42 consecutive 50-kb target segments. For each 50-kb target segment, 500 oligonucleotides were designed as template oligos. On each template oligo, the following sequences were concatenated from 5′ to 3′: (1) a 20-nucleotide (nt) forward priming sequence, (2) a 20-nt secondary probe binding sequence, (3) a 30-nt genome targeting sequence, (4, 5) two 20-nt secondary probe binding sequences, and (6) a 20-nt reverse priming sequence. The 30-nt genome targeting sequences were designed by ProbeDealer88 with an extra BLAST89 against the repetitive genome to remove repetitive target sequences. The template oligo pool for primary probes was purchased from TWIST Bioscience. The probes were synthesized as described previously49,90,91,92.
Primary probe hybridization for chromatin tracing
The experiment was modified from our published protocol49,90,91,92. The human fibroblasts derived from patients grown on a 40-mm-diameter coverslip were fixed in 4% (w/v) paraformaldehyde for 10 min at room temperature, followed by twice DPBS washes. Cells were then permeabilized with 0.5% (v/v) Triton X-100 in 1x DPBS for 10 min at room temperature, followed by two DPBS washes. Fibroblasts were treated with 0.1 M HCl for 5 min at room temperature and washed twice with DPBS, followed by a 45 min treatment with ribonuclease A (RNase A) (0.1 mg/ml) in DPBS at 37 °C and two DPBS washed. Cells were then incubated with prehybridization buffer composed of 50% (v/v) formamide and 0.1% v/v Tween-20 in 2x saline sodium citrate (SSC) for 30 min at room temperature. After carefully removing excess liquid by dipping on tissue paper, the coverslip was flipped onto a glass slide and contacted with 30 ul hybridization buffer composed of 50% (v/v) formamide, 20% (w/v) dextran sulfate, and 20 μM primary probes in 2x SSC. The samples were then heat-denatured on an 86 °C heat block (with a surface temperature of ~80 °C) for 3 min and incubated in a humid chamber overnight (>18 h) at 37 °C. Next, the samples were then washed with 0.1% (v/v) Tween 20 in 2x SSC in a 60 °C water bath twice for 15 min each and once at room temperature for 15 min.
Sequential hybridization of secondary probes for chromatin training
After primary probe hybridization, the coverslip was assembled into a Bioptech’s FCS2 flow chamber and connected to a homebuilt automated fluidics system49,90. To read out each genomic locus, we used adapter oligos and common readout oligos that were labeled with Cy3 or Cy5 dye through a disulfide bond to enable signal removal by tris(2-carboxyethyl)phosphine (TCEP) wash. Each adapter oligo is composed of one 20-nt primary probe binding sequence (that binds to the overhangs on primary probes) and two replicates of the same 20-nt common readout oligo binding sequences (Supplementary Data 1). Before each round of imaging, the sample were incubated with secondary hybridization buffer composed of 20% (v/v) ethylene carbonate (EC), two 10 nM adapter oligos, and two 15 nM Cy3- and Cy5-labeled common readout oligos in 2x SSC. To perform the imaging, we took z-stepping images with 647-nm, 560-nm, and 488-nm laser illuminations for Cy5 and Cy3 readout oligos and fiducial beads, respectively, with 200-nm step sizes and 0.4-s exposure time at each step. After each round of imaging, the signals were removed by TCEP washing buffer composed of 20% (v/v) EC, 50 mM TCEP, and 1 μM blocking oligos in 2xSSC. Blocking oligos are dye-free common readout oligos to block any unoccupied binding sites. This procedure was repeated 21 times until all 42 segments were imaged.
Chromatin tracing analysis
Before foci fitting, the color shift between the 560-nm and 647-nm laser channels was corrected with TetraSpeck bead images; the sample drift during sequential hybridization and imaging was corrected with fiducial bead images. Next, cell nucleus was segmentation with DAPI images and used as a mask to fit the 3D positions of loci only inside the cell nucleus. The fitted loci were then linked into chromatin traces based on their spatial clustering patterns. Finally, we tried to re-fit the missing loci in chromatin traces within the chromatin trace region in corresponding hybridization and added them to the chromatin traces.
