
Abstract
Variants affecting pre-mRNA splicing mechanisms are responsible for multiple monogenic disorders. However, their prioritization and interpretation remain challenging. Herein, we designed a strategy for the identification of likely spliceogenic variants in unsolved inherited retinal dystrophy (IRD) cases. We benchmarked thirteen splicing predictors on a curated training dataset, which revealed that the combination of SpliceAI and MaxEnt tools exhibited the best performance for the analysis of most splicing variants. However, for branch point variants, the BranchPoint tool (Alamut®-Batch) was the optimal choice. The proposed combination of tools was assessed using a validation cohort comprising 116 genetically diagnosed individuals with rare diseases, and subsequently applied for the analysis of 211 unsolved IRD families. The pipeline identified 30 likely pathogenic variants, 17 of which were predicted to alter splicing mechanisms. These results demonstrate an increase in diagnostic yield of up to 6.2%, reinforcing the importance of reanalysis strategies focused on identifying spliceogenic variants.
Similar content being viewed by others

Introduction
The arrival of next-generation sequencing (NGS) technologies has revolutionized the way genomic data is obtained, enabling the sequencing of gene panels, whole exomes (WES), and even whole genomes (WGS) for multiple individuals in parallel. These methods have become so cost-effective and precise that they are often applied in healthcare settings to genetically diagnose Mendelian disorders1. The identification of disease-causing variants paves the way to personalized medicine, improving patient care and facilitating future therapies. Nonetheless, although there have been significant advancements in this field, a substantial proportion of cases still cannot be diagnosed with current NGS approaches.
Inherited retinal dystrophies (IRD) are a group of clinically and genetically heterogeneous pathologies caused by the dysfunction of photoreceptors or retinal pigment epithelium cells, often resulting in severe visual impairment2. To date, mutations in more than 290 genes have been associated with these diseases (https://web.sph.uth.edu/RetNet/ accessed October 2023), hampering the achievement of a genetic diagnosis. In fact, the diagnostic yield of IRD has long remained between 52 and 74% depending on the specific phenotype and the NGS-based method used3. This suggests that certain causative variants still elude diagnostic pipelines. One possible explanation is that, while there have been advances in NGS data acquisition methods, there have not been equally rapid advances in variant interpretation and classification. As a result, a significant proportion of variants are often labeled as Variants of Uncertain Significance (VUS)4, even after applying the most widely used variant categorization guidelines5. This issue directly concerns genetic counseling for families and individuals with IRD, who should deal with unknown or incomplete molecular diagnoses.
The ambiguity in variant interpretation is even higher for rare novel variants located in the non-coding genome, outside of conserved canonical sites6. This reflects a historical bias toward covering coding exons and their intronic boundaries in genetic screening7. Consequently, online repositories such as ClinVar8 or the Human Gene Mutation Database (HGMD)9 show an enrichment of splicing variants in the vicinity of splice site regions. While there is a biological reason for this, as the sequence around splice sites is particularly dense in splicing-relevant information, it is well known that any genomic variant, regardless of its ___location, can alter pre-mRNA splicing mechanisms10,11. Therefore, it is likely that many splicing variants remain to be discovered, playing a significant role in the development of a variety of Mendelian disorders12, including IRDs13. In fact, it is estimated that at least 9–30% of causative variants may act through disruption of splicing14. Therefore, a closer look at the effect of variants on splicing would be of great benefit in improving the diagnostic yield for rare disease patients15.
Splicing dysregulation results from a variety of sequence alterations, including loss of canonical splice sites, activation of cryptic splice sites, and disruption of essential motifs such as the branch point (BP) and the polypyrimidine tract (PPT), among others. Variants at all this sites are established contributors to IRD13,16,17,18,19,20,21,22 by producing aberrant mRNA transcripts, including exon skipping16,17, intron retention or exonification13,18, pseudo-exon insertion19,20,21,22, or exon truncation.
Recent studies have shown that RNA-seq can provide a molecular diagnosis in up to 30% of WES-negative cases23. However, the tissue- and time-specific nature of splicing may cause the genes of interest to not be expressed or not expressed equally in the tissue commonly used for RNA analysis24. This is particularly relevant in the case of IRDs, where the target organ, the retina, is subject to intense alternative splicing activity25 (https://oculargenomics.meei.harvard.edu/retinal-transcriptome/). It is noteworthy that approximately 50% of the expressed genes in the retina have altered exons compared to the canonical isoforms, including several thousand novel exons13,26,27. Hence, the characteristic splicing patterns of the retina further complicate the functional assessment of splicing variants.
Numerous computational tools have been developed to assist in the interpretation and prioritization of splicing variants in the clinical setting28,29,30,31,32,33,34, circumventing the limitations of RNA studies. Recently, machine learning methods have surpassed previous approaches in predicting pathogenic splicing variants14,32. However, their performance varies depending on the studies and datasets considered35, limiting their stand-alone use in a clinical environment where accuracy is essential for decision making. In addition, whereas splicing variants at canonical sites are simple to identify computationally, other splicing variants are considerably more difficult to interpret and require the application of specific algorithms15. Therefore, there is an urgent need for benchmarking studies that independently compare the performance of different splicing methods and evaluate how they can be combined and integrated into medical workflows.
The aim of this study is to develop a pipeline to facilitate the interpretation and prioritization of splicing variants that have the potential to contribute to the diagnosis of unsolved cases of Mendelian diseases. To this end, we performed a comprehensive evaluation of thirteen in silico splicing tools using variants from online curated resources. Our benchmarking study allowed us to identify the optimal combination of tools and to redefine the recommended thresholds. Further application of this pipeline for the genetic analysis of 211 previously unsolved IRD cases resulted in an increase in diagnostic yield of up to 6.2%, confirming the importance of splicing variants in the etiopathogenesis of this rare disorder.
Results
Benchmarking study and definition of optimal thresholds
To evaluate the performance of splicing tools, we generated a training dataset comprising by 1535 pathogenic variants and 2102 benign variants (Fig. 1). This dataset was divided into five distinct categories of splicing variants (details provided in Methods): canonical splice site (CSS), non-canonical splice site (NCSS), deep intronic (DI), exonic splicing (ES), and branch point (BP) (Fig. 1).

A Schematic representation of the study design. The training dataset includes 3637 curated variants sourced from public and private databases, classified into pathogenic (P) or likely pathogenic (LP) and benign (B) or likely benign (LB) groups based on ClinVar and HGMD-pro annotations. The validation cohort consists of 116 genetically diagnosed individuals with rare diseases (RD), carriers of 118 spliceogenic causal variants. The experimental cohort comprises 238 unsolved cases from 211 families with inherited retinal disease (IRD), analyzed using either targeted sequencing (TS) or whole-genome sequencing (WGS). Patients analyzed by WGS included 19 unaffected (unaf.) relatives. B Eukaryotic gene structure showing consensus exon and intron sites for gene splicing mechanisms at DNA level. According to the distance to the splice site (ss), five distinct categories of variants were established: canonical splice site (CSS), non-canonical splice site (NCSS), exonic synonymous (ES), deep intronic (DI), and branch point (BP).
The performance of thirteen splicing predictors was assessed using the entire training dataset (overall) and across the five variant categories separately (Supplementary Fig. 1). Regarding the receiver operating characteristic (ROC) curves, the overall dataset and NCSS variant groups showed an AUC > 0.8 for most tools, except for BPP and BP Alamut. Notably, BP Alamut perfomed best when applied exclusively to BP variants, suggesting tool specialization in BP motifs (Supplementary Table 1).
As Matthews Correlation Coefficient (MCC) provides a more informative and truthful score in evaluating binary classifications36, the tool performance was assessed taken into account the MCC value for a sensitivity of 90%. In this sense, SpliceAI and MaxEnt showed the highest overall performance using the overall dataset (Supplementary Table 1). Of note, SpliceAI also displayed a MCC > 0.5 in five out of six variant groups, except for the CSS group (MCC = 0.48), reflecting the difficulty in collecting benign variants located at the canonical splice sites.
