
Abstract
Traditional carrier screening has been utilized for the detection of carriers of genetic disorders. Since a comprehensive assessment of the carrier frequencies of recessive conditions in the Southern Chinese population is not yet available, we performed a secondary analysis on the spectrum and carrier status for 315 genes causing autosomal recessive disorders in 1543 Southern Chinese individuals with next-generation sequencing data, 1116 with exome sequencing and 427 with genome sequencing data. Our data revealed that 1 in 2 people (47.8% of the population) was a carrier for one or more recessive conditions, and 1 in 12 individuals (8.30% of the population) was a carrier for treatable inherited conditions. In alignment with current American College of Obstetricians and Gynecologists (ACOG) pan-ethnic carrier recommendations, 1 in 26 individuals were identified as carriers of cystic fibrosis, thalassemia, and spinal muscular atrophy in the Southern Chinese population. When the >1% expanded carrier screening rate recommendation by ACOG was used, 11 diseases were found to meet the criteria in the Southern Chinese population. Approximately 1 in 3 individuals (35.5% of the population) were carriers of these 11 conditions. If the 1 in 200 carrier frequency threshold is used, and additional seven genes would meet the criteria, and 2 in 5 individuals (38.7% of the population) would be detected as a carrier. This study provides a comprehensive catalogue of the carrier spectrum and frequency in the Southern Chinese population and can serve as a reference for careful evaluation of the conditions to be included in expanded carrier screening for Southern Chinese people.
Similar content being viewed by others


Introduction
Carrier screening is a genetic test that allows prospective parents to determine the risk for recessive conditions in their offspring1. If both parents are carriers of the same autosomal recessive condition, there is a one in four chance they would conceive an affected child in each pregnancy2. As carriers are typically healthy and lack a family history suggestive of the disorder, most couples are unaware of their reproductive risks until they give birth to an affected child3. Prospective parents would benefit from carrier screening, to determine their risks of being carriers, their reproductive options, and to make informed decisions3.
Carrier testing as a screening test for single genetic conditions was first introduced in the 1970s for individuals with a family history of specific disease conditions or ethnicities with a higher prevalence of certain diseases4. Early implementation of screening included Tay-Sachs disease in the Ashkenazi Jewish population and β-thalassaemia in the Mediterranean population4,5. Carriers of Tay-Sachs disease and β-thalassaemia were screened via their reduced levels of hexosaminidase A enzyme and mean corpuscular volume respectively. The clinical utility of carrier screening was observed, Kaback et al. demonstrated a decrease in the incidence of Tay-Sachs disease after screening 2416 pregnant women with elevated risk4. Cost-benefit analysis of thalassaemia screening has also been previously reported in multiple studies demonstrating the economic benefit of preventing rare diseases6,7,8.
With the discovery of the CFTR gene in the 1980s, cystic fibrosis became one of the first conditions screened to undergo genetic screening9,10. Subsequent screening for autosomal recessive conditions was made possible via molecular testing methods. Organizations, such as the American College of Obstetricians and Gynecologists (ACOG) and the American College of Medical Genetics and Genomics (ACMG), then issued support to move from ethnicity-based screening to pan-ethnic screening for cystic fibrosis screening. Reasons for the change include limited access to carrier screening services for certain ethnicities, and complexities in assigning single ethnicities to individuals. Current ACOG and ACMG screening guidelines suggest pan-ethnic screening for spinal muscular atrophy and cystic fibrosis, and ACOG additionally suggests screening for haemoglobinopathies11,12,13.
The introduction of next-generation sequencing (NGS) has enabled effective simultaneous screening of multiple genes. With the use of NGS, gene-specific pan-ethnic carrier screening for recessive conditions can now be replaced by expanded carrier screening (ECS), which tests for hundreds of genetic conditions simultaneously at a reasonable cost. Multiple studies have identified carrier frequencies across various ethnicities14,15,16,17,18. The largest study carried out by Haque et al. examined carrier status in 94 disease conditions in 346,790 individuals of mixed ethnicities and demonstrated that ECS has increased the detection of carriers compared with racial- or ethnic- specific carrier screening according to recommendations from ACOG and ACMG18. The number of at-risk foetuses identified would be at least doubled compared with screening following professional guidelines, and in particular, 94 and 20% of fetuses at risk may be missed in East Asia and Southeast Asia, respectively18,19,20. This emphasizes the need for further investigation of ethnic-specific carrier data and the proportion of recessive conditions screened inside and outside of current professional guidelines.
While ECS can include as many genes as possible due to the advancement of NGS, ACOG and ACMG published a joint statement in 2015, emphasizing that proper selection of diseases to be included in the ECS panels is required20. It was suggested that the conditions included should have a well-defined phenotype with an early age of onset, which affects the quality of life detrimentally, results in cognitive/physical impairment, requires medical/surgical intervention, involves a prenatal diagnosis that results in interventions to improve perinatal/neonatal outcome/care, or necessitates prenatal education regarding special needs20. Two years later, ACOG suggested that conditions included in ECS should have, in addition to the criteria stated in 2015, a carrier frequency of at least 1 in 100. This aims to minimize anxiety associated with identifying rare conditions and the need for additional genetic testing and genetic counselling for families21. In 2021, ACMG released a practice resource outlining carrier screening guidelines for 97 autosomal recessive conditions using a tier system22. Currently, there is no consensus on which disease conditions should be included in ECS panels. The existing carrier screening research is primarily based on the Caucasian population or focused on the most prevalent conditions in different populations. Currently, information on the carrier frequency in Chinese is limited. While Zhao et al. performed ECS on 34 different Chinese ethnic groups; they evaluated carrier frequencies on 11 recessive conditions and determined a 27.49% carrier frequency which varied greatly between ethnic groups ranging from 4.15% to 81.35%14. Since a comprehensive assessment of the carrier frequencies in the Southern Chinese population is not yet available, we examined the spectrum and carrier status of 315 genes causing autosomal recessive disorders in 1543 Southern Chinese individuals; exome and genome sequencing data.