RNA fluorescence in situ hybridization (FISH) with immunofluorescence
To design the RNA FISH probes, we followed our published protocol88 (Supplementary Data 2). The human fibroblasts grown on a 40-mm-diameter coverslip were fixed in 4% PFA for 10 min at room temperature, followed by twice DPBS washes. The cells were permeabilized with 0.5% Triton in DPBS (DPBST) for 10 min and then incubated with blocking buffer (1% BSA in DPBST) for 30 min. After incubation with primary antibody (EHMT2, Millipore) and secondary antibody (Alexa Fluor 488 dye, Invitrogen) in blocking buffer for 1 h each, the cells were post-fixed with 4% PFA for 10 min at room temperature followed by three times DPBS washes. 2xSSC was used for rinse the cells, and pre-hybridization buffer (50% Formamide in 2xSSC with RNase inhibitor (1:1000)) was treated for 10 min at room temperature. Fibroblasts were incubated with hybridization buffer (50% formamide in 2xSSC with 10% dextran sulfate, 1:100 of 10 mg/ml yeast tRNA, 1:100 RNase inhibitor, probe mixture) for overnight at 37 °C. Next day, the cells were washed with 2xSSCT (2xSSC + 0.1% Tween-20) at 60 °C twice for 15 min, then at room temperature once. Washed with 2xSSC, the cells were hybridized with 3 nM cy7-labeled readout probe in secondary hybridization buffer before imaging.
Statistical analysis
We used GraphPad Prism for the statistical analyses. Differences between groups were analyzed by One-way ANOVA followed by Dunnett’s multiple comparison test, or two-way ANOVA followed by Tukey’s multiple comparisons test, or unpaired two-tailed Student’s t-test, where appropriate. *p < 0.05 was considered statistically significant (**p < 0.01, ***p < 0.001, ****p < 0.0001).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
DNA Methyl array data can be accessed at the NCBI Gene Expression Omnibus (GEO) under accession number GSE298378. All sequence data can be accessed at the NCBI Sequence Read Archive (SRA) under bioproject accession number PRJNA1187776. Previously published data were downloaded from28,93, GSE138102. Source data are provided with this paper.
References
Tucci, V., Isles, A. R., Kelsey, G., Ferguson-Smith, A. C. & Erice Imprinting, G. Genomic Imprinting and Physiological Processes in Mammals. Cell 176, 952–965 (2019).
Barlow, D. P. & Bartolomei, M. S. Genomic imprinting in mammals. Cold Spring Harb Perspect Biol. 6, 466–473 (2014).
Surani, M. A., Barton, S. C. & Norris, M. L. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 308, 548–550 (1984).
McGrath, J. & Solter, D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 37, 179–183 (1984).
Jiang, Y., Tsai, T. F., Bressler, J. & Beaudet, A. L. Imprinting in angelman and prader-willi syndromes. Curr. Opin. Genet. Dev. 8, 334–342 (1998).
Wang, S. E. & Jiang, Y. H. Novel epigenetic molecular therapies for imprinting disorders. Mol. Psychiatry 28, 3182–3193 (2023).
Eggermann, T. et al. Imprinting disorders. Nat. Rev. Dis. Prim. 9, 33 (2023).
Horsthemke, B. & Wagstaff, J. Mechanisms of imprinting of the Prader-Willi/Angelman region. Am. J. Med. Genet. A 146A, 2041–2052 (2008).
Sutcliffe, J. S. et al. Deletions of a differentially methylated CpG island at the SNRPN gene define a putative imprinting control region. Nat. Genet. 8, 52–58 (1994).
Buiting, K. et al. Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15. Nat. Genet. 9, 395–400 (1995).
Mapendano, C. K. et al. Expression of the Snurf-Snrpn IC transcript in the oocyte and its putative role in the imprinting establishment of the mouse 7C imprinting ___domain. J. Hum. Genet. 51, 236–243 (2006).
Chai, J. H. et al. Identification of four highly conserved genes between breakpoint hotspots BP1 and BP2 of the Prader-Willi/Angelman syndromes deletion region that have undergone evolutionary transposition mediated by flanking duplicons. Am. J. Hum. Genet. 73, 898–925 (2003).
Kantor, B., Kaufman, Y., Makedonski, K., Razin, A. & Shemer, R. Establishing the epigenetic status of the Prader-Willi/Angelman imprinting center in the gametes and embryo. Hum. Mol. Genet. 13, 2767–2779 (2004).
Proudhon, C. et al. Protection against de novo methylation is instrumental in maintaining parent-of-origin methylation inherited from the gametes. Mol. Cell 47, 909–920 (2012).
Bourc’his, D., Xu, G. L., Lin, C. S., Bollman, B. & Bestor, T. H. Dnmt3L and the establishment of maternal genomic imprints. Science 294, 2536–2539 (2001).
Ohta, T. et al. Imprinting-mutation mechanisms in Prader-Willi syndrome. Am. J. Hum. Genet. 64, 397–413 (1999).
