
Abstract
Somatic mosaicism produces genetic differences between cells in an individual and is an underrecognized contributor to phenotypic variability. Precise understanding of the natural history of genetic diseases, therefore, requires detection and recognition of low-level mosaicism, which remains technically challenging, particularly for X-linked genes. Here, we identify six males with mosaic X-linked adrenoleukodystrophy (X-ALD), a neurometabolic peroxisomal disorder caused by pathogenic variants in ABCD1 that is currently included in 44 state newborn screening (NBS) programs, and estimate the incidence of somatic mosaicism. Of 227 males from 2 laboratories performing ABCD1 next-generation sequencing, 1.8% (4/227) had pathogenic or likely pathogenic ABCD1 variants that were mosaic. In one mosaic male individual, allele-specific measurements across multiple tissues demonstrated ABCD1 variant allele fractions ranging from 66 to 82%. Our findings have implications for the identification of X-ALD through NBS, and additional studies could provide insight into the pathogenesis and natural history of X-ALD.
Similar content being viewed by others


Introduction
X-linked adrenoleukodystrophy (X-ALD) is a neurometabolic disorder with an estimated prevalence of 1 in 14,000–17,000 births1,2. X-ALD is caused by pathogenic loss-of-function variants in the ABCD1 gene located on the X chromosome. ABCD1 encodes an ATP-binding cassette that imports very long-chain fatty acids (VLCFA) into the peroxisome. Pathogenic ABCD1 variants cause impaired peroxisomal VLCFA β-oxidation, resulting in the accumulation of VLCFA in plasma and tissues. VLCFA levels are almost universally elevated in males with pathogenic ABCD1 variants, but there is great phenotypic variability, and biomarkers predicting disease severity, such as extent of VLCFA elevation, have only recently been identified3. Intrafamilial phenotypic variability is common, ranging from isolated adrenal insufficiency in adulthood to a rapidly progressive inflammatory demyelinating disease in early childhood (“cerebral X-ALD”). Factors contributing to X-ALD phenotypic variability are unclear, and although peroxisomal dysfunction is central, the mechanisms by which elevated VLCFA lead to disease remain an area of active inquiry. The definitive treatment for cerebral X-ALD is hematopoietic stem cell transplantation (HSCT), which, if performed early enough, can slow or arrest cerebral disease progression4. Because of the success of HSCT, as well as hormone replacement therapy for adrenal dysfunction and a recently FDA-approved gene therapy (elivaldogene autotemcel)5, 44 states now include X-ALD in universal newborn screening (NBS) programs, yielding a large, pre-symptomatic cohort of male children diagnosed with X-ALD. While promising new biomarkers have been identified3, it remains challenging to predict on an individual level which of these pre-symptomatic newborn children will progress to cerebral X-ALD. We hypothesized that somatic mosaicism for pathogenic ABCD1 variants might be present in pre-symptomatic children who were identified through NBS to be at risk for X-ALD and set out to estimate the incidence of this under-recognized factor that could potentially modify disease severity.
Results
Identification and verification of the proband with mosaic X-ALD
Our index individual (mXALD-1) was identified as having an elevated C26:0-LPC on state newborn screening, and follow-up next-generation sequencing (NGS) demonstrated the presence of a likely pathogenic missense variant (NM_000033:c.1988T>C, p.Leu663Pro) in ABCD1 in ~82% of reads (Table 1). For hemizygous XY males, X-linked variants are expected at either 0% or 100%, so the variant allele fraction (VAF) of 82% suggested somatic mosaicism in this individual. A SNP microarray established 46, XY chromosomes, and multiplex ligation-dependent probe amplification (MLPA) analysis demonstrated the presence of a single copy of ABCD1. The presence of variant and reference alleles was confirmed by Sanger sequencing (Supplementary Fig. 1), and second blood samples obtained 2 years after initial sample also found a VAF of 80%, suggesting stable persistence of the ABCD1 mutant cell line within the blood.
Variations in the variant allele fraction from different tissues in a mosaic individual
To determine the tissue distribution of this variant, 6 tissues (blood, skin, buccal swab, CSF, tonsils, and urine) were obtained, and, when possible, both genomic and cell-free DNA were isolated. Using a custom droplet digital polymerase chain reaction (ddPCR) assay, we found that the VAF ranged from 66% to 82% across these samples and was lowest in the skin (Fig. 1). These results are consistent with an asymptomatic male individual with a post-zygotic mosaic variant in ABCD1 manifesting as biochemically and molecularly confirmed X-ALD.