Results
Sequencing data characteristics
A total of 1543 unrelated, self-reported Southern Chinese individuals including 1116 with exome sequencing data (622 males and 494 females) and 427 with genome sequencing data (341 males and 86 females), passed sample-level QC procedures. This Southern Chinese cohort consisted of 963 males and 580 females and was used to evaluate the carrier status of recessive disorders in the Southern Chinese population. The Northern Chinese cohort, composed of 366 genome sequencing data with 294 males and 72 females, also passed sample-level QC procedures. This cohort was compared against the Southern Chinese cohort to reveal any Chinese subpopulation differences in carrier status.
The genes used for the evaluation of carrier frequency in this study were chosen by combining the gene list from 3 commercial companies as well as a gene list of treatable inherited diseases23. Among the 315 recessive genes evaluated in the sequencing cohort, 310 genes had at least 8 × mean coverage across the exonic region in >90% of the samples (Supplementary Fig. 1, Supplementary Data 1). The exceptions were ADGRG1, MLC1, RMRP, and ELP1 in the exome sequencing data; and CYP21A2 in both the exome and genome sequencing data. The total numbers of variants identified in the 315 recessive genes were 34,161 in exome sequencing data and 340,976 in genome sequencing data (Supplementary Table 1).
In this study, the carrier rate was estimated by combining the variants based on exome and genome sequencing data. Limited by the poor calling of copy number variations (CNVs) in the exome sequencing data, CNV calling was mainly performed on the genome sequencing data calibrated according to the CNV-JACG framework24. In addition to standard CNV calling algorithms, gene-specific bioinformatics tools were used for CNV calling of SMN1, HBA1 and HBA2 in the genome sequencing data25,26,27. Additional validation was performed for positive CNV calling cases of SMN1 copy number and HBA1/HBA2.
Carrier status of recessively inherited diseases in the Southern Chinese population
Among the 315 recessive genes, 353 variants (SNVs and small indels) and nine CNVs were classified as pathogenic or likely pathogenic (P/LP, Supplementary Data 2). In a total of 362 variants, 255 (70.4%) were loss-of-function mutations including frameshift, nonsense, splice-site, CNV, and start loss mutations. The remaining 107 (29.6%) variants included missense, in-frame, stop-loss and synonymous mutations (Supplementary Table 2). The most prevalent variant in our cohort was GJB2 c.109G>A, which is associated with autosomal recessive deafness type 1A (Table 1). This variant was observed in 22.5% of the Southern Chinese population (n = 347), with 19 individuals carrying a homozygous variant for GJB2 c.109G>A and three having a compound heterozygous mutation with another GJB2 P/LP variant (Supplementary Table 3). Other common variants in descending order included the Southeast Asian deletion (–SEA) of the alpha thalassaemia genes (4.45%), the rightward deletion (-α3.7) of the alpha thalassaemia genes (3.04%), the SMN1 exon 7 deletion (1.64%), and GALC c.1901T>C (1.43%). The top 15 most prevalent variants in the Southern Chinese population are listed in Table 2. In terms of disease condition, 11 diseases had a carrier rate of >1% in the Southern Chinese population (Table 1), and diseases that exceeded a 2% carrier rate included autosomal recessive deafness type 1A (carrier rate of 24.50%), followed by α-thalassaemia (carrier rate of 8.90%), spinal muscular atrophy type I (carrier rate of 2.11%), and systemic primary carnitine deficiency (carrier rate of 2.07%).
Half of the 1543 Southern Chinese individuals (n = 737, 47.8%) were carriers of at least one recessive disorder (Table 3). It was found that 182 individuals (11.8% of the Southern Chinese population) were carriers for multiple diseases, with 152 of them (9.9%) being carriers of two recessive disorders. There were 21 individuals (1.4%) who were carriers of three recessive disorders, seven individuals (0.5%) for four recessive disorders, and two individuals (0.1%) who were carriers for five recessive disorders.
Biallelic mutations present in the Southern Chinese population
GJB2-associated autosomal recessive deafness type 1A was the only condition in which biallelic mutations were identified in the same person (n = 22) in the study cohort. Among the 22 individuals with homozygous or compound heterozygous GJB2 mutations, 15 had a clinical summary available, and five individuals were recorded to have mild to moderate hearing loss. Hearing loss was not a primary indication of recruitment for either exome sequencing or genome sequencing.
Performance of commercial ECS panels in Southern Chinese
In this study, genes selected for evaluation were obtained from three commercial ECS panels and a list of genes that were considered treatable inherited conditions by Karnebeek et al.23. While the three commercial ECS panels have significant overlap among genes, some genes can only be identified in a specific ECS panel, ranging from three in the Myriad panel to 97 in the Invitae panel. When company-specific ECS panels were examined in the Southern Chinese population, the Invitae panel was able to detect 264 mutations in 693 carriers (94.0% of all carriers); the Baylor panel could detect 187 mutations in 643 carriers (87.2% of all carriers); and the Myriad panel was capable of detecting 200 variants in 635 carriers (86.2% of all carriers).
In Southern Chinese individuals, 1 in 12 individuals (8.30% of the population) were carriers for treatable inherited conditions according to Karnebeek et al., suggesting a high prevalence of carriers of treatable conditions in the Southern Chinese population. However, 32 genes that were associated with treatable inherited disorders were not included in any of the three commercial ECS panels, with a range of 32–53 genes absent in each ECS panel (Supplementary Fig. 2, Supplementary Data 1). When company-specific ECS panels were applied to our Southern Chinese cohort, 76 carriers (10.31% of all carriers), 69 carriers (9.36% of all carriers), and 15 carriers (2.04% of all carriers) for treatable conditions would have been missed by the Myriad panel, Baylor panel and Invitae panel, respectively.