Yang, T. et al. A mouse model for Prader-Willi syndrome imprinting-centre mutations. Nat. Genet. 19, 25–31 (1998).
Dubose, A. J., Smith, E. Y., Yang, T. P., Johnstone, K. A. & Resnick, J. L. A new deletion refines the boundaries of the murine Prader-Willi syndrome imprinting center. Hum. Mol. Genet. 20, 3461–3466 (2011).
Bressler, J. et al. The SNRPN promoter is not required for genomic imprinting of the Prader-Willi/Angelman ___domain in mice. Nat. Genet. 28, 232–240 (2001).
Xin, Z. et al. Role of histone methyltransferase G9a in CpG methylation of the Prader-Willi syndrome imprinting center. J. Biol. Chem. 278, 14996–15000 (2003).
Nakagaki, A., Osanai, H. & Kishino, T. Imprinting analysis of the mouse chromosome 7C region in DNMT1-null embryos. Gene 553, 63–68 (2014).
Kim, Y. et al. Targeting the histone methyltransferase G9a activates imprinted genes and improves survival of a mouse model of Prader-Willi syndrome. Nat. Med. 23, 213–222 (2017).
Saitoh, S. & Wada, T. Parent-of-origin specific histone acetylation and reactivation of a key imprinted gene locus in Prader-Willi syndrome. Am. J. Hum. Genet. 66, 1958–1962 (2000).
Janssen, A., Colmenares, S. U. & Karpen, G. H. Heterochromatin: guardian of the genome. Annu. Rev. Cell Dev. Biol. 34, 265–288 (2018).
Bannister, A. J. et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo ___domain. Nature 410, 120–124 (2001).
Nagano, T. et al. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 322, 1717–1720 (2008).
James, T. C. & Elgin, S. C. Identification of a nonhistone chromosomal protein associated with heterochromatin in Drosophila melanogaster and its gene. Mol. Cell Biol. 6, 3862–3872 (1986).
Jiang, Q. et al. G9a plays distinct roles in maintaining DNA methylation, retrotransposon silencing, and chromatin looping. Cell Rep. 33, 108315 (2020).
Fukuda, K. et al. Regulation of mammalian 3D genome organization and histone H3K9 dimethylation by H3K9 methyltransferases. Commun. Biol. 4, 571 (2021).
Padeken, J., Methot, S. P. & Gasser, S. M. Establishment of H3K9-methylated heterochromatin and its functions in tissue differentiation and maintenance. Nat. Rev. Mol. Cell Biol. 23, 623–640 (2022).
Zhang, T. et al. G9a/GLP complex maintains imprinted DNA methylation in embryonic stem cells. Cell Rep. 15, 77–85 (2016).
Epsztejn-Litman, S. et al. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat. Struct. Mol. Biol. 15, 1176–1183 (2008).
Tachibana, M. et al. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 16, 1779–1791 (2002).
Schaefer, A. et al. Control of cognition and adaptive behavior by the GLP/G9a epigenetic suppressor complex. Neuron 64, 678–691 (2009).
Yamada, A. et al. Derepression of inflammation-related genes link to microglia activation and neural maturation defect in a mouse model of Kleefstra syndrome. iScience 24, 102741 (2021).
Lehnertz, B. et al. Activating and inhibitory functions for the histone lysine methyltransferase G9a in T helper cell differentiation and function. J. Exp. Med. 207, 915–922 (2010).
Polvora-Brandao, D. et al. Loss of hierarchical imprinting regulation at the Prader-Willi/Angelman syndrome locus in human iPSCs. Hum. Mol. Genet. 27, 3999–4011 (2018).
Kanber, D. et al. A paternal deletion of MKRN3, MAGEL2 and NDN does not result in Prader-Willi syndrome. Eur. J. Hum. Genet 17, 582–590 (2009).
Fugón, O. J. G. et al. Integration of CTCF loops, methylome, and transcriptome in differentiating LUHMES as a model for imprinting dynamics of the 15q11-q13 locus in human neurons. Hum. Mol. Genet. 33, 1711–1725 (2024).
Hara-Isono, K. et al. Genome-wide methylation analysis in Silver-Russell syndrome, Temple syndrome, and Prader-Willi syndrome. Clin. Epigenetics 12, 159 (2020).
Dindot, S. V., Person, R., Strivens, M., Garcia, R. & Beaudet, A. L. Epigenetic profiling at mouse imprinted gene clusters reveals novel epigenetic and genetic features at differentially methylated regions. Genome Res. 19, 1374–1383 (2009).
