
Abstract
Rodents serve as natural reservoirs and vectors for a multitude of zoonotic viruses. Analyzing the viral diversity in rodents is crucial for predicting the emergence of infectious diseases. Through high-throughput sequencing, we characterized the RNA virome of nine rodent species from the families Cricetidae and Muridae, collected from suburban Beijing (n = 432). The composition of the virome exhibited significant variation at the virus family level across the nine rodent species and three types of natural habitats. A total of 142 viral species were identified, including 133 associated with vertebrates and 9 associated with invertebrates, spanning 26 families. Notably, 75 of these viruses were novel while 67 were known viruses. Twenty-five viruses were classified as high-risk, including eight zoonotic viruses and 17 spillover-risk viruses. Additionally, nine known viruses were identified for the first time in China. Thirty-three viruses demonstrated potential for cross-species transmission. Understanding the virome characterization of field-collected rodent might provide valuable insights into the potential risk of zoonotic spillover to humans.
Similar content being viewed by others

Background
Emerging and re-emerging infectious diseases pose a significant threat to public health. Over two-thirds of these diseases are zoonotic, originating from pathogens transmitted between non-human vertebrates and humans1,2. Rodents represent the most abundant and diverse group of mammals globally, comprising nearly 42% of all mammalian species and inhabiting a wide array of biotopes globally3,4. The diversity and population dynamics of rodents, coupled with their opportunistic and synanthropic behaviors, make them highly effective amplifiers and disseminators of zoonotic diseases. Notably, rodents frequently cohabit with livestock, facilitating pathogen exchange and playing a pivotal role in bridging disease hotspots between rural and urban areas. This interaction significantly increases the risk of zoonotic pathogen spillover, contributing to the emergence of infectious diseases in human populations5,6. For instance, both decreases and increases in viral richness within rodent populations have been attributed to anthropogenic alterations in host species composition and population densities7, underscoring vegetation types and landscapes as key determinants of host composition8,9. Rodents serve as reservoirs and hosts for at least 60 zoonotic diseases, including zoonotic babesiosis, Lassa fever, hemorrhagic fever with renal syndrome (HFRS), and the hantavirus cardiopulmonary syndrome (HCPS), both caused by Hantavirus infection10. Over the past few decades, there has been a notable rise in human diseases associated with rodent reservoirs. Due to the impact of human activities, such as deforestation and agricultural expansion, the frequency and intensity of human-rodent interactions are expected to increase, thereby facilitating pathogen transmission to humans. Consequently, future zoonotic spillover events from rodents are anticipated to occur more frequently. It is imperative to gain a comprehensive understanding of the composition and evolution of rodent-borne pathogens.
In China, a diverse array of rodent species has been identified due to the presence of extensive and varied habitats characterized by abundant rainfall, lush vegetation and a rich diversity of wildlife and livestock, etc. Metagenomic surveys have confirmed that wild rodents in China harbor a high diversity of viruses, including those closely related to human pathogens7,11,12. However, most previous studies were performed in sparsely populated rural areas or natural landscapes7,13. Over the past decade, rapid urbanization and ecological conservation efforts, have resulted in significant increases in population densities and the biodiversity in many cities in China, including major metropolises like Beijing. This trend is particularly evident in remote and mountainous suburban areas surrounding the city. Recent epidemiological studies in Beijing have revealed zoonotic pathogen infections among animals, including wild-caught rodents14,15,16,17. Babesia microti, the most prevalent Babesia species infecting humans, was found circulating in rodent populations across almost all districts of Beijing, indicating the widespread distribution of Babesia species in the city16. The H4N6 avian influenza virus, which circulates globally among waterfowl and has been repeatedly detected in mammalian species including humans, was isolated from mallard ducks in Beijing18. Animal-to-human transmission of Severe fever with thrombocytopenia syndrome virus (SFTSV), an emerging tick-borne Phenuiviridae causing viral hemorrhagic fever, has been recently reported in Beijing19,20. Despite these findings, the comprehensive epidemiology of mammalian viral families in small mammalian hosts in Beijing remains largely unexplored. In this study, we utilized next-generation sequencing metagenomic analysis to survey a range of mammalian viral families in rodents belonging to two prominent mammalian families (Cricetidae and Muridae) in suburban Beijing.
Results
The virome of rodent
The samples were grouped into 21 pools based on rodent species, natural habitats and sampling years and seasons, also ensuring a comparable sample size across groups. These pooled samples were then subjected to next-generation sequencing (Fig. 1 and Supplementary Tables 1, 2). Following the quality control process, we obtained more than 7.5 billion sequence reads, which were de novo assembled into 525,739 contigs. Among these, 2060 contigs were identified as viral sequences. Although bacterial, fungal, and plant-associated viruses, comprising ~1233 contigs, were also detected, they were excluded from subsequent analyses.

Circles on the map represent the geographical locations in Beijing where rodent surveys were conducted (n = 432). The colors of the circles correspond to different rodent species as indicated in the legend, while the sampled districts are highlighted in varying shades of orange.
Based on blast alignment and homology analysis of RNA-dependent RNA polymerase (RdRp) for RNA viruses and replicase protein genes for DNA viruses, we identified a total of 142 viral species from 26 viral families (Fig. 2A and Supplementary Table 3). This included nine DNA virus species belonging to the family Anelloviridae, Circoviridae, Genomoviridae, Parvoviridae, Hepadnaviridae, as well as 133 RNA virus species distributed across 21 RNA families (Astroviridae, Caliciviridae, Chuviridae, Coronaviridae, Dicistroviridae, Flaviviridae, Hantaviridae, Hepeviridae, Iflaviridae, Nairoviridae, Nodaviridae, Orthomyxoviridae, Paramyxoviridae, Peribunyaviridae, Phenuiviridae, Picobirnaviridae, Picornaviridae, Polycipiviridae, Qinviridae, Rhabdoviridae, Sedoreoviridae), along with unclassified viruses from Riboviria and the order Bunyavirales, Reovirales. The majority of the identified viruses (n = 133) were associated with vertebrates, while the remaining 9 were associated with invertebrates (Supplementary Table 3).

