
Abstract
Individuals with heritable thoracic aortic disease (HTAD) face a high risk of deadly aortic dissections, but genetic testing identifies causative variants in only a minority of cases. We explored the contribution of non-canonical splice variants (NCVAS) to thoracic aortic disease (TAD) using SpliceAI and sequencing data from diverse cohorts, including 551 early-onset sporadic dissection cases and 437 HTAD probands with exome sequencing, 57 HTAD pedigrees with whole genome sequencing, and select sporadic cases with clinical panel testing. NCVAS were identified in syndromic HTAD genes such as FBN1, SMAD3, and COL3A1, including intronic variants in FBN1 in two Marfan syndrome (MFS) families. Validation in the Penn Medicine BioBank and UK Biobank showed enrichment of NCVAS in HTAD-associated genes among dissections. These findings suggest NCVAS are an underrecognized contributor to TAD, particularly in sporadic dissection and unsolved MFS cases, highlighting the potential of advanced splice prediction tools in genetic diagnostics.
Similar content being viewed by others


Introduction
Thoracic aortic aneurysms progressively enlarge and, in the absence of surgical repair, lead to acute aortic dissections, which are a significant and preventable cause of morbidity and mortality1. Individuals with a heritable form of thoracic aortic disease (HTAD), including Marfan syndrome (MFS, MIM 154700) due to pathogenic variants in FBN1, have a significantly increased risk of acute ascending aortic dissections2,3. Elective aortic aneurysm repair can prevent such deadly dissections and allow for a near-normal life expectancy4. Thus, identifying a genetic susceptibility to aortic disease is critical to prevent the high mortality associated with dissections5,6,7. With 11 validated genes for HTAD, genetic testing has become integral to identifying patients at risk for thoracic aortic disease3. Nevertheless, the diagnostic rate of genetic testing remains low, particularly for individuals with dissections who have subtle or no syndromic features or no associated family history5,8,9.
Pre-mRNA splicing is an essential process in the production of transcripts to translate into functional proteins. This complex process involves multiple sequence motifs, including the splice donor/acceptor ± 1 and 2 nucleotide sites (i.e., canonical), the donor splice region (last two nucleotides of the exon and +3 to +6 nucleotides into intron), the acceptor splice region (first base of the exon and -3 to -20 nucleotides upstream of exon, including polypyrimidine tract), and a lariat site located 18-40 nucleotides upstream of the exon10. Aberrant splicing may result in exon skipping, in frame deletion of portions of exons, introduction of new exons, intronic or partial intron inclusion, and frameshifts that lead to unstable mRNA. The American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) variant interpretation guidelines, however, only consider the canonical ± 1,2 splice sites as having very strong evidence of pathogenicity11. Consequently, the functional effect of variants outside this small region has been difficult to predict, and such variants may not be prioritized or considered to affect splicing by bioinformatics pipelines12,13. Rare variants deep within an intron can create a new splice donor or acceptor site and create a new exon and cause disease, which has been demonstrated for FBN1 in MFS families14,15,16. We previously described such a pedigree with a variable MFS phenotype and deep intronic FBN1 variant, c.1589-1217 G > T, leading to the insertion of an out-of-frame exon in the FBN1 transcript and resultant haploinsufficiency via nonsense-mediated decay (NMD)17. We proposed that based on the efficiency of the insertion of the new exon, the associated MFS phenotype can vary by age of onset of the thoracic aortic disease and other clinical features17.
We sought to investigate sequence variants that may alter regional pre-mRNA splicing in HTAD-associated genes for which loss of function is a known disease mechanism. While RNA-sequencing (RNA-seq) and other methods are effective in detecting splicing abnormalities, they are limited by access to tissues expressing the disease-causing gene18. Advances in deep learning-based methods of predicting splicing from primary DNA sequencing data have proven effective in improving diagnostic yields and gene discovery19. We applied one such tool, SpliceAI, a deep neural network predictor of cryptic splicing developed by Illumina12, to our cohorts of unsolved thoracic aortic disease (TAD) cases to assess the role of NCVAS.
Methods
This study was approved by the institutional review board (IRB) at the University of Texas Health Science Center in Houston (UTHealth) in accordance with the ethical standards of the Declaration of Helsinki. The research was conducted using the UK Biobank (UKB) Resource under Application Number 75470. The Penn Medicine BioBank (PMBB) is approved under the University of Pennsylvania IRB protocol #813913. The baseline demographic characteristics of this study’s cohorts are shown in Table 1.
UTHealth
DNA samples from affected individuals and relevant family members were collected after obtaining informed consent and human subject research approval. The UTHealth HTAD cohort includes families with two or more members affected by thoracic aortic disease, as well as trios of probands with aneurysm surgery or dissection at ≤ 40 years of age, with unaffected parents confirmed by imaging. The ESTAD cohort focuses on sporadic dissection cases in individuals ≤ 60 years of age without syndromic features or a family history. Genetic testing reports from patients with early onset sporadic aortic dissection undergoing clinical panel testing were obtained and reviewed. Exome sequencing (ES) was performed on the full HTAD and ESTAD cohorts, with select cases from unsolved HTAD pedigrees also undergoing whole genome sequencing (WGS).