Xie, L. et al. Development of a brain-penetrant G9a methylase inhibitor to target Alzheimer’s disease-associated proteopathology. Nat. Commun. 16, 4222 (2025).
Wang, S. E. et al. Newly developed oral bioavailable EHMT2 inhibitor as a potential epigenetic therapy for Prader-Willi syndrome. Mol. Ther. 32, 2662-2675 (2024).
Wang, X. et al. KDM3B suppresses APL progression by restricting chromatin accessibility and facilitating the ATRA-mediated degradation of PML/RARalpha. Cancer Cell Int. 19, 256 (2019).
Yang, J. et al. Analysis of chromatin organization and gene expression in T cells identifies functional genes for rheumatoid arthritis. Nat. Commun. 11, 4402 (2020).
Fujimori, T. et al. Single-cell chromatin state transitions during epigenetic memory formation. Mol. Cell https://papers.ssrn.com/sol3/papers.cfm?abstract_id=5069284 (2024).
Richer, S. et al. Widespread allele-specific topological domains in the human genome are not confined to imprinted gene clusters. Genome Biol. 24, 40 (2023).
Loftus, D., Bae, B., Whilden, C. M. & Whipple, A. J. Allelic chromatin structure precedes imprinted expression of Kcnk9 during neurogenesis. Genes Dev. 37, 829–843 (2023).
Liu, M. et al. Chromatin tracing and multiplexed imaging of nucleome architectures (MINA) and RNAs in single mammalian cells and tissue. Nat. Protoc. 16, 2667–2697 (2021).
Hu, M. & Wang, S. Chromatin tracing: imaging 3D genome and nucleome. Trends Cell Biol. 31, 5–8 (2021).
Dittrich, B. et al. Imprint switching on human chromosome 15 may involve alternative transcripts of the SNRPN gene. Nat. Genet. 14, 163–170 (1996).
Fagerberg, L. et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteom. 13, 397–406 (2014).
Sledziowska, M. et al. Non-coding RNAs associated with Prader-Willi syndrome regulate transcription of neurodevelopmental genes in human induced pluripotent stem cells. Hum. Mol. Genet. 32, 608–620 (2023).
Fritsch, L. et al. A subset of the histone H3 lysine 9 methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Mol. Cell 37, 46–56 (2010).
Chin, H. G. et al. Automethylation of G9a and its implication in wider substrate specificity and HP1 binding. Nucleic Acids Res. 35, 7313–7323 (2007).
Mahmood, S. R. et al. beta-actin dependent chromatin remodeling mediates compartment level changes in 3D genome architecture. Nat. Commun. 12, 5240 (2021).
Esteve, P. O. et al. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 20, 3089–3103 (2006).
San Jose-Eneriz, E. et al. Discovery of first-in-class reversible dual small molecule inhibitors against G9a and DNMTs in hematological malignancies. Nat. Commun. 8, 15424 (2017).
Chen, Z. & Zhang, Y. Role of mammalian DNA methyltransferases in development. Annu. Rev. Biochem. 89, 135–158 (2020).
Wawrzik, M., Spiess, A. N., Herrmann, R., Buiting, K. & Horsthemke, B. Expression of SNURF-SNRPN upstream transcripts and epigenetic regulatory genes during human spermatogenesis. Eur. J. Hum. Genet. 17, 1463–1470 (2009).
Rodriguez-Jato, S., Nicholls, R. D., Driscoll, D. J. & Yang, T. P. Characterization of cis- and trans-acting elements in the imprinted human SNURF-SNRPN locus. Nucleic Acids Res. 33, 4740–4753 (2005).
Andergassen, D. et al. The Airn lncRNA does not require any DNA elements within its locus to silence distant imprinted genes. PLoS Genet. 15, e1008268 (2019).
Latos, P. A. et al. Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science 338, 1469–1472 (2012).
Wu, M. Y., Tsai, T. F. & Beaudet, A. L. Deficiency of Rbbp1/Arid4a and Rbbp1l1/Arid4b alters epigenetic modifications and suppresses an imprinting defect in the PWS/AS ___domain. Genes Dev. 20, 2859–2870 (2006).
Sanjana, N. E., Shalem, O. & Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014).
Ma, H. et al. Multicolor CRISPR labeling of chromosomal loci in human cells. Proc. Natl. Acad. Sci. USA 112, 3002–3007 (2015).
Sanson, K. R. et al. Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat. Commun. 9, 5416 (2018).
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013).
Farooq, U. & Notani, D. Optimized protocol to create deletion in adherent cell lines using CRISPR/Cas9 system. STAR Protoc. 2, 100857 (2021).