The ROC data allowed us to determine the optimal threshold for each tool, both overall and within each variant group. These threshold varied widely depending on the specific variant dataset analyzed (Supplementary Table 1). To simplify further analysis, the optimal thresholds for each tool were selected based on the ROC results from the overall dataset, except for the BP variants. Due to the difficulty in predicting branch sites, a different approach was required for this category.
The optimal thresholds of the best performing prediction tools, determined based on the maximum MCC for each variant group, are shown in Table 1.
Combinatorial analysis
As tool performance depends on the type of splice-altering variant, we hypothesize that combining multiple prediction tools could lead to a consensus on the splice-altering potential of the variant. Furthermore, these combinations could also reduce the false positive rate (%FP) of each tool compared to its sole application (Supplementary Table 1).
Bases on the ROC curve analysis of the overall dataset, the tools that met the established criteria for good performance (MCC > 0.5, %FP < 35%, and %NA < 5%) were identified and selected (Supplementary Table 1). As a result, the prediction tools SpliceAI, MaxEnt, SSF, CADD, SQUIRLS, and NNS were chosen for further combinatorial study. Out of the 63 models generated by combining the six tools, a focused analysis was conducted on the 32 models that included the SpliceAI, as this tool demonstrated strong performance in five of the six variant groups under study.
The training dataset was filtered using each of the 32 models, which were applied using the optimal threshold (Supplementary Table 1) calculated for each tool from the overall dataset. Subsequent to this, a series of statistical parameters were calculated (Supplementary Table 2) to identify the most efficient combination of tools. Among the combinatorial models, “SpliceAI+MaxEnt” combination showed the best performance with the highest area value of the radar plot polygon (Supplementary Fig. 2). Compared to using SpliceAI alone, the combination of these two tools halved the %FP, and increased the MCC and PPV values, allowing a better prioritization of the splice-altering variants (Fig. 2).

A Bar chart showing the results obtained for sensitivity, MCC, %FP and PPV in pathogenic variants when applying SpliceAI alone or the optimal SpliceAI+MaxEnt model. Statistically significant differences are indicated by asterisks (***p-value < 0.05). B Graph showing the comparison between the application of the SpliceAI+MaxEnt model using the thresholds already described (literature) or those proposed in this study (custom). The graphs were created with RStudio 2022.02.3. %FP false positive rate, MCC Matthew’s correlation coefficient, NPV Negative predictive value, PPV Positive predictive value, SEN sensitivity, SPE specificity.
In addition, we highlighted that our optimized model significantly increased sensitivity (p-value = 1.543·10−5, Chi-squared test) and MCC (p-value = 3.357·10−14, Chi-squared test) compared to the combination with the thresholds suggested by the tools developers, whereas the %FP remained similar; therefore, our model showed a higher area value of the radar plot polygon (Fig. 2).
Validation of the pipeline
To assess the effectiveness of the proposed splicing filter, a validation dataset comprising 118 previously known spliceogenic variants (Supplementary Data 1) was generated from the genomic data of 116 genetically diagnosed individuals (Fig. 1). The application of frequency-based filtering (MAF ≤ 0.01) in combination with the “SpliceAI+MaxEnt” filter successfully prioritized 88.98% of the spliceogenic, disease-causing variants (105 out of 118). The addition of the ClinVar update pathway to the workflow improved the prioritization rate of disease-causing variants to 97.46% (115 out of 118), without increasing the %FP. This “ClinVar update” step aims to prioritize variants classified as pathogenic (P), likely pathogenic (LP), or conflicting but with at least one P or LP entry in ClinVar (CIPP/LP), regardless of whether they pass the rest of the filters or not.
We also assessed the type of splice-altering variants validated by our workflow (Fig. 3). Remarkably, all CSS and synonymous variants in the validation dataset were correctly identified. However, two NCSS and one DI variants failed to pass any of the applied filters (Supplementary Data 1). It is important to note that the validation dataset lacked variants in branch sites, as none of our patients harbored disease-causing branch point variants. Therefore, the prioritization potential of BP Alamut for these variants could not be evaluated.

A The histogram shows the percentage of recovered and missing variants using the developed workflow in the validation cohort. The graph comprises annotations that indicate the number of variants corresponding to the size of each bar. The graph was created with RStudio 2022.02.3. B Overview of the 211 families of the experimental cohort analyzed using the developed pipeline. P/LP pathogenic/liley pathogenic.
The performance assessment of the optimized splicing tool combination in the validation cohort demonstrated the efficacy of this approach to prioritize spliceogenic variants. As a result, we added the “SpliceAI+MaxEnt” and “BP Alamut” pathways to our recently published in-house algorithm for analyzing NGS data37 (Fig. 4).

Genomic data from unsolved families are jointly combined, annotated and filtered. During the process, the data is also annotated with an updated ClinVar database file (2023.01.21), with the aim of identifying variants that were recently classified as pathogenic (“P”), likely pathogenic (“LP”), or as conflicting interpretations of pathogenicity (“CIP”) variants with at least one “P” or “LP” entry. In parallel, quality, recurrence, frequency, multiallelic and coding genes filterings were applied to SNVs and CNVs. After this, SNVs were filtered by CADD + MAPP+ Grantham+SIFT37, SpliceAI+MaxEnt and BP Alamut resulting in a number of likely pathogenic and spliceogenic variants. Moreover, to evaluate variants consistent with a compound heterozygous model, we also took into account other coding non-synonymous variants located in the same genes where another variant was prioritized by our filtering steps. The different paths of analysis converge into the pedigree filtering that focused on the variants consistent with the disease in each family pedigree considering the distinct modes of inheritance and the status of the different sequenced members. Subsequently, the candidate variants are manually prioritized and segregated in additional family members. In diagnosed cases, an experimental validation can be conducted, while the remaining unsolved cases, can be susceptible to re-enter in a new round of analysis. AF allele frequency, CIP conflicting interpretations of pathogenicity, Het heterozygous, Hom homozygous, LP likely pathogenic, P pathogenic, SNVs single nucleotide variations, SVs structural variants, TS targeted sequencing, WGS whole-genome sequencing.
Application of the splicing pipeline
Rare SNVs were prioritized applying the filters “CADD+MAPP+Grantham+SIFT”, “SpliceAI+MaxEnt” and BP Alamut according to the developed pipeline (Fig. 4). Variants meeting one of these filters were integrated into the pedigree filtering alongside SV variants, and the variants prioritized by the ClinVar update. Finally, a manual curation was carried out.
The application of the optimized algorithm in the experimental cohort (Fig. 1) resulted in the identification of 30 different variants (12 novel) that may explain the phenotype in 20 IRD families (Table 2). In addition, 23 monoallelic variants reported as pathogenic or likely pathogenic in the ClinVar database for recessive genes were identified in 25 families, which could contribute to the phenotype (Fig. 3). No candidate variants were found in the remaining families (n = 166).
Family segregation studies for 21 genetic variants showed segregation with the disease in 13 pedigrees (Supplementary Fig. 3). A total of 13 families (65% of prioritized families and 6.2% of all targeted/WGS-negative samples) were “solved”, “very likely solved” or “possibly solved”, while others (n = 7) were “uncertain” (Fig. 3). For clarity: i) “solved” families included all those in which the identified variants could explain the disease without the need for further studies; ii) “very likely solved” families include two families (Fam 16 and Fam 9) in which minor further studies would be required, as both families met an additional pathogenicity criterion (marked “a” in Table 2), based on the fact that the same pairs of variants were detected in trans in more than one unrelated family of our IRD cohort, strengthening their causal role; iii) “Possibly solved” families included two families (Fam 1 and Fam 15) that were harboring variants in genes previously associated with the reported phenotype in which further studies would be required; and iv) “Uncertain” families included those with a novel genotype-phenotype correlation in which major further studies are needed. Specific recommendations are shown on Table 2.