A Species richness and abundance of viruses associated with vertebrate and invertebrate in wild rodents. B The viral composition was characterized for each host species across different natural habitat. C Principal coordinate analysis illustrating the variation in virome compositions among the three distinct natural habitats.
A diverse array of viral species was identified across nine rodent species (Supplementary Fig. 1), with each rodent species hosting between 1 and 74 viruses. Notably, Rattus norvegicus harbored the highest average number of viruses (617 viruses per 100 individuals), followed by Cricetulus longicaudatus (44/100), Apodemus peninsulae (26/100), Apodemus draco (25/100), Niviventer confucianus (20/100), Myodes rufocanus (14/100), Apodemus agrarius (13/100), Allocricetulus eversmanni (12/100), and Mus musculus (6/100). The virus family composition varied significantly among the nine rodent species, and across three types of natural habitats where the rodents were collected (Fig. 2B). Principal coordinate analysis (PCoA) demonstrated that rodents inhabiting grassland had a distinct viral composition compared to those inhabit woodland and bushland (adonis test, P < 0.05; Fig. 2C).
We subsequently compared the viral composition between two rodent families, Cricetidae and Muridae (Supplementary Fig. 2). The predominant viral families differ in that Hepeviridae, Phenuiviridae and Flaviviridae predominant were in Cricetidae, while Paramyxoviridae, Picobirnaviridae and Hantaviridae were predominant in Muridae, respectively. A greater number of viral families were identified in Muridae compared to Cricetidae (25 versus 13 viral families). A statistically significant difference in species richness at the viral family level was observed between the two rodent families (Wilcoxon rank sum test, P < 0.001; Supplementary Fig. 2).
The viral composition (species richness) and alpha diversity (Shannon index) were further examined across sampling seasons and natural habitats (Supplementary Fig. 3). We identified higher viral species richness in autumn (74 viral species) and bushland (113 viral species), while more viral families in summer (23 viral families) and bushland (26 viral families). However, no statistically significant difference was observed in the Shannon index across sampling seasons and natural habitats (Wilcoxon rank sum test, P > 0.05; Supplementary Fig. 3).
In addition, a total of 67 complete viral genomes were obtained from 45 viral species across 13 viral families and unclassified Riboviria (Supplementary Table 4). Notably, 21 genomes were derived from novel viral species within seven families (Astroviridae, Caliciviridae, Dicistroviridae, Flaviviridae, Hepeviridae, Picobirnaviridae, and Picornaviridae) and unclassified Riboviria (Supplementary Table 3). Importantly, no evidence of recombination events was detected in these newly identified viruses. Simplot analysis revealed significant genetic divergence between the 16 representative novel viruses and their respective families members (Supplementary Figs. 4, 5).
Zoonotic and spillover-risk viruses
Among the identified 142 viruses, 25 were categorized as high-risk viruses from 16 families (Fig. 3A). This group included eight zoonotic viruses (marked by red dots) and 17 spillover-risk viruses (marked by blue dots) (Fig. 3B). Among the 25 high-risk viruses, Avian orthoavulavirus and Influenza A virus (H9N2) were identified across the largest number of 22 and 15 orders, respectively, followed by Rabies virus (nine orders), Rotavirus A (seven), Beiji nairovirus (four), Cardiovirus B (four) and chicken picobirnavirus (four), based on all available data (Supplementary Fig. 6). Two spillover-risk viruses, Pigeon torque teno virus and Sichuan mosquito circovirus 3, although documented only in pigeon and mosquito respectively, were found within the highest number of rodent species in our dataset (four out of nine rodent species), indicating these viruses might be primarily associated with rodents rather than other wildlife (Fig. 3B).

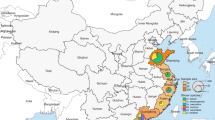
A An overview of the 25 high-risk viruses identified in rodents in this study. B The currently identified high-risk viruses in relate to the rodent species. The zoonotic viruses (n = 8) are marked in red dot; the spillover-risk viruses (n = 17) are marked in blue dot; the viruses first detected in China (n = 9) are marked in green dot. C The global distribution of nine viruses first discovered in China are illustrated. Three spillover-risk viruses, namely Chicken picobirnavirus, Lasius neglectus virus 1, and Lasius niger virus 1, are labeled with red color. D The associations between high-risk viruses and their rodent species hosts are depicted. The size of the colored circles indicates the number of zoonotic and spillover-risk viruses carried by each rodent species.
Notably, eight vertebrate viruses were identified for the first time in rodents, including Nuomin virus, Shrew hepatitis B virus, Beiji nairovirus, Influenza A virus H9N2, Avian orthoavulavirus 1, chicken picobirnavirus, Feline hunnivirus, and Pigeon torque teno virus. Additionally, nine viruses previously considered specific to invertebrates, predominantly carried by ticks (six out of nine), were also identified. These include Mukawa virus, Sichuan mosquito circovirus 3, Tick-associated circular virus-6, Tick-associated genomovirus 1, Gakugsa tick virus, Onega tick phlebovirus, Sara tick phlebovirus, Lasius neglectus virus 1, and Lasius niger virus 1 (Fig. 3B). Of particular significance is the identification of three spillover-risk viruses in China for the first time: chicken picobirnavirus, Lasius neglectus virus 1, and Lasius niger virus 1 (Fig. 3C and Supplementary Table 5). Furthermore, six other viruses—Mossman virus, rodent Paramyxovirus LR11-23, Apodemus peninsulae jeilongvirus, cardiovirus C1, Theiler’s encephalomyelitis virus, Bastrovirus BAS-3, were detected in China for the first time. Although these viruses do not belong to high-risk pathogens, their discovery remains noteworthy (Fig. 3C and Supplementary Table 5).
We performed a comprehensive analysis comparing the zoonotic pathogens carried by the nine rodent species examined in this study. Apart from M. musculus, each of the remaining eight rodent species was found to carry at least one high-risk virus (Fig. 3D). Notably, R. norvegicus exhibited the highest number of zoonotic viruses (three), followed by C. longicaudatus (two), N. confucianus (two), A. agrarius (two), A. draco (one) and A. peninsulae (one).