UKB
The UKB data release from November 2023 (v18.1) was used for this study, including WGS data from approximately 500,000 participants20. Dissection cases were identified in individuals of European ancestry with an aortic dissection International Classification of Diseases, 10th Revision (ICD10) diagnosis or cause of death code (I71.0) or surgical code for aortic dissection (L27.4, L28.4) but without rupture (I71.1), resulting in 467 cases available for analysis. Similarly, individuals of European ancestry with a thoracic aortic aneurysm (TAA) were identified using the ICD10 code for thoracic aortic aneurysm, without rupture (I71.2). After excluding individuals with dissections as previously defined, a total of 1084 TAA cases remained for further analysis. A subset of 263 TAA patients requiring surgery was identified using surgical codes for open repair of the thoracic aorta or aortic root, excluding endovascular procedures. As a control cohort, the remaining 447,570 individuals of European ancestry without any ICD10 codes for aortic disease (I71) or congenital malformations, deformations and chromosomal abnormalities (Q00-Q99) were included for comparison.
PMBB
The PMBB is a genomic and precision medicine cohort comprising Penn Medicine health system patients who consent to linkage of electronic health records with biospecimens, including 43,731 who have undergone ES21. Thoracic aortic dissection was defined as having an encounter with an ICD10 diagnosis code of I71.01 or I71.03, or International Classification of Diseases, Ninth Revision (ICD9) codes 441.01 or 441.03. TAA was defined as having an encounter with an ICD10 diagnosis code of I71.1, I71.2, I71.5, or I71.6, or an ICD9 diagnosis code of 441.1, 441.2, 441.6, or 441.7 and excluding dissection codes.
SpliceAI Analysis
We limited our search to variants in HTAD genes that cause disease when only one functional copy is present (haploinsufficiency), namely FBN1 (NM_000138.5; MIM 134797), COL3A1 (NM_000090.4; MIM 120180), SMAD3 (NM_005902.4; MIM 603109), TGFB2 (NM_003238.6; MIM 190220), LOX (NM_002317.6; MIM 153455), and MYLK (NM_053025.4; MYLK 600922)3. For MYLK variants, only variants in the short isoform expressed in the thoracic aorta were considered22. In accordance with the Clinical Genome Resource (ClinGen) Sequence Variant Interpretation (SVI) Working Group guidelines10, we used a SpliceAI12 (version 1.3.1) score of ≥ 0.2 to identify non-canonical splice site variants that were likely to disrupt normal splicing. SpliceAI generates four scores - acceptor gain (AG), acceptor loss (AL), donor gain (DG), and donor loss (DL) - that indicate the likelihood of splice site alterations caused by a particular variant. For this study, when a given variant had multiple SpliceAI scores at or above the 0.2 threshold, we selected the maximum value for further analysis. We excluded homozygous variants and those with a minor allele frequency (MAF) ≥ 5 × 10−5 in the Genome Aggregation Database (gnomAD) version 4.1.023 as well as those with only Likely Benign or Benign ClinVar24 entries after 2019, the year SpliceAI was published. Variant pathogenicity was evaluated using ACMG/AMP and ClinGen guidelines10,11,25. Statistical differences between case and control groups were calculated with the two-tailed Fisher’s exact test using the Python SciPy library26, providing p-values, odds ratios (ORs), and 95% confidence intervals (CIs).
Sequencing
UTHealth ES was performed as previously described by Genome Sciences at the University of Washington27. Contemporaneously sequenced controls without aortic disease were provided. Briefly, exonic regions were captured using the SeqCap EZ Exome Library v2.0 from Roche. Enriched libraries were then sequenced on an Illumina NextSeq2000 using manufacturer protocols. Reads were mapped to the reference human genome (hg19) with BWA (Burrows-Wheeler Aligner)28, and variant detection and genotyping were performed using GATK Haplotype Caller29, with only variants passing all quality filters and having a minimum reported total depth (DP) of 20X being considered. After liftover to hg38 coordinates, variant call files (VCFs) were annotated with ANNOVAR30 for downstream analysis, including SpliceAI12 and gnomAD23 information. Additional WGS of select HTAD pedigrees was done at the University of Washington Center for Rare Disease Research as part of the NIH Genetics of Rare Disease (GREGoR) Consortium. After PCR-free library preparation of DNA samples, genome sequencing was performed on the Illumina NovaSeq platform, targeting an average coverage of 30X with 150 bp paired-end reads. After alignment to the hg38 reference sequence with BWA28, variants were called using the GATK HaplotypeCaller tool29, annotated on the SeattleSeq Annotation Server (http://gvs.gs.washington.edu/SeattleSeqAnnotation/), and analyzed on the open-source, web-based Seqr platform31. Sanger sequencing of UTHealth probands and any available affected family members was done to confirm variants identified through ES or WGS.