According to ACMG/AMP criteria5,38, the 30 prioritized variants were classified as pathogenic (n = 9), likely pathogenic (n = 7), or variants of uncertain significance (n = 14) (Table 2). Seventeen out of the 30 variants were predicted to be spliceogenic, including 2 CSS, 4 NCSS, 3 DI, 3 BP, 4 ES and 1 missense variants (Table 3). Of these, the “SpliceAI+MaxEnt” filter prioritized 13 variants, 4 of which also passed the “CADD+MAPP+Grantham+SIFT” filter. “BP Alamut” and manual curation recovered 3 and 2 variants, respectively (Table 3). Of the 13 non-splicing variants, 9 had been previously identified but were not prioritized due to the absence of a second pathogenic allele in recessive genes or uncertain clinical significance at the time of initial analysis (Fam 7). Conversely, the duplications identified in families 11 and 12 were missed in targeted sequencing studies, as was the homozygous PLA2G5 variant in family 18 or the deletion detected in family 4, since these genomic regions were excluded from the panel design.
Interestingly, we identified two variants, one in BBS1 and one in CDH23, whose effect on the protein may vary according to the different isoforms described in Ensembl (GRCh37) (Table 2). For the canonical isoform of BBS1 (NM_024649.5), the variant is synonymous (c.339T>C; p.(Tyr113 = )), whereas for the non-canonical ENST00000537537.1 isoform, it is missense (c.50T>C; p.(Met17Thr)). Similarly, for the canonical isoform of CDH23 (NM_022124.6) the identified SV affects only the non-coding 5’ UTR region (c.-427_-30del; p.?), but for the alternative isoform NM_001171931.1 it implies the deletion of the first coding exon (c.-293_106del). RT-PCR using ready-to-use cDNA from different human tissues suggested that, in both cases, although the canonical isoform exhibits higher expression levels in the analyzed tissues, the alternative isoforms of interest are also expressed in the retina (Supplementary Fig. 4). The results indicated that the canonical isoforms display a ubiquitous expression pattern, while the non-canonical isoforms analyzed appear to exhibit a more tissue-specific expression profile. Remarkably, the second hit (c.808G>A; p.(Gly270Arg)) identified in the index patient of family 1 also affected the coding sequence of the ENST00000537537.1 isoform (c.472G>A; p.(Gly158Arg)) (Supplementary Fig. 5). The three-dimensional modeling for BBS1 alternative protein (UnitProt B4DYY3) using PyMOL Molecular Graphics System showed that Met17, a non-polar amino acid, may interact with two other amino acids through hydrogen bonding (Supplementary Fig. 5). Specifically, Met17 would form one hydrogen bond with Leu13, and with Ala14. In silico mutagenesis at position 17 to threonine, a polar amino acid, predicted the gain of a new hydrogen bonding interaction point with p.Ala14 (Supplementary Fig. 5). Likewise, the mutagenesis at position 158 from glycine to arginine showed the likely formation of a new hydrogen bonding interaction point with p.Arg156. These results suggest that the two BBS1 variants identified in family 1 may be exerting their pathogenic effect by altering the protein folding of the alternative isoform (Supplementary Fig. 5).
Genotype-phenotype correlations
The clinical characterization of the 20 families with candidate genetic variants identified in this study is summarized in Supplementary Data 2. The main clinical diagnosis was retinitis pigmentosa (n = 10), followed by Usher syndrome (n = 4), cone dystrophy (n = 3), and other 3 phenotypes (fundus albipunctatus, anophthalmia with familial exudative vitreoretinopathy, and unspecified retinal dystrophy). Of note, in six families with nonsyndromic IRD, variants were identified in genes that, according to the OMIM database (accessed October 2023), are primarily, although not exclusively, associated with syndromic human phenotypes, but which may include IRD among their associated symptoms: BBS1, MIM:*209901; CHD7, MIM:*608892; ERCC2, MIM:*126340; GRN, MIM:*138945; MED12, MIM:*300188; PRPS1, MIM:*311850. Further studies, including extended segregation analysis, are needed to evaluate the role of these variants in the etiopathogenesis of the IRD in these families.
Splicing analysis of CDH23 variants in patient RNA
Our limited availability of patient-derived blood samples and the inaccessibility of the target tissue (the retina), significantly constrain the functional validation of splicing variants. Of the 17 spliceogenic variants identified in this study, we were able to validate only two. To further characterize the potential effect on splicing for the novel branch point (c.5924-21A>C) and deep intronic (c.6713-1062G>A) variants in CDH23 gene, we performed RT-PCR experiments on patient RNA in families 2 and 5.
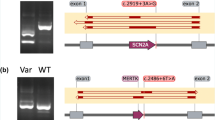
The proband of family 2 received a clinical diagnosis of Usher syndrome type I (USH1) showing a profound sensorineural hearing loss that required a cochlear implantation (Supplementary Data 2). Previous panel sequencing identified one pathogenic heterozygous variant in CDH23 (Table 2). The application of the splicing pipeline led to the identification of a second variant located in intron 45 (c.5924-21A>C), predicted to disrupt a putative BP site. For the analysis of this variant, we performed RT-PCR amplification of CDH23 exons 44–46 in individuals I:1, I:2 and II:1 from family 2. Heterozygous carriers of the c.5924-21A>C variant (I:2 and II:1) yielded, apart from the wild-type RT-PCR product (335 bp), an aberrant larger product of 540 bp (Fig. 5). Further Sanger sequencing of this product revealed the exonification of 205 bp of intron 45 (Fig. 5). The aberrant transcript would lead, if translated, to frameshift variant consisting of the insertion of three discrepant amino acids followed by a premature termination codon. This results allowed to classify the CDH23 c.5924-21A>C variant as likely pathogenic that, in combination with c.1087del, causes USH1 in family 2 (Supplementary Fig. 3). Full penetrant phenotype in this family is supported by the relative abundance of correctly (49.6%) versus aberrantly (50.4%) spliced isoforms.

A Schematic representation of CDH23 gene (NM_022124.6; GRCh37) and the ___location of the variants c.5924-21A>C and c.6713-1062G>A. B Agarose gel electrophoresis of RT-PCR products for CDH23 exons 44–46 using total RNA from frozen whole blood samples of the available individuals in family 2 (Fam 2). The individuals harboring the variant c.5924-21A>C (II:1 and I:2) showed a double band, one of which was ~200 nucleotides larger. Sanger sequencing of the reference (REF) and alternative (ALT) alleles revealed the retention of 205 intronic bases upstream of exon 46 in the ALT allele. C Agarose gel electrophoresis of RT-PCR products for CDH23 exons 48–49 using total RNA from frozen whole blood samples in negative control individual (C-) and the son (III:1) of index patient in family 5 (Fam 5), carrier of the deep intronic variant. The Sanger sequencing of CDH23 alleles in individual III:1 revealed the insertion of an intronic 70 bp region as a pseudoexon in the ALT allele. Red box indicates the predicted premature termination codon (PTC).
On the other hand, the proband of family 5 was diagnosed with atypical USH due to moderate sensorineural hearing loss (Supplementary Data 2). The analysis of WGS data with our in-house pipeline allowed us to identify a novel deep intronic variant (c.6713-1062G>A) in intron 48 of CDH23, predicted by SpliceAI to create cryptic acceptor site. This variant could explain the phenotype of the patient alongside with the previously identified c.6396del CDH23 variant. To validate the in silico prediction for the novel deep intronic CDH23 variant, RT-PCR amplification of CDH23 exons 48–49 was performed in individual III:1 from family 5 and in one control individual (Fig. 5). The RT-PCR results showed two different products (300 bp and 370 bp) in the individual carrying the heterozygous variant (c.6713-1062G>A), in contrast to the single product (300 bp) observed in the control individual (Fig. 5). Subsequent sequencing of these products unmasked the inclusion of a pseudoexon of 70 bp spliced between exons 48 and 49 of CDH23 in the mutant allele (Fig. 5). If translated, this would lead to insertion of 40 aberrant amino acids followed by a premature termination codon. Additionally, the relative band intensity of the mutant and wild-type was 26.7% and 73.3%, respectively, indicating that the mutant allele still produces a significant fraction of correctly spliced transcripts. These results suggested that the atypical Usher syndrome in family 5 could be caused by the combination of the ClinVar pathogenic allele, c.6393del, and the leaky or not fully-penetrant deep intronic variant, c.6713-1062G>A, in CDH23.