Diversification and evolution of viruses in rodents
We constructed phylogenetic trees based on the protein sequences of RdRp for RNA viruses and replicase protein for DNA viruses to validate the taxonomic phylogeny of the 67 known and 75 novel viral species (Supplementary Figs. 7–32). The 75 novel viruses, belonging to five phyla (Cossaviricota, Duplornaviricota, Kitrinoviricota, Negarnaviricota, Pisuviricota), encompass putative members of 10 unclassified viruses and 15 known viral families (Fig. 4): Anelloviridae, Astroviridae, Caliciviridae, Dicistroviridae, Flaviviridae, Hepeviridae, Iflaviridae, Nodaviridae, Parvoviridae, Peribunyaviridae, Picobirnaviridae, Picornaviridae, Polycipiviridae, Qinviridae, Sedoreoviridae. Among these families, Picobirnaviridae (41), Qinviridae (six), Parvoviridae (three), Caliciviridae (two), Hepeviridae (two), and Picornaviridae (two) are predominant, with others belonging to the Anelloviridae (Cricetulus longicaudatus anello-like virus), Astroviridae (Rattus norvegicus astro-like virus), Dicistroviridae (Apodemus draco aparavirus), Flaviviridae (Myodes rufocanus hepacivirus), Iflaviridae (Niviventer confucianus iflavirus), Nodaviridae (Apodemus agrarius nodavirus), Peribunyaviridae (Niviventer confucianus picobirna-like virus 20), Polycipiviridae (Niviventer confucianus sopolycivirus), Sedoreoviridae (Apodemus peninsulae orbi-like virus), and unclassified virus from order Bunyavirales (Rattus norvegicus bunyavirus).

Overview of 26 known viral families and three unclassified virus groups (Unclassified Bunyavirales, Unclassified Reovirales, and Unclassified Riboviria) identified in this study, including 75 novel and 67 known viruses.
Cossaviricota
A total of three novel viruses belonging to the family Parvoviridae, specifically single-stranded DNA viruses, were identified. These include a new member of the genus Aveparvovirus (Niviventer confucianus aveparvovirus) and two unclassified parvoviruses (Cricetulus longicaudatus parvo-like virus and Myodes rufocanus parvo-like virus) (Supplementary Fig. 8). Phylogenetic analysis revealed that these newly discovered parvoviruses form a distinct clade, clustering closely with the recently established genus Tetraparvovirus within the Parvoviridae family. A pairwise comparison of NS1 amino acid (aa) sequences revealed that Niviventer confucianus aveparvovirus shares 52.4% identity with Psittaciform aveparvovirus, while Cricetulus longicaudatus parvo-like virus and Myodes rufocanus parvo-like virus exhibit 65.2-74.1% aa sequence identity with Phoenicopterus roseus parvo-like hybrid virus.
Duplornaviricota
A total of six viruses, comprising four novel and two previously identified viruses were identified within the Reovirales order of the Duplornaviricota phylum (Supplementary Fig. 11). Among the four novel viruses, three exhibited significant divergence from known viruses and could not be classified into any existing viral groups. One novel virus, designated as Shidu orbi-like virus, belongs to the family Sedoreoviridae and exhibits <80% aa sequence identity with other orbiviruses. The two previously identified viruses, Rotavirus A and Rattus norvegicus rotavirus, also fall within the Sedoreoviridae family.
Kitrinoviricota
A total of eight viruses, consisting of four novel viruses and four known viruses, were identified within the phylum Kitrinoviricota, comprising three families (Flaviviridae, Hepeviridae and Nodaviridae) (Supplementary Figs. 12, 13). Within the Flaviviridae family, one novel virus, Myodes rufocanus hepacivirus, and three previously characterized viruses (Wufeng Niviventer niviventer pegivirus 1, Wufeng Niviventer fulvescens pegivirus 1, and rodent hepacivirus) were identified. The genomes of these viruses ranged from 9 to 12 kb, clustering with other species in the Pegivirus and Hepacivirus genera (Fig. 5 and Supplementary Fig. 12). Notably, the novel Myodes rufocanus hepacivirus exhibited 73.3% aa identity with Hepacivirus myodae in the RdRp sequence and encoded a full-length polyprotein encompassing viral helicase1, Peptidase, and RdRP domains (Fig. 5). Within the Hepeviridae family, two novel viruses, Allocricetulus eversmanni hepe-like virus and Cricetulus longicaudatus hepe-like virus, along with one known virus, Apodemus peninsulae hepevirus, were identified. Cricetulus longicaudatus hepe-like virus showed <75% aa identity with its closest known relatives in terms of RdRp sequence similarity, forming a distinct branch within this family (Supplementary Fig. 13). All currently identified members of the Hepeviridae family possessed a conserved RNA replication module containing a capping enzyme superfamily 1 helicase, along with an RdRp ___domain (Fig. 5). In the Nodaviridae family, a novel virus, Apodemus agrarius nodavirus, was identified. This virus shared ~53.7% aa sequence identity with Nodamura virus, and together they formed a unique clade distinct from other alphanodaviruses within the Nodaviridae family (Supplementary Fig. 14).

The genome structure of representative RNA viruses in the phyla Kitrinoviricota, Negarnaviricota, Pisuviricota, Artverviricota, Duplornaviricota, and others are shown. Red and blue colors represent the nomenclature of novel RNA viruses identified in this study and the previously reported RNA viruses, respectively. The symbols “?” and “???” represent single and multiple unknown segment(s) in the genomes of segmented RNA viruses, respectively. Regions encoding major functional proteins or protein domains are labeled, with the homologous proteins or domains across different genomes represented by the same color and shape.