Founder mutations in Southern Chinese
Within this cohort, 3.1% (n = 11) of the identified mutations were previously known or suspected East Asian founder mutations (Supplementary Data 2). The eleven founder mutations were responsible for 13.3% (n = 98) of the identified carriers in this study. There were three founder variants with a carrier rate of larger than 1% being HBA1/HBA2 –SEA deletion, GJB2 c.235delC and SLC26A4 c.919–2A>G with a carrier rate of 4.45%, 1.36% and 1.30% in the Southern Chinese population, respectively. The carrier rate for HBA1/HBA2 –SEA deletion was determined using genome sequencing samples only. Other disease conditions with founder mutations identified in this cohort includes carnitine deficiency (SLC22A5), FKRP-related disorders (FKRP), glutaric acidemia IIC (ETFDH), Pompe disease (GAA), Hb Beta chain-related hemoglobinopathy (HBB), infantile neuroaxonal dystrophy 1 (PLA2G6), phenylketonuria (PAH), and Wilson disease (ATP7B).
Northern and Southern Chinese carrier frequency comparison
In addition, using a small cohort consisting of 366 Northern Chinese individuals, we compared whether there were differences in the carrier frequency of certain recessive disorders. We calculated the 95% confidence intervals of the observed relative carrier frequencies per gene in both the Southern and Northern Chinese populations and used a two-sample z-test to compare the two proportions (Supplementary Data 3)28. Of the 55 genes with rare pathogenic variants found in both Southern and Northern Chinese individuals, GJB2 (p-value: < 0.00001) was the only gene that had a significantly higher carrier frequency Southern Chinese individuals than in Northern Chinese individuals.
Discussion
This is one of the first studies to comprehensively evaluate the carrier status in the Southern Chinese population with the secondary use of exome and genome sequencing data. In addition to the standard GATK pipeline, specific bioinformatics tools have been used to improve the deletion detection of SMN1 and HBA1/HBA2 in spinal muscular atrophy and thalassaemia, respectively. Our results demonstrated that 1 in 2 Southern Chinese individuals (47.8% of the population) was a carrier of at least one recessive disorder, with 0.1% of the population carrying up to five diseases conditions. In addition, 1 in 12 individuals (8.30% of the population) was a carrier of a treatable inherited condition, which was higher than that the rate among Singaporeans, at 1 in 1829.
The carrier rate of the most prevalent diseases in our cohort was compatible with published Southern Chinese data14,30,31,32. A direct side-by-side comparison across previously published carrier studies yields inconclusive results because of several reasons including different genomic technologies used, different reporting criteria, different disease conditions screened, and increased evidence of variant pathogenicity. A single disease condition comparison with existing literature such as the carrier rate of autosomal recessive deafness type 1A was found to be similar to that from Guangdong, which is a province of Southern China that shares population origin, culture, and language with Hong Kong33. The Southern Chinese carrier rate of pan-ethnic diseases such as spinal muscular atrophy was compatible with the 2% rate previously reported34. The carrier rate of alpha thalassaemia was found to be 8.90% in our study, which was higher than the previously reported rate of 5.0% in Hong Kong35. A possible reason for the discrepancy was that the carrier rate in our cohort was evaluated in genome sequencing data that had a small sample size (n = 427).
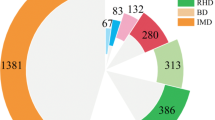
By focusing on specific disease conditions based on currently available screening guidelines, we could observe the incremental benefit of using an ECS panel (Fig. 1). Currently, antenatal mean corpuscular volume thalassaemia screening is the only form of screening employed in Hong Kong36. Using 427 genome sequencing samples, we determined the carrier rates of thalassaemia to be 1 in 11 (8.9% of the population). Cystic fibrosis, spinal muscular atrophy and thalassemia are the only conditions selected for pan-ethnic carrier screening recommended by ACOG. The three disease conditions were in the top 15 conditions with the highest carrier rate in this cohort (Table 1). We determined the carrier rates of spinal muscular atrophy at 1 in 50 individuals (2.1% of the population) using genome sequencing samples and the carrier rate of cystic fibrosis was 1 in 129 individuals (0.78% of the population) in the Southern Chinese population. If the pan-ethnic carrier screening guidelines were used, 1 in 26 individuals (73 out of 1543, 4.73% of the population) would have been identified as carriers. If carrier testing is expanded according to the ACOG recommendation that conditions included for screening should have a carrier frequency of 1 in 100 or greater21, 11 disease conditions will meet the frequency threshold (Table 1), and 1 in 3 individuals (548 out of 1543, 35.5% of the population) was a carrier for these 11 conditions. If the 1 in 200 carrier frequency threshold is used, and additional seven genes would meet the criteria, and 2 in 5 individuals (598 out of 1543, 38.7% of the population) would be detected as a carrier. If carrier testing is further expanded to all 315 genes, 1 in 2 people (47.8% of the populations) will be a carrier for any recessive condition.

The figure shows the incremental benefits of using an ECS panel. The first tier of carrier screening includes α-thalassaemia and β-thalassaemia with 3.31% of individuals identified as carriers. This is based on Hong Kong’s current antenatal screening guidelines. The second tier includes the pan-ethnic carrier screening disease conditions recommended by ACOG which accumulates spinal muscular atrophy and cystic fibrosis. Using these criteria, 4.73% of individuals were identified as carriers. The third tier is based on the 2017 ACOG guidelines on ECS with a cut-off of 1 in 100. Using these criteria, 35.5% of individuals were identified as carriers. Mutations in GJB2 were the most common, with a carrier rate of 24.5% in the Southern Chinese population (1 in 4 individuals). The fourth tier is a screening cut-off of 1 in 200, 38.7% of individuals were identified as carriers. Within this study, 47.8% of 1543 individuals were carriers of one or more recessive disease conditions.