A Various tissue samples were obtained, and the proposed germ layer of origin. B The graph shows the comparison of the variant allele fraction (VAF) from genomic DNA in six different tissues, including blood, CSF (pellet), urine (pellet), tonsil, skin, and buccal sample, and cell-free DNA derived from plasma and urine. Representative data presented as individual values of n = 4 replicates. Error bars represent the min/max “Total Error” of the mutant reads based on four merged replicates as provided by Quantasoft software. VAF is calculated by mutant concentration divided by total concentration. The figure was created using Microsoft Excel and Adobe Photoshop software.
Discovery of additional males with mosaic X-ALD
To identify additional individuals with mosaic X-ALD, we contacted large commercial sequencing laboratories (n = 5) and state newborn screening programs performing NGS sequencing (n = 7) to find males with pathogenic or likely pathogenic (P/LP) ABCD1 variants in which the VAF deviated from 100%. Five additional individuals were identified, for a total of six males, including our proband. The VAF was not consistently included in the clinical report; thus, the ordering healthcare provider was not always made aware that the ABCD1 variant was mosaic. In 4 of these individuals (mXALD-2–5) clinical information was available after contact with the ordering provider (Table 1). These individuals were 6 months–6 years old, and all were identified through newborn screening. Including our proband, all 5 males are reported as asymptomatic with normal adrenal screening labs (ACTH, cortisol) and neurologic exam (though mXALD-4 was noted to have a mild speech delay). mXALD-3 and mXALD-4 had subtle differences on brain MRI, which have remained stable. mXALD-5 was born with prenatally diagnosed arthrogryposis thought to be unrelated to X-ALD, and had a normal brain MRI at 11 months of age.
Estimated incidence of mosaic X-ALD
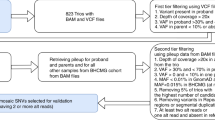
To estimate the incidence of mosaic X-ALD in a larger cohort, we examined data from two large reference labs over a 5-year period (2017–2022). During this time, 227 males with P/LP variants in ABCD1 were reported, four of which (4/227 or 1.8%) had VAFs that were less than 100%, consistent with mosaicism (Table 2). Clinical information was available for three individuals (mXALD-1–3) as described above (Table 1). We chose not to include mosaic ABCD1 variants of uncertain significance (VUS) in our estimate, since the majority of VUS are reclassified downward6, and the clinical indication for most individuals undergoing sequencing is a positive newborn screen, which by itself is only a screening test, and requires additional testing to identify false positives from true X-ALD cases.
Discussion
In summary, we estimate the frequency of mosaic X-ALD in a molecularly confirmed X-ALD cohort to be approximately 1.8%, and obtained clinical information for 5 mosaic X-ALD individuals. Although mosaicism in X-ALD has been previously reported7,8, this is the first study to measure VAF across multiple tissues and to estimate the incidence of mosaicism in a larger cohort. The presence of relatively high VAFs (66–82%) in mXALD-1 across tissues containing all three germ layers suggests that the p.Leu663Pro variant arose very early in development, prior to gastrulation, with both mutated and non-mutated cells contributing to the inner cell mass9. We cannot rule out the possibility of reversion of a mutation inherited from the egg, but since the p.Leu663Pro variant in mXALD-1 was not present in his mother’s blood, this would require two independent mutational events, which is unlikely.
Because some of the 227 individuals with P/LP ABCD1 variants had clinical testing due to a known family history of X-ALD (and thus cannot have mosaic X-ALD), our estimate of 1.8% incidence of mosaicism in X-ALD may underestimate the proportion of de novo X-ALD caused by mosaicism. At the same time, our exclusion of mosaic VUS in ABCD1 could also lead our estimate to be elevated, although we justified our decision not to include VUS in our study, as many of these variants will be downgraded, particularly in asymptomatic individuals identified on NBS. Laboratories do not routinely track or report VAF deviation from 100% in their clinical reports. VAFs between 30 and 60% are typical of heterozygous variants located on the autosomes, but caution is needed for X-linked genes, where VAFs in this range could easily be misinterpreted as heterozygous. Thus, our study suggests that mosaicism in X-ALD is underrecognized. Given the increasing number of presymptomatic males identified by expanded NBS and the increased sensitivity to mosaicism provided by NGS over Sanger sequencing, we recommend that laboratories include this information in their reports. More individuals and longer-term clinical follow-up will be required to determine if mosaic ABCD1 variants are associated with reduced phenotypic penetrance, but this will be challenging to determine unless clinical labs routinely include the presence of mosaicism in their reporting.