Negarnaviricota
A total of 31 negative-sense single-stranded RNA [ssRNA (−)] viruses were identified, all classified within the Negarnaviricota phylum (Supplementary Figs. 15–23). Among these, eight viruses were determined to be novel: six from the family Qinviridae, one from the family Peribunyaviridae, and one virus (Rattus norvegicus bunyavirus) that remains unclassified within the order Bunyavirales. The novel virus within the Peribunyaviridae family, Niviventer confucianus picobirna-like virus, exhibited <40% aa sequence identity with other members of the Peribunyaviridae family and was most closely related to Nanchang Perib tick virus 1 (Supplementary Fig. 15). The six novel viruses within the Qinviridae family exhibited significant divergence from the only Yingvirus genus, thus forming a separate branch in the Qinviridae family (Supplementary Fig. 16). For the 23 known viruses identified in the Negarnaviricota phylum, Influenza A virus belonging to the genus Alphainfluenzavirus in the family Orthomyxoviridae, and Rabies virus belonging to the genus Lyssavirus from Rhabdoviridae, displayed high diversity (Supplementary Figs. 17, 18).
Pisuviricota
A total of 49 novel viruses and 33 known viruses were identified within the Pisuviricota phylum, which were classified into eight families (Supplementary Figs. 24–32). The 49 novel viruses were distributed across seven families: Picobirnaviridae (41), Caliciviridae (two), Picornaviridae (two), Astroviridae (one), Dicistroviridae (one), Iflaviridae (one), and Polycipiviridae (one). The 33 known viruses were distributed across seven families: Picobirnaviridae (11), Picornaviridae (10), Astroviridae (five), Caliciviridae (two), Dicistroviridae (two), Polycipiviridae (two), and Coronaviride (one), respectively. One novel virus (Rattus norvegicus astro-like virus) and three known viruses were identified among the unclassified members of the Astroviridae family (Supplementary Fig. 24). The novel Rattus norvegicus astro-like virus exhibited a 58.3% aa sequence identity with Avian associated bastrovirus 2, based on RdRP comparison. Within the Caliciviridae family, two novel viruses (Rattus norvegicus sapovirus 1 and Rattus norvegicus sapovirus 2) and two known viruses (Sapovirus rat/S4-82 and Murine norovirus) were identified. These novel caliciviruses, although closely related to the Sapovirus genus, shared < 60% aa sequence identity with Sapovirus GII.8 and Murine sapovirus (Supplementary Fig. 25). Both novel viruses possess a full-length polyprotein encoding RNA_helicase and RdRP domains (Fig. 5). A total of 41 novel viruses and three known viruses belonged to the Picobirnavirus genus within the Picobirnaviridae family (Supplementary Fig. 26). The dsRNAv_Picobirnaviridae_RdRp ___domain was identified in the RdRp protein of picobirnaviruses (Fig. 5). One novel virus (Apodemus draco aparavirus) and two known viruses (Novo Mesto dicistrovirus 1 and Rattus norvegicus cripavirus) were identified within the Dicistroviridae family. The newly identified Apodemus draco aparavirus exhibits a close genetic relationship with the Aparavirus genus within the Dicistroviridae family. It encodes a full-length polyprotein that includes RNA_helicase, Peptidase_C3G, and Dicistroviridae_RdRp domains (Fig. 5 and Supplementary Fig. 27). Within the Iflaviridae family, only one novel virus was identified: Niviventer confucianus iflavirus, which clustered with Varroa destructor virus 2, sharing ~36.6% aa sequence identity (Supplementary Fig. 28). In the Polycipiviridae family, one novel virus and two previously known viruses were identified, all demonstrating a close phylogenetic relationship to the Sopolycivirus genus (Supplementary Fig. 29). Two novel viruses were also identified within family Picornaviridae, Myodes rufocanus parabovirus which belong to the Parabovirus genus; and Rattus norvegicus mischivirus categorized under the Mischivirus genus (Supplementary Fig. 30). Both viruses encode a full-length polyprotein containing rhv_like, RNA_helicase, and ps-ssRNAv_RdRp-like domains (Fig. 5).
Cross-species viruses in rodents
By integrating our dataset with viral records from the NCBI database, we identified cross-species transmission events in rodents at the species, genus, and family levels (Fig. 6A). Among the 142 identified viruses, 33 from 15 viral families were detected in two or more rodent species. Specifically, 22 viruses were found to be cross-genera, present in two or more rodent genera; seven viruses were identified across two rodent families, while the remaining 15 cross-genera viruses were confined to a single rodent family (Fig. 6A). Notably, three viral families, Picornaviridae, Hantaviridae, and Paramyxoviridae, accounted for nearly half of the cross-species viruses (48.5%, 16/33) (Fig. 6A). All five viral species within the family Hantaviridae identified in this study are known to possess cross-species transmission capabilities. Of particular interest are rodent hepacivirus and Hantaan virus, which exhibit broad host ranges and were detected in 50.9% (28/55) and 29.1% (16/55) of rodent species, respectively. Additionally, cross-species transmission appears to occur more frequently between rodent populations inhabiting different environments compared to those within the same habitat, as evidenced by a Chi-squared test (45.5% vs. 15.1%; P < 0.001). Approximately half (16/33) of the cross-species viruses were observed among individuals from different natural habitats. A. agrarius and M. musculus were more likely to share similar viruses with six species circulating within both groups, whereas A. eversmanni tended to share viruses within its own population (Fig. 6A).

A Virus transmission across our rodent data (species, genera, and families) and a tripartite network were constructed to visualize the associations among the known viruses identified in this study, rodent species, and their potential hosts using our rodent data in conjunction with virus records from the NCBI database. For known viruses, the host range was determined based on host species information retrieved from NCBI and expanded with newly identified hosts in this study. For novel viruses, the host range was established solely based on host species information obtained in this study. The red color represents the novel viruses identified in this study. B A Venn diagram illustrates the viruses shared among rodents for known viruses. C A Venn diagram illustrates the viruses shared among rodents for novel viruses. D The host-virus correlation network is presented, where node shapes denote rodent and virus species, and node colors represent rodent families. Rodent species located at center positions within the network are labeled in red.
Furthermore, we constructed a tripartite network to visually illustrated the associations among 142 viruses, 55 rodent species, and their potential non-rodent hosts derived from animals and vectors. Among these viruses, only 33 were identified as cross-species viruses, while the majority (n = 109) exhibited host specificity at the species level by exclusively infecting rodents. Among the known viral species, over 56.72% (38/67) were associated with a single rodent species (Fig. 6B); when combined with virus records from the NCBI database, this proportion increased to 88.06% (59/67). Additionally, of the novel viral species identified in our study (n = 75), the vast majority (n = 71) were also found to be associated with a single specific rodent species. (Fig. 6C). These findings collectively suggest a high degree of host specificity for these viruses.