The incremental difference between screening guidelines shows the largest leap from pan-ethnic guidelines to the ACOG 1 in 100 carrier frequency (4.73% to 35.5%). This also suggests that when a threshold cut-off is not used, a significant proportion of carriers will be identified, which may bring anxiety associated with identifying rare conditions to the couple and require extensive counselling follow-up21. Therefore, further discussions are required to reach a consensus on the frequency threshold used in ECS to include conditions that are severe but relatively common in the local population. In the 97 autosomal recessive conditions suggested by the ACMG 2021 practice protocol for screening, a few conditions are more prevalent than 1 in 200 carrier frequency in Southern Chinese yet excluded from the list22. Examples include systemic primary carnitine deficiency, citrullinemia type II, Krabbe disease, and Alstrom syndrome, which are conditions that have a carrier frequency of at least 1 in 200 in the Southern Chinese population. Their carrier frequencies were 2.07%, 1.88%, 1.56%, and 0.65% respectively. Given their clinical severity and high carrier frequency, these conditions should be included in the recommendation for screening.
Based on the proposed framework of ACOG and ACMG20, there are a few variants in the Southern Chinese population that warrant further specification in carrier testing. Two of the most common variants identified in this cohort were a non-truncating GJB2 variant c.109G>A and a truncating GJB2 variant c.235delC. The two GJB2 variants were found in 22.49% and 1.36% of Southern Chinese individuals respectively. These variants were classified as pathogenic by the ClinGen expert panel (Canonical Allele Identifier: CA172210; CA127025). All three commercial ECS panels included GJB2 for testing; however, the variable expressivity of GJB2 variants complicates the reporting of carrier status. Based on the different allelic combinations of truncating and non-truncating variants, varying levels of severity could be observed. It is known that 53% individuals carrying biallelic non-truncating variants develop mild hearing loss and 13% had profound hearing loss; 29–37% of individuals carrying biallelic truncating and non-truncating variants develop mild hearing loss and 24–30% have profound hearing loss; and 0–3% of individuals carrying biallelic truncating variants develop mild phenotypes and 59 to 64% develop profound hearing loss37. Because of a high GJB2 carrier frequency, follow-up for non-syndromic hearing loss will be required in one-fourth of Southern Chinese individuals. Huang et al. determined that 36% of hearing loss patients with the c.109G>A homozygous variant or c109G>A plus another pathogenic variant developed severe to profound hearing loss38. While it could be argued that c.109G>A is a low penetrance, mild phenotype variant, a combination with a truncating variant such as c.235delC increases the likelihood of a more severe hearing loss39. Given that GJB2 is typically screened in most carrier panels, this carrier information should be properly relayed back to the parents for them to have an informed understanding of their risks and possible future therapeutic procedures available. Therefore, genetic counselling should be included for parents to fully understand the risks of the GJB2 variants given the complex variable expressivity and penetrance. Additionally, ECS should have an opt-out option for parents in screening mild phenotypes, variable expression, and low penetrance variants to prevent unnecessary anxiety as stated by the 2013 ACMG statement40.
In this cohort, we identified a carrier rate of 1.56% in Krabbe disease. The majority of patients with Krabbe disease typically have disease onset before 1 year of age41, which fulfils the ACOG and ACMG criterion that included diseases should have early onset in life20. However, the GALC c.1901T>C identified, which had a carrier frequency of 1.43% and was the fifth most common variant in the Southern Chinese population, was known to be associated with late-onset Krabbe disease with mild phenotypes42,43. Late-onset Krabbe disease ranges from late-infantile (7 months to 3 years), juvenile (3–8 years) to adult (≥ 9 years). Bascou et al. summarized a list of previously reported in literature GALC c.1901T>C variants in combination with other GALC pathogenic variants44. The age of onset in cases with GALC c.1901T>C as one of the alleles ranged from infantile onset at 7 months to adult onset at 40 years old. The carrier rate in our study matches our current understanding of the variant allelic frequency in gnomAD East Asians with a rate of 0.0083 and a carrier frequency of 1.66%. Using Hardy-Weinberg equilibrium, an estimation of Krabbe disease prevalence based on the allelic frequency of GALC c.1901T>C in Southern Chinese would be approximately one in 20,000. This is an increased disease prevalence of Krabbe disease compared to the currently reported rate of one in 250,000 in the United States and one in 100,000 in Europe45,46. This could be explained by the late onset and less severe nature of the c.1901T>C variant and that it has only been previously reported in Asian countries47. Adult physicians should pay more attention to the possibility of late-onset Krabbe disease due to the elevated allelic frequency in the Southern Chinese population. Despite the variable onset nature of Krabbe disease, it should still be included in the ACMG 2021 practice protocol due to the high carrier frequency with previous reports of infantile and juvenile age of onset. Further recommendations by professional bodies on how to report variants known to be associated with late-onset disease phenotype and the pre-test and post-test genetic counseling will be required.
ECS is now easily accessible and offered by many commercial laboratories. We compared the three commercially available ECS panels with a list of conditions that were defined as treatable inherited conditions by Karnebeek et al.23. We found that different ECS panels included different lists of genes, and remarkably, 32 genes associated with treatable disorders were not included in all three commercial ECS panels, suggesting that there was no consensus regarding which conditions to be include in carrier screening. When applying different ECS panels to the Southern Chinese population, the number of carriers identified varies. Therefore, the choice of ECS panel may affect the report of carrier status of the tested participants; that is, a participant may not be a carrier for recessive disease in one ECS panel but could be a carrier when another ECS panel is used.
A recent study by Cheng et al. investigated the current perspective on ECS in Hong Kong and determined that 70.7% of non-pregnant women accepted ECS hypothetically compared to 61.2% of pregnant women48. The findings found in Hong Kong was consistent with previously published international studies. The study also found 94% of women perceived ECS as least as effective or superior compared to traditional screening methods. Furthermore, Johansen Taber et al. described the clinical utility of ECS and the impact on reproductive outcomes of at-risk couples defined as both partners carry pathogenic variants in the same gene49. Changes in reproductive actions were seen in both at-risk couples screened during preconception period and prenatal period. Further investigations will be required before the implementation of ECS in a clinical setting.