Our study adds to the growing body of literature detecting mosaicism in non-neoplastic, developmental diseases10,11,12. X-linked mosaicism is rare and has only been published in two other X-linked conditions, to our knowledge (hypophospatasia13 and VEXAS14). Our 1.8% estimate of mosaicism in X-ALD is consistent with other similar studies. A recent study examining autosomal dominant polycystic kidney disease (ADPKD) found that approximately 1% of all cases and up to 10% of genetically unsolved cases were mosaic, and these individuals generally had less severe disease15. We feel this is an important question that requires exploration in X-ALD, as accurate risk calculation could have direct implications for risk counseling as well as screening and management recommendations for newborns with X-ALD. Additionally, the distribution and burden of pathogenic variants across different tissues may be an independent phenotypic modifier in X-ALD, similar to engineered tissue-specific mouse mutants. In mosaic individuals with elevated tissue-specific mutation burden, stratifying the clinical impact of peroxisomal dysfunction across vulnerable tissues—such as the CNS, adrenal cortex, and testis—may yield critical insights into disease pathogenesis. Given that X-ALD pathology manifests with striking tissue selectivity despite systemic VLCFA accumulation, studying mosaicism offers a unique opportunity to dissect how localized peroxisomal deficits contribute to early cellular dysfunction and disease progression16. Although there are standards for reporting mosaicism in cancer17, no such standards currently exist for non-neoplastic conditions. We recommend that sequencing laboratories continue to report mosaicism when found and consider including the variant allele fraction and testing of other tissues when clinically indicated. Our study adds to the knowledge of X-ALD and mosaicism within common genetic conditions and opens the door for further studies determining the role of mosaicism in human disease.
Methods
Human subjects
mXALD-1 was enrolled in the study using the X-ALD Metabolomics IRB (Study #2497), which was approved (effective 10/22/2020) by the Institutional Review Board (IRB) at Seattle Children’s Hospital. mXALD-2, mXALD-3, mXALD-4, and mXALD-5 were recruited to participate in the study at their respective institutions by providing limited clinical data only, and written informed consent for publication was provided by parents or legal guardians on behalf of their children <18 years of age, as all included participants were minors. Assent was obtained from minors whenever possible. All human participants or samples are in compliance with all relevant ethical regulations, including the Declaration of Helsinki.
Sample processing
All samples were collected in conjunction with a planned surgical procedure requiring sedation. Whole blood, urine, and cerebrospinal fluid (CSF) samples were collected in cell-free DNA BCT tubes (“Streck tubes,” Streck Omaha, NE) and centrifuged at 1600g for 15 min. The resulting supernatant was centrifuged for an additional 15 min at 16,000g at 4 °C to remove remaining gDNA “pellet” debris. The supernatant was separated into aliquots and stored at −80 °C. “Pellet” gDNA was similarly aliquoted and frozen at −80 °C. cfDNA extraction was performed with QIAamp MinElute ccfDNA Kit (Qiagen, Venlo, Netherlands). Genomic DNA (gDNA) was extracted from tonsils, skin, and gDNA “pellets” of urine, CSF, and blood. using PureLink Genomic DNA Mini Kit (Invitrogen, Carlsbad, CA, USA). Buccal swabs were extracted by prepIT-L2P from DNA GenoTek, using the Oragene Saliva Protocol modified for a 2 ml sample. All DNA samples were quantified with Qubit fluorometry (Thermo Fisher Scientific, Waltham, MA, USA). cfDNA was further analyzed on an Agilent 2200 Tapestation with High Sensitivity D1000 ScreenTape (Agilent, Santa Clara, CA, USA).