Finally, we evaluated patterns of viral transmission across rodents by constructing a host-virus correlation network between the 33 cross-species transmitted viruses and their respective 55 host species within the Rodentia order (Fig. 6D). Cytoscape analysis revealed an intricate network comprising 88 nodes and 123 edges. Notably, the network highlighted that 14 rodent species carried multiple cross-species transmitting viruses, including N. confucianus (18 viruses), R. norvegicus (10 viruses), and A. draco (nine viruses), etc.
Discussion
The understanding of the virome of wild animals is essential for elucidating the potential risk of zoonotic spillover to humans posed by these viral communities. Previous studies on viral discovery in wild animals predominantly utilized cross-sectional surveys that tested individual samples. In this study, we employed next-generation sequencing to characterize the virome of rodent species captured across three habitats in suburban Beijing over a 1-year period. Significant differences in viral richness were revealed, which were associated with both host species and natural habitats. These findings suggest a complex synergistic effect from host and environmental factors on virus transmission within natural habitats.
Beijing, as a heavily urbanized and densely populated metropolis, exhibits highly diverse natural ecological habitats, forming a variety of ecosystems. Coupled with its typical continental monsoon-affected climate, these conditions provide suitable environments for a wide range of flora, invertebrates, and wildlife. In this study conducted in Beijing, we performed a sequencing and bioinformatic analysis on a diversity of wild rodents, identifying a significant number of viruses including those with single-strand or double-strand genomes that are either negative- or positive-sense, and either monopartite or segmented. The most prevalent group was found to be picobirnaviruses, followed by the families Picornaviridae, Hantaviridae, Paramyxoviridae, Astroviridae, and Nairoviridae. Compared to previous studies on rodent virome performed in China and other Asian countries11,21, we discovered numerous novel viruses, with the largest number (n = 41) belonging to the family Picobirnaviridae. Given the widespread presence of picobirnaviruses in the feces/gut contents of humans and other animals, both symptomatic and asymptomatic, these viruses have been considered opportunistic enteric pathogens affecting mammals and avian species22. Despite the current lack of clarity regarding their clinical significance and animal pathogenicity in animals, identifying novel picobirnavirus strains and their animal hosts is crucial for understanding their genetic diversity, evolution, and potential cross-species transmission. This also highlights the need to revise and update the Picobirnaviridae family classification system.
A substantial number of potential zoonotic pathogens have been identified in this study, including Pigeon torque teno virus, Murine kobuvirus 1 virus and Shrew hepatitis B. A range of zoonotic pathogens pertinent to human health, such as Influenza A virus (H9N2), Rotavirus A, Hantaan virus, Beiji nairovirus, Nuomin virus, Avian orthoavulavirus 1, Cardiovirus B, and Rabies virus have also been identified. Most of these viruses can cause non-specific clinical manifestations in humans, such as fever, cough, headache, vomiting, fatigue and flu-like symptoms. Some may progress to severe complications; for example, Hantaan virus infection has been associated with mortality rates up to 12% during certain outbreaks23. Therefore, it is imperative to include these viruses in the laboratory diagnosis of febrile patients with clear epidemiological links. The findings underscore the importance of studying wild animal virome to enhance our predictive capabilities regarding zoonotic risks.
Among zoonotic pathogens, Rabies virus is notable for its broad host range, which includes Carnivora (Canis lupus), Chiroptera, Eulipotyphla (shrews), and human24,25,26. Despite its wide distribution, Rabies virus detection in rodents has been rare, with only a few species previously reported, such as A. agrarius and Citellus undulatus were reported27,28. This study confirms the presence of Rabies virus in two additional rodent species, A. peninsulae and A. agrarius, suggesting that rodents might serve as potential hosts for this pathogen. Furthermore, our earlier research identified Rabies virus in two shrew species, Crocidura shantungensis and Anourosorex squamipes in China26. Collectively, these findings underscore the critical need to enhance surveillance of wild small mammals that carry zoonotic pathogens relevant to human health.
The identification of nine viruses from five families (Astroviridae, Paramyxoviridae, Picobirnaviridae, Picornaviridae, and Polycipiviridae) is noteworthy. These viruses were detected for the first time in China and have previously been reported in eleven countries across Asia, Africa, North America, South America, Oceania, and Europe (Fig. 3C). This finding suggests a potential global distribution and cross-border transmission of these viruses. With the exception of Theiler’s encephalomyelitis virus, which was discovered in the early 1930s, the remaining eight viruses were identified within the first two decades of the 21th century (Supplementary Table 5). All five vertebrate-associated viruses had previously been detected in rodents, including Mossman virus, Bastrovirus BAS-3, Rodent Paramyxovirus LR11-23, cardiovirus C1, and Apodemus peninsulae jeilongvirus. Two invertebrate-associated viruses, Lasius neglectus virus 1 and Lasius niger virus 1, were identified from ants in Europe in 2014.
Identifying the animal reservoirs of emerging viruses is critical for elucidating the determinants of their transmission. In addition to well-documented zoonotic pathogens such as Influenza A virus (H9N2) and Avian orthoavulavirus 1, which have a broad host range, we have identified previously presumed invertebrate-specific viruses in rodents, for instance, Mukawa virus, Sichuan mosquito circovirus 3, Tick-associated circular virus-6, Tick-associated genomovirus 1, Gakugsa tick virus, Onega tick phlebovirus, Sara tick phlebovirus, Lasius neglectus virus 1, and Lasius niger virus 1. These findings suggest the potential of these viruses to be transmitted from invertebrates to mammals. On the other hand, the detection of the virus in rodents does not necessarily indicate their role as reservoir host, rather, it may result from virus exchange during blood feeding by invertebrate. Conversely, the relatively high viral load and complete genome of certain invertebrate viruses, i.e., Mukawa virus and Sara tick phlebovirus obtained in C. longicaudatus, might imply active replication within rodents, thus suggesting a higher likelihood of the reservoir source for these tick-borne viruses.