UKB WGS data processing was done using the UKB Research Analysis Platform (RAP) hosted on DNAnexus. Briefly, Illumina DRAGEN multi-sample pVCFs (hg38) were annotated with SnpEff and SnpSift32,33 for functional impact, gnomAD frequency, and SpliceAI information, and further analyzed with custom Python scripts.
PMBB ES data was processed as previously reported by Regeneron Genomics Center (RGC)34. Patient DNA samples were sequenced on the Illumina NovaSeq 6000 (Albany, NY, USA). WeCall variant caller was utilized for sequence alignment, variant identification, and assignment of genotype21. After quality control, single nucleotide variants (SNVs) were filtered for a read depth \(\ge\) 7 and were retained if they either had one or more heterozygous variant genotype with an allele balance ratio \(\ge\) 0.15, or a homozygous variant genotype.
Splicing Assay (FBN1 c.2294-3 C > A)
Cell culture and RNA Extraction
Dermal fibroblasts from a normal control and the individual with the FBN1 c.2294-3 C > A variant were grown, each in two 100 mm plates, to near confluence in DMEM with 10% FBS. One of two culture plates was incubated in the presence of cycloheximide (100 μg/ml, Sigma-Aldrich) for 6 hours before extraction of total RNA from both plates with the RNeasy Mini kit (Qiagen).
RT-PCR and Sanger Sequencing
Complementary DNA (cDNA) was synthesized with random hexamers and SuperScript™ III reverse transcriptase (Invitrogen). The FBN1 region of interest was amplified by PCR with a sense primer in exon 17 (5’- GAATGACGTCAGCAGGCAGT) and an antisense primer in exon 21 (5’- GGAGCAGCACTGGGACTTTA). The products were separated on 7% polyacrylamide gel, stained with ethidium bromide, and visualized using the “Carestream 212PRO” camera. The normal and all abnormal products were excised from the gel. The DNA was retrieved by submersion of the gel slices, separately, in 100 μl of sterile water at room temperature overnight, and 1 μl of each was reamplified using the same primers in exons 17 and 21. The amplicons were sequenced with BigDye™ Terminator v3.1 and capillary electrophoresis on the ABI 3500 Genetic Analyzer, and the data were analyzed with the Chromas software.
Splicing Assay (FBN1 c.7820-3 C > A)
Cell Culture
Dermal fibroblasts from a patient with the FBN1 c.7820-3 C > A splicing variant and a gender and age-matched healthy control were grown in DMEM/High Glucose media (Hyclone) plus 10% FBS (Sigma) and antibiotic antimycotic solution (Sigma) in a 37°C, 5% CO2 incubator. After grown to confluence, patient and control cells were treated for 8 hours with either cycloheximide (100 µg/ml, Sigma) or DMSO (Sigma), as indicated.
RNA Extraction, RT-PCR and Sanger Sequencing
Total RNA from each plate was prepared with Trizol reagent (Thermo Fisher). cDNAs were generated using SuperScript IV VILO (Thermo Fisher). PCR products crossing the putative mutation site were amplified with primers E61-F (5’-CAGACCGGCTCCAGCTGTGAAGA-3’) and E65-R (5’-CATTGGCTTCTGTCTCAGACTG-3’) with KAPA HIFI PCR kit (Roche). The PCR products were Sanger sequenced with primers E61-Fa (5’-CCAGCTGTGAAGACGTGGAC-3’) and E64-Ra (5’-CAAGCCTCTGGGGAGAGTGA-3’).
Results
Enrichment of NCVAS in Aortic Dissection Cases
Overall, we found a significantly increased enrichment of NCVAS in the UTHealth ESTAD ES cohort compared to controls (P = 0.00045), identifying six variants (five unique) in six unrelated individuals that involved syndromic genes (Table 2). In contrast, no NCVAS were found in the HTAD families or controls on ES analysis. Four variants had additional support for their pathogenicity beyond the SpliceAI prediction. For FBN1 c.4747+5 G > C (SpliceAI = 0.95) found in this study, other variants affecting the same nucleotide, such as +5 G > A (SpliceAI = 0.89) and +5 G > T (SpliceAI = 0.9), have been reported in individuals with MFS, with cDNA analyses confirming altered splicing35,36,37,38. For example, FBN1 c.4747+5 G > T results in the exclusive use of a donor site upstream in the exon with loss of sixteen amino acids that includes two cysteines involved in the stabilization of the protein37. The FBN1 c.1837+5 G > A variant (SpliceAI = 0.83) found in two unrelated individuals in our ESTAD cohort, has also been reported elsewhere in patients with MFS39,40,41,42. This variant leads to an in-frame insertion that disrupts protein function according to the ClinGen FBN1 Variant Curation Expert Panel (VCEP, internal data). The SMAD3 c.401-6 G > A variant (SpliceAI = 1) found here has also been observed in individuals with Loeys-Dietz syndrome type 3 (LDS3, MIM 613795)43,44, and cDNA studies confirm its impact on splicing via introduction of a frameshift (p.Val134Aspfs*33)9. Lastly, the COL3A1 c.898-14 A > G variant (SpliceAI = 0.48) identified in our cohort has been seen in members of a Dutch family affected by aortic aneurysms and arterial rupture, consistent with a vascular Ehlers-Danlos syndrome (vEDS, MIM 130050) phenotype45.