Discussion
In recent years, WGS has become more readily available in research and healthcare, highlighting the need for accurate analysis of large genomic datasets39. In this sense, novel algorithms are continuously being proposed to predict the pathogenicity of variants in coding and non-coding regions37,40. Many benchmarking studies of splice prediction tools have been published with the aim of providing insight into the selection of best practices and methods to maximize successful results, however, their integration and application into medical diagnostic workflows remain unexplored and no consensus has yet been reached6,41. Similarly, the performance of branch point specific predictors were not usually evaluated in the same studies41, nor individual cutoffs according to the different variant datasets were set6. Here, we developed an efficient mode for filtering and prioritizing genomic variants based on the rational use of a set of splicing tools. In this study, the implementation of both optimized prioritization and RNA validation approaches for the splice-altering variants has improved the diagnostic yield in rare ophthalmic diseases. Through our study, 13 out of 211 (6.2%) previously unsolved IRD families have received a conclusive or possibly conclusive genetic diagnosis, with a major contribution of spliceogenic variants. Additionally, we have identified candidate variants in seven families, in which further studies are needed.
In our benchmarking study, SpliceAI demonstrated the highest performance for classifying splice variant, leading to the proposal of a revised filtering threshold (∆score-SpliceAI ≥0.235). This cutoff is significantly lower than the standard recommended cutoff (∆score-SpliceAI ≥0.5)42 and align with previous studies showing that intronic variants with a ∆score-SpliceAI <0.5 results in mis-splicing, putting the spotlight on the use of lower cutoffs6,42,43 (Supplementary Table 3). It is noteworthy that the optimal SpliceAI cutoff value varied depending on the dataset used and the type of splicing variants6,41. In our study, cutoff values ranged from 0.745 for the CSS variants to 0.015 for the ES variants (Supplementary Table 3).
This refined threshold enabled the prioritization of a novel deep intronic variant (CDH23, c.6713-1062G>A), which would have been overlooked using the standard SpliceAI threshold, even after the experimental confirmation of an associated aberrant splicing product. Of note, although a less restrictive cutoff may result in a higher %FP, the integration multiple in silico tools has been shown to mitigate this issue and enhance prediction accuracy6,42. In this work, the combinatorial model “SpliceAI+MaxEnt” identified the majority of likely spliceogenic variants in our discovery cohort, reinforzing its utility in detecting splice-altering variants from NGS data and improving diagnostic yield.
Although general cutoffs have been proposed in this work to apply to the total genomic variant burden, specific cutoffs were also defined for each variant type (CSS, NCSS, DI, BP, ES). These specific scores could be considered during manual prioritization for a better interpretation of the variants. In this regard, we recommend CADD for the analysis of CSS variants, SPiCE for NCSS variants, and SpliceAI for DI, whereas for ES variants no predictors were particularly good, which could be considered a research gap. On the other hand, our work also revealed that variants in BP sites require region-specific prediction tools, achieving BP tool (Alamut Batch® v1.11) the best performance. In fact, although further studies including lariat PCR would be needed, in this work, the use of this specific BP predictor allowed us to propose 3 candidate variants in 3 families that were considered negative in previous studies. Among these, we studied in detail the CDH23 c.5924-21A>C variant, which resulted in the retention of 205 intronic nucleotides leading to the mis-splicing of exon 46. Although a few pathogenic BP variants in the IRD genes have recently been described43,44,45,46, to our knowledge, the c.5924-21A>C variant is the first suggested BP variant within CDH23 that could result in an aberrant splicing pattern. The BP variants prioritization is of particular interest when analyzing targeted sequencing data covering exonic and flanking intronic regions (typically ±10 or ±25), as branch point sites are often captured incidentally by the design. However, variants identified in these regions, which are relatively distant from canonical splice sites, often escape conventional variant analysis protocols. The identification of likely pathogenic BP variants stresses the importance of including the assessment of this variant type in routine diagnostic genetic testing43. Likewise, the putative BP variant PRPS1: c.705-62_705-55del was located further than the theoretical branch point sites (from-18 to -44). Although more than 95% of BP sites are located within this interval, some BP sites can be located up to 400 bp upstream of the acceptor site47. Based on our results, it would be recommended to increase the number of intronic bases upstream of exons during panel design, taking into account the subsequent impact on cost-effectiveness.
Despite the usefulness of prediction tools in the analysis of genomic variants, putative splicing variants demand empirical confirmation based on expression studies from appropriate tissues48. However, one of the main limitations for variant validations in IRD is the unavailability of retinal tissue from patients. For this reason, functional validation should be often performed in other tissues, such as fresh blood2,49, fibroblasts50 or using minigene assays51 and retinal organoids52, among others. Minigenes assays are valuable tools for studying splice variants, particularly in the context of retinal dystrophies53. However, they also present several inherent limitations, which emphasize the importance of interpreting their results within the context of these limitations54. Herein, a splicing analysis of two putative spliceogenic variants in the CDH23 gene was performed using patient RNA from frozen peripheral blood samples. This approach could be an efficient alternative for future splicing assays in IRDs patients, particularly if the genes and isoforms of interest are expressed in whole blood, which is not often the case because gene expression is tissue dependent24. In summary, of the 17 putative spliceogenic variants identified in this work, four have been functionally validated (Table 3). Of these, two were validated using RNA from frozen blood (this study), one using RNA from fresh whole blood49 and the other using a minigene assay55 showing high heterogeneity in terms of the method used and results obtained. Given the limitations of the technique employed, additional studies would be required to propose a pathogenic effect for the remaining 13 spliceogenic variants.
Of note, the impact of splicing variants on disease phenotypes can vary widely depending on the specific gene, the nature of the variant and the tissues in which the variant’s effects are observed20,49. Moreover, the aberrant transcript may be degraded by the nonsense-mediated decay (NMD) process56 affecting the relative proportion of aberrant versus normal spliced mRNA underlying the phenotypic variability in some cases20,49. The evaluation of the impact on splicing of the c.5924-21A>C CDH23 variant in family 2 revealed the exonification of 205 intronic nucleotides in exon 46 in 50,4% of the transcripts suggesting a fully penetrant splicing defect underlying the USH1 phenotype. In contrast, the splicing defect exerted by the CDH23 variant identified in family 5 (c.6713-1062G>A) could not be fully penetrant (73.3% wiltype vs. 26.7% mutant). Although further studies would be needed in this case, the observed differences on transcript dosages could be explained by the NMD-process. Indeed, the proband of family 5 was diagnosed of an atypical and mild form of Usher syndrome, presenting RP (first symptom at age 10 years), moderate sensorineural hearing loss and no vestibular dysfunction. Although a wide phenotypic variability has also been described in patients with CDH23 variants, this case is reminiscent of another atypical Usher syndrome cases that harbored non-canonical splicing variants in CDH2349. These splicing variants might be better tolerated by some cell types than others or may act as not fully penetrant alleles, modulating the expression of the phenotype and being often bypassed by diagnostic clinical and genetic pipelines. However, when found in trans with a second pathogenic allele in a recessive pedigree, they can be reconsidered. In this sense, possibly spliceogenic variants in mainly syndromic genes have been identified in 6 families with suspected non-syndromic IRD resembling other cases where spliceogenic variants resulted in non-syndromic IRD phenotypes19,20,57,58. Although further studies are needed, the fact that the clinical features of affected individuals in these families were milder than those associated with fully penetrant variants may be explained by the heterogeneous nature of splicing variants.