The study also revealed significant variation in viral composition and prevalence across different host species and ecological contexts. Notably, a higher viral diversity was determined for R. norvegicus, N. confucianus, A. draco, C. longicaudatus and A. peninsulae compared to the other four species examined. These five rodent species are widely distributed throughout Northern China29, making their viral community in Beijing particularly informative for understanding potential zoonotic pathogens in other regions as well. Additionally, by comparing Shannon index across natural habitats, we revealed less important role of habitat in shaping viral communities at the viral family level. Still, future studies should focus on individual rodent species, and conduct more intensive sampling across various landscape types, in order to test the influence of habitat alone on rodent-associated virome.
Emerging infectious diseases frequently originate from the spillover or cross-species transmission of zoonotic viruses from their natural reservoir hosts. A thorough understanding of virus–host interactions is crucial for predicting potential future emergence events, particularly for viruses with pathogenic potential in humans. Our data provide robust evidence of frequent interspecies transmission of rodent-borne viruses across different host levels, including species, genus, and even family. Approximately 23% of the identified viruses were detected in more than two rodent species, representing nearly half of all observed cross-species transmission events observed in our study. This suggests a substantial level of viral sharing among rodent species. Notably, all five identified species within the Hantaviridae family are cross-species viruses, which exhibit a higher propensity for cross-species transmission compared to other viral families. Successful interspecies transmission is influenced by a complex interplay of biological, ecological, and epidemiological factors. Although our current analysis does not permit definitive conclusions regarding transmissibility competence, these findings might assist in identify priority viruses for further investigation into their potential emergence risks in humans.
Our study has limitations that need to be acknowledged. Firstly, the current surveys are generally of cross-sectional design, capturing snapshots of rodent population at a given time point. Due to low sampling frequency and insufficient sequence data, some low-abundance or high-risk viruses may have been overlooked. It is not feasible to conduct a more systematic comparison of viral diversity and abundance based on seasonal changes. A comprehensive understanding of the virome requires temporally dynamic analysis since environmental and meteorological factors might also influence viral communities in rodents. Secondly, current surveillance efforts primarily rely on metagenomic sequencing and epidemiological investigations. Further studies are required to isolate the newly identified viruses and to assess their potential pathogenicity to humans by performing experimental assays in animal models. However, virus isolation and culture can be particularly challenging. In such cases, synthetic virology, which leverages complete sequence data to engineer and assemble viruses, can serve as a viable alternative when tradition isolation and culture methods are not feasible. Even when critical genomic segments are missing, recombinant viruses could still provide valuable insights into viral characteristics30,31. Thirdly, this study utilized only spleen samples for NGS analysis. Previous studies have indicated significant variations in viral composition and abundance across different tissues in wild mammals7,32. Therefore, future research should aim to investigate and compare the distribution and abundance of the viruses within different organs. Lastly, the limited availability of sequence data complicates the accurate assessment of zoonotic risks for some identified known viruses, such as Mossman virus, for which only two sequences are available in the NCBI nucleotide database.
In conclusion, our research reveals that rodents in Beijing host an extensive and highly diverse array of viruses, offering significant insights into the remarkable diversity of RNA viruses within the two largest families of the Rodentia order. Cross-species analysis utilizing extensive sequencing data establishes a foundation for evaluating the risk of future emergence of rodent-borne zoonotic diseases in other wildlife or humans. These findings enhance our understanding of the virome of diverse rodent species, and underscore the potential threat from undiscovered viruses that could spill over to humans.
Methods
Field survey and sample collections
From May 2017 to October 2018, a field investigation was performed in three suburban districts of Beijing (Fangshan, Mentougou, and Miyun) to capture wild rodents (Supplementary Table 1). Altogether 32 sampling sites were selected across these districts (8–15 sites per district), representing three distinct habitat types: grassland, bushland and woodland. These sampling sites were located ~70.4–77.5 miles from the city center of Beijing. Despite their mountainous terrain, the sampling locations have experienced varying degree of urbanization and are involved in poultry and livestock farming activities. Rodents were captured using snap traps and identified to the species level through mitochondrial cytochrome b (mt-cyt b) gene sequencing33. During the study period, a total of 432 wild rodents were captured, classified into nine species belonging to seven genera within the two largest mammalian families (Cricetidae and Muridae) under the order Rodentia (Fig. 1 and Supplementary Fig. 1). Within each suburban district, 3–8 species of rodents were sampled, indicating moderate host diversity, with no significant difference in rodent numbers across the three suburban districts (Supplementary Table 1; Wilcoxon rank sum test, P > 0.05). In each natural habitat, four to six species were identified, with over two-thirds having sample sizes >20 individuals (Supplementary Table 1). All small animals captured by snap traps were dead and dissections were performed in a BSL-2 laboratory setting. Dissections instruments were sterilized with 75% ethanol prior to use. Five organ samples (heart, lung, liver, kidney, and spleen) were collected from each rodent and stored at −80 °C until further analysis.
Next-generation sequencing (NGS) and virus confirmation
The viral metagenomics analysis of 432 spleen samples from nine rodent species (A. eversmanni, A. agrarius, A. draco, A. peninsulae, C. longicaudatus, M. musculus, M. rufocanus, N. confucianus, R. norvegicus) was conducted using NGS as previously described34 (Supplementary Table 2).
Spleen samples from each pool were homogenized in Buffer RLT solution and subsequently processed for DNA/RNA extraction using the AllPrep DNA/RNA Mini Kit (Qiagen, Germany) following the manufacturer’s protocol, Extraction negative controls (RNase-free water) were included in parallel with all RNA/DNA extractions. The rRNA depletion was performed using the MGIEasy rRNA Depletion Kit (BGI, China). High-throughput sequencing libraries were constructed using the MGIEasy RNA Library Prep Kit (BGI) and sequenced on the MGI2000 platform (BGI), generating 150 bp paired-end reads. One extraction negative control and one library construction negative control using RNase-free water were included alongside the tested samples during nucleic acid extraction and sequencing library preparation. Sequencing reads were quality-trimmed using Trimmomatic program (v0.38). After data filtering, trimming, and error removal, high-quality reads were obtained and mapped to reference sequences using BWA (Version: 0.7.15). De novo assembly was performed using MEGAHIT (v1.2.9) software.