With the advancement of bioinformatics tools, it is now possible to detect CNVs from NGS data, or even distinguish a targeted gene from genes with high sequence homology. First, NGS4THAL was used to detect the deletions of HBA1 and HBA227. As a result, in addition to the four carriers who had SNVs that were detected by the standard bioinformatics pipeline, 34 carriers with HBA1/HBA2 deletions were identified. Second, SMN1 deletions were detected by SMAca and SMNCopyNumberCaller25,26. Because of the high sequence similarity between SMN1 and SMN2 and the complexity of the SMN locus, the detection of SMN1 CNVs from NGS data is difficult. SMAca and SMNCopyNumberCaller are tools optimized to overcome the technical difficulties in detecting SMN1 deletion from NGS, and the use of these tools allowed the detection of 33 carriers who had SMN1 deletions, which were all missed by the standard GATK workflow. Furthermore, SMAca and SMNCopyNumberCaller were able to call the SMN1 silent carrier through utilizing the detection of the highly correlated SMN1 c.*3+80T>G variant. In the future, CNVs and structural variations could be more reliably identified from exome data with updated bioinformatics tools.
The study cohort was comprised of individuals enrolled for rare disease or complex disease research in exome sequencing and Hirschsprung’s disease in genome sequencing samples. To reduce potential biases of the recruitment cohort, the study design removed individuals with pathogenic variants as carriers in recessive condition congruent with their primary indications. Due to the nature of conducting secondary findings on next-generation sequencing samples with primary indications, the carrier frequencies identified in this study will only provide an estimate of the genuine carrier frequency of the Southern Chinese population. Further studies on a large healthy control population cohort will be required to estimate the genuine carrier frequency.
In this study, CNVs were evaluated in the genome sequencing data but not the exome sequencing data since the detection of CNVs in exome data is inconsistent among different methods50. Carrier rates of X-linked conditions were also not studied due to high proportion of males in the cohort. In addition, with the advancement of bioinformatics, we were able to detect large deletions in SMN1, HBA1, and HBA2 in the genome sequencing data, which was not possible using standard CNV calling algorithms. The carrier rate of spinal muscular atrophy detected in this study, a pan-ethnic disease, was compatible with the 2% previously reported34. However, there were limitations in validating the silent carrier mutations of SMN1 using conventional MLPA methods. The carrier rate of alpha thalassaemia was found to be 8.90% in this study, which was higher than the previously reported rate of 5.0% in Hong Kong35. Since the sample size of the genome sequencing data is small (n = 427), the carrier rates of thalassaemia and spinal muscular atrophy should be interpreted with caution. In the future, the advancement of exome CNV calling algorithms may facilitate the detection of CNVs and structural variations from exome data.
Third, we compared whether there were differences in the carrier frequency of certain recessive disorders between Southern and Northern Chinese individuals and found that GJB2 was the only gene found to have significant difference (Supplementary Data 3). Since the sample size of the Northern Chinese population was small (n = 366) compared to that of the Southern Chinese population (n = 1543), we may not have enough statistical power to detect other genes with significant differences. For example, thalassaemia has a much higher prevalence in Southern Chinese individuals than in Northern Chinese individuals due to the presence of the –SEA founder mutation51, whereas phenylketonuria is more common in Northern Chinese individuals52. Therefore, it is important to carry out population-based studies in Northern Chinese individuals to evaluate their carrier spectrum.
With the use of exome and genome sequencing data from 1543 Southern Chinese individuals, the carrier status for 315 recessive genes was evaluated. It was found that 1 in 2 people (47.8% of the population) was a carrier for any recessive condition, and 1 in 12 individuals (8.30% of the population) was a carrier for treatable inherited conditions. Our results could serve as a reference to evaluate the conditions to be included in expanded carrier screening in Southern Chinese individuals.
Methods
Subjects and sequencing
A total of 2095 unrelated, self-reported Chinese individuals were enrolled for rare disease diagnosis or complex disease research from 2012 to 2019. Exome sequencing was performed in 1141 individuals, whereas genome sequencing was performed in 956 of them. Sequencing was performed on genomic DNA derived from peripheral blood or buccal mucosa by Illumina sequencing platforms. The details of the exome sequencing cohort have been previously described and used to evaluate the spectrum and prevalence of incidental findings and pharmacogenetics in the Southern Chinese population53,54. The genome sequencing cohort consisted of 956 Asian individuals and was used to study the genetics of Hirschsprung’s disease in Asia55. If an individual was identified as a carrier for a recessive condition congruent with the primary indications for referral, they were not counted as carrier in this study.
The paired-end sequencing reads were processed by a pipeline based on the Genome Analysis Toolkit (GATK) version 3.456. Briefly, reads were aligned to the University of California Santa Cruz (UCSC) hg19 reference genome assembly by the Burrows-Wheeler Aligner, and duplicated reads were removed by Picard57,58. Local realignment around indels, base quality score recalibration, and cohort-based multisample variant calling were performed using the GATK toolset. Both exome and genome sequencing datasets were subjected to stringent quality control (QC) procedures. At the sample level, a sample check was performed using PLINK 1.90 beta 5.3 or Peddy and duplicated or first-degree related samples were removed from further analysis59,60. Principal component analysis was performed using Peddy to compare with 1000 Genomes Project reference data to remove samples not clustered with the East Asian population61,62. As a result, 1116 Southern Chinese exome sequencing data samples passed sample QC, whereas for genome sequencing, 427 Southern Chinese and 366 Northern Chinese samples remained for further analysis. Following variant level QC, variants with genotype quality <20 and read depth <8× were removed by KGGseq63, and variants also had to pass Variant Quality Score Recalibration (VQSR) annotated by GATK with a SNP tranche sensitivity threshold of 99.5% and INDEL tranche sensitivity threshold of 99.0%. The variants were annotated using ANNOVAR (Build 20200223)64 with the dbNSFPv4.0a database.