In addition, our work also highlights the need to analyze the differential effects of a genomic variant in all known transcripts of the gene, not just in the canonical isoforms, which may not be the predominant ones in the target tissues. These are the cases of families 1 and 4, which carried variants that may or may not alter the protein sequence, depending on the isoform involved of BBS1 (canonical transcript: NM_024649.5 and alternative transcript: ENST00000537537.1) and CDH23 (canonical transcript: NM_022124.6 and alternative transcript: NM_001171931.1), respectively. Indeed, our results showed that both canonical and alternative isoforms affected by the variants are expressed in the retina, suggesting the presence of likely aberrant isoforms that could explain the isolated ocular phenotype observed in the affected individuals. However, further studies are necessary to substantiate this hypothesis. Notably, this is particularly relevant in the case of pleiotropic genes that can cause a wide spectrum of systemic manifestations depending on the variant59. In this scenario, as the same genomic variant may result in different protein impacts depending on the isoforms involved, it becomes evident that ACMG/AMP classification should also take this into account. Therefore, looking more closely at variants in syndromic genes, that are not strictly associated with the initial clinical diagnosis, and considering all known transcripts, would improve the diagnostic yield for patients with rare diseases.
Therefore, our study contributes to expand the mutational landscape of IRDs caused by splicing variants, and highlights the importance of these mechanisms as key players in photoreceptor degeneration. Herein, we report 17 likely spliceogenic variants, including the recurrent CNGB3 deep-intronic variant (c.1663-1205G>A) that was identified in two independent families of our cohort and is one of the major causes of achromatopsia in certain populations55,60. In this sense, the identification of prevalent causative splicing variants may also have relevant clinical implications, as it opens the door to promising therapeutic strategies, such as antisense oligonucleotides (AONs), capable of modulating aberrant splicing and increasing functional protein production52,61.
In conclusion, the application of a comprehensive algorithm, based on the combined use of prediction tools with optimized cutoffs, has facilitated the establishment of a definitive genetic diagnosis in nine families and identified a highly likely molecular cause in four additional families. This approach has improved the diagnostic rate to 6.2%. Moreover, it allowed the identification of potential causal variants in seven more families could further increase the diagnostic yield to 9.5%, pending confirmation of the proposed diagnoses. This study will thus reinforze the contribution of spliceogenic variants in the etiopathogenesis of IRDs.
Methods
Splicing variants definition and training dataset
The training dataset was composed of 3637 genomic variants, of which 1535 were pathogenic and 2102 were benign (Fig. 1). All of them were classified into five categories of splicing variants according to the distance to the splice site41,47: canonical splice site (CSS) located in the intronic positions ±1 and ±2; non-canonical splice site (NCSS) located in the intronic positions +3 to +6 in the donor site, −3 to −14 in the acceptor site, and variants in positions ±1 and ±2 of the exons; deep intronic (DI) including variants in intronic positions +7 to −15; branch point (BP), which is embedded in the DI group, with variants in positions −18 to −44; and exonic synonymous variants (ES) located in exonic positions other than ±1 and ±2 (Fig. 1).
The pathogenic group included variants classified as pathogenic or likely pathogenic in both ClinVar and HGMD professional (“DM” tag), known to impact the splicing mechanisms. The benign group included variants classified as benign or likely benign in ClinVar. Both pathogenic and benign variants were located in genes associated with Mendelian disorders (https://www.omim.org/) and had a MAF ≤ 0.01 in gnomAD. As deep-intronic variants can be found in a wide range of genomic positions, and considering the influence of the genomic context, we restricted the selection of benign DI variants to the vicinity (±1000 bp) of a pathogenic DI variant (Fig. 1). Additionally, to reduce the imbalance between benign and pathogenic variants in the BP and ES groups, 21 and 172 pathogenic variants from the HGMD-Pro database, but absent in ClinVar, were included respectively (Fig. 1).
Splicing prediction tools
Thirteen in silico splice prediction tools were tested: Combined Annotation Dependent Depletion v1.662 (CADD), IntSplice263, MaxEntScan28 (MaxEnt), Modular Modeling of Splicing33 (MMSplice), NNSplice30 (NNS), Splice Site Finder-like29 (SSF), SpliceAI32, Splicing Prediction Pipeline64 (SPiP), Branch Point Prediction65 (BPP), Splicing Predictions in Consensus Elements31 (SPiCE), Super Quick Information-content Random-forest Learning of Splice variants66 (SQUIRLS), branchPointChange (BP Alamut) and nearestSSChange (Avg. Alamut). The training dataset was annotated using Alamut® Batch v1.11 software (Interactive Biosoftware) to obtain scores for the MaxEnt, NNS, SSF, SPiCE, BP Alamut and Avg Alamut tools, and CADD Webserver for the CADD v1.6, SpliceAI and MMSplice tools. For the rest, we used the standalone versions for each software: the python script for BPP, the Windows software for SPiP, the java script for SQUIRLS, and the pre-processed genome-wide scores for IntSplice2. The association between each tool and the specific type of variants that can be evaluated is listed in Supplementary Table 4. To compare the performance of these prediction methods, the scores of Avg. Alamut, MMSplice and BP Alamut tools have been transformed so that a higher score indicates a higher risk of deleteriousness (Supplementary Table 4). On the other hand, the scores of the SSF, MaxEnt and NNS predictors were converted into the percentage variation between the scores for the wild-type sequence and the mutated sequences, as described previously41. Missing values provided by Alamut® Batch were considered as zero. In addition, when multiple splice sites close to the investigated variant were scored, the maximum value was chosen. For all annotation tools, the input file was variant calling format (VCF), except for the BPP tool, which used a 76 bp FASTA sequence as input.
Statistical analysis
The statistical analysis was performed using RStudio 2022.02.3 and R version 4.1.3. The chi-square test was conducted considering a significant value of p < 0.05.
The performance of each tool was assessed by receiver operating characteristic (ROC) and precision-recall (PR) curves analyses using the R packages pROC and ROCR67,68. ROC analysis was used to calculate the Matthews Correlation Coefficient (MCC) and the optimal threshold for a sensitivity of 90%, or alternatively, the highest sensitivity. This analysis was conducted both for the overall training dataset and, separately, for the five distinct categories of variants. Moreover, the area under the curve (AUC) of ROC and PR curves were also calculated in the six datasets in order to benchmark the performance of each predictor with the different groups of splicing variants. MCC and AUC values ≤ 0.5 indicate random classification, while values close to 1 reflects a better tool performance with improved discrimination between benign and pathogenic variants. Other useful calculated parameters for the evaluation of the tools were the false positive (%FP) and missing value (%NA) rates.
The prediction tools with a MCC > 0.5, %FP < 35% and %NA < 5% were selected for a combinatorial analysis. A total of six tools were combined, and 32 different models were analyzed. The sensitivity (SEN), specificity (SPE), MCC, positive predictive value (PPV) and negative predictive value (NPV) were calculated by applying each model with the recommended threshold to the training dataset (Supplementary Table 2). The optimal combinatorial model was identified based on the polygon area of the radar plot derived from the highest values of MCC, sensitivity, PPV, NPV and specificity measurements (R package “fmsb”).
Subjects, clinical evaluation and previous studies
All individuals included in this study have signed written informed consent to be studied in the Department of Maternal-Fetal Medicine, Genetics and Reproduction of the University Hospital Virgen del Rocio in Seville during the period from 2016 to 2023. The research was conducted in accordance with the tenets of the Declaration of Helsinki (Edinburgh, 2000)69, and all experimental protocols were approved by the University Hospitals Virgen Macarena-Virgen del Rocio Institutional Review Board (Comité de Ética de la Investigación de los Hospitales Universitarios Virgen Macarena-Virgen del Rocío de Sevilla, reference number A01037780). Genomic DNA of all subjects was isolated from whole blood using standard procedures.