To identify viral contigs, all assembled contigs were subjected to comparison against the non-redundant (nr) protein database from GenBank available until July 2023 using Diamond Blastx (v2.0.14), with an e value threshold of 1 × 10−5. Contigs that exhibited significant similarity to viral proteins within the superkingdom “Viruses” through top blast hits were initially identified as potential virus sequences. Virus contigs assigned to Retroviridae were excluded from further analysis. In cases where no specific family could be assigned to a given viral contig based on the best match, it was labeled as unclassified virus.
Potential host associations for the virus contigs were initially identified using the taxonomic information derived from Blastx results and subsequently confirmed through their phylogenetic relationships with viruses that have known host associations. Specifically, viral contigs that clustered within known vertebrate and/or invertebrate-associated virus groups were retained, while those clustering with bacterial, fungal or plant virus groups were excluded. To quantify virus abundance, Ribosomal RNA reads were subtracted from each library by aligning them against the SILVA rRNA database (https://www.arb-silva.de/) using Bowtie2 (v2.3.5.1). The remaining reads were then aligned end-to-end to the potential viral contigs using alignment tools. SAMtools (v1.10) was used for sorting and indexing these alignments, from which read counts for each contig were obtained. Viral abundance in each sample was evaluated by calculating the number of viral reads per million non-rRNA reads in each library (RPM). To reduce false positives, only viral contigs with an RPM ≥ 1 were considered. Contigs with overlapping sequences that had not been previously assembled were merged using Geneious Prime (v2021.1.1) to generate longer viral contigs. During this process, viral contigs containing RNA-dependent RNA polymerase (RdRp) for RNA viruses or replicase for DNA viruses were retained Species assignment of the resulting viral contigs was performed according to species demarcation criteria established by ICTV (https://ictv.global/) for each virus genus (Supplementary Table 6). In cases where a genus lacked clear species demarcation criteria, a stringent threshold of 80% amino acid identity to RdRp or replicase of known virus species was applied (Supplementary Table 6). These criteria were also used to identify both novel and identical virus species across sequencing libraries and rodent species (Supplementary Table 6). Specifically, if a virus species was found in more than one rodent species, it was inferred to have the potential for cross- species transmission.
The presence of the identified novel viruses was validated through PCR-/RT-PCR-based sequencing using primers designed from assembled viral contigs, with each virus being targeted by a specific contig (Supplementary Table 7). PCR amplification was performed using the PrimeScript™ one-step RT-PCR kit version 2 (TaKaRa, Japan) and Ex Taq™ Version 2.0 plus dye (TaKaRa) following the manufacturer’s instructions. The amplified products were analyzed by agarose gel electrophoresis and then subjected to Sanger sequencing. All PCR test were conducted alongside PCR negative controls (RNase-free water) and extraction negative controls. For comprehensive genome characterization of significant and newly discovered viruses, RT-PCR and the 5′and 3′ RACE Kit (Invitrogen) were utilized as appropriate.
Zoonotic risk assessment
Zoonotic risk assessment was conducted based on host information retrieved from the NCBI/GenBank version released on December 30, 2023 (https://ftp.ncbi.nlm.nih.gov/genbank). We have compiled and summarized the host range of 67 known viruses identified in our study. A “zoonotic virus” is defined as one that has been detected in humans at least once. A “spillover-risk virus”, with potential for zoonotic transmission refers to a virus that, while not reported to infect humans, has demonstrated cross-species transmission across more than two taxonomic orders. A “high-risk virus” encompasses both zoonotic virus and spillover-risk virus35.
Host-virus correlation network analysis
The Cytoscape software (version 3.9.1) was utilized to construct an active component/target gene/enrichment host-virus correlation network using the prefuse force-directed layout option36. The “Analyze Network” function was employed to conduct a topological analysis of host-virus correlation network. Node shapes were used to differentiate between virus species (circles) and rodent species (squares), while node colors distinguished different families of rodents.
Phylogenetic and recombination analyses
Phylogenetic analyses were conducted using the most conserved RNA-dependent RNA polymerase (RdRp) protein for RNA viruses and replicase protein genes for DNA viruses. Viral amino acid sequences were aligned with their respective virus families or genera based on Diamond BLASTX results using ClustalW (v2.1). Phylogenetic trees were constructed using IQ-TREE (v1.6.12), employing LG + G as the best-fit substitution model with 1,000 bootstrap replicates. To determine the accurate rodent species, a phylogenetic tree was estimated with mt-cyt b gene sequences using IQ-TREE, employing GTR + G as the best-fit substitution model and 1,000 bootstrap replicates. Potential recombination events within viral genomes and possible recombination breakpoints were detected through Simplot (v3.5.1)37 and RDP (v4.97)38.
Ethics statement
All animal experiment protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the Academy of Military Medical Sciences (Permit number: IACUC-DWZX-22-060). All animals were treated in accordance with the guidelines of the Regulations for the Administration of Laboratory Animals.
Data availability
All available viral metagenomic data are available through the NCBI SRA (reads). SRA identifiers of all viral metagenomics, along with detailed information for each library, are available in Supplementary Table 2. The virus sequences obtained from the study were submitted to NCBI with the GenBank Accession Numbers PP211199-PP211444.
References
Jones, K. E. et al. Global trends in emerging infectious diseases. Nature 451, 990–993 (2008).
Woolhouse, M. E. & Gowtage-Sequeria, S. Host range and emerging and reemerging pathogens. Emerg. Infect. Dis. 11, 1842–1847 (2005).
Blanga-Kanfi, S. et al. Rodent phylogeny revised: analysis of six nuclear genes from all major rodent clades. BMC Evol. Biol. 9, 71 (2009).
Milholland, M. T. et al. Global diversity and distribution of hantaviruses and their hosts. Ecohealth 15, 163–208 (2018).