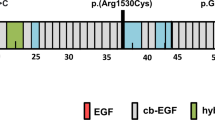
In addition, two non-canonical FBN1 variants predicted by SpliceAI to affect splicing were ascertained through clinical panel testing of individuals presenting with sporadic dissection (Table 2). The first, FBN1 c.2294-3 C > A (SpliceAI = 0.45), was identified post-mortem in a 49-year-old male who died of an acute type A aortic dissection. Cascade testing identified several additional carriers of the variant in the family, including an older brother (Fig. 1a). Interestingly, both the proband and his brother were evaluated for MFS as teenagers due to their Marfanoid habitus but did not meet diagnostic criteria, as they lacked aortic dilatation and eye findings at the time. A comprehensive genetic evaluation of the proband was not possible postmortem; however, he was noted to have tall stature (height: 188 cm), an elongated face, and long fingers. In addition to the aortic dissection, the autopsy revealed an ascending aortic aneurysm measuring 6 cm. After the proband’s death, a clinical genetics evaluation of the brother revealed a similarly tall stature (height: 191 cm), an arm span of 193 cm, a high-arched palate, mild myopia, mild skin striae, mild thoracolumbar scoliosis, positive thumb and wrist signs, a history of pectus excavatum requiring surgical repair, and a subsequent pneumothorax after heavy lifting. According to the revised Ghent nosology for MFS46, his systemic score was calculated to be 8, based on the presence of positive wrist and thumb signs, pectus excavatum, scoliosis, skin striae, and pneumothorax. Echocardiography in him revealed a normal aortic diameter (3.4 cm at the sinuses of Valsalva, Z-Score: -0.7547), with additional imaging showing no evidence of aneurysms or dissections in the remainder of the aorta. Other variant carriers were also found to have normal aortic root diameters (additional clinical information unavailable). Targeted pre-mRNA splicing analysis in cultured dermal fibroblasts from the proband’s brother showed very low-level use of two cryptic acceptor sites in the downstream exon, but only after the cells were incubated with cycloheximide (CHX) for 6 hours before RNA extraction to inhibit NMD (Fig. 1b). Sanger sequence analysis across a heterozygous site in the 3’ untranslated region (c.*314 C/T) showed similar peak heights for “C” and “T” in genomic DNA and CHX-untreated RNA/cDNA (Supplementary Fig. 1), which indicated that the cryptic acceptor sites were used in only a very small number of variant transcripts. According to the ClinGen FBN1 VCEP guidelines25, this variant meets the criteria for classification as a variant of uncertain significance (VUS) due to its absence in the general population and its predicted impact on splicing (PM2_supp, PP3). The second FBN1 variant identified on clinical testing, c.7820-3 C > A (SpliceAI = 0.88), was found in a 40-year-old male presenting with an acute aortic dissection who lacked syndromic features or relevant family history. Notably, this variant has been reported in two individuals with classic MFS48,49, including one confirmed de novo case. Nevertheless, RNA studies using the patient’s dermal fibroblasts failed to show definitive evidence of an impact on regional pre-mRNA splicing (Supplementary Fig. 2). This variant is classified as a VUS due to its absence in the general population, its predicted impact on splicing, and reported association with MFS (PM2_supp, PP3, PP4, PS2_mod).

a Family members carrying the FBN1 variant are marked with a ‘+’, including the proband’s brother with a systemic score (SS) of 8. Individuals with normal aortic imaging are marked with a ‘*’. b Gel electrophoresis image of RT-PCR from skin fibroblasts of proband’s brother. Band 1 (blue) represents a small amount of two abnormal splice products generated by the rare use of two cryptic acceptor sites in the exon. Bands 2-5 (blue) are two heteroduplex pairs formed between normal and abnormal products. Ao: Aortic diameter at the sinuses of Valsalva; Z-score: normalized aortic diameter. CHX – sample treated with cycloheximide, an inhibitor of nonsense-mediated decay (NMD).
Analysis of additional TAD cohorts replicated our finding of an increased number of NCVAS in dissection cases (Fig. 2, Table 3). In ES data from the PMBB, there was an enrichment of NCVAS in the six HTAD-associated genes among 262 dissection cases versus 41,614 controls, with an OR of 7.4 (95% CI: 2.3, 23.7) and a p-value of 0.009. Similarly, genome data from the UKB revealed an increased NCVAS number in 467 dissection patients compared to 447,570 controls, with an OR of 5.87 (95% CI: 3.03, 11.4) and a p-value of 3.61 × 10⁻⁵. Notably, there was no such enrichment in the PMBB or UKB thoracic aortic aneurysm (TAA) cases (p > 0.05), including UKB cases that underwent surgical repair of a root or ascending aneurysm (Fig. 2).