This study included a total of 354 individuals with rare diseases divided into two cohorts, the validation cohort and the experimental cohort (Fig. 1). The validation cohort consisted of 116 genetically diagnosed individuals, including 52 IRD patients and 64 individuals with different rare diseases, with the main phenotypes represented being hereditary cancer, neurofibromatosis and cystic fibrosis. All samples in the validation cohort were analyzed using a custom targeted sequencing of 1166 genes associated with different rare diseases, as previously described70. The genomic data from this cohort was used to optimize the pipeline developed with the training dataset. The validation dataset included 118 pathogenic splicing variants (Supplementary Data 1), 85% of which were not included in the training dataset. These variants were reported to the patient as the potentially causal variant according to the available clinical data, the co-segregation results, and the presence of additional pathogenic variants in trans in recessive genes. The experimental cohort consisted of 238 unsolved IRD individuals belonging to 211 families to whom the validated strategy was applied. The experimental cohort dataset included both NGS data from 193 IRD probands analyzed by targeted sequencing comprising 146 IRD-associated genes (unpublished data) following the pipeline described in Puppo-Moreno et al.71 and 45 individuals from 18 IRD families analyzed by WGS, five of which were studied as previously described37.
Clinical diagnosis of IRD was based on a comprehensive ophthalmic examination including best corrected visual acuity (BCVA), fundus photographs, fundus fluorescein angiography (FA), optical coherence tomography (OCT), visual field, and electroretinography (ERG). In addition, clinical data relevant to the disease was obtained from the electronic health record (EHR).
In addition, an in-house database containing WGS data from 14 additional individuals from 5 solved IRD families was used as pseudo-controls to obtain local-frequency data and to evaluate the recurrence of novel candidate variants.
Next-generation sequencing and data analysis
Genomic DNA libraries from the individuals of the experimental cohort were constructed using the SeqCap EZ Library SR version 5.1 (Roche, Indianapolis, IN, USA), or the TruSeq Nano DNA Library Prep Kit (Illumina, CA, USA) according to the manufacturer’s instructions. Sequencing was performed using 2 ×150 bases paired-end reads on Illumina NextSeq500 for gene panel sequencing, or HiSeqX and NovaSeq 6000 platforms for WGS. After sequencing, trimmed reads were mapped to the hg19 human reference genome using BWA-MEM (v. 0.7.17). BAM files were sorted, and duplicates were removed using Picard (v. 2.18.2). GATK (v. 4.0.5.1) was used for base quality recalibration and variant calling of single-nucleotide variants (SNVs). Multi-sample vcf files were obtained using VCF combine (Vcflib)72. Annotation was performed using Slivar v.0.2.873, SnpEff v.5.174,75 and Alamut® Batch v1.11 software (Interactive Biosoftware).
Depending on the type of NGS data, the variant calling of structural variants (SVs) and copy number variations (CNVs) were perfomed using Manta (v. 1.5.0), Control-FREEC (v.11.5), PennCNV (v. 1.0.5), MELT (v2.2.2) or ERDS (v.1.1) for WGS data and an in-house script based on the coverage and statistical studies for panel sequencing cases71. The resulting call sets were annotated using AnnotSV 3.3.6 online software76 and filtered according to the quality status and the recurrence between samples when possible.
Filtering and prioritization of genomic data was conducted using the developed pipeline (Fig. 4). First, an annotation of the original vcf file with an updated ClinVar file (version 2023.01.21) was performed to directly prioritize known variants classified as pathogenic (P), likely pathogenic (LP) or conflicting but with at least one P or LP entry (CIPP/LP). In parallel to this ClinVar update step, all variants were prioritized based on quality (PASS only), internal recurrence (present in less than 30% of samples and not found in homozygous state in unnaffected individuals), population frequency (MAF ≤ 0.01 in gnomAD), excluding multiallelic variants or those variants located in non-coding genes or pseudogenes. SNVs were specifically prioritized in parallel by three types of filters: 1) “CADD+MAPP+ Grantham+SIFT” (CADD ≥ 22.25, MAPP ≤ 0.098 or absent, Grantham ≥28 or absent, and SIFT ≤ 0.175 or absent)37; 2) “SpliceAI+MaxEnt” (SpliceAI ≥ 0.235 or absent, MaxEnt ≥ 15.583); and 3) BP Alamut (BP Alamut ≤ −17.072). Variants passing at least one of these filters were integrated into the pedigree filtering together with SVs variants, and the variants prioritized by the ClinVar update.
Finally, a manual curation was performed based on the American College of Medical Genetics/Association for Molecular Pathology (ACMG/AMP) guidelines5,38 using the Franklin Genoox platform (https://franklin.genoox.com/), the consistency with the clinical diagnosis, the clinical significance in other databases such as HGMD-pro or LOVD, the number of homozygotes, heterozygotes and hemizygotes in gnomAD, and expert criteria. Moreover, for recessive genes for which only a single likely causative heterozygous variant was prioritized by our filters, additional exonic non-synonymous variants in the same genes were also considered during manual filtering.
Family segregation of candidate variants in the available family members was conducted by Sanger sequencing according to the manufacturer’s protocols (3730 DNA Analyzer, Applied Biosystems, Foster City, CA, USA). In cases of suspected compound heterozygous variants without parental confirmation, phasing studies were performed in probands using long-read sequencing by Oxford Nanopore Technologies on a MinION device (LongSeq Applications, Murcia, Spain) as described elsewhere77.
Splicing analysis of CDH23 variants in patient RNA
Based on the availability of patient blood RNA and gene expression profiles, only two novel intronic variants, including one BP and one DI, could be selected for splicing assays. Frozen blood samples from 4 individuals belonging to two IRD families (Fam2-I:1, I:2 and II:1; and Fam5-III:1) (Supplementary Fig. 3), and 3 control individuals were used to analyze the splicing pattern of CDH23 by RT-PCR. Thawing of frozen blood was conducted on aluminium blocks at room temperature for 15–20 min to minimize RNA degradation78. Total RNA was extracted using the NucleoSpin RNA Blood Midi kit (Macherey-Nagel), and first-strand cDNA was synthesized using RT2 First Strand Kit (Qiagen) according to the manufacturer’s protocols. For the analysis of splicing events, cDNA templates were used to amplify the region of interest (exons 44–46 and 48–49 of CDH23 gene) with specific primers (Exon44F: 5′-GAAGGACAACCCGGAGAATC-3′, Exon46R: 5′-CGGTGCTGCTGTTGATGTC-3′, Exon48F: 5′-CACAGCCATTGTCACCATTC-3′, Exon49R: 5′-ACCTCATAGGTGGCAACCAG-3′). The amplification reaction was performed using HotStarTaq DNA Polymerase and Multiplex PCR Master Mix (Qiagen) (94 °C for 30 s, 60 °C for 90 s, and 72 °C for 90 s x40 cycles). PCR products were visualized on 1.5% agarose gels and purified using QIAquick Gel Extraction Kit (Qiagen). Unprocessed gel images are shown in Supplementary Fig. 6. DNA bands were quantified using the ImageJ Gel Analysis program (version 1.54 d, March 2023), and analyzed by Sanger sequencing.
Expression of alternative isoforms of BBS1 and CDH23
In order to assess the expression of the alternative isoforms of BBS1 (ENST00000537537.1) and CDH23 (NM_001171931.1) in the human retina, RT-PCR was conducted using ready-to-use cDNA (QUICK-Clone™ Clontech Laboratories, Inc., CA, USA). Moreover, this evaluation was also conducted in cDNA from different tissues: liver, kidney, placenta, and skeletal muscle (Zyagen, CA, USA). Specific primers for each transcripts were designed (Supplementary Fig. 4): 5′-GCAGATTTACATGGGGATGG-3′ (BBS1 exon 3, forward), 5′-CAGCTGGAGCTAAGTGAAATGGAGGC-3′ (BBS1 exon 7, reverse), 5′-GGCCAAAGAGGGAGACGGCAGAGG-3′ (BBS1 exon 4, forward), 5′-AGTGTTCCTCCCAGAGACTCCAAGCA-3′ (BBS1 exon 9, reverse) 5′-GCTCACCATCCATGTGCTGGATGTGA-3′ (CDH23 exon 26, forward), 5′-CCCCAAGTCTGGGTGACAGAGCAG-3′ (CDH23 exon 26, reverse), 5′-CTTCATCACAGGTGGCAACGTGGATG-3′ (CDH23 exon 27, foward), 5′-AGCTCCGTCAGGGTGATTGTGTACGT-3′ (CDH23 exon 31, reverse). The amplification reaction was done using Multiplex PCR Master Mix (Qiagen) (94 °C for 30 s, 60 °C for 90 s, and 72 °C for 90 s x35 cycles). PCR products were analyzed using the QIAxcel capillary electrophoresis system and QIAxcel ScreenGel software (Qiagen)79. Unprocessed gel images are shown in Supplementary Fig. 6.