For genome sequencing data, variant calling of CNVs was performed with four complementary tools, which included CNVnator, Delly, Lumpy, and Seeksv65,66,67,68. This was calibrated according to the CNV-JACG framework with CNVs passing all filters with the “PASS” mark24. Because of the high sequence homology of pseudogenes and locus complexity, gene-specific bioinformatic tools were used for CNV calling for SMN1 and HBA1/HBA2, which are the disease-causing genes of spinal muscular atrophy and alpha thalassaemia, respectively. CNV calling for SMN1 was performed on genome sequencing data by both SMAca and SMNCopyNumberCaller which utilizes differentiating bases between SMN1 and SMN225,26; whereas CNV calling for HBA1 and HBA2 was performed by NGS4THAL on genome sequencing data69.
Selection of recessive genes for evaluation of carrier status
To examine the spectrum of pathogenic variants in the Chinese population, genes included in three commercially available expanded carrier screening panels were selected. In addition, 104 genes that were associated with treatable inherited diseases, which were defined by Karnebeek et al., were also included23,29. This resulted in a list of 315 genes (Supplementary Data 1), where genes related to mitochondrial inheritance, X-linked conditions and autosomal dominant diseases were excluded from our analysis.
Variant and CNV classification
Manual review and classification of the variants identified in the 315 genes were performed regardless of the pathogenicity stated in the ClinVar (version 20190305) and Human Gene Mutation Database (HGMD) (version 201701). Variants in the noncoding regions were excluded from analysis unless they have previously been reported as pathogenic/likely pathogenic in ClinVar or DM/DM? on HGMD. In-silico prediction tools used for the computational evidence of deleterious effects include CADD70, REVEL71, SIFT72, PolyPhen2 HDIV73, MutationAssessor74, FATHMM75, PROVEAN76, and LOFTEE77. Variants (single nucleotide variants and small indels) were classified according to the ACMG and the Association for Molecular Pathology (AMP) guidelines for sequence variant interpretation as the framework78. Disease-specific ClinGen variant interpretation guidelines were used for variant classification in hearing loss, phenylketonuria, and lysosomal storage disorders79,80,81,82. In addition, loss of function variants were classified according to the ClinGen recommendations for interpreting the loss of function PVS1 criterion79,83. Finally, CNVs were classified according to the ACMG and ClinGen guidelines for constitutional CNVs84.
SMN1 validation
Validation of SMN1 deletions was performed using the SALSA® MLPA® probemix P060 SMA Carrier (MRC Holland) according to the manufacturer’s protocol. The results of the validation are described in the supplementary information.
HBA1/HBA2 validation
While –SEA variants were called consistently by both NGS4THAL and CNV-JACG, validation was performed only on -α3.7 and -α4.2 deletions using the SALSA® MLPA® probemix P140 HBA (MRC Holland) according to the manufacturer’s protocol. The results of the validation are described in the supplementary information.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author. Data available from the corresponding author will be de-identified before the data is handed over to qualified researchers.
Code availability
Processing of raw sequence files, variant calling, variant annotation, and all other analyses described in this study were performed using available and open source software, as described in the “Methods” section. No custom code was therefore used, apart from simple scripts integrating commands for such programs.
References
Gameiro, G. R. et al. Precision medicine: changing the way we think about healthcare. Clinics (Sao Paulo) 73, e723 (2018).
Neha Kumar, S. A. R. Reproductomics 63–75 (Academic Press, 2018).
Henneman, L. et al. Responsible implementation of expanded carrier screening. Eur. J. Hum. Genet. 24, e1–e12 (2016).
Kaback, M. et al. Tay-Sachs disease-carrier screening, prenatal diagnosis, and the molecular era. An international perspective, 1970 to 1993. The International TSD Data Collection Network. JAMA 270, 2307–2315 (1993).
Cao, A. et al. Molecular diagnosis and carrier screening for beta thalassemia. JAMA 278, 1273–1277 (1997).
Ostrowsky, J. T., Lippman, A. & Scriver, C. R. Cost-benefit analysis of a thalassemia disease prevention program. Am. J. Public Health. 75, 732–736 (1985).
Cronin, E. K. et al. Organisation and cost-effectiveness of antenatal haemoglobinopathy screening and follow up in a community-based programme. BJOG 107, 486–491 (2000).
Leung, K. Y. et al. Cost-effectiveness of prenatal screening for thalassaemia in Hong Kong. Prenat. Diagn. 24, 899–907 (2004).
Riordan, J. R. et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245, 1066–1073 (1989).
Ioannou, L. et al. Population-based carrier screening for cystic fibrosis: a systematic review of 23 years of research. Genet. Med. 16, 207–216 (2014).
Committee Opinion No. 691: Carrier Screening for Genetic Conditions. Obstet. Gynecol. 129, e41–e55 (2017).
Prior, T. W. Carrier screening for spinal muscular atrophy. Genet. Med. 10, 840–842 (2008).
Grody, W. W. et al. Laboratory standards and guidelines for population-based cystic fibrosis carrier screening. Genet. Med. 3, 149–154 (2001).
Zhao, S. et al. Pilot study of expanded carrier screening for 11 recessive diseases in China: results from 10,476 ethnically diverse couples. Eur. J. Hum. Genet. 27, 254–262 (2019).
Kaseniit, K. E. et al. Genetic ancestry analysis on >93,000 individuals undergoing expanded carrier screening reveals limitations of ethnicity-based medical guidelines. Genet. Med. 22, 1694–1702 (2020).
Lazarin, G. A. et al. An empirical estimate of carrier frequencies for 400+ causal Mendelian variants: results from an ethnically diverse clinical sample of 23,453 individuals. Genet. Med. 15, 178–86. (2013).
Hogan, G. J. et al. Validation of an expanded carrier screen that optimizes sensitivity via full-exon sequencing and panel-wide copy number variant identification. Clin. Chem. 64, 1063–1073 (2018).
Haque, I. S. et al. Modeled fetal risk of genetic diseases identified by expanded carrier screening. JAMA 316, 734–742 (2016).
Obstetrics, A.C.o. ACOG Practice Bulletin No. 78: hemoglobinopathies in pregnancy. Obstet. Gynecol. 109, 229–237 (2007).