ES exome sequencing; WGS whole genome sequencing; ESTAD sporadic aortic dissection cohort ( < 60 years of age without family history or syndromic features); HTAD Families with multiple members affected by heritable thoracic aortic disease; TAA thoracic aortic aneurysm; HI haploinsufficiency; MAF minor allele frequency; LB/B likely benign or benign; NS not significant (p > 0.05) *number of HTAD pedigrees, 97 total individuals sequenced.
MFS Pedigrees with Negative Prior Genetic Testing
Applying SpliceAI to a cohort of 57 unsolved HTAD families undergoing whole genome sequencing (WGS), we identified two additional NCVAS (Table 4). In one family with extensive aortic disease and multiple members having a clinical diagnosis of MFS but negative genetic testing (Fig. 3a), two affected brothers shared a novel FBN1 variant, c.6872-1003 C > T (SpliceAI = 0.39), that was predicted to activate a cryptic splice site deep within intron 56, possibly leading to a new exon insertion and NMD. The proband in this MFS family underwent a mechanical composite valve graft replacement of the aortic root and ascending aorta at age 31 following a type A aortic dissection. He subsequently experienced a type B aortic dissection at age 53. Based on the available information, his systemic score was calculated as 7, considering his history of mitral valve prolapse, scoliosis, dural ectasia, pneumothorax, and characteristic facial features. This variant is classified as a VUS given its absence in the general population, its predicted impact on splicing, and the proband meeting revised Ghent criteria (PM2_supp, PP3, PP4). In a second family with clinical MFS and aortic disease in multiple members but negative molecular workup (Fig. 3b), the proband carried the novel FBN1 c.1148-16 T > A variant (SpliceAI = 0.9). This variant is predicted to create a new splice acceptor site that results in partial intron retention, a reading frame shift, NMD, and FBN1 haploinsufficiency. The proband in this family was diagnosed clinically with MFS at birth shortly after her father with MFS died of an aortic dissection. Her systemic score was calculated as 8, based on a history of a pectus deformity, pes planus, skin striae, mitral valve prolapse, positive thumb and wrist signs, and an increased arm span-to-height ratio. Echocardiography revealed a dilated aortic root measuring 4.4 cm at the sinuses of Valsalva (Z-score: +5.647). The proband’s FBN1 variant is classified as a VUS due to its absence in the general population, predicted impact on splicing, and the patient’s fulfillment of the revised Ghent criteria (PM2_supp, PP3, PP4). Unfortunately, tissue samples were not available to perform splicing assays on either pedigree’s FBN1 variant.

Pedigree 1 (a) had extensive aortic disease and multiple individuals with a clinical diagnosis of MFS but negative genetic testing. WGS identified a novel FBN1 variant, c.6872-1003 C > T (SpliceAI – 0.39), in two affected brothers, predicted to activate a cryptic splice site within intron 56. Pedigree 2 (b) was similarly affected with aortic disease and clinical MFS diagnoses but negative molecular testing. WGS of the proband revealed a novel FBN1 variant, c.1148-16 T > A (SpliceAI – 0.9), predicted to cause intron retention and extension of exon 11 of the mRNA transcript. Systemic scores (SS), aortic diameters at the sinuses of Valsalva (Ao), and normalized aortic diameter Z-scores are shown for individuals who underwent clinical assessment. MFS – Marfan syndrome; WGS – whole genome sequencing.
Discussion
The study assessed the contribution of non-canonical splice site variation in the spectrum of TAD, including cases presenting with dissections, thoracic aortic aneurysms, sporadic and heritable disease, and genetic testing-negative MFS. Using the SpliceAI computational tool to identify variants predicted to alter splicing, we demonstrated an enrichment of these variants in sporadic aortic dissection cases based on ES data, in which 1% of patients harbored a NCVAS. We further replicated this finding in dissection cases from two additional cohorts (PMBB and UKB). Interestingly, NCVAS were not enriched in patients with TAA in these validation cohorts. We also identified NCVAS in two sporadic dissection cases ascertained through clinical panel testing. These findings suggest that NCVASs identified by SpliceAI may play an underrecognized role in aortic dissection, particularly in sporadic dissection cases. Importantly, these variants were found in syndromic HTAD genes, though the cases lacked documented clinical features to trigger the diagnosis of a genetic syndrome. Furthermore, some variants had been previously seen in documented cases of syndromic HTAD, including MFS and LDS, suggesting that NCVAS may be associated with variable expressivity of systemic characteristics. This principle also extends to TAD, as we previously demonstrated for a deep intronic FBN1 rare variant that led to pseudoexon insertion and NMD in a large five-generation family that included members with dissection as young as 19 year of age and others with normal aortic root dimensions up to the age of 84 years17. A possible contributor to the varied phenotype associated with NCVAS is the efficiency of aberrant splicing, which may lead to varied levels of the wild-type transcript in different tissues. The effect of the variant also depends on whether a new donor or acceptor site is created and how many nucleotides are inserted or deleted. If the number is a multiple of three, the translational reading frame is preserved, but the protein structure may be altered, potentially impacting its function. If the insertion or deletion of nucleotides is not a multiple of three, a frameshift results, leading to an unstable mRNA molecule that may be degraded via NMD. While bioinformatic computational tools can predict such events, only RNA-splicing assays can functionally validate the impact and extent of splicing changes.