To analyze the impact of mutagenesis in the BBS1 alternative protein (UnitProt B4DYY3), PyMOL Molecular Graphics System© v.3.0.3 was used from PDB model of AlphaFold (AF-B4DYY3-F1).
Data availability
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding authors.
References
Sullivan, J. A., Schoch, K., Spillmann, R. C. & Shashi, V. Exome/genome sequencing in undiagnosed syndromes. Annu. Rev. Med. 74, 489–502 (2023).
Toulis, V. et al. Increasing the genetic diagnosis yield in inherited retinal dystrophies: assigning pathogenicity to novel non-canonical splice site variants. Genes 11, https://doi.org/10.3390/genes11040378 (2020).
Britten-Jones, A. C. et al. The diagnostic yield of next generation sequencing in inherited retinal diseases: a systematic review and meta-analysis. Am. J. Ophthalmol. 249, 57–73 (2023).
Iancu, I. F. et al. Prioritizing variants of uncertain significance for reclassification using a rule-based algorithm in inherited retinal dystrophies. NPJ Genom. Med. 6, 18 (2021).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Rowlands, C. et al. Comparison of in silico strategies to prioritize rare genomic variants impacting RNA splicing for the diagnosis of genomic disorders. Sci. Rep. 11, 20607 (2021).
Lionel, A. C. et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet. Med. 20, 435–443 (2018).
Landrum, M. J. et al. ClinVar: improvements to accessing data. Nucleic Acids Res. 48, D835–D844 (2020).
Stenson, P. D. et al. The Human Gene Mutation Database (HGMD((R))): optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 139, 1197–1207 (2020).
Soemedi, R. et al. Pathogenic variants that alter protein code often disrupt splicing. Nat. Genet. 49, 848–855 (2017).
Vaz-Drago, R., Custodio, N. & Carmo-Fonseca, M. Deep intronic mutations and human disease. Hum. Genet. 136, 1093–1111 (2017).
Scotti, M. M. & Swanson, M. S. RNA mis-splicing in disease. Nat. Rev. Genet. 17, 19–32 (2016).
Weisschuh, N., Buena-Atienza, E. & Wissinger, B. Splicing mutations in inherited retinal diseases. Prog. Retin Eye Res. 80, 100874 (2021).
Rowlands, C. F., Baralle, D. & Ellingford, J. M. Machine learning approaches for the prioritization of genomic variants impacting Pre-mRNA splicing. Cells 8, https://doi.org/10.3390/cells8121513 (2019).
Blakes, A. J. M. et al. A systematic analysis of splicing variants identifies new diagnoses in the 100,000 Genomes Project. Genome Med. 14, 79 (2022).
Stingl, K. et al. CDHR1 mutations in retinal dystrophies. Sci. Rep. 7, 6992 (2017).
Sangermano, R. et al. Photoreceptor progenitor mRNA analysis reveals exon skipping resulting from the ABCA4 c.5461-10T->C mutation in Stargardt disease. Ophthalmology 123, 1375–1385 (2016).
Tucker, B. A. et al. Using patient-specific induced pluripotent stem cells to interrogate the pathogenicity of a novel retinal pigment epithelium-specific 65 kDa cryptic splice site mutation and confirm eligibility for enrollment into a clinical gene augmentation trial. Transl. Res. 166, 740–749.e741 (2015).
den Hollander, A. I. et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 79, 556–561 (2006).
Webb, T. R. et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23). Hum. Mol. Genet. 21, 3647–3654 (2012).
Vache, C. et al. Usher syndrome type 2 caused by activation of an USH2A pseudoexon: implications for diagnosis and therapy. Hum. Mutat. 33, 104–108 (2012).
Cremers, F. P. M., Lee, W., Collin, R. W. J. & Allikmets, R. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog. Retin Eye Res. 79, 100861 (2020).
Lee, H. et al. Diagnostic utility of transcriptome sequencing for rare Mendelian diseases. Genet. Med. 22, 490–499 (2020).
Aicher, J. K., Jewell, P., Vaquero-Garcia, J., Barash, Y. & Bhoj, E. J. Mapping RNA splicing variations in clinically accessible and nonaccessible tissues to facilitate Mendelian disease diagnosis using RNA-seq. Genet. Med. 22, 1181–1190 (2020).
Aisa-Marin, I., Garcia-Arroyo, R., Mirra, S. & Marfany, G. The alter retina: alternative splicing of retinal genes in health and disease. Int. J. Mol. Sci. 22, https://doi.org/10.3390/ijms22041855 (2021).
Ray, T. A. et al. Comprehensive identification of mRNA isoforms reveals the diversity of neural cell-surface molecules with roles in retinal development and disease. Nat. Commun. 11, 3328 (2020).
Ruiz-Ceja, K. A. et al. Definition of the transcriptional units of inherited retinal disease genes by meta-analysis of human retinal transcriptome data. BMC Genomics 24, 206 (2023).
Yeo, G. & Burge, C. B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput Biol. 11, 377–394 (2004).
Shapiro, M. B. & Senapathy, P. RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res. 15, 7155–7174 (1987).
Reese, M. G., Eeckman, F. H., Kulp, D. & Haussler, D. Improved splice site detection in Genie. J. Comput Biol. 4, 311–323 (1997).
Leman, R. et al. Novel diagnostic tool for prediction of variant spliceogenicity derived from a set of 395 combined in silico/in vitro studies: an international collaborative effort. Nucleic Acids Res. 46, 7913–7923 (2018).
Jaganathan, K. et al. Predicting splicing from primary sequence with deep learning. Cell 176, 535–548.e524 (2019).
Cheng, J. et al. MMSplice: modular modeling improves the predictions of genetic variant effects on splicing. Genome Biol. 20, 48 (2019).
Leman, R. et al. SPiP: Splicing Prediction Pipeline, a machine learning tool for massive detection of exonic and intronic variant effects on mRNA splicing. Hum. Mutat. 43, 2308–2323 (2022).
Lord, J. & Baralle, D. Splicing in the diagnosis of rare disease: advances and challenges. Front. Genet. 12, 689892 (2021).
Chicco, D. & Jurman, G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genomics 21, 6 (2020).
Gonzalez-Del Pozo, M. et al. A comprehensive WGS-based pipeline for the identification of new candidate genes in inherited retinal dystrophies. NPJ Genom. Med. 7, 17 (2022).
Riggs, E. R. et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 22, 245–257 (2020).
Li, Z. et al. A framework for detecting noncoding rare-variant associations of large-scale whole-genome sequencing studies. Nat. Methods 19, 1599–1611 (2022).
Gonzalez-Del Pozo, M. et al. Unmasking Retinitis Pigmentosa complex cases by a whole genome sequencing algorithm based on open-access tools: hidden recessive inheritance and potential oligogenic variants. J. Transl. Med. 18, 73 (2020).
Riepe, T. V., Khan, M., Roosing, S., Cremers, F. P. M. & 't Hoen, P. A. C. Benchmarking deep learning splice prediction tools using functional splice assays. Hum. Mutat. 42, 799–810 (2021).
Walker, L. C. et al. Using the ACMG/AMP framework to capture evidence related to predicted and observed impact on splicing: Recommendations from the ClinGen SVI Splicing Subgroup. Am. J. Hum. Genet. 110, 1046–1067 (2023).
Reurink, J. et al. Whole genome sequencing for USH2A-associated disease reveals several pathogenic deep-intronic variants that are amenable to splice correction. HGG Adv. 4, 100181 (2023).
Fadaie, Z. et al. BBS1 branchpoint variant is associated with non-syndromic retinitis pigmentosa. J. Med. Genet. 59, 438–444 (2022).