Edwards, J. G. et al. Expanded carrier screening in reproductive medicine-points to consider: a joint statement of the American College of Medical Genetics and Genomics, American College of Obstetricians and Gynecologists, National Society of Genetic Counselors, Perinatal Quality Foundation, and Society for Maternal-Fetal Medicine. Obstet. Gynecol. 125, 653–62. (2015).
Committee Opinion No. 690 Summary: Carrier Screening in the Age of Genomic Medicine. Obstet. Gynecol. 129, 595–596 (2017).
Gregg, A. R. et al., Screening for autosomal recessive and X-linked conditions during pregnancy and preconception: a practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 10, 1793–1806 (2021).
van Karnebeek, C. D. & Stockler, S. Treatable inborn errors of metabolism causing intellectual disability: a systematic literature review. Mol. Genet Metab. 105, 368–381 (2012).
Zhuang, X. et al. A random forest-based framework for genotyping and accuracy assessment of copy number variations. NAR Genomics Bioinform. 2, lqaa071 (2020).
Chen, X. et al. Spinal muscular atrophy diagnosis and carrier screening from genome sequencing data. Genet. Med. 22, 945–953 (2020).
Lopez-Lopez, D. et al. SMN1 copy-number and sequence variant analysis from next-generation sequencing data. Hum. Mutat. 41, 2073–2077 (2020).
Yujie, C. et al. NGS4THAL, a one-stop molecular diagnosis and carrier screening tool for thalassemia and other hemoglobinopathies by next-generation sequencing. Research Square https://doi.org/10.21203/rs.3.rs-542196/v1 (2021).
Newcombe, R. G. Two-sided confidence intervals for the single proportion: comparison of seven methods. Stat. Med. 17, 857–72. (1998).
Bylstra, Y. et al. Population genomics in South East Asia captures unexpectedly high carrier frequency for treatable inherited disorders. Genet. Med. 21, 207–212 (2019).
Chan, O. Y. M. et al. Expanded carrier screening using next-generation sequencing of 123 Hong Kong Chinese families: a pilot study. Hong Kong Med. J. 27, 177–183 (2021).
Xi, Y. et al. Expanded carrier screening in Chinese patients seeking the help of assisted reproductive technology. Mol. Genet. Genom. Med. 8, e1340 (2020).
Wei, C.-Y. et al. Genetic profiles of 103,106 individuals in the Taiwan Biobank provide insights into the health and history of Han Chinese. npj Genom. Med. 6, 10 (2021).
Dai, P. et al. The prevalence of the 235delC GJB2 mutation in a Chinese deaf population. Genet. Med. 9, 283–289 (2007).
Muralidharan, K. et al. Population carrier screening for spinal muscular atrophy a position statement of the association for molecular pathology. J. Mol. Diagn. 13, 3–6 (2011).
Lau, Y. L. et al. Prevalence and genotypes of alpha- and beta-thalassemia carriers in Hong Kong - implications for population screening. N. Engl. J. Med. 336, 1298–1301 (1997).
Sin, S. Y. et al. Ten years’ experience of antenatal mean corpuscular volume screening and prenatal diagnosis for thalassaemias in Hong Kong. J. Obstet. Gynaecol. Res. 26, 203–208 (2000).
Snoeckx, R. L. et al. GJB2 mutations and degree of hearing loss: a multicenter study. Am. J. Hum. Genet. 77, 945–957 (2005).
Huang, S. et al. The relationship between the p.V37I mutation in GJB2 and hearing phenotypes in Chinese individuals. PLoS ONE 10, e0129662 (2015).
Shen, N. et al. Association between the p.V37I variant of GJB2 and hearing loss: a pedigree and meta-analysis. Oncotarget 8, 46681–46690 (2017).
Grody, W. W. et al. ACMG position statement on prenatal/preconception expanded carrier screening. Genet. Med. 15, 482–483 (2013).
Orsini, et al. in GeneReviews® (eds. Adam, M. P. et al.) (University of Washington, 1993). Copyright © 1993-2020, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.: Seattle (WA).
Hossain, M. A. et al. Late-onset Krabbe disease is predominant in Japan and its mutant precursor protein undergoes more effective processing than the infantile-onset form. Gene 534, 144–154 (2014).
Xu, C. et al. Six novel mutations detected in the GALC gene in 17 Japanese patients with Krabbe disease, and new genotype-phenotype correlation. J. Hum. Genet. 51, 548–554 (2006).
Bascou, N. A., Beltran-Quintero, M. L. & Escolar, M. L. Pathogenic variants in GALC gene correlate with late onset Krabbe disease and vision loss: case series and review of literature. Front. Neurol. 11, 563724 (2020).
Barczykowski, A. L. et al. Death rates in the U.S. due to Krabbe disease and related leukodystrophy and lysosomal storage diseases. Am. J. Med. Genet. A 158a, 2835–2842 (2012).
Tappino, B. et al. Identification and characterization of 15 novel GALC gene mutations causing Krabbe disease. Hum. Mutat. 31, E1894–E1914 (2010).
Xia, Z. et al. Adult-onset Krabbe disease due to a homozygous GALC mutation without abnormal signals on an MRI in a consanguineous family: a case report. Mol. Genet. Genomic Med. 8, e1407 (2020).
Cheng, H. Y. H. et al. Expanded carrier screening in Chinese population—a survey on views and acceptance of pregnant and non-pregnant women. Front. Genet. 11, 594091 (2020).
Johansen Taber, K. A. et al. Clinical utility of expanded carrier screening: results-guided actionability and outcomes. Genet. Med. 21, 1041–1048 (2019).
Hong, C. S. et al. Assessing the reproducibility of exome copy number variations predictions. Genome Med. 8, 82 (2016).
Lai, K. et al. The prevalence of thalassemia in mainland China: evidence from epidemiological surveys. Sci. Rep. 7, 920 (2017).
Xiang, L. et al. Phenylketonuria incidence in China between 2013 and 2017 based on data from the Chinese newborn screening information system: a descriptive study. BMJ Open 9, e031474 (2019).