Importantly, SpliceAI scores do not correlate with phenotype penetrance, as recently noted in families with variable penetrance of MFS harboring deep intronic FBN1 pathogenic variants16. In this study, for example, the FBN1 c.6872-1003 C > T variant identified on WGS in two brothers with MFS had a lower score (SpliceAI = 0.39) than the c.2294-3 C > A variant (SpliceAI = 0.45) found in a sporadic dissection case and family members without any aortic enlargement. Thus, additional genetic and non-genetic factors, such as dissection-specific polygenic risk due to common variants and hypertension, may also augment the likelihood of aortic dissection in carriers of NCVAS. The absence of these variants in TAA cases suggests that alternative mechanisms, such as unidentified genes, other polygenic risk, or environmental factors, may contribute to aneurysm formation in the general population rather than rare variants disrupting HTAD genes.
Through a combination of WGS and splice prediction analysis, we also identified NCVAS in two MFS pedigrees with negative prior genetic testing. Given previous reports of deep intronic variants in FBN1 as a cause of MFS14,15,16,17, these results indicate that WGS should be considered for individuals and families who meet the diagnostic criteria for MFS but a causative variant is not identified with clinical genetic testing.
In our previous splicing assays to assess NCVAS, we determined that treatment of samples with CHX to inhibit NMD significantly improves the ability to detect abnormally spliced mRNA transcripts17. However, such studies can be time-consuming, require a tissue sample, and are limited by gene expression levels in available tissues. Interestingly, for the FBN1 c.2294-3 C > A variant described here, though there was a small amount of detectable abnormal splice products observed after CHX treatment (Fig. 1b), the majority of the variant FBN1 pre-mRNA was normally spliced. Furthermore, we could not confirm abnormal splicing for the FBN1 c.7820-3 C > A variant, even after CHX treatment. These studies were performed in dermal fibroblasts, however, and so it is unclear if the proportion of misspliced FBN1 mRNA is greater in aortic smooth muscle cells, a finding that would then support these variants’ roles in TAD. Nevertheless, SpliceAI has demonstrated a sensitivity of 78% when using a threshold of ≥0.2 to predict spliceogenicity for variants located outside the canonical ± 1,2 splice sites, rising to 91% within the donor/acceptor splice region10. Therefore, in the absence of RNA-based assays, improving splice prediction methods will be important to aid in the assessment of the impact of variants identified through genomic sequencing for TAD. However, while SpliceAI is currently considered the gold standard for splice prediction, it is not tissue-specific. Newer algorithms such as AbSplice50 and Pangolin51 that take into account tissue-specific splicing may further improve these predictions, allowing more accurate assessments of aberrant splicing and its role in disease.
There are several limitations involved in this work. The ~1% solve rate in ESTAD probands may be underestimated due to the inherently limited intronic coverage of ES. Supporting this assertion, of the nine NCVAS identified in the UKB WGS dissection cases, three are deep intronic and would not likely be detected by ES. In addition, potential age-related issues should be considered given the use of biobank validation cohorts. Compared to the younger UTHealth ESTAD cases, it is plausible that the older UKB and PMBB dissection cohorts might exhibit fewer pronounced cases of dissection, possibly leading to an underestimation of NCVAS prevalence. Additionally, different rates of hypertension among the various dissection groups could indicate underlying baseline differences between these populations that may be contributing to our findings. Lastly, by selecting the maximum SpliceAI score, our analysis focused on the most significant predicted splicing effect of the variant. However, as demonstrated in FBN1 for example16,52,53, multiple splice products are possible and should be considered due to the potential impact on pathogenicity and phenotype.
In summary, variants outside the canonical ± 1,2 splice sites may be an underrecognized contributor to TAD, specifically for early-onset sporadic dissection cases and MFS patients meeting diagnostic criteria but with negative molecular testing. Modern splice prediction tools, such as SpliceAI, show promise in identifying such variants that have been excluded or not identified in bioinformatic analyses. Despite the observed overall enrichment of these variants in dissection cases, further investigation is required to predict the penetrance of disease in carriers, elucidate other factors influencing TAD, and improve predictive models overall.
Data availability
The UTHealth datasets are available in dbGaP Study Accession: phs000693.v7.p3. The PMBB dataset is not publicly available due to IRB restrictions requiring a collaboration with a Penn investigator to access PMBB data. The UKB data is available to researchers upon approval by an expert access committee.