Raab, J. R. et al. SWI/SNF remains localized to chromatin in the presence of SCHLAP1. Nat. Genet. 51, 26–29 (2019).
Wang, S. E. et al. TRPV1 regulates stress responses through HDAC2. Cell Rep. 19, 401–412 (2017).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 (2005).
Kent, W. J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002).
Corces, M. R. et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. J Bioinform. Action 17, 3 (2011).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Ramirez, F., Dundar, F., Diehl, S., Gruning, B. A. & Manke, T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Xu, K. et al. Epigenome-wide DNA methylation association analysis identified novel loci in peripheral cells for alcohol consumption among European American male veterans. Alcohol Clin. Exp. Res. 43, 2111–2121 (2019).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Robinson, J. T. et al. Juicebox.js provides a cloud-based visualization system for Hi-C Data. Cell Syst. 6, 256–258.e1 (2018).
Zhou, X. et al. Epigenomic annotation of genetic variants using the Roadmap Epigenome Browser. Nat. Biotechnol. 33, 345–346 (2015).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Hu, M. et al. ProbeDealer is a convenient tool for designing probes for highly multiplexed fluorescence in situ hybridization. Sci. Rep. 10, 22031 (2020).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinform. 10, 421 (2009).
Wang, S. et al. Spatial organization of chromatin domains and compartments in single chromosomes. Science 353, 598–602 (2016).
Cheng, Y., Liu, M., Hu, M. & Wang, S. TAD-like single-cell ___domain structures exist on both active and inactive X chromosomes and persist under epigenetic perturbations. Genome Biol. 22, 309 (2021).
Patterson, B. et al. Female naive human pluripotent stem cells carry X chromosomes with Xa-like and Xi-like folding conformations. Sci. Adv. 9, eadf2245 (2023).
Bernstein, B. E. et al. The NIH roadmap epigenomics mapping consortium. Nat. Biotechnol. 28, 1045–1048 (2010).
Acknowledgements
This work was supported in part by the grant R01HD088626 (to J.J.), R01AG084184 (to J.J.), DP2GM137414 (to S.W.), R01HG011245 (to S.W.), U01CA260701 (to S.W.), R01HD088626 (to Y-H.J), HD077197 (to Y-H.J), MH117289 (to Y-H.J.), and HD088007 (to Y-H.J.) from the U.S. National Institutes of Health and AWD0009884-GR120668 from Critical Path Institute (to Y-H.J) and Foundation for Prader-Willi Research (to Y-H.J.), 2021R1C1C1005725 (to M-A.J.), RS-2024-00335057 (to J.L.), 2021R1A6A1A03039503 (to S.J), Yonsei Education and Research Center for Biosystems Brain Korea 21 Four program, and Brain Pool program RS-2024-00409006 (to S.E.W and J.Y.H) funded by the Ministry of science and ICT through the National Research Foundation of Korea (NRF). This research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number 1S10OD030363-01A1. We would like to thank Dr. Fabio M.V. Rossi for providing Ehmt2f/f mice used to establish our colony.
Author information
Authors and Affiliations
Contributions
S.E.W., Y.C., and J.L. designed the experiments, collected and analyzed the data. S.E.W., Y.C., and Y-H.J. wrote the manuscript. S.E.W1., M-A.J., Y.K., M.D., S.J., and S-N.Q. performed in vivo experiments and visualized the data. D.E.L. and J.Y.H. curated the data. J.J. and Y.X. synthesized the EHMT2/G9a inhibitor MS1262. Y-H.J. and S.W. designed and supervised the work, as well as revising the manuscripts. E.N.F. gave comments and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
Y-H.J. is a co-founder of, have an equity interest in, a scientific advisor to, and have an unpaid seat on the board of directors to Couragene, Inc., a biotechnology company that has licensed his intellectual property. However, Couragene did not have any direct role in this paper. J.J., Y.X., Y-H.J., and S.E.W. are described in a patent application for MS1262 as inventors, titled “G9A/GLP INHIBITORS AND METHODS OF USE” which was filed in the United States Patent and Trademark Office as application 63/515,010 (the “Application”). S.W. is an inventor on patent applications related to the chromatin tracing technology applied for by Harvard University and Yale University. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Shinji Saitoh, Chaochun Zou, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, S.E., Cheng, Y., Lim, J. et al. Mechanism of EHMT2-mediated genomic imprinting associated with Prader-Willi syndrome. Nat Commun 16, 6125 (2025). https://doi.org/10.1038/s41467-025-61156-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-61156-8