Rodríguez-Hidalgo, M. et al. ABCA4 c.6480-35A>G, a novel branchpoint variant associated with Stargardt disease. Front. Genet. 14, https://doi.org/10.3389/fgene.2023.1234032 (2023).
Corradi, Z. et al. ABCA4 c.859-25A>G, a frequent palestinian founder mutation affecting the intron 7 branchpoint, is associated with early-onset stargardt disease. Invest. Ophthalmol. Vis. Sci. 63, 20 (2022).
Leman, R. et al. Assessment of branch point prediction tools to predict physiological branch points and their alteration by variants. BMC Genomics 21, 86 (2020).
Viner, C., Dorman, S. N., Shirley, B. C. & Rogan, P. K. Validation of predicted mRNA splicing mutations using high-throughput transcriptome data. F1000Res. 3, 8 (2014).
Valero, R. et al. Aberrant splicing events associated to CDH23 noncanonical splice site mutations in a proband with atypical usher syndrome 1. Genes 10, https://doi.org/10.3390/genes10100732 (2019).
Garanto, A. et al. In vitro and in vivo rescue of aberrant splicing in CEP290-associated LCA by antisense oligonucleotide delivery. Hum. Mol. Genet. 25, 2552–2563 (2016).
Rodriguez-Munoz, A. et al. Functional assays of non-canonical splice-site variants in inherited retinal dystrophies genes. Sci. Rep. 12, 68 (2022).
Kaltak, M. et al. Antisense oligonucleotide therapy corrects splicing in the common Stargardt disease type 1-causing variant ABCA4 c.5461-10T>C. Mol. Ther. Nucleic acids 31, 674–688 (2023).
Gamundi, M. J. et al. Transcriptional expression of cis-acting and trans-acting splicing mutations cause autosomal dominant retinitis pigmentosa. Hum. Mutat. 29, 869–878 (2008).
Anna, A. & Monika, G. Splicing mutations in human genetic disorders: examples, detection, and confirmation. J. Appl. Genet. 59, 253–268 (2018).
Weisschuh, N. et al. Deep-intronic variants in CNGB3 cause achromatopsia by pseudoexon activation. Hum. Mutat. 41, 255–264 (2020).
Frischmeyer, P. A. & Dietz, H. C. Nonsense-mediated mRNA decay in health and disease. Hum. Mol. Genet. 8, 1893–1900 (1999).
Dvaladze, A. et al. Deep intronic variant in MVK as a cause for mevalonic aciduria initially presenting as non-syndromic retinitis pigmentosa. Clin. Genet. 102, 524–529 (2022).
Fernandez-Suarez, E. et al. Expanding the phenotype of THRB: a range of macular dystrophies as the major clinical manifestations in patients with a dominant splicing variant. Front. Cell Dev. Biol. 11, 1197744 (2023).
Estrada-Cuzcano, A. et al. BBS1 mutations in a wide spectrum of phenotypes ranging from nonsyndromic retinitis pigmentosa to Bardet-Biedl syndrome. Arch. Ophthalmol. 130, 1425–1432 (2012).
Aweidah, H. et al. A deep intronic substitution in CNGB3 is one of the major causes of achromatopsia among Jewish patients. Mol. Vis. 27, 588–600 (2021).
Amato, A. et al. Gene therapy in inherited retinal diseases: an update on current state of the art. Front. Med. 8, 750586 (2021).
Rentzsch, P., Schubach, M., Shendure, J. & Kircher, M. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 13, 31 (2021).
Takeda, J. I., Fukami, S., Tamura, A., Shibata, A. & Ohno, K. IntSplice2: prediction of the splicing effects of intronic single-nucleotide variants using LightGBM modeling. Front. Genet. 12, 701076 (2021).
Leman, R. et al. SPiP: splicing prediction pipeline, a machine learning tool for massive detection of exonic and intronic variant effect on mRNA splicing. Hum. Mutat. https://doi.org/10.1002/humu.24491 (2022).
Zhang, Q. et al. BPP: a sequence-based algorithm for branch point prediction. Bioinformatics 33, 3166–3172 (2017).
Danis, D. et al. Interpretable prioritization of splice variants in diagnostic next-generation sequencing. Am. J. Hum. Genet. 108, 2205 (2021).
Robin, X. et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinforma. 12, 77 (2011).
Sing, T., Sander, O., Beerenwinkel, N. & Lengauer, T. ROCR: visualizing classifier performance in R. Bioinformatics 21, 3940–3941 (2005).
World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA 310, 2191–2194, (2013).
Méndez-Vidal, C. et al. A genomic strategy for precision medicine in rare diseases: integrating customized algorithms into clinical practice. J. Transl. Med. 23, 86 (2025).
Puppo Moreno, A. M. et al. Genetic profile in patients with complicated acute aortic syndrome: the GEN-AOR study. Rev. Esp. Cardiol. https://doi.org/10.1016/j.rec.2022.10.005 (2022).
Garrison, E., Kronenberg, Z. N., Dawson, E. T., Pedersen, B. S. & Prins, P. A spectrum of free software tools for processing the VCF variant call format: vcflib, bio-vcf, cyvcf2, hts-nim and slivar. PLoS Computational Biol. 18, e1009123 (2022).
Pedersen, B. S. et al. Effective variant filtering and expected candidate variant yield in studies of rare human disease. NPJ Genom. Med. 6, 60 (2021).
Cingolani, P. Variant annotation and functional prediction: SnpEff. Methods Mol. Biol. 2493, 289–314 (2022).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012).
Geoffroy, V. et al. AnnotSV: an integrated tool for structural variations annotation. Bioinformatics 34, 3572–3574 (2018).
McClinton, B. et al. Haplotyping using long-range PCR and nanopore sequencing to phase variants: lessons learned from the ABCA4 Locus. Lab. Invest. 103, 100160 (2023).
Yamagata, H. et al. Optimized protocol for the extraction of RNA and DNA from frozen whole blood sample stored in a single EDTA tube. Sci. Rep. 11, 17075 (2021).
McMurray, C. L., Hardy, K. J. & Hawkey, P. M. Rapid, automated epidemiological typing of methicillin-resistant Staphylococcus aureus. J. Microbiol. Methods 80, 109–111 (2010).
Acknowledgements
The authors thank the families who participated in this study, and the Andalusian Association of Retinitis Pigmentosa. This work was supported by the Instituto de Salud Carlos III (ISCIII), Spanish Ministry of Science and Innovation, Spain and co-funded by ERDF (“A way to make Europe”) [PI21-00244; PI24-02129]; The strategic plan for the Precision Medicine Infrastructure associated with Science and Technology - IMPaCT [IMP-0009], Regional Ministry of Health and Families of the Autonomous Government of Andalusia [PEER-0501-2019] and the Foundation Isabel Gemio/Foundation Cajasol [FGEMIO-2019-01]. E.F.S. is supported by fellowship FI19/00091 from ISCIII (ESF, “Investing in your future”). M.M.S. [RH-0049-2021] are supported by a fellowship funded by the Regional Ministry of Health and Families of the Autonomous Government of Andalusia.
Author information
Authors and Affiliations
Contributions
E.F.-S. and M.G.-P. are co-first authors. G.A. and S.B. conceived and designed the study. E.R.-R. and M.J.M.-S. performed the ophthalmic evaluations. E.F.-S., M.G.-P. and A.G.-N. conducted the experiments. E.F.-S., M.G.-P., C.M.-V., M.M.-S., M.M. and N.B-G. analyzed and interpreted the generated data. E.F.-S. and M.G.-P. wrote the manuscript with the collaboration of all co-authors. G.A., S.B, C.M.-V, M.M.-S., N.B-G. and M.M. revised the paper critically for important intellectual content. All authors approved the final version to be published.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fernández-Suárez, E., González-del Pozo, M., Méndez-Vidal, C. et al. New genetic diagnoses for inherited retinal dystrophies by integrating splicing tools into NGS pipelines. npj Genom. Med. 10, 52 (2025). https://doi.org/10.1038/s41525-025-00500-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-025-00500-9