Code Availability
The underlying code for this study is not publicly available but may be made available to qualified researchers on reasonable request from the corresponding author.
References
Isselbacher, E. M. et al. 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation 146, e334–e482 (2022).
Dietz, H. C. et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 352, 337–339 (1991).
Milewicz, D. M. et al. Update on the genetic risk for thoracic aortic aneurysms and acute aortic dissections: implications for clinical care. J. Cardiovasc. Surg. 62, 203–210 (2021).
Silverman, D. I. et al. Life expectancy in the Marfan syndrome. Am. J. Cardiol. 75, 157–160 (1995).
Harris, S. L. & Lindsay, M. E. Role of Clinical Genetic Testing in the Management of Aortopathies. Curr. Cardiol. Rep. 23, 10 (2021).
Milewicz, D. M. & Regalado, E. S. Use of genetics for personalized management of heritable thoracic aortic disease: how do we get there? J. Thorac. Cardiovasc. Surg. 149, S3–S5 (2015).
De Backer, J., Campens, L. & De Paepe, A. Genes in thoracic aortic aneurysms/dissections - do they matter? Ann. Cardiothorac. Surg. 2, 73–82 (2013).
Renner, S. et al. Next-generation sequencing of 32 genes associated with hereditary aortopathies and related disorders of connective tissue in a cohort of 199 patients. Genet. Med. 21, 1832–1841 (2019).
Arnaud, P. et al. Genetic diversity and pathogenic variants as possible predictors of severity in a French sample of nonsyndromic heritable thoracic aortic aneurysms and dissections (nshTAAD). Genet. Med. 21, 2015–2024 (2019).
Walker, L. C. et al. Using the ACMG/AMP framework to capture evidence related to predicted and observed impact on splicing: Recommendations from the ClinGen SVI Splicing Subgroup. Am. J. Hum. Genet. 110, 1046–1067 (2023).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Jaganathan, K. et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 176, 535–548.e24 (2019).
Lord, J. et al. Predicting the impact of rare variants on RNA splicing in CAGI6. Hum. Genet. https://doi.org/10.1007/s00439-023-02624-3 (2024).
Guo, D.-C. et al. An FBN1 pseudoexon mutation in a patient with Marfan syndrome: confirmation of cryptic mutations leading to disease. J. Hum. Genet. 53, 1007–1011 (2008).
Gillis, E. et al. An FBN1 deep intronic mutation in a familial case of Marfan syndrome: an explanation for genetically unsolved cases? Hum. Mutat. 35, 571–574 (2014).
Kim, J. A. et al. Overcoming challenges associated with identifying FBN1 deep intronic variants through whole-genome sequencing. J. Clin. Lab. Anal. 38, e25009 (2024).
Guo, D.-C. et al. An FBN1 deep intronic variant is associated with pseudoexon formation and a variable Marfan phenotype in a five generation family. Clin. Genet. 103, 704–708, https://doi.org/10.1111/cge.14322 (2023).
Murdock, D. R. et al. Transcriptome-directed analysis for Mendelian disease diagnosis overcomes limitations of conventional genomic testing. J. Clin. Invest. 131, e141500 (2021).
Pagnamenta, A. T. et al. Structural and non-coding variants increase the diagnostic yield of clinical whole genome sequencing for rare diseases. Genome Med 15, 94 (2023).
Sudlow, C. et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 12, e1001779 (2015).
Verma, A. et al. The Penn Medicine BioBank: Towards a genomics-enabled learning healthcare system to accelerate precision medicine in a diverse population. J. Pers. Med. 12, 1974 (2022).
Wallace, S. E. et al. MYLK pathogenic variants aortic disease presentation, pregnancy risk, and characterization of pathogenic missense variants. Genet. Med. 21, 144–151 (2019).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Landrum, M. J. et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 42, D980–D985 (2014).
ClinGen FBN1 Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines Version 1. Available at https://www.clinicalgenome.org/site/assets/files/7445/clingen_fbn1_acmg_specifications_v1.pdf.
Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272 (2020).
Duan, X.-Y. et al. SMAD4 rare variants in individuals and families with thoracic aortic aneurysms and dissections. Eur. J. Hum. Genet. 27, 1054–1060 (2019).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Poplin, R. et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 201178. https://doi.org/10.1101/201178 (2018).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164 (2010).
Pais, L. S. et al. seqr: A web-based analysis and collaboration tool for rare disease genomics. Hum. Mutat. 43, 698–707 (2022).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. (Austin) 6, 80–92 (2012).
Cingolani, P. et al. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program. SnpSift. Front. Genet. 3, 35 (2012).
Dewey, F. E. et al. Inactivating variants in ANGPTL4 and risk of coronary artery disease. N. Engl. J. Med. 374, 1123–1133 (2016).
Comeglio, P. et al. The importance of mutation detection in Marfan syndrome and Marfan-related disorders: report of 193 FBN1 mutations. Hum. Mutat. 28, 928 (2007).