Digital droplet PCR (ddPCR)
Primers, Affinity Plus® Iowa Black™ FQ Probes, and positive control gene block were designed for the patient-specific variant ABCD1 c.1988T>C (p.Leu663Pro) following previously used assay design guidelines18 and purchased from IDT (Integrative DNA Technologies, Coralville, IA). The reverse primer used in our assay is a previously published sequence known to avoid amplifying notorious ABCD1 pseudogenes3. Primers were designed based on published sequences in order to avoid known ABCD1 pseudogenes (F-ATTGCCCTGCTCTCCATCAC, R-TGCTGCTGCCGGGCCCGC) and amplified a 121 bp fragment surrounding the known variant. 20 ng of DNA was used per ddPCR reaction for male patients and male controls in order to match the amount of coverage that 10 ng input achieves in non-sex chromosome assays. All controls and samples were run in quadruplicate. The variant allele fraction (VAF) was calculated as the mutant concentration divided by the mutant concentration plus wild-type concentration. Controls included a no-template control (NTC) (VAF 0%), WT cfDNA (VAF 0%), WT gDNA (VAF 0%), ABCD1 L663P gblock (VAF 100%), and a second patient with a different, non-mosaic ABCD1 variant (1553 T > C) (VAF 0%).
Data compilation
mXALD-1 was identified through clinical care at our institution. To identify other mosaic individuals (mXALD-2 through mXALD-4), contact was made with multiple commercial ABCD1 sequencing laboratories and state NBS labs from across the United States. Some labs that were approached that used NGS-sequencing platforms were able to provide aggregate deidentified information about ABCD1 variants that were able to be extracted from large internal databases after performing a query. The requested data included all pathogenic and likely pathogenic ABCD1 variants identified from male individuals from each laboratory, and if any of those pathogenic and likely pathogenic ABCD1 variants that had been reported in clinical reports had actually been mosaic. If mosaic variants were identified, we requested any additional information on confirmatory Sanger sequencing. Communication with the clinical provider was made only if they responded to the lab’s inquiry on our behalf. Where clinical information was made available, we have included that in Table 1.
Identifying the frequency of mosaicism
Two large commercial labs identified all males who had pathogenic or likely pathogenic (P/LP) ABCD1 variants identified and reported out on clinical sequencing. The majority of these patients were infant males who were referred for ABCD1 sequencing after presenting with elevations of C26:0-LPC on newborn screening. Some of these males were older individuals who had a clinical suspicion for X-ALD either because of positive family members or because of symptomatology. We asked each lab if they were able to identify if any of the males with P or LP ABCD1 variants had actually had mosaic variants identified. For each lab, the number of males with mosaic P/LP ABCD1 variants was divided by the total number of males with identified P/LP ABCD1 variants. Both commercial labs used NGS sequencing, which had been verified by Sanger sequencing. Variants of uncertain significance were excluded, as the clinical relevance of these variants is more challenging to interpret. The four mosaic variants identified were unique from one another, indicating four separate individuals. Three of the four variants are included in Table 1. For the remaining variant, we obtained the level of mosaicism, but the ordering provider was not reachable to provide clinical information.
Data availability
Information on the pathogenicity of reported ABCD1 variants is publicly available on “The ABCD1 Variant Registry” (https://adrenoleukodystrophy.info/mutations-and-variants-in-abcd1). Clinical data are available from the authors on reasonable request.
References
Bezman, L. et al. Adrenoleukodystrophy: incidence, new mutation rate, and results of extended family screening. Ann. Neurol. 49, 512–517 (2001).
Turk, B. R. et al. X-linked adrenoleukodystrophy: Pathology, pathophysiology, diagnostic testing, newborn screening and therapies. Int J. Dev. Neurosci. 80, 52–72 (2020).
Jaspers, Y. R. J. et al. Lipidomic biomarkers in plasma correlate with disease severity in adrenoleukodystrophy. Commun. Med. (Lond.). 4, 175 (2024).
Bonkowsky, J. L. & Wilkes, J. Time to transplant in X-linked adrenoleukodystrophy. J. Child Neurol. 37, 397–400 (2022).
Keam, S. J. Elivaldogene autotemcel: first approval. Mol. Diagn. Ther. 25, 803–809 (2021).
Bennett, G. et al. Distinct rates of VUS reclassification are observed when subclassifying VUS by evidence level. medRxiv https://doi.org/10.1101/2024.11.13.24317242 (2024).
Wang, Y. et al. X-linked adrenoleukodystrophy: ABCD1 de novo mutations and mosaicism. Mol. Genet Metab. 104, 160–166 (2011).