Yu, M. H. C. et al. Actionable secondary findings in 1116 Hong Kong Chinese based on exome sequencing data. J. Hum. Genet. 66, 637–641 (2020).
Yu, M. H. C. et al. Actionable pharmacogenetic variants in Hong Kong Chinese exome sequencing data and projected prescription impact in the Hong Kong population. PLoS Genet. 17, e1009323 (2021).
Tang, C. S. et al. Identification of genes associated with hirschsprung disease, based on whole-genome sequence analysis, and potential effects on enteric nervous system development. Gastroenterology 155, 1908–1922.e5 (2018).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Broad Institute. “Picard Tools.” Broad Institute, GitHub repository. (Accessed: 21 feb 2018; version 2.17.8).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Pedersen, B. S. & Quinlan, A. R. Who’s who? detecting and resolving sample anomalies in human DNA sequencing studies with peddy. Am. J. Hum. Genet. 100, 406–413 (2017).
Jun, G. et al. Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. Am. J. Hum. Genet. 91, 839–848 (2012).
Genomes Project, C. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Li, M. X. et al. A comprehensive framework for prioritizing variants in exome sequencing studies of Mendelian diseases. Nucleic Acids Res. 40, e53 (2012).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164–e164 (2010).
Abyzov, A. et al. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 21, 974–984 (2011).
Rausch, T. et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339 (2012).
Layer, R. M. et al. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 15, R84 (2014).
Liang, Y. et al. Seeksv: an accurate tool for somatic structural variation and virus integration detection. Bioinformatics 33, 184–191 (2017).
Cao, W. Y. Y., Method of thalassemia molecular diagnosis and screening and compositions and systems therefore, in Washington, DC: U.S. Patent and Trademark Office, U.S.P.P. Application, Editor (2020).
Rentzsch, P. et al. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 13, 31 (2021).
Ioannidis, N. M. et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99, 877–885 (2016).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081 (2009).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. Chapter 7, Unit7.20 (2013).
Reva, B., Antipin, Y. & Sander, C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic acids Res. 39, e118–e118 (2011).
Shihab, H. A. et al. Ranking non-synonymous single nucleotide polymorphisms based on disease concepts. Hum. Genomics 8, 11 (2014).
Choi, Y. & Chan, A. P. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747 (2015).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Rivera-Muñoz, E. A. et al. ClinGen variant curation expert panel experiences and standardized processes for disease and gene-level specification of the ACMG/AMP guidelines for sequence variant interpretation. Hum. Mutat. 39, 1614–1622 (2018).
Oza, A. M. et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 39, 1593–1613 (2018).
Zastrow, D. B. et al. Unique aspects of sequence variant interpretation for inborn errors of metabolism (IEM): The ClinGen IEM Working Group and the Phenylalanine Hydroxylase Gene. Hum. Mutat. 39, 1569–1580 (2018).
Panel, C. E. ClinGen Lysosomal Storage Disorders Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines Version 1 (2019).
Abou Tayoun, A. N. et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 39, 1517–1524 (2018).
Riggs, E. R. et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 22, 245–257 (2020).
Acknowledgements
This study was supported by the Society for the Relief of Disabled Children, the Health and Medical Research Fund (HMRF), Li Ka Shing Donation Account: Enhanced New Staff Start up Packages, the Children’s Heart Foundation, the Edward and Yolanda Wong Fund, and T12C-714/14-R.
Author information
Authors and Affiliations
Contributions
J.F.T.C. and M.H.C.Y. are co-first authors of this manuscript. J.F.T.C. and M.H.C.Y. drafted the manuscript. J.F.T.C., M.M.C.C., C.C.W.Y., A.W.C.K., X.Z., R.L., J.L.F.F., M.L., K.S.Y., and C.S.M.T. performed variant classification and analysis on cohort with technical support from M.H.C.Y. Further analysis was performed by J.F.T.C. and M.H.C.Y. with the advice from C.C.Y.M., M.C.Y.C., J.C.K.C., and M.H.Y.T. Literature review was performed by J.F.T.C., M.M.C.C., A.W.C.K., R.L., C.C.Y.C., and N.Y.T.N., K.Y.K.C., A.S.Y.K., and W.Y. provided molecular validation of the data set. K.Y.K.C., A.S.Y.K., P.H.Y.C., W.Y., S.L.L., G.C.F.C., P.K.H.T., and Y.L.L. gave advice and provided intellectual input to the manuscript. B.H.Y.C., K.S.Y., and C.S.M.T. critically reviewed the manuscripts with suggestions for improvement and revision. K.S.Y., B.H.Y.C., and C.S.M.T. conceptualized, oversaw and supervised this project and provided guidance and support. They contributed equally and should be considered as co-corresponding authors. All authors contributed to the discussion, overall data interpretation and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
All of the authors have reviewed and approved the manuscript for submission and we confirm that the content of this manuscript has not been published or submitted elsewhere. The authors do not have any potential competing interests.
Ethics approval
Ethics approval was granted by the Institutional Review Board, the University of Hong Kong/Hospital Authority Hong Kong West Cluster (UW12-211, UW12-383, UW 05–282T/945, UW 12–382, UW 12–469). Written informed consent was obtained from the parents of the participants.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chau, J.F.T., Yu, M.H.C., Chui, M.M.C. et al. Comprehensive analysis of recessive carrier status using exome and genome sequencing data in 1543 Southern Chinese. npj Genom. Med. 7, 23 (2022). https://doi.org/10.1038/s41525-022-00287-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-022-00287-z
This article is cited by
-
Estimating at-risk couple rates across 1000 exome sequencing data cohort for 176 genes and its importance relevance for health policies
European Journal of Human Genetics (2025)
-
Comprehensive analysis of NGS-based expanded carrier screening and follow-up in southern and southwestern China: results from 3024 Chinese individuals
Human Genomics (2024)
-
Carrier frequency estimation of pathogenic variants of autosomal recessive and X-linked recessive mendelian disorders using exome sequencing data in 1,642 Thais
BMC Medical Genomics (2024)