Han, B. A., Schmidt, J. P., Bowden, S. E. & Drake, J. M. Rodent reservoirs of future zoonotic diseases. Proc. Natl. Acad. Sci. USA 112, 7039–7044 (2015).
Meerburg, B. G., Singleton, G. R. & Kijlstra, A. Rodent-borne diseases and their risks for public health. Crit. Rev. Microbiol. 35, 221–270 (2009).
Chen, Y. M. et al. Host traits shape virome composition and virus transmission in wild small mammals. Cell 186, 4662–4675.e4612 (2023).
Escutenaire, S. et al. Behavioral, physiologic, and habitat influences on the dynamics of Puumala virus infection in bank voles (Clethrionomys glareolus). Emerg. Infect. Dis. 8, 930–936 (2002).
Guterres, A. & de Lemos, E. R. S. Hantaviruses and a neglected environmental determinant. One Health 5, 27–33 (2018).
Dahmana, H. et al. Rodents as hosts of pathogens and related zoonotic disease risk. Pathogens 9, 202 (2020).
Wu, Z. et al. Comparative analysis of rodent and small mammal viromes to better understand the wildlife origin of emerging infectious diseases. Microbiome 6, 178 (2018).
Kane, Y. et al. Viral diversity in wild and urban rodents of Yunnan Province, China. Emerg. Microbes Infect. 13, 2290842 (2024).
Yin, H. C., Wan, D. C. & Chen, H. Y. Metagenomic analysis of viral diversity and a novel astroviruse of forest rodent. Virol. J. 19, 138 (2022).
Zhang, X., Zhao, C., Si, X., Hu, Q. & Zheng, A. Natural circulation of tick-borne severe fever with thrombocytopenia syndrome virus in the city ecosystem, China. Virol. Sin. 38, 832–835 (2023).
Tang, Y. et al. Molecular characteristics and genetic evolutionary analyses of circulating parvoviruses derived from cats in Beijing. BMC Vet. Res. 18, 195 (2022).
Wei, C. Y. et al. High prevalence of Babesia microti in small mammals in Beijing. Infect. Dis. Poverty 9, 155 (2020).
Zhang, X. et al. Study on the genetic difference of SEO type Hantaviruses. Zhonghua Liu Xing Bing Xue Za Zhi 21, 349–351 (2000).
Hu, J. et al. Isolation and characterization of H4N6 avian influenza viruses from mallard ducks in Beijing, China. PLoS One 12, e0184437 (2017).
Zhang, S. et al. Severe fever with thrombocytopenia syndrome virus infection attributed to cat contact: A case report in Beijing, China. Virol. Sin. 39, 513–515 (2024).
Sun, Y. et al. The first reported cases of severe fever with thrombocytopenia syndrome virus from domestic sick camel to humans in China. Emerg. Microbes Infect. 13, 2309990 (2024).
Wu, Z. et al. Decoding the RNA viromes in rodent lungs provides new insight into the origin and evolutionary patterns of rodent-borne pathogens in Mainland Southeast Asia. Microbiome 9, 18 (2021).
Ghosh, S. & Malik, Y. S. The true host/s of picobirnaviruses. Front. Vet. Sci. 7, 615293 (2020).
Jonsson, C. B., Figueiredo, L. T. & Vapalahti, O. A global perspective on hantavirus ecology, epidemiology, and disease. Clin. Microbiol Rev. 23, 412–441 (2010).
Hampson, K. et al. Estimating the global burden of endemic canine rabies. PLoS Negl. Trop. Dis. 9, e0003709 (2015).
Wang, L., Tang, Q. & Liang, G. Rabies and rabies virus in wildlife in mainland China, 1990-2013. Int J. Infect. Dis. 25, 122–129 (2014).
Zhang, J. T. et al. Decoding the RNA viromes in shrew lungs along the eastern coast of China. NPJ Biofilms Microbiomes 10, 68 (2024).
Kuzmin, I. V. et al. Molecular epidemiology of terrestrial rabies in the former Soviet Union. J. Wildl. Dis. 40, 617–631 (2004).
Lei, Y. L. et al. [New animal hosts of rabies virus in mountain areas in Zhejiang province]. Zhonghua Liu Xing Bing Xue Za Zhi 30, 344–347 (2009).
Chen, J. J. et al. A dataset of diversity and distribution of rodents and shrews in China. Sci. Data 9, 304 (2022).
White, R. A. 3rd The future of virology is synthetic. mSystems 6, e0077021 (2021).
tenOever B. R. Synthetic virology: building viruses to better understand them. Cold Spring Harb. Perspect. Med. 10, a038703 (2020).
He, X. et al. Metagenomic analysis of viromes in tissues of wild Qinghai vole from the eastern Tibetan Plateau. Sci. Rep. 12, 17239 (2022).
Nicolas, V. et al. Mitochondrial phylogeny of African wood mice, genus Hylomyscus (Rodentia, Muridae): implications for their taxonomy and biogeography. Mol. Phylogenetics Evol. 38, 779–793 (2006).
Zhang, X. A. et al. A zoonotic henipavirus in febrile patients in China. N. Engl. J. Med. 387, 470–472 (2022).
Lu, M. et al. Zoonotic risk assessment among farmed mammals. Cell 186, 2040–2040.e2041 (2023).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Lole, K. S. et al. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73, 152–160 (1999).
Martin, D. P., Murrell, B., Golden, M., Khoosal, A. & Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 1, vev003 (2015).
Author information
Authors and Affiliations
Contributions
Z.H., F.T., M.Z., and J.Z. contributed equally to this work. P.B., X.Z., and W.L. designed and supervised the project. Z.H., F.T., M.Z., J.Z., P.B., X.Z., and W.L. wrote the manuscript. Z.H., J.Z., Y.Z., B.O., C.P., and M.Z. performed the bioinformatic analysis. G.S., G.W., and S.L. performed the experiments. F.T., L.Z., X.M., Y.L., H.S., and P.B. contributed to the rodent collection and sampling treatments. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hu, ZY., Tang, F., Zhang, MQ. et al. Virome characterization of field-collected rodents in suburban city. npj Biofilms Microbiomes 11, 103 (2025). https://doi.org/10.1038/s41522-025-00740-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41522-025-00740-8