Ruiz, M. et al. X-linked adrenoleukodystrophy: phenotype distribution and expression of ALDP in Spanish kindreds. Am. J. Med Genet. 76, 424–427 (1998).
Bischoff, M., Parfitt, D. E. & Zernicka-Goetz, M. Formation of the embryonic-abembryonic axis of the mouse blastocyst: relationships between orientation of early cleavage divisions and pattern of symmetric/asymmetric divisions. Development 135, 953–962 (2008).
Poduri, A., Evrony, G. D., Cai, X. & Walsh, C. A. Somatic mutation, genomic variation, and neurological disease. Science 341, 1237758 (2013).
Bennett, J. T. et al. Mosaic activating mutations in FGFR1 cause encephalocraniocutaneous lipomatosis. Am. J. Hum. Genet. 98, 579–587 (2016).
Truty, R. et al. Patterns of mosaicism for sequence and copy-number variants discovered through clinical deep sequencing of disease-related genes in one million individuals. Am. J. Hum. Genet. 110, 551–564 (2023).
Broseta, J. J., López-Romero, L. C., Cerón, J. A., Mendizábal, S. & Hernández-Jaras, J. Mosaicism in 2 cases of X-linked hypophosphatemia. Endocrinol. Diabetes Nutr. 67, 70–71 (2020).
Beck, D. B. et al. Somatic Mutations in UBA1 and severe adult-onset autoinflammatory disease. N. Engl. J. Med. 383, 2628–2638 (2020).
Hopp, K. et al. Detection and characterization of mosaicism in autosomal dominant polycystic kidney disease. Kidney Int. 97, 370–382 (2020).
Yska, H. A. F., Engelen, M. & Bugiani, M. The pathology of X-linked adrenoleukodystrophy: tissue specific changes as a clue to pathophysiology. Orphanet J Rare Dis. 19, 138 (2024).
Li, M. M. et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 19, 4–23 (2017).
Rowlands, V. et al. Optimization of robust singleplex and multiplex droplet digital PCR assays for high confidence mutation detection in circulating tumour DNA. Sci. Rep. 9, 12620 (2019).
Acknowledgements
We would like to thank the newborn screening laboratories and commercial sequencing laboratories that provided internal data. We would like to thank the individuals and their families for their participation in this study.
Author information
Authors and Affiliations
Contributions
A.C.K. designed the study, initiated this work, supervised the conduct of the study, and drafted the paper. A.C.K., A.S., and M.P.P. collected and analyzed patient samples. D.M.J. designed ddPCR assay. A.C.K., D.M.J., M.P., and N.A. performed and analyzed molecular experiments. R.B., M.M., I.S., and P.K. analyzed and interpreted genetic data. E.B., L.O.G., F.S.E., B.E.R., D.W., A.R., E.S., D.V., E.C., K.W., E.S., B.E.H., R.T., T.C.L., and A.S. recruited and evaluated the study participants. J.T.B. supervised the study. All authors revised the paper.
Corresponding author
Ethics declarations
Competing interests
Authors P.K. and M.M.M. are employees of GeneDx, LLC, but declare no financial or non-financial competing interests. A.R. is an employee at Natera but declares no financial or non-financial competing interests. F.S.E. is PI of an in vivo gene therapy trial in Canavan disease (Sponsor: ASPA Therapeutics), Co-PI of ex vivo lentiviral gene therapy trial in cerebral adrenoleukodystrophy (Sponsor: bluebird bio), Co-PI of PI of in vivo gene therapy trial in GM2 (Former Sponsor: Sio Therapeutics), Site-PI of Alexander Disease trial of ASOs (Sponsor: Ionis Pharmaceuticals), Consultant to Atlas Venture, Acadia Pharmaceuticals, Leal Therapeutics, Vigil, Takeda Therapeutics, Sanofi, SwanBio Therapeutics and UpToDate, founder of SwanBio Therapeutics, and is a NINDS Advisory Council Member but declares no financial or non-financial competing interests. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Keefe, A.C., Jensen, D.M., Pham, M.M. et al. Mosaic X-linked adrenoleukodystrophy in males identified by newborn screening and next-generation sequencing. npj Genom. Med. 10, 38 (2025). https://doi.org/10.1038/s41525-025-00497-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-025-00497-1