Groth, K. A. et al. Evaluating the quality of Marfan genotype-phenotype correlations in existing FBN1 databases. Genet. Med. 19, 772–777 (2017).
McGrory, J. & Cole, W. G. Alternative splicing of exon 37 of FBN1 deletes part of an “eight-cysteine” ___domain resulting in the Marfan syndrome. Clin. Genet. 55, 118–121 (1999).
Wai, H. A. et al. Blood RNA analysis can increase clinical diagnostic rate and resolve variants of uncertain significance. Genet. Med. 22, 1005–1014 (2020).
Béroud, C., Collod-Béroud, G., Boileau, C., Soussi, T. & Junien, C. UMD (Universal mutation database): a generic software to build and analyze locus-specific databases. Hum. Mutat. 15, 86–94 (2000).
Biggin, A., Holman, K., Brett, M., Bennetts, B. & Adès, L. Detection of thirty novel FBN1 mutations in patients with Marfan syndrome or a related fibrillinopathy. Hum. Mutat. 23, 99 (2004).
Hung, C.-C. et al. Mutation spectrum of the fibrillin-1 (FBN1) gene in Taiwanese patients with Marfan syndrome. Ann. Hum. Genet. 73, 559–567 (2009).
Overwater, E. et al. NGS panel analysis in 24 ectopia lentis patients; a clinically relevant test with a high diagnostic yield. Eur. J. Med. Genet. 60, 465–473 (2017).
Campens, L. et al. Gene panel sequencing in heritable thoracic aortic disorders and related entities - results of comprehensive testing in a cohort of 264 patients. Orphanet J. Rare Dis. 10, 9 (2015).
Koppen, H., Baars, M. J. H., van Gils, A. & Vis, J. C. Spontaneous intracranial hypotension as first symptom of aneurysms-osteoarthritis syndrome: a case report. Headache 55, 711–712 (2015).
van de Luijtgaarden, K. M. et al. First genetic analysis of aneurysm genes in familial and sporadic abdominal aortic aneurysm. Hum. Genet. 134, 881–893 (2015).
Loeys, B. L. et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 47, 476–485 (2010).
Z-score for adults. Marfan Foundation https://marfan.org/dx/z-score-adults/ (2021).
Stheneur, C. et al. Identification of the minimal combination of clinical features in probands for efficient mutation detection in the FBN1 gene. Eur. J. Hum. Genet. 17, 1121–1128 (2009).
Yoo, E.-H. et al. Clinical and genetic analysis of Korean patients with Marfan syndrome: possible ethnic differences in clinical manifestation. Clin. Genet. 77, 177–182 (2010).
Wagner, N. et al. Aberrant splicing prediction across human tissues. Nat. Genet. 55, 861–870 (2023).
Zeng, T. & Li, Y. I. Predicting RNA splicing from DNA sequence using Pangolin. Genome Biol. 23, 103 (2022).
Dougarem, D., Chen, Y.-X., Sun, Y.-N., Huang, H.-F. & Luo, Q. A novel heterozygous intronic FBN1 variant contributes to aberrant RNA splicing in marfan syndrome. Mol. Genet. Genom. Med. 12, e70004 (2024).
Hu, K. et al. Functional Analysis of an Intronic FBN1 Pathogenic Gene Variant in a Family With Marfan Syndrome. Front. Genet. 13, 857095 (2022).
Nijbroek, G. et al. Fifteen novel FBN1 mutations causing Marfan syndrome detected by heteroduplex analysis of genomic amplicons. Am. J. Hum. Genet. 57, 8–21 (1995).
Acknowledgements
This work was supported by NHLBI R01HL109942 (D.M.M), the Remebrin’ Benjamin and John Ritter Foundations (D.M.M) and funds from the American Heart Association 23POST1011251 (J.D.), and the Collagen Diagnostic Laboratory, University of Washington (U.S. and P.H.B.). Sequencing and data analysis were provided by the University of Washington Center for Rare Disease Research (UW-CRDR), with support from NHGRI grants U01 HG011744 and U24 HG011746. The PMBB is supported by the Perelman School of Medicine at the University of Pennsylvania, a gift from the Smilow family, and the National Center for Advancing Translational Sciences of the National Institutes of Health under CTSA award number UL1TR001878.
Author information
Authors and Affiliations
Contributions
D.R.M., S.M.D., and D.M.M. conceived the project. D.R.M. performed the research. D.R.M., D.G., and J.S.D. analyzed the data. J.X.C., M.J.B., U.S., and X.D. performed sequencing. A.C.C., I.S.M., Y.T., K.A.L., and P.H.B. contributed patient information. D.R.M. wrote the manuscript with input from other co-authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Murdock, D.R., Guo, Dc., DePaolo, J.S. et al. Non-canonical splice variants in thoracic aortic dissection cases and Marfan syndrome with negative genetic testing. npj Genom. Med. 10, 25 (2025). https://doi.org/10.1038/s41525-025-00472-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-025-00472-w
